

Abstract
The synthesis of O‐doped polyaromatic hydro‐ carbons in which two polycyclic aromatic hydrocarbon sub units are bridged through one or two O atoms has been achieved. This includes high‐yield ring‐closure key steps that, depending on the reaction conditions, result in the formation of furanyl or pyranopyranyl linkages through intramolecular C−O bond formation. Comprehensive photophysical measurements in solution showed that these compounds have exceptionally high emission yields and tunable absorption properties throughout the UV/Vis spectral region. Electrochemical investigations showed that in all cases O annulation increases the electron‐donor capabilities by raising the HOMO energy level, whereas the LUMO energy level is less affected. Moreover, third‐order nonlinear optical (NLO) measurements on solutions or thin films containing the dyes showed very good values of the second hyperpolarizability. Importantly, poly(methyl methacrylate) films containing the pyranopyranyl derivatives exhibited weak linear absorption and NLO absorption compared to the nonlinearity and NLO refraction, respectively, and thus revealed them to be exceptional organic materials for photonic devices.
Keywords: annulation, chromophores, doping, nonlinear optics, polycyclic aromatic hydrocarbons
Introduction
Amongst the plethora of organic semiconductors available, polycyclic aromatic hydrocarbons (PAHs) have attracted increasing attention.1, 2, 3, 4, 5, 6 With respect to infinite graphene, PAHs show nonzero tunable bandgaps and are thus of use as chromophores in antennae7, 8, 9, 10, 11, 12 or emissive molecular architectures13, 14, 15, 16, 17, 18, 19 and in general in all optoelectronic applications requiring a tunable semiconducting material.6, 20 By exploiting organic synthetic tools,21, 22 one can tune the molecular HOMO–LUMO gap8 by 1) changing the size and edge of the carbon‐based aromatic framework; 2) varying the molecular planarity upon insertion of bulky substituents or bridging chains; 3) changing the aromatic properties of the constituent monomeric units; 4) varying the peripheral functionalization through the insertion of electron‐donating or electron‐ withdrawing substituents; 5) enclosing structural defects; 6) promoting supramolecular interactions between individual molecules governing their organization into a condensed phase, and 7) replacing selected carbon atoms by isostructural and isoelectronic analogues (i.e., doping). In particular, the heteroatom‐doping approach23, 24 is increasingly becoming important, as significant perturbation of the optoelectronic properties can be obtained without a substantial structural modification.25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36
Bottom‐up covalent synthesis can be exploited to access structurally defined heteroatom‐doped graphene fragments with precise control over the size, periphery, substitution pattern, doping ratio, and position.36, 37 In this respect, peri‐xanthenoxanthene (PXX),38, 39 the O‐doped analogue of anthanthrene, can be conceptualized as a building unit for engineering a new class of O‐doped PAHs. Substituted PXX derivatives are characterized by excellent carrier‐transport and injection properties, as well as easy processability, chemical inertness, and high thermal stability.40, 41 Due to these properties, PXX has shown exceptional performance when used as active organic semiconductor in transistors for rollable OLEDs.42, 43 In particular, it has been proven that the good performance is triggered by 1) the good charge‐transport properties and 2) the good thermal and chemical stability against parasitic oxidation occurring at the periphery of the π‐ conjugated system.41, 44
Capitalizing on these heterodoped structures, we have reported recently the synthesis of a series of O‐doped isosteres of benzorylenes in which peripheral carbon atoms are replaced by oxygen atoms at the armchair edges by exploiting the Pummerer‐modified CuI‐catalyzed ring‐closure reaction, which involves intramolecular C−O bond formation as the planarization reaction.45 With the aim of studying the effect of the π extension of the carbon framework, here we report on the preparation of π‐extended PXX derivatives through the fusion of two PAH subunits (Figure 1). In our approach, we considered extended PXX derivatives based on bis‐hydroxy PAHs with 2‐hydroxyperylene and 2‐hydroxynaphthalene moieties as the key constituent units. At the synthetic planning level, we contemplated the oxidative CuI‐catalyzed planarization reaction developed by us45 to form the pyranopyranyl motif (Figure 1). In this synthetic protocol, the easily prepared bis‐hydroxy PAH precursors46, 47, 48, 49, 50, 51, 52, 53, 54, 55 give us the opportunity to alternatively fuse the PAHs through furanyl linkages56 by an acid‐catalyzed cyclization strategy (Figure 1). Photophysical and electrochemical characterization showed that complementary spectroscopic and redox properties can be tailored through fine tuning of both the π extension of the carbon scaffold and the oxygen linkages (i.e., furanyl versus pyranopyranyl rings), with coverage of the visible absorption and emission spectral region. All derivatives exhibit high absorption and strong emission. Nonlinear optical (NLO) responses of all O‐doped polyaromatics were also investigated, both in solution and as thin films, by the Z‐scan technique with 35 ps, 532 nm (Vis) laser excitation. All showed large second hyperpolarizabilities, as predicted by theoretical calculations at the CAM‐B3LYP/6‐31+G** level of theory. To simplify the description of the different substitution patterns around the two fundamental furanyl and pyrano pyranyl cores, the nomenclature depicted in Figure 1 is used throughout this paper. As the fundamental cores of the two molecular families composed of binaphthofuran or PXX, each naphthyl sub‐ring can be differently π‐extended. In accordance with this general scheme, a labeling nomenclature is proposed in which n and m indicate the π extension of the carbon framework, expressed as the number of C−C‐fused naphthyl rings.
Figure 1.
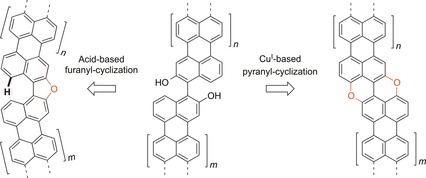
Fusion of PAHs through furanyl (left) and pyranopyranyl (right) cyclization strategies (n and m indexes indicate the π extension of the carbon framework expressed as a number of C−C fused naphthyl rings).
Results and Discussion
Synthesis
The synthesis commenced with the preparation of hydroxyl perylene 2 (Scheme 1). Friedel–Crafts alkylation of perylene by the protocol of Pillow et al.57 in the presence of a large excess of tBuCl58, 59, 60 leads to an inseparable mixture of di‐, tri‐, and tetrasubstituted tBu‐perylene derivatives. The unpurified perylene mixture was subsequently submitted to a selective C−H borylation reaction in the presence of 10 mol % of [{Ir(COD)(OMe)}2] (COD=1,5‐cyclooctadiene) catalyst and 20 mol % of 4,4′‐di‐tert‐butyl‐2,2′‐bipyridyl (dtbpy) and B2pin2 (pin=pinacolato) in n‐hexane at 80 °C for 24 h,61 which allowed the isolation of tetra‐tert‐butylperylene 1 a as well as mono‐ and bis‐boronic esters 1 b and 1 c (for X‐ray structure, see Supporting Information) in 33, 20 and 30 % yield, respectively. Oxidation of boronic ester 1 b with H2O2 and NaOH in THF at RT62 yielded perylenol derivative 2 in 80–85 % yield. Following the literature synthetic routes for preparing BINOLs,46, 47, 48, 49, 50, 51, 52, 54, 55, 63 we turned our attention toward Cu/TMEDA‐based (TMEDA=N,N,N′,N′‐tetramethylethylenediamine) oxidative C−C bond formation64, 65 as dimerization reaction. Thus, homodimerization of perylenol 2 was performed in the presence of [Cu(OH)Cl⋅TMEDA] catalyst under air at 20 °C in CH2Cl2 to give bis‐perylenol 4 in 71 % yield. Small transparent crystals of 4 were obtained by vapor diffusion of MeOH into a solution of 4 in CH2Br2. The asymmetric unit of the crystals contains one independent molecule, which is H‐bonded to two MeOH molecules (Figure 2). The molecular structure depicted in Figure 2 a reveals a nonplanar arrangement of the two perylene scaffolds with an interplanar angle of 62.8° and the two hydroxyl groups adopting a syn conformation. The crystal packing shows the formation of dimeric species by H‐bonding interactions bridged by solvent (MeOH) molecules. Similarly, perylenol 2 was cross‐coupled with 2‐naphthol to give naphthalenylperylene derivative 3 in 26 % yield (homodimers bis‐perylenol 4 and BINOL were also formed and thus separated by column chromatography). Bis‐hydroxy PAHs 3 and 4 were used as scalemic mixtures. Following the oxidative protocol recently developed by us,45 the dihydroxy species were subsequently cyclized to the relevant pyranopyran derivatives. Specifically, Cu‐catalyzed oxidative intramolecular etherification of dihydroxy derivatives 3 and 4 (CuI and PivOH in DMSO at 140 °C in air) afforded pyranopyran derivatives 5Pp and 6Pp in 57 and 84 % yield, respectively. The structures of all intermediates and products were unambiguously identified by HR‐MALDI MS through the detection of the peak corresponding to the molecular mass and by 1H and 13C NMR, UV/Vis, and IR spectroscopy (see Supporting Information). In particular, HR‐MALDI MS showed molecular peaks at m/z 574.2889 (C42H38O2, calcd: 574.2872) and 866.5059 (C64H66O2, calcd: 866.5063) for extended pyranopyrans 5Pp and 6Pp, respectively. Unfortunately, 13C NMR spectra could not be recorded, as both pyranopyranyl derivatives seem to exhibit limited solubility, likely triggered by their pronounced tendency to undergo strong aggregation.
Scheme 1.
