

ABSTRACT
We investigated the mutation spectrum of the TANK‐Binding Kinase 1 (TBK1) gene and its associated phenotypic spectrum by exonic resequencing of TBK1 in a cohort of 2,538 patients with frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), or FTD plus ALS, ascertained within the European Early‐Onset Dementia Consortium. We assessed pathogenicity of predicted protein‐truncating mutations by measuring loss of RNA expression. Functional effect of in‐frame amino acid deletions and missense mutations was further explored in vivo on protein level and in vitro by an NFκB‐induced luciferase reporter assay and measuring phosphorylated TBK1. The protein‐truncating mutations led to the loss of transcript through nonsense‐mediated mRNA decay. For the in‐frame amino acid deletions, we demonstrated loss of TBK1 or phosphorylated TBK1 protein. An important fraction of the missense mutations compromised NFκB activation indicating that at least some functions of TBK1 are lost. Although missense mutations were also present in controls, over three times more mutations affecting TBK1 functioning were found in the mutation fraction observed in patients only, suggesting high‐risk alleles (P = 0.03). Total mutation frequency for confirmed TBK1 LoF mutations in the European cohort was 0.7%, with frequencies in the clinical subgroups of 0.4% in FTD, 1.3% in ALS, and 3.6% in FTD‐ALS.
Keywords: TANK‐Binding Kinase 1, TBK1, frontotemporal dementia, FTD, amyotrophic lateral sclerosis, ALS, mutations, NFκB luciferase reporter assay
Introduction
Multiple lines of evidence have now strongly established that frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) share a common molecular etiology and are part of a disease continuum. Up to 50% of frontotemporal dementia (FTD) patients develop signs of motor neuron disease (MND) at some stage in the disease course with about 15% meeting the diagnostic criteria of ALS; conversely, over 30% of ALS patients show signs of FTD [Lomen‐Hoerth et al., 2002]. Neuropathologically, the majority of these patients display accumulation of TDP‐43 aggregates in affected brain regions and motor neurons. Furthermore, common genetic factors underlying FTLD and ALS, such as the chromosome 9 open‐reading frame 72 (C9orf72, MIM# 614260) [DeJesus‐Hernandez et al., 2011; Renton et al., 2011; Gijselinck et al., 2012], valosin containing protein (VCP, MIM# 601023) [Watts et al., 2004; Johnson et al., 2010], TAR DNA‐binding protein (TARDBP, MIM# 605078) [Kabashi et al., 2008; Sreedharan et al., 2008; Borroni et al., 2009; Gelpi et al., 2014], and Fused in sarcoma RNA‐binding protein (FUS, MIM# 137070) [Kwiatkowski et al., 2009; Van Langenhove et al., 2010] genes, are key pathological genes in both diseases. Of these, the G4C2 repeat expansion in C9orf72 is the most frequent genetic cause of the FTD–ALS spectrum, accounting for up to 29%, 50%, and 88% of FTD, ALS, and FTD‐ALS patient series, respectively [Cruts et al., 2013].
Also, loss‐of‐function (LoF) of the TANK‐binding kinase 1 (TBK1, MIM# 604834) was causally associated with ALS and FTD [Cirulli et al., 2015; Freischmidt et al., 2015; Gijselinck et al., 2015; Le Ber et al., 2015; Pottier et al., 2015; Williams et al., 2015]. TBK1 is a multifunctional kinase regulating a number of cellular processes, including the innate immune system and inflammation, autophagy, and cell proliferation, by phosphorylating a wide range of substrates [Clément et al., 2008; Pilli et al., 2012; Larabi et al., 2013]. Of interest, these substrates include optineurin (OPTN) and p62, two autophagic proteins that are also genetically implicated in the FTD‐ALS spectrum. Furthermore, TBK1 homodimerization and autophosphorylation at the serine 172 residue is necessary for its activation. One of the downstream effects of TBK1 activation is the upregulation of interferon (IFN)‐stimulated genes by activation of the nuclear factor of the kappa light polypeptide gene enhancer in B‐cells (NFκB) complex.
Frameshift, out‐frame, splice‐site, and nonsense mutations generating premature termination codons (PTC) in TBK1 have been demonstrated to result in LoF through loss of mutant transcript and protein [Freischmidt et al., 2015; Gijselinck et al., 2015; Pottier et al., 2015]. In addition to these clear pathogenic mutations, a small number of in‐frame single amino acid deletions have been found, some of which cosegregated with disease that led to loss of protein, whereas others did not [Freischmidt et al., 2015; Gijselinck et al., 2015]. Furthermore, numerous rare missense mutations have been identified, in both patient and control subjects [Cirulli et al., 2015; Freischmidt et al., 2015; Gijselinck et al., 2015; Le Ber et al., 2015; Pottier et al., 2015; Williams et al., 2015]. Prediction of their pathogenic effect may be ambiguous, certainly in the absence of supportive cosegregation. One report was able to demonstrate functional deficits for a number of missense mutations by testing their effect in vitro on the IFNβ pathway and on the interaction with adaptor protein OPTN [Freischmidt et al., 2015], indicating that at least some missense mutations may be disease‐causing.
In the previous study, we reported the identification of TBK1 LoF mutations in a Belgian discovery cohort of FTD (n = 460), FTD‐ALS (n = 22), and ALS (n = 147) patients, yielding mutation frequencies of 1.1% in FTD, 3.4% in ALS patients, and 4.5% in FTD‐ALS [Gijselinck et al., 2015]. In the present study, we expanded the TBK1 genetic screen with a European replication cohort of 1,755 patients with FTD (n = 1,271), ALS (n = 407), or FTD‐ALS (n = 77). In addition to the protein‐truncating mutations that led to loss of transcript, we also assessed the pathogenic effect of in‐frame deletions and missense mutations on protein level and function, the latter using an in vitro luciferase assay measuring the effect of mutant TBK1 on NFκB activation in the IFN pathway. Taken together, this study reports on the mutation frequency and mutation spectrum of TBK1 in an extended European study population of 2,538 patients with FTD (n = 1,873), ALS (n = 554), or FTD‐ALS (n = 111).
Materials and Methods
European Study Population
The patient and control cohorts under study were ascertained through the Belgian Neurology (BELNEU) consortium or the European Early‐Onset Dementia (EU EOD) consortium, as described in previous studies [van der Zee et al., 2013, 2014; Gijselinck et al., 2015; Van Mossevelde et al., 2015]. DNA and medical/demographic information was included on 2,538 patients, comprising 1,873 patients diagnosed with FTD, 111 with concomitant FTD‐ALS, and 554 patients with ALS. Patients originated from Austria, Belgium, Bulgaria, Czech Republic, Germany, Greece, Italy, Portugal, Spain, and Sweden (Table 1). Patients were evaluated and diagnosed with FTD according to the Lund and Manchester group criteria [Neary et al., 1998], and for ALS, according to the revised El Escorial criteria [Brooks et al., 2000]. Clinical diagnoses of behavioral variant FTD (bvFTD) was based on the international consensus criteria by Rascovsky et al. (2011), and of primary progressive aphasia (PPA) on the classification of Gorno‐Tempini et al. (2011). A positive family history was defined for index patients with first‐ or second‐degree relatives with symptoms of dementia or MND. In the FTD group, 34% (635/1,873) had a positive family history, in the FTD‐ALS group 29% (32/111) and in the ALS group 8% (46/554). As control group, 2,183 age‐ and origin‐matched European control individuals, with no personal or family history of neurodegenerative or psychiatric diseases, were included.
Table 1.
Descriptives of the EU EOD Patient Cohorts
| FTD | FTD‐ALS | ALS | Total | |
|---|---|---|---|---|
| EU EOD cohorts and PI | 1,873 | 111 | 554 | 2,538 |
| Belgium (n = 783) | ||||
| Van Broeckhoven ( = Belgian discovery cohort) | 460 | 22 | 147 | 629 |
| Van Broeckhoven | 142 | 12 | 0 | 154 |
| Spain (n = 548) | ||||
| Clarimon | 76 | 7 | 92 | 175 |
| Pastor | 126 | 3 | 12 | 141 |
| Ruiz | 95 | 0 | 0 | 95 |
| Sanchez | 53 | 8 | 0 | 61 |
| Gelpi | 12 | 8 | 16 | 36 |
| Gómez‐Tortosa | 38 | 2 | 0 | 40 |
| Italy (n = 442) | ||||
| Borroni | 165 | 12 | 24 | 201 |
| IRCCS Brescia 01 | 110 | 0 | 0 | 110 |
| Nacmias | 74 | 5 | 0 | 79 |
| Frisoni | 48 | 1 | 0 | 49 |
| Fabrizi | 0 | 2 | 1 | 3 |
| Germany (n = 317) | ||||
| Diehl‐Schmid | 153 | 3 | 0 | 156 |
| Ramirez | 60 | 0 | 21 | 81 |
| Synofzik | 0 | 0 | 78 | 78 |
| Danek | 1 | 1 | 0 | 2 |
| Portugal (n = 195) | ||||
| Mendonça | 130 | 4 | 0 | 134 |
| Santana | 57 | 4 | 0 | 61 |
| Bulgaria (n = 134) | ||||
| Jordanova | 0 | 2 | 132 | 134 |
| Sweden (n = 60) | ||||
| Graff | 55 | 4 | 1 | 60 |
| Czech Republic (n = 30) | ||||
| Matěj | 18 | 8 | 4 | 30 |
| Greece (n = 26) | ||||
| Fraidakis | 0 | 1 | 25 | 26 |
| Austria (n = 3) | ||||
| Kovacs | 0 | 2 | 1 | 3 |
Of the 783 Belgian patients and 1,074 Belgian controls included in the present European study population, TBK1 mutation screening data on 629 patients and 1,044 control individuals were previously published as part of the Belgian discovery cohort [Gijselinck et al., 2015; Van Mossevelde et al., 2015]. Novel patients and control subjects reported in this study are part of the European replication cohort. Together, the Belgian discovery cohort and the European replication cohort constitute the European study population. P.I., principle investigator.
Of the 783 Belgian patients and 1,074 Belgian controls included in the present European study population (Table 1), TBK1 mutation screening data on 629 patients and 1,044 control individuals was generated and published as part of the Belgian discovery cohort [Gijselinck et al., 2015; Van Mossevelde et al., 2015]. Novel patients (n = 1,755) and control subjects (n = 1,109) reported in this study are part of the European replication cohort. Together, the Belgian discovery cohort and the European replication cohort constitute the European study population (Table 1).
For all participants, informed consent for participation in the genetic studies was obtained according to sampling protocols that were approved by the local Ethics Committees of the collaborating medical centers. The protocols for the genetic studies were approved by the Ethics Committee of the University of Antwerp, Belgium.
Exonic Resequencing
All coding exons of TBK1 (NM_013254.3) were sequenced on a MiSeq platform using the MiSeq V2 chemistry (Illumina, San Diego, CA), except for exon 4, which was sequenced by Sanger sequencing using the BigDye® Terminator Cycle Sequencing kit v3.1 (Applied Biosystems, Foster City, CA). Detailed technical procedures were previously described [Gijselinck et al., 2015].
Reported variants follow cDNA numbering according to reference sequence NM_013254.3. In addition, for intronic variants, the genomic reference sequence NC_000012.12 was used. Nucleotide positions refer to cDNA sequence and nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. Protein numbering is given according to reference sequence NP_037386.1. Variants identified in this study have been submitted to the Alzheimer Disease & Frontotemporal Dementia Mutation Database (AD&FTDMDB, http://www.molgen.vib-ua.be/FTDMutations) and Locus Specific Database [Cruts et al., 2012].
Transcript Analysis
When the required biomaterials were available, transcript analysis was performed for patients with a TBK1 mutation (Fig. 1). For the c.288delT (p.Val97Phefs*2) and the c.1340+1G>A (p.Ala417*) carriers, total RNA was isolated from whole blood using the PAXgene Blood RNA kit (PreAnalytiX; Qiagen, Valencia, CA) in accordance with the manufacturer instructions. First‐strand cDNA was synthesized from total RNA with random hexamers primers using the Invitrogen ThermoScript RT‐PCR kit (Invitrogen, Carlsbad, CA). For transcript analysis of the c.235_237delACA (p.Thr79del) and c.379C>T (p.Arg127*) carriers, total RNA was extracted from blood mononuclear cells (p.Arg127*) or frontal cortical brain tissue (p.Thr79del) from the carriers. First‐strand cDNA was synthesized with oligodT and random hexamers primers, using the SuperScript III First‐Strand Synthesis System for RT‐PCR kit (Invitrogen). cDNA of these samples was amplified using the universal amplification protocol (Applied Biosystems). For p.Val97Phefs*2, exon 4 was amplified with flanking PCR primers positioned in exons 3 and 5. For p.Ala417*, predicted to lead to exon 11 skipping, cDNA was amplified with primers positioned in exons 10 and 13. For p.Arg127*, exon 5 was amplified with PCR primers in exon 3 and exon 6. All PCR products were Sanger sequenced to evaluate whether the mutant transcript was present based on the coding mutation and whether alternative transcripts were formed. Genotypes of cDNA sequences were compared with genomic DNA sequences. Primer sequences will be provided upon request.
Figure 1.
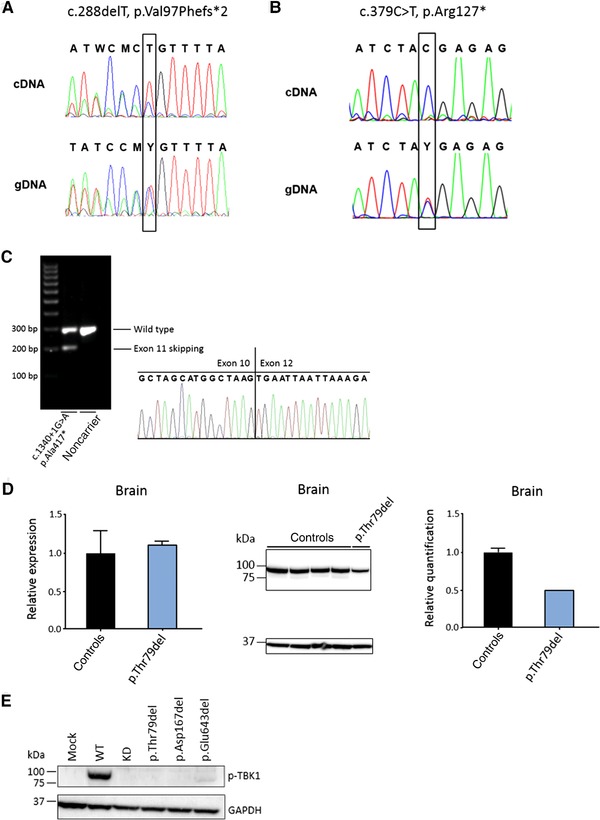
Transcript and protein analysis of TBK1 LoF and single amino acid deletion mutations. A: gDNA and cDNA sequence traces around the c.288delT (p. Val97Phefs*2) mutation, showing reduced expression of the mutant transcript on cDNA extracted from blood. B: gDNA and cDNA sequence traces around the c.379C>T (p.Arg127*) mutation, showing the absence of the mutant transcript on cDNA extracted from blood. C: Sizing of cDNA fragments generated with primers in TBK1 exon 10 and exon 13 of the c.1340+1G>A (p.Ala417*) carrier on cDNA extracted from blood. Sequence traces from the low‐expressed aberrant transcript demonstrates skipping of exon 11. D: Transcript and protein analysis on brain frontal cortex from the c.235_237delACA (p.Thr79del) carrier and four age‐matched control brains. The graph on the left shows the relative expression in the patient sample (blue) compared with the control samples (black) measured by quantitative real‐time PCR (qRT‐PCR). In the middle, Western blot analysis is shown of protein extracts from the patient carrier compared with control individuals. The upper band represents TBK1 (84 kDa) and the lower band represents the housekeeping protein GAPDH (37 kDa). The graph on the right shows the quantification in the patient sample (blue) and control samples (black) of the TBK1 signal normalized to the signal of GAPDH. Error bars represent the SD. E: Western blot analysis of phosphorylated TBK1 (Ser172, p‐TBK1) (upper band, 84 kDa) in HEK293T cells overexpressing the in‐frame single amino acid deletions (p.Thr79del, p.Asp167del, and p.Glu643del) compared with wild type, relative to GAPDH (lower band, 37 kDa). Mock and kinase dead (p.Ser172Ala, KD) were used as negative control. cDNA numbering according to reference sequence NM_013254.3, in addition, for intronic variants, the genomic reference sequence NC_000012.12 was used. Nucleotide positions refer to cDNA sequence and nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. Protein numbering according to reference sequence NP_037386.1.
For quantification of the transcripts created by the p.Thr79del mutation, semiquantitative real‐time PCR (qPCR) was performed using SYBR Green assays on the ViiA™ 7 Real‐Time PCR System (Applied Biosystems). A qPCR amplicon was designed, spanning exons 19 and 20 (primers 5’‐ CATGACCCCAATTTATCCAAGTTC‐3’ and 5’‐ CATCTCTTCCTTTAATTTCTTCATACCA‐3'), detecting the TBK1 refgene transcript variant NM_013254 using PrimerExpress Software (Applied Biosystems) and quantified against three housekeeping genes, HPRT, GAPDH, and SDHA. Relative expression levels were calculated by comparing normalized quantities between patient and control samples using qbase+ software (Biogazelle, Ghent, Belgium).
Protein Analysis
Protein lysates from the TBK1 p.Thr79del carrier and control frontal cortex tissue were prepared for Western blot with 0.1% RIPA and sonicated on ice, cleared at 20,000 g for 15 min at 4°C and supernatants used for immunoblotting (Fig. 1). Protein concentrations were measured by a BCA assay (Pierce, Rockford, IL), and 30 μg of protein were separated on 4%–12% Nupage Bis–Tris gels (Invitrogen), electroblotted onto a PVDF membrane (Hybond P; Amersham Biosciences, GE Helthcare, Little Chalfont, UK), and probed with a monoclonal antibody against TBK1 (Abcam, Cambridge, MA; 1:1,000, 84 kDa). Immuno‐detection was performed with specific secondary antibodies conjugated to horseradish peroxidase and the ECL‐plus chemiluminescent detection system (Amersham Biosciences). TBK1 signal intensities were quantified against GAPDH (Genetex, Irvine, CA; 1:10,000, 37 kDa), using ImageQuantTL software (GE Healthcare Life Sciences, Little Chalfont, UK).
TBK1 Cloning and Overexpression in HEK293T Cells
TBK1 plasmids containing a missense mutation or a single amino acid deletion were generated by in vitro mutagenesis using the KAPA HiFi HotStart DNA polymerase (Kapa Biosystems, Wilmington, MA) starting from the TBK1 wild‐type gateway pDONR vector (NM_013254.3) (GeneCopoeia, Rockville, MD). The p.Ser172Ala mutant (kinase dead) generating a TBK1 protein lacking kinase activity, and the mock plasmid containing no TBK1, were used as negative controls. The gateway‐compatible pCR3 vector (Life Technologies, Carlsbad, CA) was used as expression vector.
Each TBK1 plasmid was transiently transfected in HEK293T cells using Lipofectamine 2000 (Life Technologies). Protein lysates of these TBK1 overexpression cells were used for Western blotting with an antibody against TBK1 (Abcam; 1:1,000, 84 kDa) and GAPDH (Genetex; 1:10,000, 37 kDa). Autophosphorylation activity of the single amino acid deletions was determined by Western blot analysis with p‐TBK1 antibody (phospho S172) (Abcam; 1:500, 84 kDa).