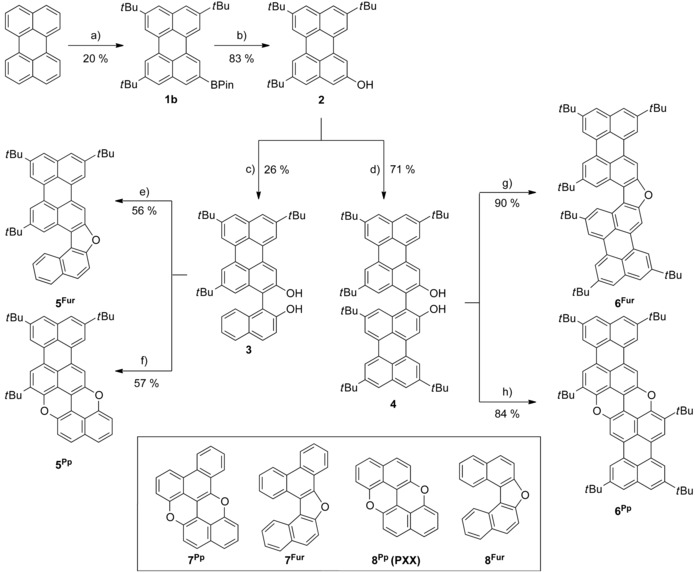
Synthetic pathways for compounds 1–6Fur/Pp. Reagents and conditions: a) 1. AlCl3, tBuCl, ODCB, 0 °C to RT, 24 h; 2. 10 mol % [{Ir(COD)(OMe)}2], 20 mol % dtbpy, B2Pin2, n‐hexane, 80 °C, 24 h; b) NaOH, H2O2 aq. 35 wt %, THF, RT, 2 h; c) 2‐Naphthol, [Cu(OH)(Cl)(TMEDA)], air, CH2Cl2, 20 °C, 2 h; d) [Cu(OH)(Cl)(TMEDA)], air, CH2Cl2, 20 °C, 1 h; e, g) p‐TsOH, toluene, reflux, Ar, 4 h; f, h) CuI, (CH3)3CCOOH, DMSO, 140 °C, 2 h.
Figure 2.
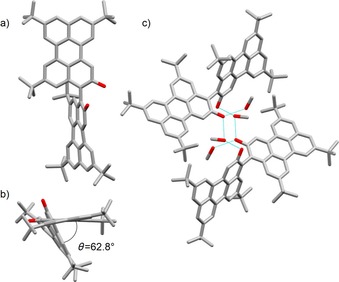
a) Top view, and b, c) side views of the crystal structure and packing of bis‐perynol derivative 4 (space group: P ; atom colors: red O, gray C; H omitted for clarity). Notably, the hydroxyl groups of the bis‐perynols are engaged in H‐bonding interactions through two bridging MeOH solvent molecules with formation of dimeric species in the solid state.
To qualitatively support this assumption, a 2 mm toluene solution of 6Pp was drop cast on a silicon wafer. After solvent evaporation, SEM imaging of the remaining powder showed the presence of microscale, brittle, sticklike morphologies (Figure S28, Supporting Information). Alternatively, when di hydroxy precursors 3 and 4 were treated with p‐TsOH in refluxing toluene solution,66, 67 extended furanyl derivatives 5Fur and 6Fur could be obtained in 56 and 90 % yield. Again, their structures were unambiguously identified by 1H and 13C NMR, UV/Vis, and IR spectroscopy, and the HR‐MALDI mass spectra showed peaks corresponding to the molecular mass at m/z 560.3079 (C42H40O, calcd: 560.3079) and 852.5268 (C64H68O, calcd: 852.5270) for extended furans 5Fur and 6Fur, respectively. Reference compounds 7Fur/Pp and 8Fur/Pp (Scheme 1) were also prepared by following similar synthetic strategies to those developed for compounds 5 and 6 (Scheme S1, Supporting Information). To further corroborate the chemical structure of the cyclized derivatives, crystals suitable for X‐ray diffraction analysis were obtained by vapor diffusion or slow solvent evaporation of solutions containing the relevant product (Figures 3, 4, 5; for the detailed crystallization procedures, see the Supporting Information). Despite many attempts, no suitable crystals were obtained for the pyranopyran derivatives, and only the structures obtained for the furanyl derivatives are discussed in this paper.
Figure 3.
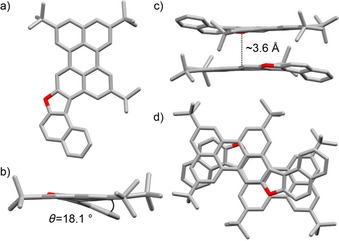
a, d) Top views, and b, c) side views of the crystal structure and π–π stacking arrangement of furanyl derivative 5Fur (space group: P21/c; atom colors: red O and grey C; H omitted for clarity). Crystals were obtained by slow vapor diffusion of MeOH to a C6D6 solution.
Figure 4.
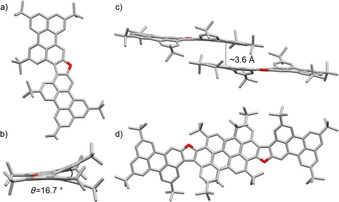
a, d) Top views, and b, c) side views of the crystal structure and π–π stacking arrangement of furanyl derivative 6Fur (space group: P2/c; atom colors: red O and gray C; H omitted for clarity). Crystals were obtained by slow evaporation of a C6D6/hexane solution.
Figure 5.
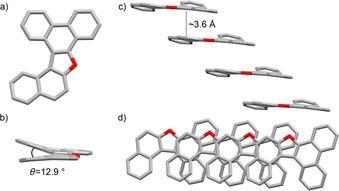
a, d) Top views, and b, c) side views of the crystal structure and π–π stacking arrangement of furanyl derivative 7Fur (space group: P212121; atom colors: red O and gray C; H omitted for clarity). Crystals were obtained by slow evaporation of a CD2Cl2 solution.
The X‐ray structures of 5Fur–7Fur all confirm the presence of the furanyl ring. Looking at their organization at the solid state (Figures 3, 4, 5 c, d), one can clearly evidence the presence of columnar arrangements in which the molecules are organized in π–π stacks, with a similar average interplanar spacing of about 3.6 Å for 5Fur, 6Fur, and 7Fur. As expected, the presence of the five‐membered ring induces a significant distortion of the PAH moieties due to C−H⋅⋅⋅H−C repulsive interactions (see also Figure 1), which results in interplanar angles of about 18.1, 16.7, and 12.9° for 5Fur, 6Fur, and 7Fur, respectively. Notably, the presence of the sterically bulky tBu groups in 5Fur and 6Fur further increases the interplanar angle. While furan 5Fur undergoes antiparallel stacking into a columnar arrangement with no lateral offset, 6Fur and 7Fur form offset solid‐state arrangements. In particular, bis‐perylene furan 6Fur organizes through perylene–perylene stacking with a lateral offset of 3.5(1) Å, whereas phenanthrenenaphthyl furan 7Fur undergoes offset π–π stacking involving the naphthalene and phenanthrene moieties. The thermal stability of compounds 4, 6Fur, and 6Pp was investigated by thermogravimetric analysis (TGA) and compared with that of reference compound 8Pp. While 8Pp starts to sublimate at around 188 °C and reaches its maximum at 265 °C (see also DTG profile in Figure S35a, Supporting Information), the TGA profiles of compounds 4, 6Fur, and 6Pp showed very high thermal stability under N2, with sublimation temperatures above 320, 340, and 400 °C, respectively (Figure S35 b–d, Supporting Information). The thermal stability of extended pyranopyran derivative 6Pp was also evaluated in air at a scan rate of 10 °C min−1. The TGA profile (Figure S36, Supporting Information) only exhibits a significant weight loss above 370 °C, which suggests that 6Pp can stand the critical environmental conditions typical of a fully operative device, namely, under O2 at high temperatures.
Absorption and emission spectroscopy
The effects of increasing the conjugation for both furanyl and pyranopyranyl derivatives were evaluated by steady‐state UV/Vis absorption and emission spectroscopy (Table 1). In general, all compounds show high molar absorption coefficients (up to 105 m −1 cm−1) spanning from the blue to the red region of the visible spectrum, with luminescence lifetimes consistent with singlet radiative deactivation (τ=2–6 ns). Quantum yields are remarkably high across the visible spectrum, with the pyranopyranyl derivatives showing lower emission quantum efficiencies (average Φ value of ≈0.5) with respect to the homo logous furanyl molecules (average Φ value of ≈0.8).
Table 1.
Optical properties for O‐doped PAHs in toluene solution.
| Compound | λ [nm],[a] ϵ [m −1 cm−1] | λ em [nm][b] | Φ | τ [ns] |
|---|---|---|---|---|
| 7Fur | 352, 26 900 | 371 | 0.43 | 5.7 |
| 7Pp | 446, 14 100 | 452 | 0.48 | 4.8 |
| 2 | 445, 25 600 | 463 | 0.55 | 4.9 |
| 3 | 452, 33 700 | 508 | 0.66 | 3.7 |
| 4 | 463, 65 000 | 472 | 0.88 | 2.7 |
| 5Fur | 477, 47 300 | 494 | 0.84 | 3.0 |
| 5Pp | 556, 36 300 | 569 | 0.50 | 3.8 |
| 6Fur | 534, 97 400 | 547 | 0.80 | 3.0 |
| 6Pp | 639, 66 400 | 649 | 0.52 | 2.2 |
[a] UV/Vis absorption maximum of the lowest‐energy band in toluene. [b] Emission maximum in toluene at 25 °C.