NFκB Reporter Assay
Each TBK1 plasmid was transiently transfected in human embryonic kidney cells (HEK293T) using X‐tremeGENE 9 DNA Transfection Reagent (Sigma–Aldrich, St Louis, MO), together with the NFκB luciferase reporter vector (Affymetrix, Santa Clara, CA) containing an inducible Firefly luciferase reporter gene to monitor the activation of the NFκB signal transduction pathway and the pTK‐GLuc plasmid that encodes the Gaussia luciferase gene (New England Biolabs, Ipswich, MA) for normalization. Firefly luciferase activities (LAF) and Gaussia luciferase activities (LAG) were measured in fivefold by the use of a Dual‐Glo Luciferase Assay System (Promega, Madison, WI) and a BioLux Gaussia Luciferase Assay Kit (New England Biolabs) on a Veritas Microplate Luminometer (Promega). To correct for transfection efficiency and DNA uptake, the relative luciferase activity (RLA) was calculated as RLA = LAF/LAG. This experiment was repeated three times. Differences in relative luciferase activities between mutant and wild‐type TBK1 plasmids were calculated by a linear mixed model using the statistical packages lme4 and nlme in R and corrected for multiple testing using Bonferroni correction.
Neuropathology of TBK1 p.Thr79del Carrier
Neuropathological work‐up was performed at the Neurological Tissue Bank of the Biobanc‐Hospital Clinic‐IDIBAPS (Barcelona, Spain), according to standardized procedures. In brief, fragments of frontal cortex and cerebellum were immediately frozen at −80°C, whereas the remaining brain tissue was fixed in 10% buffered formaldehyde solution for 4 weeks. For histopathological evaluation, 5 μm thick sections were cut from formalin‐fixed and paraffin‐embedded tissue from multiple brain areas including frontal, temporal, parietal and occipital cortices, motor cortex, anterior cingulate gyrus, anterior and posterior basal ganglia, anterior, medial and posterior thalamic nuclei, hippocampus and parahippocampal gyrus, amygdala, n. basalis Meynert, midbrain, pons, medulla oblongata, cerebellar vermis, and dentate nucleus, as well as cervical, thoracic, and lumbar segments of spinal cord.
Sections were stained with hematoxylin‐eosin, Luxol fast blue, and for immunohistochemistry using the following monoclonal (mc) and polyclonal (pc) primary antibodies on an automated immunostainer (DAKO autostainer plus; DAKO, Glostrup, Denmark) after heat‐ or chemically induced epitope retrieval with formic acid: anti‐bA4‐amyloid (DAKO; mc, clone 6F/3D, dilution 1:400), antiphosphorylated tau (Thermo Scientific, Rockford, IL; mc, clone AT8, dilution 1:200), antiubiquitin (DAKO; pc, dilution 1:400), anti‐alpha‐synuclein (Novocastra, Newcastle, UK; mc, clone KM51, dilution 1:500), anti‐TDP‐43 (Abnova, Taipei, Taiwan; mc, clone 2E2‐D3, dilution 1:500), antineurofilaments (Novocastra; clone RT97, dilution 1:800), anti‐RD3 (Millipore, Temecula, CA; mc, clone 8E6/C11, dilution 1:1,000), anti‐RD4 (Millipore; mc, clone 1E1/A6, dilution 1:50), anti‐alpha‐internexin (Invitrogen; mc, clone 2E3, dilution 1:800), and anti‐alpha‐B‐crystallin (Novocastra; mc, clone G2JF, dilution 1:100).
Statistical Analysis
Missense mutation frequencies were compared between patients and controls using a Fisher exact test. Significance level was set at P < 0.05.
Results
Protein‐Truncating Mutations
In the Belgian discovery cohort, we have reported four TBK1 LoF mutations (Table 2) [Gijselinck et al., 2015]. In this study, massive parallel exonic resequencing of the TBK1 coding region was performed in the EU replication cohort of 1,755 patients, among which 1,271 FTD, 407 ALS patients, and 77 FTD‐ALS, together with 1,109 European control individuals. In the patients, seven additional predicted LoF mutations were identified, including three small out‐frame deletions, three nonsense mutations, and one splice donor‐site mutation (Table 2). These mutations introduce a PTC and are expected to result in loss of transcript due to nonsense‐mediated mRNA decay (NMD). No PTC or splice‐site mutations were observed in the control subjects. We had already confirmed NMD for two PTC mutations, c.1192delT (p.Ser398Profs*11) and c.1551_1552insTT (p.Ser518Leufs*32) [Gijselinck et al., 2015]. In addition, for the predicted in‐frame exon‐skipping mutation c.992+1G>T (p.Gly272_Thr331del) affecting exon 8, we demonstrated cryptic splicing activation and reduction of transcript and protein in brain [Gijselinck et al., 2015]. In the present study, we demonstrated NMD for three more mutations. For c.288delT (p.Val97Phefs*2), we showed a strong but incomplete reduction of the mutant transcript, for c.379C>T (p.Arg127*) a complete absence of the mutant transcript (Fig. 1A and B). For the previously reported splice donor‐site mutation in intron 11 (c.1340+1G>A), we confirmed out‐of‐frame skipping of exon 11 leading to a PTC (p.Ala417*) (Fig. 1C) [Freischmidt et al., 2015]. For three of the mutation carriers in the EU replication cohort, c.288delT (p.Val97Phefs*2), c.1340+1G>A (p.Ala417*), and c.1385_1388delCAGA (p.Thr462Lysfs*3), we identified an additional affected relative with the mutation.
Table 2.
TBK1 Predicted LoF Mutations in the European Study Population
| FTD | FTD‐ALS | ALS | |||
|---|---|---|---|---|---|
| cDNA | Predicted protein | n = 1,873 | n = 111 | n = 554 | Cohorts of the European study population |
| Protein‐truncating mutations leading to loss of transcript | |||||
| c.4C>T | p.Gln*2 | 1 | Belgian discovery cohort | ||
| c.86delA | p.Lys29Argfs*15 | 1 | European replication cohort | ||
| c.288delT | p.Val97Phefs*2 | 1 | European replication cohort | ||
| c.349C>T | p.Arg117* | 1 | European replication cohort | ||
| c.379C>T | p.Arg127* | 1 | European replication cohort | ||
| c.992+1G>T | p.Gly272_Thr331del | 1 | Belgian discovery cohort | ||
| c.1192delT | p.Ser398Profs*11 | 1 | Belgian discovery cohort | ||
| c.1335G>A | p.Trp445* | 1 | European replication cohort | ||
| c.1340+1G>A | p.Ala417* | 1 | European replication cohort | ||
| c.1385_1388delCAGA | p.Thr462Lysfs*3 | 1 | European replication cohort | ||
| c.1551_1552insTT | p.Ser518Leufs*32 | 1 | Belgian discovery cohort | ||
| In‐frame deletions leading to loss‐of‐protein or protein function | |||||
| c.235_237delACA | p.Thr79del | 1 | European replication cohort | ||
| c.499_501delGAT | p.Asp167del | 1 | Belgian discovery cohort | ||
| c.1927_1929delGAA | p.Glu643del | 3 | 1 | 2 | Belgian discovery cohort |
| Predicted in‐frame deletions with unknown effect | |||||
| c.228+1G>A | p.Lys30_Glu76del | 1 | European replication cohort | ||
| c.992+4_992+7delAGTA | p.Gly272_Thr331del | 1 | European replication cohort | ||
All listed mutations were absent from 2,183 screened control individuals and dbSNP build 138. The respective cohorts of the European study population in which mutations were identified are indicated in the last column. Mutations and carriers identified in the Belgian discovery cohort were previously published by our group [Gijselinck et al., 2015; Van Mossevelde et al., 2015]. Novel mutations and carriers reported in this study are part of the European replication cohort. cDNA numbering was according to the reference sequence NM_013254.3. In addition, for intronic variants, the genomic reference sequence NC_000012.12 was used. Nucleotide positions refer to cDNA sequence and nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. Protein numbering according to reference sequence NP_037386.1.
In addition to these proven loss‐of‐transcript mutations, we identified two novel predicted in‐frame exon‐skipping mutations. These splice donor‐site mutations in intron 3 (c.228+1G>A) and intron 8 (c.992+4_7delAGTA) are predicted to result in a deletion of 47 (p.Lys30_Glu76del) and 60 amino acids (p.Gly272_Thr331del). No cells or tissues from the carriers were available for transcript and protein analysis; therefore, we were unable to demonstrate the effect of these two mutations, and as a consequence, they were not considered further when calculating TBK1 mutation frequencies.
In‐Frame Deletions
We have observed two in‐frame, single amino acid deletions in the BE discovery cohort and one additional in‐frame deletion in the EU replication cohort (Table 2). All three, c.235_237delACA (p.Thr79del), c.499_501delGAT (p.Asp167del), and c.1927_1929delGAA (p.Glu643del), were absent from control subjects, and no other amino acid deletion mutations were observed in the control cohort. The p.Thr79del mutation was identified in a Spanish patient. The two other mutations, p.Asp167del and p.Glu643del, had been detected in Belgian patients [Gijselinck et al., 2015]. The p.Glu643del mutation was present in one index patient and five affected relatives and an additional five unrelated index patients [Gijselinck et al., 2015; Van Mossevelde et al., 2015]. In contrast to the protein‐truncating mutations, the p.Glu643del mutation did not affect transcript levels but produced 50% loss of TBK1 protein in vivo [Gijselinck et al., 2015]. In this study, we obtained comparable results for the Spanish mutation, p.Thr79del, with loss of expression only at the protein level observed in brain (Fig. 1D). The p.Asp167del behaved differently. Here, TBK1 protein expression in brain was preserved [Gijselinck et al., 2015]. In vitro TBK1 protein expression of p.Thr79del and p.Glu643del mutations overexpressed in HEK293T cells was reduced but not completely absent (Supp. Fig. S1), whereas phospho‐TBK1 was completely absent for p.Thr79del and p.Asp167del, and almost completely absent for p.Glu643del (Fig. 1E).
We further investigated the effect of the three in‐frame deletions on NFκB activation, using an NFκB in vitro luciferase reporter assay. Results showed that all three amino acid deletion mutations severely disrupted NFκB activation (Fig. 2). The two single amino acid deletions located in the KD, p.Thr79del and p.Asp167del, resulted in a complete loss of NFκB activation. The p.Glu643del mutation that maps to the scaffold dimerization domain (SDD; residues 408–657) [Larabi et al., 2013], showed a highly significant reduction of about 70%.
Figure 2.
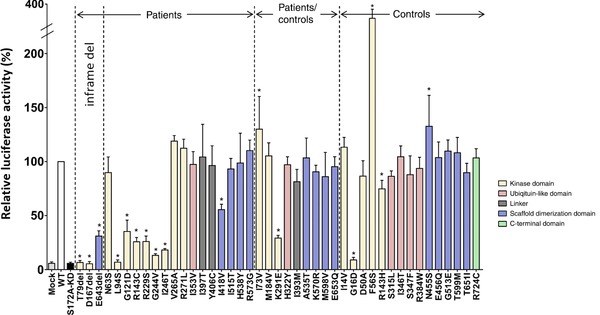
Impact of mutant TBK1 on NFκB activity in the IFN pathway. Graphical representation of the mean NFκB‐induced luciferase activity of identified in‐frame amino acid deletions and missense mutations found in patients‐only, shared by patients and controls, and in controls‐only, normalized to the mean signal from wild type. Luciferase activities were measured in at least three independent experiments and measured five times per experiment. The different domains are indicated in different colors as shown in the figure legend. WT, wild type TBK1 vector; Mock, empty vector containing no TBK1; S172A‐KD, p.Ser172Ala TBK1 kinase dead mutation. Mock and S172A‐KD were used as negative controls. Error bars depict standard deviation and asterisks above the bars indicate significant difference from the wild‐type level after Bonferroni correction (P<0.001). Protein numbering according to reference sequence NP_037386.1.
Taken together, the in vivo protein expression and in vitro experiments demonstrated that, in addition to the protein‐truncating LoF mutations, in‐frame deletions also lead to TBK1 LoF through loss‐of‐protein and/or protein function.
In conclusion, in the European replication cohort, we identified seven LoF protein‐truncating mutations and one LoF in‐frame deletion mutation in a total of eight index patients, resulting in a mutation frequency of TBK1 LoF mutations of 0.5% (8/1,755) overall and 0.2% in FTD patients (3/1,271), 0.5% in ALS patients (2/407), and 3.9% in FTD‐ALS patients (3/77). Meta‐analysis of the Belgian discovery cohort and the EU replication cohort resulted in a total mutation frequency for TBK1 LoF mutations in the overall European study population of 0.7% (19/2,538), comprising 0.4% FTD patients (8/1,873), 1.3% ALS patients (7/554), and 3.6% FTD‐ALS patients (4/111).
TBK1 Missense Mutations
In the present study, we also investigated the prevalence and functional effect of missense mutations in the European study population. In the patient cohort, we identified 25 missense mutations in 26 patients, of which 16 were found only in patients (Table 3). In the control cohort, we identified 15 missense mutations in 16 individuals that were absent in patients (Table 3). We tested all 40 missense mutations using the NFκB reporter assay (Table 3; Fig. 2).
Table 3.
TBK1 Missense Mutations in the European Study Population
| FTD | FTD‐ALS | ALS | Controls | |||||
|---|---|---|---|---|---|---|---|---|
| cDNA | Predicted protein | Domain | dbSNP | NFκB activity | n = 1,873 | n = 111 | n = 554 | n = 2,183 |
| Missense mutations found in patients only | ||||||||
| c.188A>G | p.Asn63Ser | KD | ‐ | n.s. | 1 | |||
| c.281T>C | p.Leu94Ser | KD | ‐ | Reduced | 1 | |||
| c.362G>A | p.Gly121Asp | KD | ‐ | Reduced | 1 | |||
| c.427C>T | p.Arg143Cys | KD | ‐ | Reduced | 1 | |||
| c.687G>T | p.Arg229Ser | KD | ‐ | Reduced | 1 | |||
| c.731G>T | p.Gly244Val | KD | ‐ | Reduced | 1 | |||
| c.737T>C | p.Ile246Thr | KD | ‐ | Reduced | 1 | |||
| c.794T>C | p.Val265Ala | KD | ‐ | n.s. | 1 | |||
| c.812G>T | p.Arg271Leu | KD | ‐ | n.s. | 1a | |||
| c.1057A>G | p.Ile353Val | ULD | ‐ | n.s. | 1 | |||
| c.1190T>C | p.Ile397Thr | linker | ‐ | n.s. | 1 | |||
| c.1217A>G | p.Tyr406Cys | linker | ‐ | n.s. | 1 | |||
| c.1252A>G | p.Ile418Val | SDD | rs138839127 | Reduced | 1 | |||
| c.1544T>C | p.Ile515Thr | SDD | rs151225287 | n.s. | 1a | |||
| c.1612C>T | p.His538Tyr | SDD | ‐ | n.s. | 1 | |||
| c.1717C>G | p.Arg573Gly | SDD | ‐ | n.s. | 1 | |||
| Missense mutations found in patients and control subjects | ||||||||
| c.217A>G | p.Ile73Val | KD | ‐ | Increased | 2 | 1 | ||
| c.550A>G | p.Met184Val | KD | ‐ | n.s. | 1 | 1 | ||
| c.871A>G | p.Lys291Glu | KD | rs34774243 | Reduced | 1a | 2 | ||
| c.964C>T | p.His322Tyr | ULD | rs145905497 | n.s. | 1a | 1 | ||
| c.1179A>G | p.Ile393Met | linker | ‐ | n.s. | 1 | 1 | ||
| c.1603G>A | p.Ala535Thr | SDD | rs199905735 | n.s. | 1a | 1 | ||
| c.1709A>G | p.Lys570Arg | SDD | ‐ | n.s. | 1 | 2 | ||
| c.1792A>G | p.Met598Val | SDD | ‐ | n.s. | 1 | 2 | ||
| c.1957G>C | p.Glu653Gln | SDD | rs144370662 | n.s. | 1 | 4 | ||
| Missense mutations found in control subjects only | ||||||||
| c.40A>G | p.Ile14Val | KD | ‐ | n.s. | 1 | |||
| c.47G>A | p.Gly16Asp | KD | ‐ | Reduced | 1 | |||
| c.149A>C | p.Asp50Ala | KD | ‐ | Increased | 1 | |||
| c.167T>C | p.Phe56Ser | KD | ‐ | n.s. | 1 | |||
| c.428G>A | p.Arg143His | KD | ‐ | Reduced | 1 | |||
| c.944C>T | p.Ser315Leu | ULD | rs369620088 | n.s. | 1 | |||
| c.1037T>C | p.Ile346Thr | ULD | ‐ | n.s. | 1 | |||
| c.1040C>T | p.Ser347Phe | ULD | ‐ | n.s. | 1 | |||
| c.1150C>T | p.Arg384Trp | ULD | ‐ | n.s. | 1 | |||
| c.1364A>G | p.Asn455Ser | SDD | ‐ | Increased | 1 | |||
| c.1366G>C | p.Glu456Gln | SDD | ‐ | n.s. | 1 | |||
| c.1538G>A | p.Gly513Glu | SDD | ‐ | n.s. | 1 | |||
| c.1796C>T | p.Thr599Met | SDD | ‐ | n.s. | 1 | |||
| c.1952C>T | p.Thr651Ile | SDD | ‐ | n.s. | 1 | |||
| c.2170C>T | p.Arg724Cys | CTD | rs185524052 | n.s. | 2 | |||
TBK1 functional domains according to Larabi et al. (2013): KD, kinase domain (residues 1–307); ULD, ubiquitin‐like domain (residues 309–384); linker; linker (residues 385–407); SDD, scaffold dimerization domain (residues 408–657); CTD, C‐terminal domain (residues 657–745). Results of the in vitro NFkB luciferase reporter assay measuring the effect of mutant TBK1 on NFκB activity are listed (see also Fig. 2). Reduced indicates that a significant reduction in luciferase activity was measured; increased indicates that a significant increase in luciferase activity was measured. n.s., no significant change in luciferase activity was measured.
aRefers to mutations and carriers identified in the Belgian discovery cohort that were published earlier [Gijselinck et al., 2015]. cDNA numbering according to reference sequence NM_013254.3. Nucleotide positions refer to cDNA sequence and nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. Protein numbering was according to the reference sequence NP_037386.1.
In the patient‐only group, we found seven missense mutations in seven patients that showed an effect on NFκB induction (7/16 carriers, 44%). Six missense mutations showed a ≥70% decrease in NFκB activation as compared with wild‐type TBK1, of which the c.281T>C (p.Leu94Ser) mutation showed a complete loss. These six missense mutations were located in the KD of TBK1. To a lesser extent but still significant, the c.1252A>G (p.Ile418Val) mutant in the SDD decreased NFκB induction by 45%.
In the group of missense mutations present in both patients and controls, just one missense mutation c.871A>G (p.Lys291Glu), also located in the KD domain and present in one patient and two control subjects, showed a significant decrease in NFκB induction (3/25 carriers, 12%).
Of the 15 missense mutations that were observed in control subjects only, two mutations located in the KD protein domain showed a significant reduction, with a complete loss for c.47G>A (p.Gly16Asp) and a ∼25% reduction for c.428G>A (p.Arg143His) (2/16 carriers, 13%).