For singly bonded naphthol perylenol 3 and conjugated derivatives 5Fur and 5Pp, clear bathochromic shifts both in the absorption and in the emission spectra are evidenced on passing from the nonplanar to the planarized derivatives (Figure 6 a), with the relevant pyranopyranyl molecule displaying the larger redshifts compared to the furanyl analogue. The same trend is also observed for perylenol 2, its singly linked dimer 4, and compounds 6Fur and 6Pp, which show largely tunable absorption and emission energies that lie in the red spectral region for the O‐annulated derivatives (Figure 6 b). Comparing with reports in the literature on all‐carbon PAHs exhibiting comparable visible absorption energies,68, 69, 70, 71 the results of our steady‐state studies suggest that, although it provides significant bathochromic shifts, C−O planarization does not dramatically suppress the emission quantum yields, as opposed to the formation of the C−C bonds in planar rylenes, which usually show strong absorptivities but faint luminescence.72, 73
Figure 6.
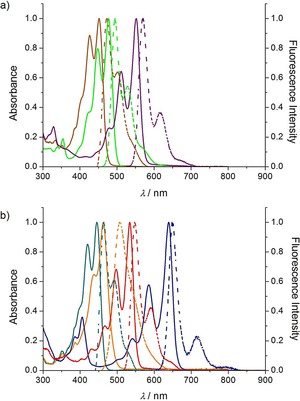
a) Normalized absorption (solid lines) and emission (dashed lines) spectra in toluene at 25 °C of: a) naphthol‐perylenol series: 3 (dark yellow), 5Fur (green), and 5Pp (purple), and b) bis‐perylenol series: 2 (cyan), 4 (orange), 6Fur (red), and 6Pp (blue).
To probe the solid‐state emissive properties of all conjugates,56 different molding solvents74 were screened to reproducibly obtain solid morphologies of defined shape. Furans 5Fur and 6Fur and pyranopyrans 5Pp and 6Pp were initially investigated. Among the different conditions, we found that slow addition of MeOH to THF solutions of the dye gives rise to cloudy colloidal solutions, which in time undergo precipitation leading to crystalline powdery solids. SEM images (Figure 7) of the dried powders show the formation of well‐defined and reproducible structures on the microscale with different morphologies depending on the chemical structure of the crystallizing molecules. Specifically, compound 5Fur leads to the formation of elongated hexagonal prisms (Figure 7 a and b), 1 μm wide with lengths of 5–10 μm. Needle‐like structures, longer than the average prisms, were also present in the sample. On the other hand, 5Pp forms 3–10 μm‐long sticklike morphologies with thinner diameters (Figure 7 c and d). Bis‐peryleno derivative 6Fur arranges in disklike structures with diameters in the micrometer range (Figure 7 e and f), while 6Pp forms rodlike structures (Figure 7 g and h), which also have microscopic dimensions (for more images, see Figure S29, Supporting Information).
Figure 7.

SEM images of the organic nanostructures obtained from THF solution on addition of MeOH for: a, b) 5Fur, c, d) 5Pp, e, f) 6Fur, and g, h) 6Pp. Scale bars: 1 μm (a–d, f, h); 2 μm (e, g).
The solid‐state emission spectra of compounds 5Fur, 6Fur and 7Fur are shown in Figure 8. Both model compounds 8Fur and 8Pp show appreciable luminescence on excitation of the powders at room temperature. While 8Fur shows blue emission with a structured spectrum featuring a maximum around 400 nm, pyranopyranyl derivative 8Pp is a yellow‐green emitter with a broad spectrum having a maximum around 545 nm (Figure S44, Supporting Information). Owing to the narrowing effect of the pyrano conjugation, the solid‐state emission spectrum of 8Pp shows a markedly redshifted profile compared to that of 8Fur. The extension of the π surface of 8Pp negatively affects the solid‐state emissive properties. Thus the luminescence outputs of solids obtained from compounds 5Pp and 6Pp are hardly detectable, whereas increasing the conjugation length of furanyl compounds 5Fur and 6Fur does not drama tically affect the solid‐state luminescence. Specifically, compound 5Fur intensely emits a yellow‐orange color and shows a structured emission spectrum with a maximum around 560 nm, whereas 6Fur strongly luminesces and exhibits vibrational structure and a sharp peak at 645 nm. Although the emission spectra of the amorphous powders of 5Fur, 6Fur, and 7Fur were to some extent sensitive to prolonged UV excitation, and we cannot therefore determine precisely the value of each luminescence quantum yield, these solid‐state emission findings are in agreement with literature data reporting high quantum yield for the naphthofuran derivatives.56, 67 Differences in the emissive properties between the naphthofurans and pyranopyranyl derivatives in the solid are probably correlated with the structural properties of the respective crystal lattices (see above). For instance, considering that 8Pp (PXX) is self‐ organized at the solid‐state through π–π stacking,44 we can hypothesize that all pyranopyranyl derivatives likely form similar face‐to‐face arrangements, which favor nonradiative decay of the emissive excited states.15, 75, 76
Figure 8.
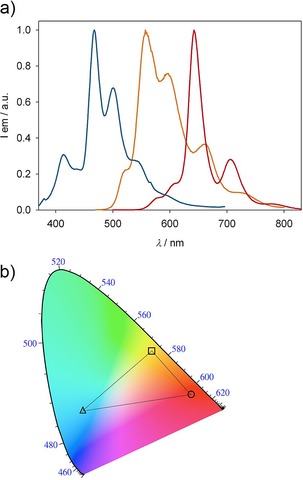
a) Emission spectra of 7Fur (blue line), 5Fur (orange line), and 6Fur (red line) in solid samples at RT. Excitation wavelength: 360 nm. b) Calculated CIE diagram for solid‐state emissions of 7Fur (triangle), 5Fur (square), and 6Fur (circle). Dotted lines are plotted to display the color gamut accessible by combination of the three solid emitters.
Electrochemical investigations
Cyclic voltammetry (CV) in 1,2‐dichlorobenzene (ODCB) was used to get further information about the redox properties of compounds 5Fur/Pp–8Fur/Pp. Their redox potentials versus ferro‐ cenium /ferrocene (Fc+/Fc) are summarized in Table 2.
Table 2.
Half‐wave potentials calculated versus the Fc+/Fc couple.[a]
| Compound | [V] | [V] | [V] | [V] |
|---|---|---|---|---|
| 8Fur | 0.89 (117) | n.d. | n.d. | n.d. |
| 8Pp (PXX) | 0.30 (111) | n.d. | n.d. | n.d. |
| 7Fur | 0.84 (100) | n.d. | n.d. | n.d. |
| 7Pp | 0.25 (81) | n.d. | n.d. | n.d. |
| 4 b | 0.30 (133) | 0.52 (128) | −2.37[b] (irr) | −2.50[b] (irr) |
| 5Fur | 0.37 (128) | 0.90 (140) | −2.20 (90) | n.d. |
| 5Pp | 0.00 (133) | 0.56 (122) | −2.25 (101) | n.d. |
| 6Fur | 0.20 (127) | 0.57 (127) | −2.15 (112) | −2.32 (123) |
| 6Pp | −0.15 (100) | 0.32 (107) | −2.16 (100) | −2.26 (120) |
[a] Half‐wave potentials were calculated as E 1/2=(E pa+E pc)/2 by considering anodic (E pa) and cathodic (E pc) peak potentials, unless otherwise specified. Values in parentheses are referred to the peak separation (E pa−E pc [mV]) of each reversible process; n.d.: not detected; irr: irreversible. [b] Determined by considering the cathodic peak of an irreversible reduction process.
As depicted in Figures 9 a and b, reference compounds 7Fur, 8Fur, and 7Pp, 8Pp each show only a 1e reversible redox couple, which is at similar positive potentials for the furanyl (0.89 and 0.84 V for 8Fur and 7Fur) and pyranopyranyl derivatives (0.30 and 0.25 V for 8Pp and 7Pp), and suggests that addition of an extra benzo[a] ring in the molecular structure marginally affects the oxidation potential of the fundamental O‐annulated bis‐naphthyl derivatives. Notably, no reductions were observed in the electrochemical window of investigation in ODCB. Interestingly, direct comparison of voltammetric behaviors of compounds 8Fur and 8Pp evidences the strong electron‐donor character of the pyranopyranyl moiety compared to that of the furanyl ring, with a significant lowering of the value of about 0.60 V. In the naphthofuran family, expansion of the carbon‐based π surface, as in 5Fur (m=0, n=1) and 6Fur (m=1, n=1) dramatically affects the voltammetric behavior compared to that of the reference compounds (Figure 10 a and b). Specifically, two one‐electron, reversible oxidation peaks for 5Fur appear at 0.37 and 0.90 V as a consequence of the Coulombic interactions between the first and second oxidation holes. Curiously, a first 1e reduction wave also appears as a separated, reversible couple at −2.20 V. As expected, with respect to 8Fur, the lateral extension of the π surface through the introduction of a naphthyl unit (m=0, n=1) makes 5Fur a stronger electron donor. This trend is further evidenced by 6Fur, featuring symmetrical substitution with two perylenyl units (m=n=1), which exhibits even lower oxidation potentials (0.20 and 0.57 V for the first and the second wave, respectively); at the same time, two reversible reductions are recorded at similar potentials to those observed for reducing compound 5Fur.
Figure 9.
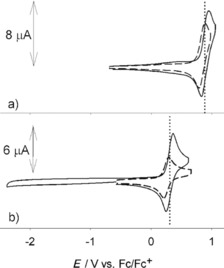
CV of: a) 8Fur (0.80 mm, solid line) and 7Fur (0.75 mm, dashed line). b) 8Pp (PXX) (0.81 mm, solid line) and 7Pp (0.43 mm, dashed line). Half‐wave oxidation potentials of 8Fur and 8Pp (PXX) are indicated by vertical dotted lines in a) and b), respectively, for comparison purposes. Scan rate: 50 mV s−1. Supporting electrolyte: TBAPF6. Ferrocene was used as internal reference standard.
Figure 10.