When comparing the prevalence of missense mutations with a compromised NFκB activation capacity (= functional missense mutations) in patients and controls, we observed about twice as many in patients. In the patients, eight carriers of a functional and therefore potentially pathogenic missense mutation were identified (8/2,538, 0.32%) as compared to four in control subjects (4/2,183, 0.18%) (P = 0.40). Although functional missense mutations were identified in the patient‐only, the patient and control, as well as the control‐only group, over three times as many carriers of functional missense mutations were counted in the patient‐only group (7/16, 44%) versus the group of carriers of missense mutations that were also observed in the control population (5/41, 12%) (P = 0.025).
Phenotypic Characteristics of TBK1 Mutation Carriers
Demographic, genetic, and clinical features of the pathogenic and possibly pathogenic TBK1 mutation carriers (protein‐truncating mutations, in‐frame deletions, and functional missense mutations) are summarized in Table 4.
Table 4.
Clinical and Pathological Phenotype of Patients Carrying a TBK1 LoF Mutation or Possibly Pathogenic Missense Mutation
| Mutation | Origin | Gender | Clinical diagnosisa | Subtypeb | Family history | Age at onset (years) | Age at last examination/death (years) | Disease duration (months) | Additional information |
|---|---|---|---|---|---|---|---|---|---|
| Truncating mutations | |||||||||
| p.Gln*2c | Spanish | Female | FTD | bvFTD | 56 | 60 | >53 | ||
| p.Lys29Argfs*15 | German | Male | FTD | PNFA + PSP | 73 | †77 | 48 | ||
| p.Val97Phefs*2 | Swedish | Male | ALS | + | 62 | †63 | MND‐TDP | ||
| p.Arg117* | Italian | Male | FTD | bvFTD | + | 67 | †74 | 86 | |
| p.Arg127* | German | Female | ALS | Bulbar | 70 | Alive | |||
| p.Trp445* | Spanish | Female | FTD + CBS | PNFA/agramatic variant | 78 | 84 | >72 | ||
| p.Gly272_Thr331delc | Belgian | Male | FTD | bvFTD | + | 48 | †50 | 29 | |
| p.Ser398Profs*11c | Belgian | Male | ALS | Bulbar | + | 59 | Alive | >75 | |
| p.Ala417* | Swedish | Female | FTD | bvFTD | + | 68 | †71 | 27 | FTLD‐TDP type B |
| p.Thr462Lysfs*3 | German | Male | ALS + D | + | 74 | †75 | 11 | LMN>>UMN | |
| p.Ser518Leufs*32c | Belgian | Female | ALS | + | 64 | †64 | 6 | ||
| In‐frame deletions | |||||||||
| p.Thr79del | Spanish | Male | FTD + ALS | bvFTD + bulbar | 56 | †58 | 18 | FTLD‐MND‐TDP type B + argyrophylic grain disease (stage III) | |
| p.Asp167delc | Belgian | Male | ALS | 60 | †61 | ||||
| p.Glu643delc | Belgian | Female | FTD + ALS | bvFTD + spinal | + | 62 | †74 | 136 | |
| p.Glu643delc | Belgian | Female | FTD | bvFTD | + | 64 | Alive | >109 | |
| p.Glu643delc | Belgian | Male | ALS | Bulbar | + | 51 | †53 | 20 | |
| p.Glu643delc | Belgian | Male | ALS | 63 | †66 | ||||
| p.Glu643delc | Belgian | Male | FTD | PPA | + | 70 | †73 | 42 | |
| p.Glu643delc | Belgian | Female | FTD | bvFTD | 69 | Alive | >99 | ||
| Functional missense mutations | |||||||||
| p.Leu94Ser | Bulgarian | Male | ALS | Spinal | + | 44 | 55 | >120 | Slow disease progression |
| p.Gly121Aspc | Spanish | Male | ALS | Spinal | 34 | 39 | >60 | Slow disease progression | |
| p.Arg143Cys | German | Male | FTD | bvFTD | 45 | ||||
| p.Arg229Ser | German | Male | ALS | 47 | |||||
| p.Gly244Val | Portuguese | Female | FTD + ALS | Bulbar | + | 41 | †43 | C9orf72 repeat expansion carrier | |
| p.Ile246Thr | German | Female | ALS | Bulbar | 57 | †59 | |||
| p.Lys291Gluc | Belgian | Male | FTD | bvFTD | + | 52 | †61 | ||
| p.Ile418Val | Portuguese | Female | FTD | bvFTD | + | 53 | 54 | ||
aPresenting diagnosis or symptoms are listed first.
bClinical subtype is given where documented. In FTD, the subtypes behavioral variant FTD (bvFTD), primary progressive aphasia (PPA), progressive nonfluent aphasia (PNFA), semantic dementia (SD), and progressive supranuclear palsy (PSP) are specified where documented. In ALS, spinal or bulbar onset is specified where documented.
cRefers to mutation carriers identified in the Belgian discovery cohort that were published earlier [Gijselinck et al., 2015; Van Mossevelde et al., 2015].
CBS, corticobasal syndrome; MND, motor neuron disease; LMN, lower motor neuron symptoms; UMN, upper motor neuron symptoms; D, unspecified dementia. +, a positive family history was documented.
Eleven of the 19 index patients with a TBK1 LoF mutation had FTD as presenting clinical phenotype. In this subset of patients, nine developed FTD without clinical signs of MND. In the years following first diagnosis, two patients developed ALS and another patient progressed into a corticobasal syndrome (CBS). Eight patients presented first with ALS, of which one showed concomitant cognitive deterioration suffering from a severe memory disorder and visuoconstructional deficits but no specification of the dementia diagnosis was possible. Of all patients diagnosed with ALS (n = 10), four had bulbar onset and one had spinal onset. In five ALS patients, the site of onset was not specified. Of all FTD (or unspecified dementia) patients (n = 12), the majority was diagnosed with bvFTD (8/12). Three others presented with a language variant of FTD, progressive nonfluent aphasia (PNFA) or PPA. The remaining patient received a diagnosis of unspecified dementia (together with ALS). Mean age at onset in all 19 TBK1 LoF carriers was 63.9 ± 7.8 years (range 48–78). Of the 12 patients that died, mean age at death was 65.5 ± 8.9 years (range 50–77) with an average disease duration of 42 ± 40 months (range 6–136).
Neuropathological examination was performed in three of the deceased patients. The patient with the p.Val97Phefs*2 mutation displayed pure MND with TDP‐43 inclusions. The carrier of the p.Ala417* mutation showed mild TDP‐43 proteinopathy in affected brain regions (type B). Neuropathological investigation in the p.Thr79del carrier confirmed the diagnosis of FTLD‐MND‐TDP (type B), but additional argyrophilic grain disease stage III was noted (Fig. 3).
Figure 3.
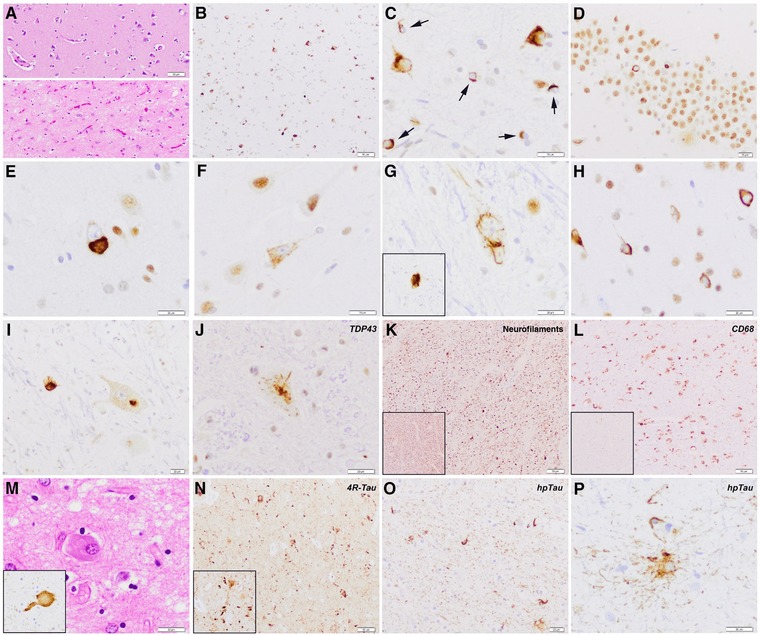
Neuropathology features of the FTD‐ALS patient with the TBK1 p.Thr79del mutation. Severe neuronal loss, gliosis, and loosening of the neuropil are observed in entorhinal and transentorhinal region (lower image) as compared with relatively well‐preserved frontal cortex (upper image) (HE) (A). Abundant TDP‐43 protein aggregates in neurons and oligodendroglial cells (B), better seen in (C) at higher magnification (arrows). Hippocampal dentate gyrus shows some granular neurons lacking physiological nuclear immunoreactivity but shifting toward pathological inclusions in the cytoplasm (D). Morphological spectrum of neuronal cytoplasmic inclusions (E–J), seen as compact bodies (E), diffuse granular cytoplasmic immunoreactivity or “preinclusion” type (F), skein‐like inclusions (G, inset), compact ring‐like inclusions (H), or a combination of diffuse cytoplasmic and compact in the same motor neuron (I). Signs of corticospinal tract degeneration at the level of the spinal cord with marked reduction of axonal density as shown by antineurofilament immunohistochemistry (K, inset shows regular density of axons for comparison), and increased macrophagic activity (L, anti‐CD68 immunohistochemistry, inset shows regular density of CD68+ cells in the spinal cord for comparison). Neuropathological features of concomitant argyrophilic grain pathology (M–P). Ballooned cells are seen in amygdala (M) and are nicely stained by hyperphosphorylated tau (AT8, inset). Moreover, frequent hpTau‐positive grains, mainly composed of four‐repeat tau isoforms, are detected in the limbic system (N, CA1 sector is shown), and represent enlargements/verrucosities of dendritic spines (N, inset). Oligodendroglial coiled bodies (O) and bush‐like astrocytes (P) accompany the full picture.
In the eight functional missense mutation carriers, four presented first with FTD, of whom one developed concomitant ALS, and four presented with ALS only. Again, in the FTD patients, bvFTD was the predominant subtype (3/4). In the ALS patients, two had spinal onset and two had bulbar onset. Average onset age was low at 46.6 ± 7.4 years (range 34–57). The three patients that passed away died at relatively young ages of 43, 59, and 61 years (average age at death 54.3 ± 9.9 years).
Neuropathology of the p.Thr79del Mutation Carrier
Macroscopic evaluation showed a moderate and temporally accentuated brain atrophy with an unfixed brain weight of 1,340 g. On histological examination (Fig. 3), neuronal loss, reactive astrogliosis, and microglial activation were observed predominantly involving the medial temporal lobe. Abundant and widespread neuronal TDP‐43 protein inclusions were detected in neurons of the limbic system including amygdala, entorhinal, and transentorhinal region, hippocampus (dentate gyrus and CA1 sector), cingulate cortex, inferior temporal cortex, and in the motor cortex, with only few inclusions in frontal and parietal cortex, in basal ganglia and thalamus, and brainstem nuclei. Type of inclusions varied and comprised compact and ring‐like neuronal cytoplasmic inclusions that were observed predominantly, but not exclusively, in superficial cortical layers and also throughout the cortex. Only very isolated dystrophic neurites were detected. Diffuse granular cytoplasmic immunoreactivity (neuronal "preinclusions") was frequently seen in middle‐sized neurons of frontal and temporal cortex, limbic system, basal ganglia, brainstem, and spinal cord. Skein‐like inclusions were evident in motor neurons of the anterior horn of the spinal cord, but also in motor neurons of the hypoglossal nucleus, pigmented neurons of the substantia nigra, and few striatal neurons. Frequent oligodendroglial inclusions were detected both in white matter and gray matter (arrows). Involvement of upper motor neurons was appreciated, and there was florid corticospinal tract degeneration showing myelin and axonal loss, and abundant CD68‐positive macrophages, whereas lower motor neurons of anterior horns of spinal cord were moderately affected. The distribution pattern of pathology was most concordant with FTLD type B according to the current FTLD consensus classification [Mackenzie et al., 2011]. In addition, concomitant argyrophilic grain pathology (AgD) was observed. This was characterized by the presence of hyperphosphorylated tau (AT8) and four‐repeat tau‐positive grains in amygdala, entorhinal and transentorhinal cortex, CA1 sector of the hippocampus, and cingulum, along with frequent pretangles and neuropil threads, tau‐positive oligodendroglial coiled bodies in medial temporal white matter, bush‐like astrocytes, and ballooned neurons in amygdala corresponding to stage III according to Saito et al. (2004).
Discussion
In the pursuit of the missing heritability of the FTD‐ALS spectrum, TBK1 haploinsufficiency was recently put forward as a novel disease mechanism. Classical LoF mutations include nonsense and frameshift mutations that lead to mutant transcripts containing a PTC, which are degraded by NMD resulting in a 50% loss of protein. In addition to loss of transcript, several other possibilities can lead to loss–of‐protein or protein function. For example, deletions of one or more key amino acids due to small in‐frame indels or in‐frame exon skipping and missense mutations can affect the catalytic function or the stability of the protein. In the present study, we investigated the full mutation spectrum of TBK1 and its associated phenotypic spectrum in a large study population of 2,538 European FTD and ALS patients. In addition to protein‐truncating LoF mutations, the functional effect of in‐frame amino acid deletions and missense mutations was further explored in vivo on protein level and in vitro by an NFκB‐induced luciferase reporter assay as functional readout for TBK1 activity.
In total, we identified 11 index patients carrying a TBK1 protein‐truncating mutation (Table 2). Nine mutations were nonsense or frameshift mutations, of which five, (c.86delA (p.Lys29Argfs*15), p.Val97Phefs*2, p.Arg127*, c.1335G>A (p.Trp445*), p.Thr462Lysfs*3), are reported here for the first time. Multiple studies have demonstrated that PTC mutations in TBK1 lead to loss or severe reduction of mutant transcript levels by NMD [Freischmidt et al., 2015; Gijselinck et al., 2015; Pottier et al., 2015], which we additionally confirmed in the present study (Fig. 1A and B). Further, we detected two splice donor‐site mutations. The c.1340+1G>A in intron 11 leads to out‐frame skipping of exon 11 and the introduction of a PTC (p.Ala417*). Exon 11 skipping by this mutation was previously demonstrated by Freischmidt et al. (2015) and was identified in five Swedish individuals: three sporadic ALS patients, one familial ALS patient, and one unaffected individual with a family history of ALS. Interestingly, the FTD patient in whom we identified the same p.Ala417* mutation was also of Swedish origin, potentially indicative of a founder mutation. In a Belgian FTD patient, we identified the c.992+1G>T mutation in the consensus splice donor sequence of intron 8 and demonstrated activation of a cryptic splice‐site upstream in exon 8, resulting in a frameshift and 30%–50% reduced expression in blood and brain [Gijselinck et al., 2015]. In addition to these 11 proven protein‐truncating mutations, we detected two additional splice donor‐site mutations in intron 3 (c.228+1G>A) and intron 8 (c.992+4_7delAGTA) (Table 2). Both are predicted to lead to in‐frame skipping of their respective exons. Unfortunately, no biomaterials of the carriers were available to investigate potential cryptic splicing or the formation of unstable/nonfunctional protein due to the loss of conserved amino acid sequence; therefore, their pathogenic effect remains speculative.
In addition to TBK1 protein‐truncating mutations, we identified eight carriers of three in‐frame amino acid deletions: p.Thr79del, p.Asp167del, and p.Glu643del (Table 2). For p.Thr79del and p.Glu643del, we demonstrated a near total loss of mutant protein in vivo and partial loss in vitro, likely due to protein instability [Gijselinck et al., 2015] (Fig. 1D; Supp. Fig. S1). In contrast, this was not the case for p.Asp167del for which TBK1 protein expression was preserved [Gijselinck et al., 2015] (Supp. Fig. S1). Yet, total (p.Thr79del and p.Asp167del) or highly reduced (p.Glu643del) loss of TBK1 autophosphorylation and NFκB activation was measured for all three mutations. Therefore, the observed effects of p.Thr79del and p.Glu643del on autophosphorylation and on NFκB activation might also result from reduced activity of the remaining mutant protein, in addition to reduced protein expression. The deleterious effect of p.Asp167del was to be expected since Asp167 is located in the activation loop of the KD, of which phosphorylation is required for TBK1 activation [Larabi et al., 2013]. Also, p.Thr79del and p.Glu643del affect highly conserved amino acids and map to the catalytic KD and the SDD, located further downstream. TBK1 is a multifunctional kinase, phosphorylating a wide range of substrates, including the NFkB complex. Importantly, as indicated above, TBK1 itself is activated by homodimerization and subsequent autophosphorylation. Although it is currently not known which function(s) of TBK1 contribute to the disease mechanism(s) in FTD and ALS, measuring NFκB induction by observed TBK1 mutations compared with wild‐type TBK1 can serve as a first functional readout for their effect on TBK1 function related to the NFkB pathway, awaiting more tailored functional pathogenicity assays. In vivo, the two mechanisms of LoF through loss‐of‐protein and protein function may very well complement each other.
The largest group of mutations that we identified were missense mutations. In total, screening of 2,538 patients and 2,183 control individuals revealed 40 missense mutations, spread over the different protein domains and observed both in patients and controls (Table 3; Fig. 2). Using the NFκB reporter assay, we were able to discriminate 10 functional missense mutations that abolished or significantly reduced NFκB activation. Functional missense mutations mainly clustered in the KD, with only one exception for the p.Ile418val mutation located in the SDD, and most likely exert their effect through a defect in the catalytic kinase activity of TBK1. These functional missense mutations are possibly pathogenic, although their degree of pathogenicity may be variable, correlating with a complete absence of TBK1 activity for some to a significant reduction of ∼50%, or more for others (Fig. 2). The p.Leu94Ser mutation was a bit particular compared to the other functional missense mutations as it showed reduced protein expression, probably due to unstable protein (Supp. Fig. S1). The loss of NFκB activation by this mutation might therefore result from a combination of both loss‐of‐protein and protein function, similar to what was observed for some in‐frame single amino acid deletions. For three missense mutations, one patient‐specific c.217A>G (p.Ile73Val) and two control‐specific c.149A>C (p.Asp50Ala) and c.1364A>G (p.Asn455Ser), the NFκB reporter assay measured a significant increase in activity (Table 3; Fig. 2). The biological relevance of these observations is not clear; however, an increase in TBK1 activity does not seem relevant in the context of a LoF disease mechanism, and most likely these missense mutations are neutral with respect to ALS and FTD.
Even though occasional functional missense mutations were observed in the healthy study population (4/2,183, 0.18%), twice as many were present in patients (8/2,538, 0.32%). In other words, of the 12 carriers of a functional missense mutation, eight (67%) were diagnosed with ALS or FTD. When we looked specifically at missense mutations that were only observed in patients, 44% (7/16) had a compromised NFκB activation capacity compared with 12% (5/41) of the missense mutations observed in controls, which was statistically significant (P = 0.025). The observed enrichment of functional TBK1 missense mutations in patients suggests that they may function as high‐risk alleles through various degrees of loss‐of‐protein function.