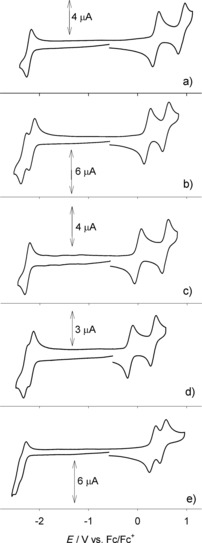
CV of: a) 5Fur (0.64 mm), b) 6Fur (0.63 mm), c) 5Pp (0.61 mm), d) 6Pp (0.63 mm), and e) 4 b (0.53 mm). Scan rate: 50 mV s−1. Supporting electrolyte: TBAPF6. Ferrocene was used as internal reference standard.
As expected, in the case of the PXX‐derivatives, the trend is very similar to that described for the naphthofuran family: compound 5Pp (m=0, n=1) shows lower oxidation potentials (at ca. 0 V vs. Fc+/Fc) compared to model compound 8Pp, while a one‐electron reduction process appears at −2.25 V (Figure 10 c). The symmetrical substitution on both sides of the PXX core by two perylenyl units (m=n=1) makes 6Pp electron‐richer and easier to oxidize than 8Pp, and shifts the first oxidation potential to −0.15 V (Figure 10 d). To gain insight into the electronic role of oxygen atoms in the conjugated system of the PXX‐based derivatives, CV of dimethoxy biperylene 4 b (see Supporting Information) was also performed (Figure 10 e). Two reversible oxidation processes are detected at 0.30 and 0.52 V, reflecting the weaker electron‐donating nature of this structure compared to that featuring the two oxygen atoms in the π‐conjugated pyranopyranyl motif, as in the case of 6Pp. Furthermore, the inclusion of the two oxygen atoms in the aromatic system affects significantly the HOMO–LUMO energy gap (Table 3), which was experimentally estimated to be 2.67 and 2.01 eV for 4 b and 6Pp, respectively.
Table 3.
Estimated HOMO–LUMO energy gaps E g for compounds 5Fur/Pp–8Fur/Pp, as determined from the optical (E 00),[a] electrochemical ( ), and theoretical ( )[b] studies.
| Compound | E 00 [nm, eV] | [eV] | [eV] | [eV] | [eV] | E 00 [nm, eV] | Compound | |
|---|---|---|---|---|---|---|---|---|
| 8Fur | 361, 3.43 | – | 3.95 |
|
3.32 | – | 449, 2.76 | 8Pp |
| 7Fur | 371, 3.34 | – | – | – | – | 452, 2.74 | 7Pp | |
| 5Fur | 494, 2.51 | 2.57 | 2.83 | 2.50 | 2.25 | 569, 2.18 | 5Pp | |
| 6Fur | 547, 2.27 | 2.35 | 2.48 | 2.20 | 2.01 | 649, 1.91 | 6Pp |
[a] Calculated from the wavelength of the emission maximum in toluene. [b] Calculated bandgap value at the B3LYP/6‐31G** gas‐phase optimized geometry.
From these data it becomes apparent that the lateral π extension of the PAH substructures with a naphthyl unit accounts for a decrease of the value of approximately 0.30 V (cf. the furanyl (left) and pyranopyranyl (right) families in Table 3). Similarly, O cyclization of the bis‐hydroxy PAHs strongly affects the value, with systematic decreases of about 0.30 and 0.6 V for the furanyl and pyranopyryl ring, respectively (cf. the furanyl and pyranopyranyl analogues in Table 3 and Figure 11). Notably, excellent accord between the electrochemical and optical E g values is clearly observable, whereby the latter was calculated from λ max of the lowest‐energy electronic transition. The rainbow collection in Figure 11 perfectly illustrates how the HOMO–LUMO gaps decrease, mainly because of the increase of the HOMO energy levels as a consequence of the progressive π extension and the inclusion of donating furanyl and pyranopyranyl cores. By playing with the type of O‐containing ring and the number of fused naphthyl rings one can cover the primary colors, moving from yellow furanyl 5Fur to orange bis‐perylenyl furan 6Fur to pink 5Pp and blue 6Pp pyranopyranyl derivatives. Compared to the tunable core‐substituted naphthalenediimides (cNDIs),77, 78 color tailoring of which is achieved by a combination of electron‐donating substituents in the core leading to push–pull chromophores with the electron‐withdrawing imide groups,79 the O‐doped PAHs investigated in this work can be considered to be valid alternatives for applications in which electron‐rich (i.e., high HOMO energy levels) and strongly emissive chromophores are required.80, 81, 82
Figure 11.
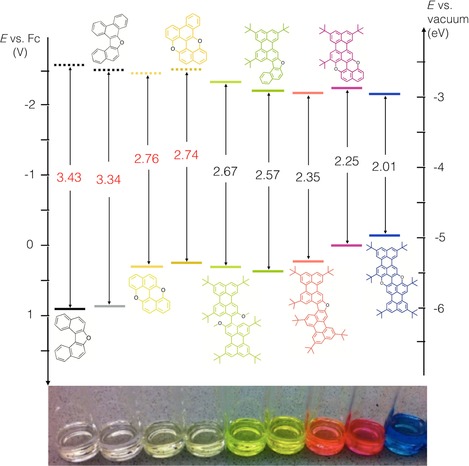
Frontier orbital energies for compounds analyzed by CV in 1,2‐dichlorobenzene (dashed lines corresponding to energies calculated by using the optical E 00 value in red). The formal potential of the Fc+/Fc redox couple, taken as a reference, is assumed to be −5.1 eV versus vacuum.
Theoretical modeling
To shed further light on the electronic structure and optical properties of the O‐annulated derivatives, the electronic properties of the HOMO and LUMO levels were calculated by means of DFT calculations with the Gaussian 09 package83 (Tables 3 and 4). As the tBu substituents have a small effect on E HOMO and E LUMO, some of the calculations were performed without the alkyl substituents. Each molecule was modeled in its neutral state by performing a geometry optimization and a single‐point calculation with the restricted Becke three‐ parameter exchange functional85 and the Lee–Yang–Parr correlation functional84 (B3LYP/6‐31G** level of theory). The crystal structures, when available, were considered as starting geometries. The HOMOs and LUMOs were plotted with the Avogadro software.86 Other orbitals up to HOMO−4 and LUMO+4 were also calculated (Figures S49–56, Supporting Information). The molecular HOMOs and LUMOs are located on the entire π surface of the molecules, with the oxygen atoms contributing to the given orbitals differently, depending on the nature of the cyclic linkage. In particular, for the furanyl derivatives the O atom does not significantly contribute to the HOMO, whereas a significant involvement of the O atoms in the HOMO of the pyranopyranyl derivatives is clearly observ able (Figure 12). On the contrary, a non‐negligible contribution of the O atoms to the LUMOs is noticeable for both O‐annulated derivatives. As observed with the electrochemical characterization, O cyclization greatly affects E HOMO, with the pyrano pyranyl ring inducing the greatest enhancement of the E HOMO values and thus having the greatest impact on both optical and electrochemical E g values. On the other hand, π extension of the all‐carbon aromatic units (m=0, n=1 and m=n=1) affects both E HOMO and E LUMO, and ultimately triggers a decrease of the E g value, as typically observed for π‐extended PAHs.87, 88, 89 Notably, the solvent effect (toluene) on the wavelength λ for the first allowed electronic transition was also considered in the simulation (Table 4).
Table 4.
Computed E HOMO,[a] E LUMO,[b] E HOMO−E LUMO ( ), absorption wavelength λ [nm], and excitation energy E exc [eV] for the first allowed electronic transitions. The values were calculated with the CAM‐B3LYP/6–31+G**method with explicit solvation (toluene).
| Molecule | E HOMO, E LUMO [eV] | [eV] | λ [nm] | E exc [eV] | Type of excitation |
|---|---|---|---|---|---|
| 8Fur | −5.36, −1.42 | 3.95 | 323.09 | 3.84 | HOMO→LUMO (92 %) |
| 5Fur | −4.87, −2.07 | 2.80 | 446.22 | 2.78 | HOMO→LUMO (96.9 %) |
| −4.73,[b] −1.90[b] | 2.83 | 444.87[b] | 2.79 | ||
| 6Fur | −4.71, −2.23 | 2.48 | 496.40 | 2.50 | HOMO→LUMO (90.3 %) |
| −4.54,[b] −2.07[b] | 2.48 | 494.02[b] | 2.51[a] | ||
| 8Pp | −4.71, −1.39 | 3.32 | 394.9 | 3.14 | HOMO→LUMO (95 %) |
| 5Pp | −4.49, −2.01 | 2.48 | 502.67 | 2.47 | HOMO→LUMO (95.8 %) |
| −4.38,[b] −1.88[b] | 2.50 | 499.96[b] | 2.48[a] | ||
| 6Pp | −4.38, −2.20 | 2.18 | 565.23 | 2.19 | HOMO→LUMO (93.0 %) |
| −4.25,[b] −2.04[b] | 2.20[b] | 566.72[b] | 2.19[a] | ||
| 4 | −4.71,[b] −1.96[b] | 2.75[b] | 438.05[b] | 2.83 | HOMO→LUMO (50 %) |
| HOMO→LUMO+1 (20 %) |
[a] B3LYP/6‐31G**. [b] with tBu group.
Figure 12.
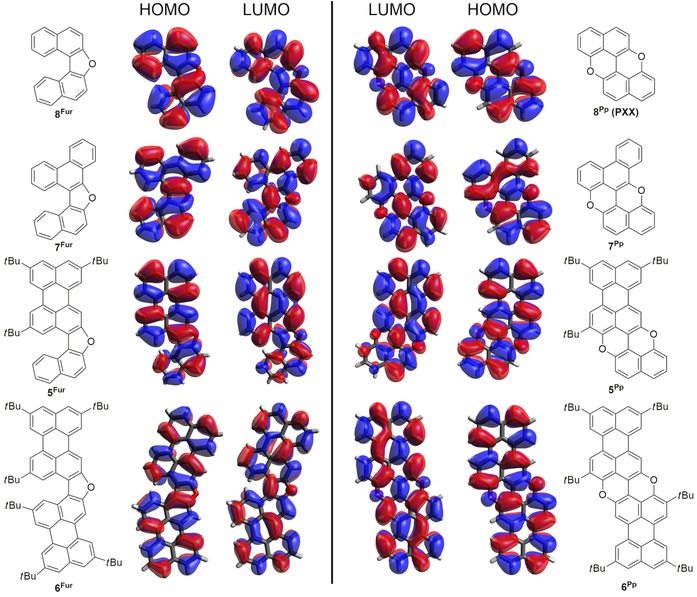
Molecular orbitals for the furanyl (left) and pyranopyranyl (right) derivatives calculated at the B3LYP/6‐31G** level of theory.
For both furanyl and pyranopyranyl derivatives, λ increases with n, and the absorption of the pyranopyranyl derivatives is more red‐shifted with respect to the furanyl analogues (λ Pp>λ Fur). For 8Pp a maximum is observed at 400 nm, which lies close to the absorption (λ=394.90 nm), whereas the maximum of 8Fur is located at λ≈330 nm, and is also close to the absorption (λ=323.09 nm). Figure S65 (Supporting Information) shows first allowed electronic transitions computed with the aid of natural transition orbital pairs90 for compounds 8Fur, 8Pp, 5Fur, 5Pp, 6Fur, and 6Pp. All electronic transitions occur through π→π* excitations, and the lowest‐energy absorption band is assigned to the HOMO→LUMO transition (see Supporting Information for the simulated UV/Vis spectra and the computed bands of the allowed electronic transitions for 8Fur and 8Pp).
NLO studies
The NLO properties of 4, 5Fur/Pp, 6Fur/Pp, and 8Fur/Pp were investigated by the Z‐scan technique, by employing visible (532 nm), 35 ps laser pulses from a mode‐locked Nd:YAG laser. By performing measurements on solutions at different concentrations under various incident laser excitation energies, the nonlinear absorption coefficient β and the nonlinear refractive parameter γ′ were determined (Supporting Information, Table S2; for more details on the dependence between β, γ′, and the third‐order nonlinear susceptibility χ (3), see Supporting Information). Most of the O‐doped PAHs exhibit negligible NLO absorption and significant NLO refraction. Specifically, only 8Pp and 5Pp, which exhibit significant NLO absorption, show reverse saturable absorption (RSA, β>0) and saturable absorption (SA, β<0) behaviors, respectively (Figure 13). As RSA materials show lower transmission with increasing incident laser intensity (i.e., the material becomes less transparent) and SA materials become progressively more transparent at higher incident laser intensities, both types of NLO absorption behaviors are of great interest for a variety of photonic and opto electronic applications (e.g., optical limiters, saturable absorbers). Concerning the NLO refraction, toluene solutions of most of the O‐annulated PAHs exhibited self‐defocusing (i.e., negative sign of the nonlinear refractive‐index parameter γ′, see Table 5), with the exception of pyranyl derivatives 5Pp and 6Pp, which exhibited self‐focusing behavior (i.e., positive γ′ values, see Figure S57, Supporting Information). To better rationalize these findings, the UV/Vis/NIR absorption spectra of toluene solutions containing the relevant O‐annulated PAHs were compared to those obtained from calculations (Figures S60 and S61, Supporting Information). Thus, from the comparison between the energy of the lower‐energy electronic transitions of the different O‐doped PAHs studied here and that of the laser excitation photons, it becomes evident that different degrees of resonant enhancement are expected to occur. In particular, going from 8Fur to 5Fur and then to 6Fur, the lower‐energy electronic transitions shift to longer wavelengths, get closer to the laser excitation wavelength, and result in more efficient resonance enhancement, as shown in Table S2 of the Supporting Information. Specifically, the lowest‐energy electronic transition of 8Fur, which occurs at 357 nm, shifts to 477 nm in the case of 5Fur and to 534 nm in the case of 6Fur. Similar observations have been made for the pyranyl counterparts as well. These energy shifts of the lower‐energy electronic transition give rise to significant resonant enhancement of the NLO response, in line with the general rule according to which the closer the lowest‐energy electronic transition to the energy of the laser excitation photon (532 nm), the higher the degree of resonant enhancement of the NLO response. Therefore, considering the lowest‐energy transitions centered at 357, 477, and 534 nm for 8Fur, 5Fur, and 6Fur, respectively, the excitation of furanyl derivative 6Fur gives rise to the largest NLO response. Furthermore, as the sign of the NLO refraction depends on the relative position of the excitation wavelength with respect to the molecular absorption band,91, 92, 93 one can obtain NLO refractions with opposite signs. In particular, whereas for 5Fur excitation occurs at a longer wavelength compared to its lowest‐energy absorption band, compounds 5Fur and 5Pp exhibit opposite NLO refractions. Along this line, compounds 8Fur, 8Pp, and 4 were also found to exhibit negative NLO refractions, as the excitations take place at longer wavelengths than the lowest‐energy absorption bands of the relevant colorant.
Figure 13.
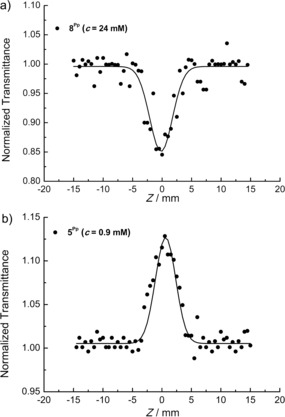
Open‐aperture Z‐scans of solutions containing: a) 8Pp, and b) 5Pp measured with 35 ps, 532 nm laser excitation.
Table 5.
Second hyperpolarizability γ values of O‐doped PAHs dissolved in toluene, determined with 35 ps, 532 nm laser excitation.
| Compound (λ max)[a] | Reγ [10−31 esu] | Imγ [10−31 esu] | γ [10−31 esu] | γ [10−31 esu] | Imγ [10−31 esu] | Reγ [10−31 esu] | Compound (λ max)[a] | |
|---|---|---|---|---|---|---|---|---|
| 8Fur (357 nm) | −0.0015±0.0003 | – | 0.0015±0.0003 |
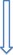
|
0.013±0.002 | 0.0058±0.002 | −0.012±0.002 | 8Pp (444 nm) |
| 5Fur (477 nm) | −0.11±0.05 | – | 0.11±0.05 | 0.14±0.04 | −0.099±0.003 | 0.094±0.001 | 5Pp (556 nm) | |
| 6Fur (534 nm) | −0.14±0.05 | – | 0.14±0.05 | 1.34±0.20 | – | 1.34±0.20 | 6Pp (639 nm) | |
| 4 (463 nm) | −0.025±0.005 | – | 0.025±0.005 |
[a] Wavelength of the lowest‐energy electronic transition in toluene.
The third‐order susceptibility χ (3) and the second hyperpolarizability γ were then deduced (see Table 5 and Table S2, Supporting Information), and a trend within each family of O‐annulated compounds was identified. Specifically, starting from model compounds 8Fur and 8Pp, we can observe an increase of the γ values by two orders of magnitude on extending the molecular π surface, that is, for compounds 5Fur/Pp and 6Fur/Pp. To shed further light on this NLO response, we calculated the electronic static (i.e., when the excitation frequency of the incoming laser beam tends to zero, with ω→0) and the frequency‐dependent (i.e., ω≠0) second hyperpolarizabilities (Table 6), denoted γ(0;0,0,0) and γ(−ω;ω,−ω,ω) respectively. As shown in Table 6, γ(0;0,0,0) and γ(−ω;ω,−ω,ω) of the pyranopyranyl derivatives are larger than those of the furanyl compounds. This is in full agreement with the experimental observations. An exception to this response was found for γ(0;0,0,0) of 8Pp and 8Fur, for which an opposite behavior was observed, namely, γ Fur(0;0,0,0)>γ Pp(0;0,0,0). As shown in Figure 14 a, in the spectral region in which the excitation occurs (532 nm), the theoretical γ(−ω;ω,−ω,ω) value for pyranopyranyl derivative 8Pp is significantly larger than that of its furanyl analogue 8Fur, again in agreement with the experimental findings (see above); furthermore, the large increase of the γ(−ω;ω,−ω,ω) value for pyranopyranyl compound 8Pp at about 250 nm is associated with its significant light absorption in the given spectral region. Similar results were found for 5Pp, which shows a clear enhancement of the γyyyy(−ω;ω,−ω,ω) value at ca. 500 nm (see Figure 14 b). Corroborating the experimental findings, theoretical calculations indicate that γ(0;0,0,0) and γ(−ω;ω,−ω,ω) are strongly affected by the type of O annulation, with the pyranyl derivatives showing the larger values. Finally, the NLO response of the O‐doped PAHs studied herein was found to be of comparable magnitude to those of some other organic conjugated materials that were reported recently and are also promising for photonic and optoelectronic applications.98, 99
Table 6.