Mutations in the KD affect the catalytic domain and likely affect phosphorylation of all targets. As such, the NFκB activation assay may be considered a valid test of pathogenicity because it measures the kinase activity, independent of substrate. However, we cannot exclude that additional missense mutations with no assessable effect on the NFκB/IFN pathway may be pathogenic. Other pathways may be affected by hampered binding or phosphorylation of the respective substrates. Indeed, for at least one additional missense mutation, c.1717C>G (p.Arg573Gly) located in the SDD, we have evidence for cosegregation in four affected sibs, suggestive of an autosomal‐dominant inheritance. This sib‐ship presented with a heterogeneous disease presentation of FTD, AD‐type dementia, and MND (Gómez‐Tortosa, Submitted). Mutations in other domains, such as the C‐terminal substrate‐binding domain, may selectively affect specific kinase activities, and therefore the NFκB assay may not be a good readout for pathogenicity for these mutations, depending on which TBK1 functions are central to disease. As proposed by Freischmidt (2015), loss of OPTN binding may be sufficient to cause ALS and FTD [Freischmidt et al., 2015]. To further investigate the possibility that some of the observed missense mutations had an effect on OPTN binding, we modeled the missense mutations of the patient and control fraction outside the KD by OPTN coimmunoprecipitation. The missense mutations under study did not show any evidence for reduced OPTN binding (data not shown). Since TBK1 operates as a homodimer, pathogenic TBK1 missense mutations, regardless of which domain they are located in, may in addition also have a dominant‐negative effect by competing with wild‐type TBK1.
Mean onset age in the 19 TBK1 LoF mutation carriers (protein‐truncating mutations + in‐frame deletions) was 63.9 ± 7.8 years (range 48–78). Remarkable in terms of survival was the huge range in disease duration from 6 to 136 months. In terms of phenotypic heterogeneity, one patient (p.Lys29Argfs*15) showed a clinically mixed picture of behavioral disturbances (bvFTD), PNFA, and symmetric Parkinsonian syndrome of bilateral rigidity, hypokinesia, vertical gaze paresis, and gait disorder, suggestive for progressive supranuclear palsy. Patient (p.Trp445*) was diagnosed with CBS and nonfluent/agrammatic variant PNFA. Neuropathological examination of the FTLD‐ALS patient with the p.Thr79del mutation showed a temporal accentuated brain atrophy with abundant neuronal and glial TDP‐43 protein aggregates, with upper and lower motor neuron involvement, and AgD in the medial temporal lobe. The concomitant AgD probably contributed to the lobar atrophy and to the clinical picture of this patient. AgD has clinically been related to mild forms of dementia with mood and behavioral disturbances, personality changes, aggressiveness, and even psychosis. AgD pathology is considered to occur more frequently in aged than in younger individuals [Pham et al., 2011], and has also been detected in cognitively normal subjects. Moreover, AgD has been described in association with several other neurodegenerative conditions. Coincidental TDP‐43 protein aggregates in the medial temporal lobe have been repeatedly reported to occur relatively frequently in association with AgD [Fujishiro et al., 2009; Arnold et al., 2013]. Therefore, the co‐occurrence of FTLD‐TDP and AgD in this relatively young (age at death 58 years) patient may suggest some—sporadic or mutation‐driven—link between abnormal Tau and TDP‐43 protein deposition.
The absence of a documented family history in 42% (8/19) of the LoF mutation carriers underscores the marked reduced clinical penetrance of TBK1 mutations. This reduces penetrance, which has also been confirmed in multigenerational families segregating a TBK1 LoF mutation [Freischmidt et al., 2015; Gijselinck et al., 2015; Van Mossevelde et al., 2015], explains why apparently sporadic patients carry a Mendelian TBK1 mutation. In these patients, family history may be obscured due to small family size or pleiotropic manifestation of TBK1 mutations as FTD or ALS. Inquiry of family history in isolated FTD patients may not take into account relatives with motor neuron symptoms and, vice versa, in ALS patient's relatives with cognitive problems. Reduced penetrance and observation of Mendelian mutations in apparently sporadic patients or isolated patients is also a recurring theme in other major Mendelian genes in FTD and ALS, such as GRN (MIM# 138945) and C9orf72. We previously showed that 50% of TBK1 carriers are affected by the age of 70 [Van Mossevelde et al., 2015]. In the extended set of 19 TBK1 LoF carriers from this study, just one patient was affected by the age of 50 (5%), six by the age of 60 (32%), and 16 by the age of 70 years (84%). It is noteworthy that in the carriers of a functional missense high‐risk variant, average onset age of 46.6 ± 7.4 years (range 34–57) was markedly lower than in the group of LoF mutations. Two of the TBK1 missense variant carriers also carried a C9orf72 repeat expansion. Interestingly, the patient with the TBK1 c.731G>T (p.Gly244Val) mutation shown to attenuate NFκB induction in combination with a C9orf72 repeat expansion had a pronounced early disease onset of 41 years. We already described this patient when we identified the C9orf72 repeat expansion in FTD‐ALS spectrum patients [van der Zee et al., 2013]. Her disease course was characterized by rapidly progressive FTD with bulbar ALS and death after 2 years [Chester et al., 2013]. In contrast, the other C9orf72 expansion carrier with the TBK1 c.550A>G (p.Met184Val) mutation without effect on the NFκB activation had an onset age of 70 years. Considering the high prevalence of C9orf72 repeat expansions in FTD‐ALS spectrum patients, it is not surprising that in some patients a mutation in TBK1, GRN, or another gene can co‐occur with a C9orf72 repeat expansion that may influence disease penetrance.
In conclusion, we investigated a large European study population of 2,538 European FTD‐ALS spectrum patients to get a deeper appreciation of the mutation frequency, mutation spectrum, and the genotype–phenotype profile of TBK1 patient carriers. In addition to the typical LoF protein‐truncating mutations, we demonstrated that in‐frame amino acid deletion mutations and a significant fraction of the missense mutations, particularly those mapping to the kinase domain, also lead to loss‐of‐protein and/or protein function. We observed a mutation frequency for TBK1 LoF protein‐truncating and in‐frame deletion mutations of 0.7%, representing 0.4% in the FTD, 1.3% in the ALS, and 3.6% in the FTD‐ALS clinical subgroups. Taking also the functional missense high‐risk alleles into account, mutation frequencies increased to 1.1% overall and 0.6% in FTD, 2.0% in ALS, and 4.5% in FTD‐ALS patients. These mutation frequencies are relatively low compared with other FTD‐ALS spectrum genes like C9orf72. Nevertheless, the higher mutation frequencies in FTD‐ALS and in ALS patients warrant diagnostic testing. Furthermore, as indicated above, calculated TBK1 mutation frequencies may still be an underestimation with a substantial fraction of missense mutations remaining unresolved. Although TBK1 is a well‐characterized, multifunctional kinase, which of the TBK1 functions that specifically contribute to the disease mechanism in FTD and ALS are poorly understood. Hopefully, in the years to come, more functional data will become available, and based on this information, better informed and targeted pathogenicity assays or biomarkers can be developed for more reliable risk prediction and counseling in TBK1 mutation carriers.
Belgian Neurology (BELNEU) consortium side author list
The following members of the BELNEU consortium contributed to the clinical and pathological phenotyping and follow‐up of the Belgian patient cohorts: Johan Goeman and Dirk Nuytten (Hospital Network Antwerp, Antwerp); Katrien Smets (Antwerp University Hospital, Edegem); Wim Robberecht and Philip Van Damme (University Hospitals Leuven Gasthuisberg, Leuven); Jan De Bleecker, Patrick Santens, and Bart Dermaut (University Hospital Ghent, Ghent); Jan Versijpt and Alex Michotte (University Hospital Brussels, Brussels); Adrian Ivanoiu (Saint‐Luc University Hospital, Brussels); Olivier Deryck and Bruno Bergmans (General Hospital Sint‐Jan Brugge, Bruges); Jean Delbeck (General Hospital Sint‐Maria, Halle); Marc Bruyland (General Hospital Glorieux Ronse); Christiana Willems (Jessa Hospital, Hasselt); and Eric Salmon (University of Liège and Memory Clinic, CHU Liège, Liège).
European Early‐Onset Dementia (EU EOD) consortium side author list
The following members of the EU EOD consortium contributed to the clinical and pathological phenotyping and follow‐up of the patients at their site that were included in the EU EOD cohort: Pau Pastor (University Hospital Mútua de Terrassa and Fundació Docència i Recerca Mútua Terrassa, University of Barcelona School of Medicine, Terrassa, Barcelona, Spain); Pau Pastor and Sara Ortega‐Cubero (CIBERNED Instituto de Salud Carlos III, Madrid, Spain); Sara Ortega‐Cubero (Deparment of Neurology, Complejo Asistencial Universitario de Palencia, Palencia, Spain); Luisa Benussi and Roberta Ghidoni (Molecular Markers Laboratory, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy); Giuliano Binetti (MAC Memory Center and Molecular Markers Laboratory, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy); Isabel Hernández, Mercè Boada, and Agustín Ruiz (Fundació ACE, Institut Català de Neurociències Aplicades, Barcelona, Spain); Sandro Sorbi, Benedetta Nacmias, and Silvia Bagnoli (Department of Neurosciences, Psychology, Drug Research and Child Health, University of Florence, Florence, Italy); Sandro Sorbi (IRCCS Don Carlo Gnocchi Scandicci, Florence, Italy); Raquel Sanchez‐Valle and Albert Llado (Hospital Clínic, IDIBAPS, Barcelona, Spain); Isabel Santana and Maria Rosário Almeida (University of Coimbra, Coimbra, Portugal); Giovanni B Frisoni (Hôpitaux Universitaires de Genève et Université de Genève, Genève, Switzerland and IRCCS Fatebenefratelli, Brescia, Italy); Walter Maetzler (Hertie Institute for Clinical Brain Research, Tübingen, Germany); Radoslav Matej (Thomayer Hospital, Prague and Charles University, Prague, Czech Republic); Matthew J. Fraidakis (NeuroRARE Centre for Rare and Genetic Neurological & Neuromuscular Diseases & Neurogenetics Athens, Greece); Gabor G. Kovacs (Medical University of Vienna, Vienna, Austria); and Gian Maria Fabrizi and Silvia Testi (University of Verona, Verona, Italy).