Average values of the static [γ(0)] and dynamic second hyperpolarizabilities [γ(−ω;ω,−ω,ω)] of pyranopyranyl and furanyl derivatives. The reported data were calculated at the B3LYP/6‐31G**gas‐phase optimized geometry, by using the CAM‐B3LYP/6‐31+G** method.
| Property | 8Fur | 8Pp | 5Fur [b] | 5Pp [b] | 6Fur [b] | 6Pp [b] | 4 [b] |
|---|---|---|---|---|---|---|---|
| ρ=γ sol/γ gas [d] | 1.68 | 1.72 | 1.94 | 2.07 | 2.09 | 2.18 | 1.74 |
| γ(0;0,0,0) | 104 | 96.4 | 457.5 | 476.6 | 1712 | 1842 | 627 |
| [103 a.u.] | 175[d] | 166[d] | 889[d] | 989[d] | 3578[e] | 4010[e] | 1094[d] |
| γ(−ω;ω,−ω,ω | 140 | 2010 | −4264 | −15 770 | −11 6933 | NC[f] | 9110 |
| [103 a.u.][a] | 235[e] | 3457[e] | 8272[e] | −32 640[e] | 24 5559[e] | – | 15 860[e] |
| γ(−ω;ω,−ω,ω) | 0.00145±0.00030[g] | 0.0133±0.0020[g] | 0.113±0.050[g] | 0.14±0.04[i] | 0.138±0.050[g] | 1.335±0.200[g] | 0.025±0.005[g] |
| [10−31 esu] | 0.0012[e,h] | 0.017[e,h] | 0.04[e,h] | −0.16[e,h] | −1.2[e,h] | – | 0.079[e,h] |
[a] Frequency‐dependent value, λ=532 nm. [b] The tBu groups were substituted by H. [c] γ sol: second hyperpolarizability computed in the presence of the solvent (toluene); γ gas: second hyperpolarizability computed in the gas phase. [d] Value computed in the presence of toluene. [e] Computed by multiplying the gas‐phase value by the scaling factor ρ, in order to get an estimation of the property in solution. [f] Non‐convergence. [g] The experimental value was measured by using the Z‐scan technique, with 35 ps laser excitation at 532 nm (solvent: toluene). [h] The computed value was converted to esu by using the conversion factor 1 a.u.=5.0367×10−40 esu.
Figure 14.
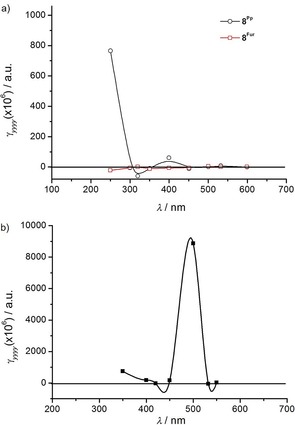
Calculated wavelength‐dependent γyyyy(−ω;ω,−ω,ω) profile for: a) 8Pp and 8Fur, and b) 5Pp. The values were computed with the CAM‐B3LYP/6‐31+G** method in the gas phase.
To study the effect of the dielectric environment (solvent effect) on the NLO response, we also computed the ρ value, defined as the ratio between the static second hyperpolarizability values in solution γ(0;0,0,0)sol and in the gas phase γ(0;0,0,0)gas (Table 6). As shown in Figure 15 a, the ρ value increases with the extension of the molecular π surface; also, larger ρ values are found for the planar pyranopyranyl derivatives (ρ Pp>ρ Fur). These data suggest that the more extended the π surface and the planarity of the molecule, the greater the effect of the solvent on the NLO response. Moreover, in Figure 15 b the dependence between γ(0;0,0,0) and for 5Fur/Pp, 6Fur/Pp, and 8Fur/Pp is depicted. Clearly, a decrease of leads to an increase of γ(0;0,0,0), which suggests that for these molecular scaffolds the tuning of the HOMO and LUMO energy levels greatly affects their NLO response. Note that the difference of one order of magnitude between the NLO responses of 8Fur and 8Pp can be attributed to the change in the degree of molecular planarity, as their lower absorption bands lie at 357 and 444 nm, respectively, well below the excitation wavelength of 532 nm.
Figure 15.
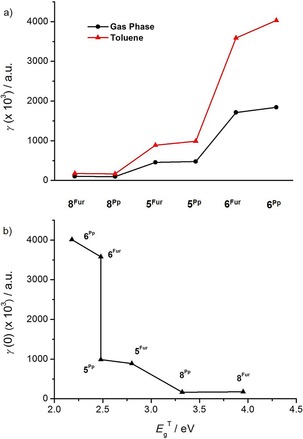
a) The average value of the static second‐hyperpolarizability of the studied compounds computed in the gas phase and in toluene solution. b) Variation of the static average second hyperpolarizability γ(0;0,0,0) for the O‐annulated PAHs with . The CAM‐B3LYP/6‐31+G** method was used in the presence of toluene as solvent.
NLO responses in thin films: towards devices
Given the promising NLO results obtained in solution, thin films of poly(methyl methacrylate) (PMMA) containing the relevant dyes were prepared by spin coating (thickness of 350–550 nm, as measured by a Dektak XT stylus profilometer). The results are gathered in Table 7 (for details, see Supporting Information).94, 95 As thin films have a different dielectric environment compared to a liquid solution, different behaviors of NLO responses are observed.96 The Z‐scan measurements were performed with 532 nm, 35 ps laser pulses. The UV/Vis/NIR optical absorption spectra of the prepared thin films are shown in Figures S62 and S63 of the Supporting Information. While the absorption spectra of the thin films containing the furanyl derivatives were found to closely match those recorded from toluene solutions, the absorption profiles of the films containing the pyranopyranyl derivatives (with the exception of those containing 8Pp) exhibited significant broadening of the main absorption bands, which suggests non‐negligible aggregation of the molecules in PMMA. Concerning the general properties of the thin films, it is important to note that the NLO responses of all films are exclusively dominated by NLO refraction (for the range of incident laser intensities used, i.e., up to 30–35 GW cm−2). No evidence of nonlinear absorption was found for laser intensities as high as the damage threshold of the films. These results are very promising for practical applications, as they show high damage thresholds, while the absence of absorption reduces significantly the drawbacks of any thermal effects.97 As for the magnitude of the NLO refraction (i.e., the real part of the third‐order susceptibility Reχ (3)), thin films containing furanyl derivatives 8Fur and 5Fur show very similar values to those obtained from the corresponding toluene solutions (i.e., ca. 10−13 esu). On the other hand, the values of pyranyl derivatives 5Pp, 6Pp, and 8Pp are lower, most probably because of aggregation (see broadening of the UV/Vis absorption profile for the pyranyl molecules in PMMA in Figures S62 and S63, Supporting Information). To assess the exploitability and suitability of the thin‐film NLO response for engineering optoelectronic devices, two figures of merit are usually considered: T and W. These parameters are defined as follows [Eq. (1)]:94, 96
| (1) |
Table 7.
Thickness (L), nonlinear refractive index parameters γ′ and Reχ (3) and figure of merit W of PMMA thin films containing the relevant O‐doped PAHs on glass.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | L [nm] | γ′ [10−18 m2 W−1] | Reχ (3) [10−13 esu] | W | W | Reχ (3) [10−13 esu] | γ′ [10−18 m2 W−1] | L [nm] | Compound |
| 8Fur | 367 | −0.43±0.02 | −0.62±0.03 | 340 | 390 | −0.37±0.,01 | −0.26±0.01 | 361 | 8Pp |
| 5Fur | 377 | −1.12±0.02 | −1.59±0.03 | 200 | 20 | −0.94±0.04 | −0.66±0.03 | 391 | 5Pp |
| 6Fur | 380 | – | – | – | 10 | −0.70±0.03 | −0.49±0.02 | 383 | 6Pp |
where β is the nonlinear absorption coefficient, λ the excitation wavelength, γ′ the nonlinear refractive parameter, Δn=γ′I the induced index change, and α the linear absorption coefficient [cm−1]. The first parameter (T<1) infers that the NLO absorption must be weak compared to the NLO refraction, while the second (W>1) suggests that the linear absorption must be relatively weak compared to the nonlinearity. In the present case, the T value is always <1, and thus fulfils the necessary requirements of a negligible NLO absorption of the films (i.e., β≈0). High W values ranging between about 390 and 10 are estimated for the less π‐extended derivatives 8Fur/Pp and most π‐extended 6Pp, respectively, assuming an intermediate incident laser from those employed. These W values are very high and thus very encouraging to further exploit thin films containing pyranopyranyl derivatives in waveguiding devices and optical couplers.94, 95, 96
Conclusions
We have described the synthesis of O‐doped polyaromatic hydrocarbons in which two polycyclic aromatic hydrocarbon substructures are fused through furanyl or pyranopyranyl rings. Starting from bis‐hydroxy PAHs, acid‐ and Cu‐catalyzed O‐annulation reactions allowed the planarization of the molecules through the formation of furanyl or pyranopyranyl rings. Comprehensive photophysical measurements in solution showed that these compounds have high emission yields (Φ=0.5–0.9) and tunable absorption properties throughout the UV/Vis spectral region. Complementary solid‐state photophysical studies on the dyes organized in microscopic morphologies showed that only those prepared from the furanyl derivatives retain the emissive molecular properties. Electrochemical investigations showed that in all cases O annulation increases the electron‐donor capabilities by raising the HOMO energy level, whereas the LUMO energy level is less affected. This ultimately causes shrinking of HOMO–LUMO energy gaps, whereby pyranopyranyl planarization triggers narrower gaps and thus the lowest‐energy emissive species. Finally, third‐order NLO measurements on solutions containing the relevant dyes showed significant second hyperpolarizability, the extent of which depends on 1) the molecular planarity and 2) the HOMO–LUMO energy gap. Theoretical computation of the optoelectronic properties performed with the CAM‐B3LYP/6‐31+G** method provided reliable data for predicting the excitation spectra, energy gaps, and second hyperpolarizability values, whereby the pyranopyranyl derivatives show the larger second hyperpolarizability values. In this respect, PMMA films containing the pyranopyranyl derivatives showed weak linear absorption and NLO absorption compared to the nonlinearity and NLO refraction, respectively, and thus are prime materials for engineering photonic devices, such as waveguiding and optical couplers or fluorescent probes in lipid bilayer membranes.100 From these results, the potential to build a broad variety of new colorful molecules is apparent. Thus, further investigations will be now centered on studying different heteroatoms, for example, other chalcogens such S, Se, and Te, the polarizability of which is expected to further affect the NLO response and finely tune the HOMO–LUMO gap.