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supp. Figure S1. Western blot analysis of all identified single amino acid deletions and missense mutations overexpressed in HEK293T cells. TBK1 plasmids containing a missense mutation or a single‐amino acid deletion were transiently transfected in HEK293T cells. The p.Ser172Ala mutant (kinase dead, KD), generating a TBK1 protein lacking kinase activity and the Mock plasmid containing no TBK1, were used as negative controls. Protein lysates of the TBK1 overexpression cells were used for western blotting with an antibody against TBK1 (Abcam, 1:1000, 84 kDa) and GAPDH (Genetex, 1:10000, 37 kDa) as a reference protein. TBK1 signals of the mutant TBK1 plasmids were compared to wild type TBK1 (WT). p.Thr79del, p.Asp643del and p.Lys94Ser showed near 50% reduced protein expression, probably due to protein instability. All other TBK1 mutations were equally expressed as the wild type TBK1.
Acknowledgments
The authors thank the personnel of the Neuromics Support Facility of the VIB Center for Molecular Neurology (http://www.vibgeneticservicefacility.be) and the Antwerp Biobank of the Institute Born‐Bunge for their expert support. The Neurological Tissue Bank of the Biobanc‐Hospital Clinic‐IDIBAPS thanks all brain donors and families for generous brain donation for research and the Neurological Tissue Bank of the IDIBAPS Biobank for data and sample procurement. Caroline Graff wishes to express her acknowledgements to Inger Nennesmo (for the neuropathological assessments), Huei‐Hsin Chiang, Jenny Björkström, Lena Lilius, Charlotte Forsell, Marie Fallström (Department of Neurobiology, Care Sciences and Society [NVS], Center for Alzheimer Research, Division of Neurogeriatrics, Karolinska Institutet and Department of Geriatric Medicine, Genetics Unit, Karolinska University Hospital, Stockholm, Sweden), and The Brain Bank at Karolinska Institutet. The LMU Munich site acknowledges the essential support of the team at Zentrum für Neuropathologie und Prionforschung, Ludwig‐Maximilians‐Universität Munich (Thomas Arzberger, Sigrun Roeber, Manuela Neumann, Armin Giese, and Hans Kretzschmar) for their neuropathological work and the safekeeping of DNA samples.
Disclosure statement: The authors declare no conflict of interest.
Contract grant sponsors: Belgian Science Policy Office Interuniversity Attraction Poles Program; Flemish Government (Flanders Impulse Program on Networks for Dementia Research, Methusalem Excellence Program); the Research Foundation Flanders (FWO); the University of Antwerp Research Fund; Fondazione Cassa di Risparmio di Pistoia e Pescia (grants 2014.0365, 2011.0264, and 2013.0347); the Cassa di Risparmio di Firenze (grant 2014.0310); Fondo di Ateno 2014; Ricerca Corrente; Italian Ministry of Health; Else Kröner‐Fresenius‐Stiftung (EKMS 018); Swedish Brain Power; Swedish Research Council (grant numbers 521‐2010‐3134, 2015‐02926); Gun and Bertil Stohne; Gamla tjänarinnor; Demensfonden; Sweden Alzheimer Foundation (AF‐556561); King Gustaf V and Queen Victoria's Foundation of Freemasons; StratNeuro at Karolinska Institute (KI).
Communicated by Lars Bertram
References
- Arnold SJ, Dugger BN, Beach TG. 2013. TDP‐43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol 126:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ber I, De Septenville A, Millecamps S, Camuzat A, Caroppo P, Couratier P, Blanc F, Lacomblez L, Sellal F, Fleury M‐C, Meininger V, Cazeneuve C, et al. 2015. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging 36:3116.e5–3116.e8. [DOI] [PubMed] [Google Scholar]
- Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, Papetti A, Stuani C, Di Luca M, Gennarelli M, Padovani A. 2009. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat 30:E974–E983. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Miller RG, Swash M, Munsat TL. 2000. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299. [DOI] [PubMed] [Google Scholar]
- Chester C, de Carvalho M, Miltenberger G, Pereira S, Dillen L, van der Zee J, Van Broeckhoven C, de Mendonça A. 2013. Rapidly progressive frontotemporal dementia and bulbar amyotrophic lateral sclerosis in Portuguese patients with C9orf72 mutation. Amyotroph Lateral Scler Frontotemporal Degener 14:70–72. [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion A, Leblond CS, Couthouis J, Lu Y, Wang Q, Brian J, Ren Z, Keebler J, et al. 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clément J‐F, Meloche S, Servant MJ. 2008. The IKK‐related kinases: from innate immunity to oncogenesis. Cell Res 18:889–899. [DOI] [PubMed] [Google Scholar]
- Cruts M, Theuns J, Van Broeckhoven C. 2012. Locus‐specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33:1340–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, Van Langenhove T, van der Zee J, Van Broeckhoven C. 2013. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci 36:450–459. [DOI] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, et al. 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, Pinto S, Press R, et al. 2015. Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nat Neurosci 18:631–636. [DOI] [PubMed] [Google Scholar]
- Fujishiro H, Uchikado H, Arai T, Hasegawa M, Akiyama H, Yokota O, Tsuchiya K, Togo T, Iseki E, Hirayasu Y. 2009. Accumulation of phosphorylated TDP‐43 in brains of patients with argyrophilic grain disease. Acta Neuropathol 117:151–158. [DOI] [PubMed] [Google Scholar]
- Gelpi E, van der Zee J, Turon Estrada A, Van Broeckhoven C, Sanchez‐Valle R. 2014. TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol Appl Neurobiol 40:225–230. [DOI] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, et al. 2012. A C9orf72 promoter repeat expansion in a Flanders‐Belgian cohort with disorders of the frontotemporal lobar degeneration‐amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11:54–65. [DOI] [PubMed] [Google Scholar]
- Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Philtjens S, Heeman B, Engelborghs S, Vandenbulcke M, De Baets G, Bäumer V, Cuijt I, Van den Broeck M, et al. 2015. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology 85:2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno‐Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, et al. 2011. Classification of primary progressive aphasia and its variants. Neurology 76:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, et al. 2010. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard J‐P, Lacomblez L, Pochigaeva K, Salachas F, Pradat P‐F, Camu W, et al. 2008. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, et al. 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208. [DOI] [PubMed] [Google Scholar]
- Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, Van den Broeck M, Mattheijssens M, Peeters K, De Deyn PP, Cruts M, Van Broeckhoven C. 2010. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74:366–371. [DOI] [PubMed] [Google Scholar]
- Larabi A, Devos JM, Ng S‐L, Nanao MH, Round A, Maniatis T, Panne D. 2013. Crystal structure and mechanism of activation of TANK‐binding kinase 1. Cell Rep 3:734–746. [DOI] [PubMed] [Google Scholar]
- Lomen‐Hoerth C, Anderson T, Miller B. 2002. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59:1077–1079. [DOI] [PubMed] [Google Scholar]
- Mackenzie IRA, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DMA, Lee VMY. 2011. A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol 122:111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Mossevelde S, van der Zee J, Gijselinck I, Engelborghs S, Sieben A, Van Langenhove T, De Bleecker J, Baets J, Vandenbulcke M, Van Laere K, Ceyssens S, Van den Broeck M, et al. 2015. Clinical features of TBK1 carriers compared with C9orf72, GRN and non‐mutation carriers in a Belgian cohort. Brain 139:452–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, et al. 1998. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554. [DOI] [PubMed] [Google Scholar]
- Pham CT, de Silva R, Haïk S, Verny M, Sachet A, Forette B, Lees A, Hauw JJ, Duyckaerts C. 2011. Tau‐positive grains are constant in centenarians’ hippocampus. Neurobiol Aging 32:1296–1303. [DOI] [PubMed] [Google Scholar]
- Pilli M, Arko‐Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun JA, Hansen TE, et al. 2012. TBK‐1 promotes autophagy‐mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, Brown P, Ravenscroft T, van Blitterswijk M, Nicholson AM, DeTure M, Knopman DS, et al. 2015. Whole‐genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 130:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EGP, Onyike CU, Hillis AE, Josephs KA, et al. 2011. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón‐Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, et al. 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y, Yamanouchi H, Murayama S. 2004. Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Exp Neurol 63: 911–918. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, et al. 2008. TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. 2004. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet 36:377–381. [DOI] [PubMed] [Google Scholar]
- Williams KL, McCann EP, Fifita JA., Zhang K, Duncan EL, Leo PJ, Marshall M, Rowe DB, Nicholson GA., Blair IP. 2015. Novel TBK1 truncating mutation in a familial amyotrophic lateral sclerosis patient of Chinese origin. Neurobiol Aging 36:3334.e1–3334e.5. [DOI] [PubMed] [Google Scholar]
- van der Zee J, Gijselinck I, Dillen L, Van Langenhove T, Theuns J, Engelborghs S, Philtjens S, Vandenbulcke M, Sleegers K, Sieben A, Bäumer V, Maes G, et al. 2013. A pan‐European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 34:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee J, Van Langenhove T, Kovacs GG, Dillen L, Deschamps W, Engelborghs S, Matěj R, Vandenbulcke M, Sieben A, Dermaut B, Smets K, Van Damme P, et al. 2014. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol 128:397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supp. Figure S1. Western blot analysis of all identified single amino acid deletions and missense mutations overexpressed in HEK293T cells. TBK1 plasmids containing a missense mutation or a single‐amino acid deletion were transiently transfected in HEK293T cells. The p.Ser172Ala mutant (kinase dead, KD), generating a TBK1 protein lacking kinase activity and the Mock plasmid containing no TBK1, were used as negative controls. Protein lysates of the TBK1 overexpression cells were used for western blotting with an antibody against TBK1 (Abcam, 1:1000, 84 kDa) and GAPDH (Genetex, 1:10000, 37 kDa) as a reference protein. TBK1 signals of the mutant TBK1 plasmids were compared to wild type TBK1 (WT). p.Thr79del, p.Asp643del and p.Lys94Ser showed near 50% reduced protein expression, probably due to protein instability. All other TBK1 mutations were equally expressed as the wild type TBK1.