Experimental Section
Full experimental details and characterization data, spectroscopic measurements, cyclic voltammograms, computational studies, and NLO measurements are gathered in the Supporting Information. CCDC 1424424 (1 c), 1424425 (4), 1424426 (5Fur), 1424427 (6Fur), and 1424428 (7Fur) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
D.B. gratefully acknowledges the EU through the ERC Starting Grant “COLORLANDS” project, the Science Policy Office of the Belgian Federal Government (BELSPO‐IAP 7/05 project), MIUR through the FIRB Futuro in Ricerca “SUPRACARBON” (contract no. RBFR10DAK6), the “SACS” project (grant number 310651), and Cardiff University. We thank Ms. Francesca Vita for the SEM analyses, Ms. Maria Mercedes Lorenzo Garcia for the help with ATR analyses, Dr. Caroline A. Ahad Hadad for some TGA measurements and Dr. Domenico Milano for the artwork used in the table of contents image. The authors also acknowledge Dr. Simon J. A. Pope for help with some of the fluorescence lifetime measurements and Panagiotis Aloukos for the precious suggestions and help with the NLO analyses on the thin films.
T. Miletić, A. Fermi, I. Orfanos, A. Avramopoulos, F. De Leo, N. Demitri, G. Bergamini, P. Ceroni, M. G. Papadopoulos, S. Couris, D. Bonifazi, Chem. Eur. J. 2017, 23, 2363.
References
- 1. Liu J., Li B.-W., Tan Y.-Z., Giannakopoulos A., Sanchez-Sanchez C., Beljonne D., Ruffieux P., Fasel R., Feng X., Müllen K., J. Am. Chem. Soc. 2015, 137, 6097–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li X., Wang X., Zhang L., Lee S., Dai H., Science 2008, 319, 1229–1232. [DOI] [PubMed] [Google Scholar]
- 3. Chen L., Hernandez Y., Feng X., Müllen K., Angew. Chem. Int. Ed. 2012, 51, 7640–7654; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7758–7773. [Google Scholar]
- 4. Feng X., Pisula W., Müllen K., Pure Appl. Chem. 2009, 81, 2203–2224. [Google Scholar]
- 5. Anthony J. E., Angew. Chem. Int. Ed. 2008, 47, 452–483; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 460–492. [Google Scholar]
- 6. Wu J., Pisula W., Müllen K., Chem. Rev. 2007, 107, 718–747. [DOI] [PubMed] [Google Scholar]
- 7. Cotlet M., Vosch T., Habuchi S., Weil T., Müllen K., Hofkens J., De Schryver F., J. Am. Chem. Soc. 2005, 127, 9760–9768. [DOI] [PubMed] [Google Scholar]
- 8. Lewandowska U., Zajaczkowski W., Chen L., Bouillière F., Wang D., Koynov K., Pisula W., Müllen K., Wennemers H., Angew. Chem. Int. Ed. 2014, 53, 12537–12541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12745–12749. [Google Scholar]
- 9. Balzani V., Bergamini G., Ceroni P., Marchi E., New J. Chem. 2011, 35, 1944–1954. [Google Scholar]
- 10. Rocard L., Berezin A., De Leo F., Bonifazi D., Angew. Chem. Int. Ed. 2015, 54, 15739–15743; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15965–15969. [Google Scholar]
- 11. Kishore R. S. K., Kel O., Banerji N., Emery D., Bollot G., Mareda J., Gomez-Casado A., Jonkheijm P., Huskens J., Maroni P., Borkovec M., Vauthey E., Sakai N., Matile S., J. Am. Chem. Soc. 2009, 131, 11106–11116. [DOI] [PubMed] [Google Scholar]
- 12. Wilson A., Gasparini G., Matile S., Chem. Soc. Rev. 2014, 43, 1948–1962. [DOI] [PubMed] [Google Scholar]
- 13. Zhylitskaya H., Cybińska J., Chmielewski P., Lis T., Stępień M., J. Am. Chem. Soc. 2016, 138, 11390–11398. [DOI] [PubMed] [Google Scholar]
- 14. Maggini L., Bonifazi D., Chem. Soc. Rev. 2012, 41, 211–241. [DOI] [PubMed] [Google Scholar]
- 15. Würthner F., Chem. Commun. 2004, 1564–1579. [DOI] [PubMed] [Google Scholar]
- 16. Ajayaghosh A., Praveen V. K., Vijayakumar C., Chem. Soc. Rev. 2008, 37, 109–122. [DOI] [PubMed] [Google Scholar]
- 17. Babu S. S., Aimi J., Ozawa H., Shirahata N., Saeki A., Seki S., Ajayaghosh A., Möhwald H., Nakanishi T., Angew. Chem. Int. Ed. 2012, 51, 3391–3395; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3447–3451. [Google Scholar]
- 18. Babu S. S., Phys. Chem. Chem. Phys. 2015, 17, 3950–3953. [DOI] [PubMed] [Google Scholar]
- 19. Babu S. S., Praveen V. K., Ajayaghosh A., Chem. Rev. 2014, 114, 1973–2129. [DOI] [PubMed] [Google Scholar]
- 20. Bhosale R., Míšek J., Sakai N., Matile S., Chem. Soc. Rev. 2010, 39, 138–149. [DOI] [PubMed] [Google Scholar]
- 21. Roncali J., Chem. Rev. 1997, 97, 173–206. [DOI] [PubMed] [Google Scholar]
- 22. Roncali J., Macromol. Rapid Commun. 2007, 28, 1761–1775. [Google Scholar]
- 23. Wang X., Sun G., Routh P., Kim D.-H., Huang W., Chen P., Chem. Soc. Rev. 2014, 43, 7067–7098. [DOI] [PubMed] [Google Scholar]
- 24. Maiti U. N., Lee W. J., Lee J. M., Oh Y., Kim J. Y., Kim J. E., Shim J., Han T. H., Kim S. O., Adv. Mater. 2014, 26, 40–67. [DOI] [PubMed] [Google Scholar]
- 25. Jiang W., Li Y., Wang Z., Chem. Soc. Rev. 2013, 42, 6113–6127. [DOI] [PubMed] [Google Scholar]
- 26. Mateo-Alonso A., Chem. Soc. Rev. 2014, 43, 6311–6324. [DOI] [PubMed] [Google Scholar]
- 27. Wang X.-Y., Wang J.-Y., Pei J., Chem. Eur. J. 2015, 21, 3528–3539. [DOI] [PubMed] [Google Scholar]
- 28. Bosdet M. J. D., Piers W. E., Can. J. Chem. 2009, 87, 8–29. [Google Scholar]
- 29. Campbell P. G., Marwitz A. J. V., Liu S.-Y., Angew. Chem. Int. Ed. 2012, 51, 6074–6092; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6178–6197. [Google Scholar]
- 30. Bonifazi D., Fasano F., Lorenzo-Garcia M. M., Marinelli D., Oubaha H., Tasseroul J., Chem. Commun. 2015, 51, 15222–15236. [DOI] [PubMed] [Google Scholar]
- 31. Wang X.-Y., Narita A., Feng X., Müllen K., J. Am. Chem. Soc., 2015, 137, 7668–7671. [DOI] [PubMed] [Google Scholar]
- 32. Matano Y., Imahori H., Org. Biomol. Chem. 2009, 7, 1258–1271. [DOI] [PubMed] [Google Scholar]
- 33. Baumgartner T., Réau R., Chem. Rev. 2006, 106, 4681–4727. [DOI] [PubMed] [Google Scholar]
- 34. Baumgartner T., Acc. Chem. Res. 2014, 47, 1613–1622. [DOI] [PubMed] [Google Scholar]
- 35. Liu Z., Marder T. B., Angew. Chem. Int. Ed. 2008, 47, 242–244; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 248–250. [Google Scholar]
- 36.M. Stępień, E. Gońka, M. Żyła, N. Sprutta, Chem. Rev 2016, DOI: 10.1021/acs.chemrev.6b00076. [DOI] [PubMed]
- 37. Narita A., Wang X.-Y., Feng X., Müllen K., Chem. Soc. Rev. 2015, 44, 6616–6643. [DOI] [PubMed] [Google Scholar]
- 38. Pummerer R., Prell E., Rieche A., Chem. Ber. 1926, 59, 2159–2161. [Google Scholar]
- 39. Pummerer R., Rieche A., Chem. Ber. 1926, 59, 2161–2175. [Google Scholar]
- 40. Li H., Zhang F., Qiu S., Lv N., Zhao Z., Li Q., Cui Z., Chem. Commun. 2013, 49, 10492–10494. [DOI] [PubMed] [Google Scholar]
- 41. Lv N., Xie M., Gu W., Ruan H., Qiu S., Zhou C., Cui Z., Org. Lett. 2013, 15, 2382–2385. [DOI] [PubMed] [Google Scholar]
- 42. Semiconductor Device, Method of Manufacturing the Same, and Method of Forming Multilayer Semiconductor Thin Film, N. Kobayashi, M. Sasaki, T. Ohe, US Patent 8399288 B2, 2013.
- 43. Novel Materials for Organic Electroluminescent Devices, P. Stoessel, A. Buesing, H. Heil, US Patent 2010/0013381 A1, 2010.
- 44. Kobayashi N., Sasaki M., Nomoto K., Chem. Mater. 2009, 21, 552–556. [Google Scholar]
- 45. Stassen D., Demitri N., Bonifazi D., Angew. Chem. Int. Ed. 2016, 55, 5947–5951; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6051–6055. [Google Scholar]
- 46. Hovorka M., Ščigel R., Gunterová J., Tichý M., Závada J., Tetrahedron 1992, 48, 9503–9516. [Google Scholar]
- 47. Hovorka M., Závada J., Tetrahedron 1992, 48, 9517–9530. [Google Scholar]
- 48. Feringa B., Wynberg H., Bioorg. Chem. 1978, 7, 397–408. [Google Scholar]
- 49. Brussee J., Groenendijk J. L. G., te Koppele J. M., Jansen A. C. A., Tetrahedron 1985, 41, 3313–3319. [Google Scholar]
- 50. Smrcina M., Polakova J., Vyskocil S., Kocovsky P., J. Org. Chem. 1993, 58, 4534–4538. [Google Scholar]
- 51. Noji M., Nakajima M., Koga K., Tetrahedron Lett. 1994, 35, 7983–7984. [Google Scholar]
- 52. Furuta T., Tanaka K., Tsubaki K., Fuji K., Tetrahedron 2004, 60, 4431–4441. [Google Scholar]
- 53. Tsubaki K., Tanaka H., Takaishi K., Miura M., Morikawa H., Furuta T., Tanaka K., Fuji K., Sasamori T., Tokitoh N., Kawabata T., J. Org. Chem. 2006, 71, 6579–6587. [DOI] [PubMed] [Google Scholar]
- 54. Egami H., Matsumoto K., Oguma T., Kunisu T., Katsuki T., J. Am. Chem. Soc. 2010, 132, 13633–13635. [DOI] [PubMed] [Google Scholar]
- 55. Tanaka K., Furuta T., Fuji K., Miwa Y., Taga T., Tetrahedron: Asymmetry 1996, 7, 2199–2202. [Google Scholar]
- 56. Wang S., Lv B., Cui Q., Ma X., Ba X., Xiao J., Chem. Eur. J. 2015, 21, 14791–14796. [DOI] [PubMed] [Google Scholar]
- 57. Blue light emitting material, J. Pillow, S. Kobayashi, M. Humphries, WO2010/013006 A2, 2010.
- 58. Zimmermann C., Willig F., Ramakrishna S., Burfeindt B., Pettinger B., Eichberger R., Storck W., J. Phys. Chem. B 2001, 105, 9245–9253. [Google Scholar]
- 59. Al-Kaysi R. O., Sang Ahn T., Müller A. M., Bardeen C. J., Phys. Chem. Chem. Phys. 2006, 8, 3453–3459. [DOI] [PubMed] [Google Scholar]
- 60. Johansson L. B. A., Molotkovsky Y. G., Bergelson L. D., J. Am. Chem. Soc. 1987, 109, 7374–7381. [Google Scholar]
- 61. Coventry D. N., Batsanov A. S., Goeta A. E., Howard J. A. K., Marder T. B., Perutz R. N., Chem. Commun. 2005, 2172–2174. [DOI] [PubMed] [Google Scholar]
- 62. Crawford A. G., Liu Z., Mkhalid I. A. I., Thibault M.-H., Schwarz N., Alcaraz G., Steffen A., Collings J. C., Batsanov A. S., Howard J. A. K., Marder T. B., Chem. Eur. J. 2012, 18, 5022–5035. [DOI] [PubMed] [Google Scholar]
- 63. Allen S. E., Walvoord R. R., Padilla-Salinas R., Kozlowski M. C., Chem. Rev. 2013, 113, 6234–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Aydin J., Kumar K. S., Sayah M. J., Wallner O. A., Szabó K. J., J. Org. Chem. 2007, 72, 4689–4697. [DOI] [PubMed] [Google Scholar]
- 65. Nakajima M., Miyoshi I., Kanayama K., Hashimoto S., Noji M., Koga K., J. Org. Chem. 1999, 64, 2264–2271. [Google Scholar]
- 66. Areephong J., Ruangsupapichart N., Thongpanchang T., Tetrahedron Lett. 2004, 45, 3067–3070. [Google Scholar]
- 67. Nakanishi K., Fukatsu D., Takaishi K., Tsuji T., Uenaka K., Kuramochi K., Kawabata T., Tsubaki K., J. Am. Chem. Soc. 2014, 136, 7101–7109. [DOI] [PubMed] [Google Scholar]
- 68. Weil T., Vosch T., Hofkens J., Peneva K., Müllen K., Angew. Chem. Int. Ed. 2010, 49, 9068–9093; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9252–9278. [Google Scholar]
- 69. Chen L., Li C., Müllen K., J. Mater. Chem. C 2014, 2, 1938–1956. [Google Scholar]
- 70. Yuan Z., Lee S.-L., Chen L., Li C., Mali K. S., De Feyter S., Müllen K., Chem. Eur. J. 2013, 19, 11842–11846. [DOI] [PubMed] [Google Scholar]
- 71. Pschirer N. G., Kohl C., Nolde F., Qu J., Müllen K., Angew. Chem. Int. Ed. 2006, 45, 1401–1404; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1429–1432. [Google Scholar]
- 72. Bohnen A., Koch K.-H., Lüttke W., Müllen K., Angew. Chem. Int. Ed. Engl. 1990, 29, 525–527; [Google Scholar]; Angew. Chem. 1990, 102, 548–550. [Google Scholar]
- 73. Karabunarliev S., Gherghel L., Koch K. H., Baumgarten M., Chem. Phys. 1994, 189, 53. [Google Scholar]
- 74. Đorđević L., Marangoni T., Miletić T., Rubio-Magnieto J., Mohanraj J., Amenitsch H., Pasini D., Liaros N., Couris S., Armaroli N., Surin M., Bonifazi D., J. Am. Chem. Soc. 2015, 137, 8150–8160. [DOI] [PubMed] [Google Scholar]
- 75. Würthner F., Sautter A., Thalacker C., Angew. Chem. Int. Ed. 2000, 39, 1243–1245; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 1298–1301. [Google Scholar]
- 76. Würthner F., Kaiser T. E., Saha-Möller C. R., Angew. Chem. Int. Ed. 2011, 50, 3376–3410; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3436–3473. [Google Scholar]
- 77. Sakai N., Mareda J., Vauthey E., Matile S., Chem. Commun. 2010, 46, 4225–4237. [DOI] [PubMed] [Google Scholar]
- 78. Suraru S.-L., Würthner F., Angew. Chem. Int. Ed. 2014, 53, 7428–7448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7558–7578. [Google Scholar]
- 79. Miros F. N., Matile S., ChemistryOpen 2016, 5, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bolag A., López-Andarias J., Lascano S., Soleimanpour S., Atienza C., Sakai N., Martín N., Matile S., Angew. Chem. Int. Ed. 2014, 53, 4890–4895; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4990–4995. [Google Scholar]
- 81. Hwang D.-H., Kim S.-K., Park M.-J., Lee J.-H., Koo B.-W., Kang I.-N., Kim S.-H., Taehyoung Z., Chem. Mater. 2004, 16, 1298–1303. [Google Scholar]
- 82. Iyoda M., Shimizu H., Chem. Soc. Rev. 2015, 44, 6411–6424. [DOI] [PubMed] [Google Scholar]
- 83.Gaussian 09, Revision A1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian, Inc., Wallingford CT 2009.
- 84. Becke A. D., Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- 85. Lee C., Yang W., Parr R., Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 86. Hanwell M. D., Curtis D. E., Lonie D. C., Vandermeerschd T., Zurek E., Hutchison G. R., J. Cheminf. 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Martin R. E., Diederich F., Angew. Chem. Int. Ed. 1999, 38, 1350–1377; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 1440–1469. [Google Scholar]
- 88. Berresheim A. J., Müller M., Müllen K., Chem. Rev. 1999, 99, 1747–1786. [DOI] [PubMed] [Google Scholar]
- 89. Figueira-Duarte T. M., Müllen K., Chem. Rev. 2011, 111, 7260–7314. [DOI] [PubMed] [Google Scholar]
- 90. Peterson K. A., Figgen D., Goll E., Stoll H., Dolg M., J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar]
- 91. Boyd R. W., Nonlinear Optics, Academic Press, San Diego, 1992, p. 176. [Google Scholar]
- 92. Xenogiannopoulou E., Medved M., Iliopoulos K., Couris S., Papadopoulos M. G., Bonifazi D., Sooambar C., Mateo-Alonso A., Prato M., ChemPhysChem 2007, 8, 1056–1064. [DOI] [PubMed] [Google Scholar]
- 93. Đorđević L., Marangoni T., De Leo F., Papagiannouli I., Aloukos P., Couris S., Pavoni E., Monti F., Armaroli N., Prato M., Bonifazi D., Phys. Chem. Chem. Phys. 2016, 18, 11858–11868. [DOI] [PubMed] [Google Scholar]
- 94. Brzozowski L., Sargent E. H., J. Mater. Sci. Mater. Electron. 2001, 12, 483–489. [Google Scholar]
- 95. Smith P. W., Bell Syst. Tech. J. 1982, 61, 1975–1993. [Google Scholar]
- 96. Stegeman G. I., in Nonlinear Optical Properties of Advanced Materials, Proc. SPIE, Vol. 1852, 1993, p. 75. [Google Scholar]
- 97. Bredas J. L., Adant C., Tackx P., Persoons A., Pierce B. M., Chem. Rev. 1994, 94, 243–278. [Google Scholar]
- 98. Fuks-Janczarek I., Nunzi J.-M., Sahraoui B., Kityk I. V., Berdowski J., Caminade A. M., Majoral J.-P., Martineau A. C., Frere P., Roncali J., Optics Commun. 2002, 209, 461–466. [Google Scholar]
- 99. Sahraoui B., Luc J., Meghea A., Czaplicki R., Fillaut J. L., Migalska-Zalas A., J. Opt. A 2009, 11, 024005. [Google Scholar]
- 100. Takeuchi T., Matile S., Chem. Commun. 2013, 49, 19–29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary