

ABSTRACT
Cytochrome P450 2D6 (CYP2D6) is among the most important genes involved in drug metabolism. Specific variants are associated with changes in the enzyme's amount and activity. Multiple technologies exist to determine these variants, like the AmpliChip CYP450 test, Taqman qPCR, or Second‐Generation Sequencing, however, sequence homology between cytochrome P450 genes and pseudogene CYP2D7 impairs reliable CYP2D6 genotyping, and variant phasing cannot accurately be determined using these assays. To circumvent this, we sequenced CYP2D6 using the Pacific Biosciences RSII and obtained high‐quality, full‐length, phased CYP2D6 sequences, enabling accurate variant calling and haplotyping of the entire gene‐locus including exonic, intronic, and upstream and downstream regions. Unphased diplotypes (Roche AmpliChip CYP450 test) were confirmed for 24 of the 25 samples, including gene duplications. Cases with gene deletions required additional specific assays to resolve. In total, 61 unique variants were detected, including variants that had not previously been associated with specific haplotypes. To further aid genomic analysis using standard reference sequences, we have established an LOVD‐powered CYP2D6 gene‐variant database, and added all reference haplotypes and data reported here. We conclude that our CYP2D6 genotyping approach produces reliable CYP2D6 diplotypes and reveals information about additional variants, including phasing and copy‐number variation.
Keywords: pharmacogenomics, CYP2D6, variant phasing, copy‐number variation, PacBio long‐read sequencing
Introduction
Cytochrome P450 2D6 (CYP2D6) is one of the most important genes in pharmacogenetics [Mahgoub et al., 1977; Eichelbaum et al., 1979]. The enzyme metabolizes about 25% of all prescription drugs [Owen et al., 2009] and the CYP2D6 gene is highly polymorphic, with over 100 genetic variants reported, including copy‐number variation (CNV) and gene rearrangements [Gaedigk, 2013]. Different CYP2D6 genotyping technologies are available, including Taqman qPCR assays, microarrays, classical Sanger sequencing, and next‐generation sequencing (e.g., exome‐ and whole‐genome sequencing). The AmpliChip CYP450 test from Roche Diagnostics was the first US FDA approved array to genotype CYP2D6 in a diagnostic setting by profiling a preselected number of variants [Rebsamen et al., 2008]. However, due to the high costs and inability to add novel SNVs to the AmpliChip, this array was discontinued. Multiple alternative platforms exist, such as the xTAG CYP2D6 kit (Luminex), AutoGenomics [Vairavan, 2004], and the Genochip CYP2D6 [Bank et al., 2015] (Pharmgenomics). Also these assays cannot detect which allele is duplicated, determine the copy number and are unable to detect complex structural variants. Moreover, sequence homology between several CYP450 genes and pseudogenes CYP2D7/2D8 impairs reliable CYP2D6 genotyping, especially for second‐generation sequencing [Drögemöller et al., 2013]. In addition, variant phasing, that is, to identify the linkage of SNVs or haplotypes present in a subject, cannot be accurately determined with these assays.
Targeted long‐amplicon sequencing using the PacBio single‐molecule real‐time (SMRT) sequencing platform offers many advantages over these routinely used assays for genotyping. The main advantage comes from the ability to handle and generate continuous sequence reads from template molecules of multiple kilobases in length without the need for DNA fragmentation steps. The current P6‐C4 polymerase and chemistry release yields average read lengths of 15 kb. Long sequence reads are pivotal to accurately identify and exclude off‐target signals from homologous sequences in the genome of interest, such as pseudogenes. In addition, the PacBio platform has a context‐free error profile, allowing for high‐quality consensus reads of >QV50 (one error in 105 bases) to be generated from the relatively high‐error single‐pass data [Travers et al., 2010; Carneiro et al., 2012]. This combination of long reads and high‐quality sequences allows for accurate variant calling and phasing of multiple heterozygous variants, potentially separated by several kilobases on the genome, into separate haplogroup sequences.
The current output of the PacBio system could accommodate sequencing of multiple samples on a single SMRT cell, depending on the size of the target region of interest. Options for multiplexing are needed in order to make CYP2D6 genotyping with PacBio cost‐efficient, flexible, and scalable. In this paper, we present a two‐step PCR‐based barcoding scheme for PacBio‐targeted long‐amplicon sequencing. Compared with the recent publication of Qiao et al. (2016) that describes PacBio sequencing for CYP2D6 haplotyping covering only the coding sequences, our setup targets a larger gene region that also includes the promoter and downstream gene regions. Moreover, the PCR setup we have applied is less laborious compared to that described in Qiao et al. (2016), and our approach is universally applicable due to the use of generic M13 sequences. After applying our method, we found that full‐length CYP2D6 could be sequenced reliably. We detected 61 unique variants across all samples, while retaining accurate phasing information. With the exception of one sample, the previously established diplotypes by the Roche AmpliChip CYP450 test were confirmed by the PacBio data. We conclude that this approach is cost‐efficient and reveals complete and reliable information about all variants in CYP2D6, including phasing and CNVs.
Methods
Long‐Range PCR, SMRT Library Prep, and PacBio Sequencing
All work described in this paper is subject to the LUMC Good Research Practice & Integrity guidelines and Ethical requirements. Samples were selected to represent a clinically relevant set of CYP2D6 haplogroups from the CYPTAM study (The Netherlands Trial Register 1509) and other anonymized samples. CYP2D6 genotypes were established by Roche AmpliChip CYP450 test (Roche, Almere, The Netherlands) as previously described [Dezentjé et al., 2015]. PCR primers used in this study (Supp. Table S1) were obtained from IDT‐DNA Technologies Leuven, Belgium. All oligos with barcode sequences were ordered as HPLC‐purified. Other primers were ordered as standard desalted. The CYP2D6‐specific primer sequences, used to generate a 6.6‐kb fragment covering the CYP2D6 gene locus including up and downstream regions, were based on Gaedigk et al. (2007). These primers exclude the CYP2D7 and CYP2D8 pseudogenes from downstream analysis.
Direct barcoding: sample‐specific barcode sequences were introduced during a single PCR reaction using barcoded fusion primers. The target regions were amplified using the Takara‐v2 kit in a 25‐μl reaction volume containing 400 nM gene‐specific primers, 50–100 ng genomic DNA, 400 mM of each dNTP, 1x PCR buffer with 2.5 mM MgCl2, and 1 U Takara LA Taq. Cycle parameters were 3' at 95°C, followed by 30 cycles of 10'' at 98°C and 15' at 68°C, and a final extension of 15' at 68°C. The length of the products was confirmed on a 1% agarose gel or Bioanalyzer 12000 chip (Agilent Amstelveen, the Netherlands). All samples were pooled in equimolar amounts and the sample pool was purified with 0.5x volume AMPure XP Beads (Beckman‐Coulter Woerden, the Netherlands) and eluted in 30 μl 10 mM Tris–HCl, pH 8.5 prior to PacBio library preparation.
Two‐step barcoding: the CYP2D6‐specific amplification, QC, and pooling were performed with M13‐tailed primers using identical conditions as the PCR of the direct barcoding scheme. Sample‐specific barcodes were introduced in a second PCR with identical conditions as the first, but using 3 μl of the purified product from PCR #1 and #5 cycles of amplification. The direct and two‐step barcode schemes are summarized in Figure 1A and B, respectively.
Figure 1.
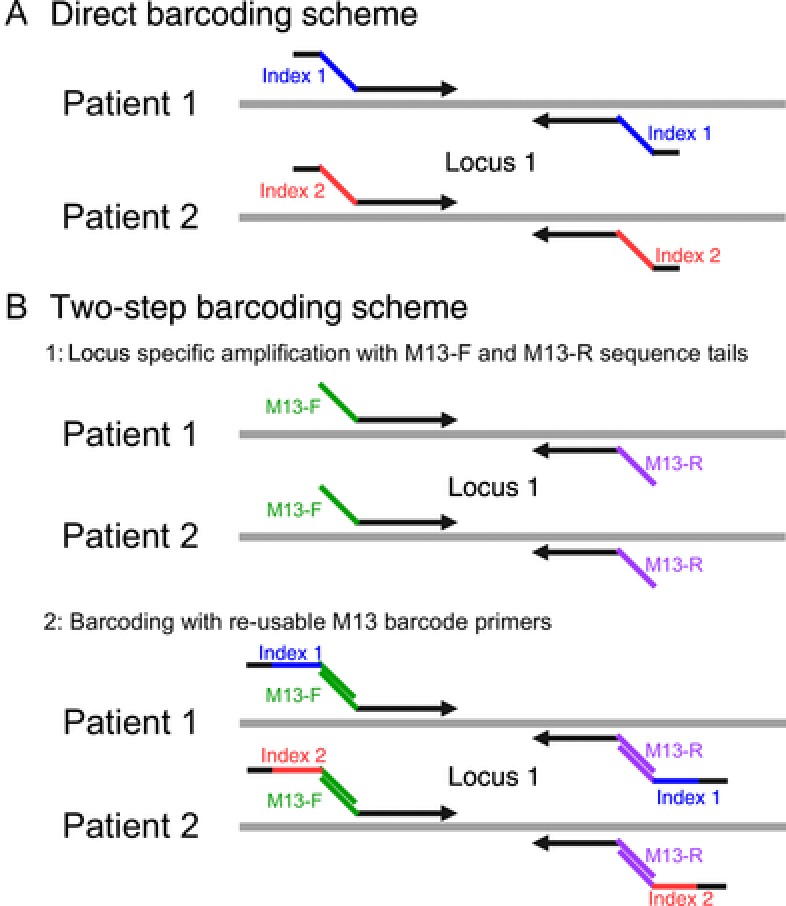
Barcoding schemes. Direct versus two‐step sample barcoding. A: In the direct barcoding scheme the sample specific barcodes, indicated by the blue and red for patients 1 and 2, respectively, are attached to the gene‐specific sequences (black arrows) and are introduced in a single PCR reaction. B: For the two‐step procedure for each individual, the region of interest is first amplified with a pair of gene‐specific primers with M13 forward (green) and reverse (purple) sequence tails. A symmetrical sample barcode, indicated by blue and red for patients 1 and 2, respectively, is introduced in a second PCR using a set of M13 barcode primers. A 5' padding sequence (black) is present on the index primers for both the direct and two‐step barcoding schemes to give all fragment identical end sequences to avoid ligation biases during the SMRT bell library preparation.
Sequence libraries were prepared from the pooled amplicons following the standard procedures for SMRT‐loop adapter ligation, starting from 500 ng of the pooled fragments. Sequencing of the libraries was performed with standard procedures using either the P4 or P6 enzyme.
PCR Detection for CYP2D6 Gene Deletion, Gene Duplication, and CYP2D6‐7 Fusion Genes
CYP2D6*5 gene deletion events were detected using a duplex PCR assay [Gaedigk et al., 2008], and CYP2D6 gene duplications and CYP2D6‐7 fusion events were assayed by a triplex PCR [Gaedigk et al., 2007; Gaedigk and Coetsee, 2008] using KAPA LongRange HotStart reagents. Reactions (25 μl) contained 1x reaction buffer, 1.75 mM MgCl2, 0.3 mM dNTP (each), 0.625 U KAPA LongRange HotStart DNA Polymerase, 10 ng human genomic DNA, and 1.25 μl of primer mix (Supp. Table S1). Triplex reactions were supplemented with 5% DMSO. Cycle parameters were 3' at 95°C, followed by 35 cycles of 15'' at 95°C and 10' at 68°C, with a final extension of 15' at 68°C. PCR products were visualized using a Genomic DNA ScreenTape assay on the Agilent 4200 TapeStation system.
PacBio Sequence Data Processing
For the CYP2D6 data, phased haplogroup sequences were retrieved for each individual using the long‐amplicon analysis (v2) protocol in the PacBio SMRT portal (v2.3.0). Analysis settings for both the two‐step barcoding and direct barcoding setup were: minimum subread length = 6,000; maximum number of subreads = 800; ignore primer sequence = 21; trim ends = 21; only most supported = 0; cluster per gene fam = y; phase alleles = y; split results = n; MinSnr = 4.5. The resulting haplogroup sequences were manually inspected on length and subread coverage to exclude spurious artefact sequences. All sequences were orientated to the plus strand and gene‐specific and M13 primer sequences were removed (cutadapt v1.7.1) [Martin, 2011] prior to aligning the reads to chr22 of the human genome (GRCh38) with BWA MEM (v1.7.1) [Li et al., 2009]. Bam and pileup files were generated using Samtools (v1.2) [Li et al., 2009; Li, 2011], and variants were called with bcftools (v1.2; bcftools call ‐mv ‐Ov ‐P 0.99 ‐p 0.99 | bcftools norm ‐m ‐both). All variants were merged into one vcf file, and described in HGVS format. A fasta file with all full‐length CYP2D6 sequences is available in Supp. Information. All variants have been submitted to the LOVD [Fokkema et al., 2011] CYP2D6 database (www.LOVD.nl/CYP2D6).
CYP2D6 Genotype Calling
The translation table listing the allele to genotype information was obtained from the Pharmacogenomics Knowledgebase (PharmGKB) [Whirl‐Carrillo et al., 2012] (http://www.pharmgkb.org/; version February 4, 2016). All variants were lifted from the M33388 format to GRCh38 genomic positions in HGVS format and checked with Mutalyzer v2.0.7 [Wildeman et al., 2008]. For future use and to support variant descriptions using different reference sequences, all CYP2D6 reference haplotypes were submitted to the LOVD database (http://www.LOVD.nl/CYP2D6). The haplogroup sequences per individual were processed separately to assign genotype by matching the individual variants to genotypes via the PharmGKB translation table.
Results
Full‐Length CYP2D6 Sequencing Using Direct Barcoding
Barcoded fusion primers for amplification of full‐length CYP2D6 were designed based on the PacBio multiplex PCR primer guidelines. Initial analysis of one individual was performed using two technical replicate libraries with unique barcodes, prepared and sequenced together on one PacBio SMRT cell. Full‐length CYP2D6 sequences resulted after barcode demultiplexing and processing the data with the Long‐Amplicon Analysis software. Two different sequences with approximately equal subread coverage (226 vs. 200 reads) were identified for this individual, indicating the presence of two distinct CYP2D6 haplogroup sequences (Table 1; Supp. Table S2). No chimeric long‐range PCR fragments were evident in the data. Variant calling resulted in 24 single‐nucleotide substitutions, one insertion, and two deletions, all of which were heterozygous with one of the deletions located on one haplogroup and the remaining variants located within the other haplogroup (Fig. 2A and B). Genotype calling for the two separate haplogroup sequences indicated the CYP2D6*1/*35A diplotype for this individual (CYP2D6*35A was based on variant rs769258 and s1135840; rs16947; rs1058164; rs1080985; Fig. 2B). It should be noted that since the entire gene was sequenced, including intronic and direct flanking regions, we generated a complete haplotype including a range of variants that have so far not been reported to be associated with the CYP2D6*35 haplotype. CYP2D6*1/*35A predicts a normal metabolizer phenotype, consistent with the AmpliChip array result. The technical replicate was in full agreement with haplogroup 2 for barcode 1 except for an one nucleotide length difference in a 22‐bp T‐homopolymer located in the upstream region of CYP2D6 (22:g.42132029delT) for haplogroup 2. Different settings for the long‐amplicon analysis could not resolve this discrepancy.
Table 1.
PacBio Haplogroup and Genotype Data
| Sample | Haplo group | Coverage ratio | SUB–INS–DEL | Call het/hom | PacBio‐based genotype | PacBio metabolizer group | AmpliChip CYP450 test genotype | AmpliChip metabolizer group |
|---|---|---|---|---|---|---|---|---|
| db1 index1 | 1 | 1:1 | 0–0–1 | Het 27/hom 0 | CYP2D6 *1 | Normal | CYP2D6 *1/*35 | Normal |
| 2 | 1 | 24–1–1 | CYP2D6 *35A | |||||
| db1 index2 | 1 | 1:3 | 0–0–0 | Het 26/hom 0 | CYP2D6 *1 | Normal | CYP2D6 *1/*35 | Normal |
| 2 | 1 | 24–1–1 | CYP2D6 *35A | |||||
| 1 | 1 | 1 | 0–0–1 | het 28/hom 0 | CYP2D6 *1 | Normal | CYP2D6 *1/*35 | Normal |
| 2 | 1:3 | 25–1–1 | CYP2D6 *35A | |||||
| 2 | 1 | 1:1 | 0–0–0 | het 25/hom 0 | CYP2D6 *1 | Normal | CYP2D6 *1/*2 | Normal |
| 2 | 1 | 23–1–1 | CYP2D6 *2A | |||||
| 3 | 1 | 1 | 1–0–0 | het 27/hom 0 | CYP2D6 *1 | Normal | CYP2D6 *1/*2 | Normal |
| 2 | 1:3 | 24–1–1 | CYP2D6 *2A | |||||
| 4 | 1 | 1 | 1–0–1 | het 27/hom 0 | CYP2D6 *1B | Ultrarapid | CYP2D6 *1/*2XN | Ultrarapid |
| 2 | 2:3 | 23–1–1 | CYP2D6 *2AXN | |||||
| 5 | 1 | 1 | 1–0–0 | het 26/hom 0 | CYP2D6 *1 | Intermediate | CYP2D6 *1/*41 | Intermediate |
| 2 | 1:0 | 23–1–1 | CYP2D6 *41 | |||||
| 6 | 1 | 1 | 0–1–0 | het 3/hom 0 | CYP2D6 *1 | Normal | CYP2D6 *1/*1 | Normal |
| 2 | 1:0 | 1–0–1 | CYP2D6 *1 | |||||
| 7 | 1 | 1 | 0–0–1 | het 27/hom 0 | CYP2D6 *1 | Normal | CYP2D6 *1/*2 | Normal |
| 2 | 1:2 | 24–1–1 | CYP2D6 *2A | |||||
| 8 | 1 | 1 | 19–1–0 | het 30/hom 8 | CYP2D6 *4A | Intermediate | CYP2D6 *4/*35 | Intermediate |
| 2 | 1:1 | 24–1–1 | CYP2D6 *35A | |||||
| 9 | 1 | 1 | 18–1–0 | het 28/hom 8 | CYP2D6 *4A | Intermediate | CYP2D6 *2/*4 | Intermediate |
| 2 | 1:0 | 23–1–1 | CYP2D6 *2A | |||||
| 10 | 1 | 1:1 | 23–1–1 | het 5/hom 23 | CYP2D6 *41 | Intermediate | CYP2D6 *2/*41 | Intermediate |
| 2 | 1 | 24–1–1 | CYP2D6 *2A | |||||
| 11 | 1 | 1:0 | 1–0–1 | het 27/hom 0 | CYP2D6 *1 | Intermediate | CYP2D6 *1/*41 | Intermediate |
| 2 | 1 | 23–1–1 | CYP2D6 *41 | |||||
| 12 | 1 | 5:6 | 18–1–0 | het 0/hom 19 | CYP2D6 *4A/*5 | Poor | CYP2D6 *4/*4 | Poor |
| 13 | 1 | 1 | 0–0–2 | het 27/hom 0 | CYP2D6 *3A | Intermediate | CYP2D6 *2/*3 | Intermediate |
| 2 | 1:0 | 23–1–1 | CYP2D6 *2A | |||||
| 14 | 1 | 1 | 24–1–1 | het 0/hom 26 | CYP2D6 *5/*35A | Intermediate | CYP2D6 *5/*35A | Intermediate |
| 15 | 1 | 1 | 14–1–0 | het 0/hom 15 | CYP2D6 *10D/*10D | Intermediate | CYP2D6 *10D/*10D | Intermediate |
| 16 | 1 | 1 | 2–0–2 | het 30/hom 0 | CYP2D6 *9 | Intermediate | CYP2D6 *9/*35 | Intermediate |
| 2 | 1:7 | 24–1–1 | CYP2D6 *35A | |||||
| 17 | 1 | 1:4 | 2–0–1 | het 22/hom 0 | CYP2D6 *6B | Poor | CYP2D6 *4/*6 | Poor |
| 2 | 1 | 18–1–0 | CYP2D6 *4A | |||||
| 18 | 1 | 1 | 2–0–2 | het 23/hom 1 | CYP2D6 *1 | Intermediate | CYP2D6 *1/*17 | Intermediate |
| 2 | 1:2 | 20–1–0 | CYP2D6 *17 | |||||
| 19 | 1 | 1 | 23–1–1 | het 0/hom 25 | CYP2D6 *2A/*5 | Intermediate | CYP2D6 *2/*5 | Intermediate |
| 20 | 1 | 1 | 23–1–1 | het 0/hom 25 | CYP2D6 *2A/*2AXN | Ultrarapid | CYP2D6 *2/*2XN | Ultrarapid |
| 21 | 1 | 1 | 0–0–1 | het 26/hom 0 | CYP2D6 *9 | Intermediate | CYP2D6 *9/*41XN | Intermediate |
| 2 | 2:1 | 23–1–1 | CYP2D6 *41XN | |||||
| 22 | 1 | 2:2 | 1–0–1 | het 28/hom 0 | CYP2D6 *1XN | Ultrarapid | CYP2D6 *1XN/*35 | Ultrarapid |
| 2 | 1 | 24–1–1 | CYP2D6 *35A | |||||
| 23 | 1 | 1:2 | 1–0–1 | het 21/hom 0 | CYP2D6 *1 | Intermediate | CYP2D6 *1/*4 | Intermediate |
| 2 | 1 | 18–1–0 | CYP2D6 *4A | |||||
| 24 | 1 | 1 | 1–0–2 | het 22/hom 0 | CYP2D6 *1 | Intermediate | CYP2D6 *1/*4 | Intermediate |
| 2 | 1:1 | 18–1–0 | CYP2D6 *4A |
Summary of the CYP2D6 genotyping data describing the number of variants found for the separate haplogroup sequences for each sample. Indicated from left to right for the duplicate direct barcoded sample and each of the 24 two‐step barcoded samples are the haplogroup number; coverage ratio between haplogroups; the number of single‐nucleotide substitutions, insertions, and deletions (SUB—INS–DEL) per haplogroup; and the number of heterozygous and homozygous variants. The last four columns describe the CYP2D6 genotype and the predicted metabolizer group derived from the PacBio data and RocheAmpliChip CYP450 test.
Figure 2.
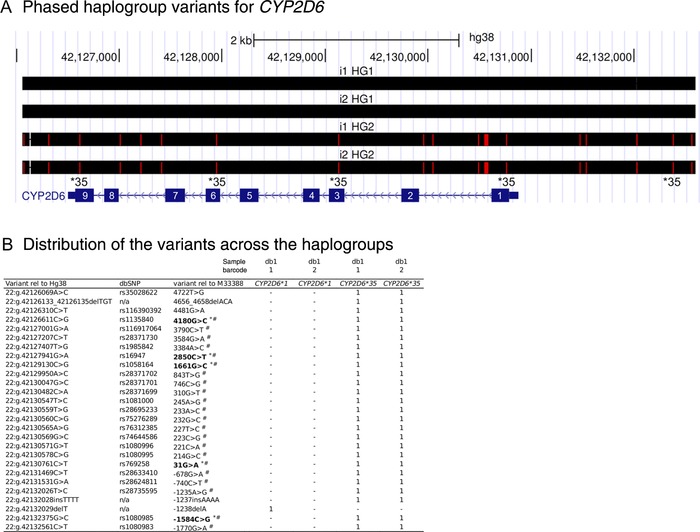
Direct barcoding results. Direct barcoding results for the technical replicate with different barcodes. A: UCSC browser screenshot illustrating the detected variants (red lines) of the two fully‐phased haplogroup sequences for each sample barcode relative to the GRCh38 reference. The CYP2D6 gene is located on the negative strand. Exon numbers are indicated in white based on the NM_000106.5 transcript sequence. For the haplogroup 2 sequences, the variants determining the CYP2D6*35 call are indicated. B: Identity and distribution of all 27 variants across the haplogroup sequences. Order in this table (top to bottom) is identical to those in Figure 2A (left to right). Variants in bold determine the CYP2D6*35 haplotype; * indicates that the variant was included on the Roche AmpliChip CYP450 test; # indicates that the variant was included in PharmGKB (February 4, 2016); and “HG” and “i” denote haplogroup and index, respectively.
M13 Sequence‐Based Two‐Step Barcoding Scheme For Multiplexing of CYP2D6
Although the direct barcoding scheme is able to deliver high‐quality results and accurate CYP2D6 phenotype predictions, the setup is rigid. Extending the existing setup to cover more targets would require a complete set of new barcoded fusion primer pairs for each additional target. In parallel, for each additional individual to be sequenced on the same SMRT cell, two additional, expensive, HPLC‐purified barcoded primers for each target in the experiment design are needed. Therefore, we set up a generally applicable, versatile, and cost‐efficient PCR‐based multiplexing strategy for long‐amplicon sequencing based on a two‐step barcoding system. In this scheme, the ∼6.6‐kb CYP2D6 gene locus, including the upstream and downstream regions, is first amplified with a pair of gene‐specific primers with forward and reverse M13 sequence tails for each individual sample separately. A symmetrical sample barcode is subsequently introduced in a second PCR using a set of generic M13‐tailed barcode primers.
Using this setup, we sequenced CYP2D6 for 24 individuals with different predicted CYP2D6 phenotypes based on the Roche AmpliChip CYP450 test assay representing the CYP2D6 *1‐6, *9, *10, *17, *35, and *41 haplogroups. Samples received a unique barcode during the second round of PCR, and all barcodes could be identified in the data with approximately equal numbers of subread coverage in a multiplex of 12 samples per SMRT cell (Supp. Table S2). Full‐length CYP2D6 sequences were generated for all samples without evidence for chimeric sequences. For five individuals, a single haplogroup sequence was obtained. For the remaining samples, two separate haplogroup sequences were found, 15 of which contained only heterozygous variants across the two haplogroups. The other four individuals showed the presence of both heterozygous and homozygous variants (Table 1).
In total, 695 variants were detected for the 24 individuals, representing 61 unique variants, comprising 49 single‐nucleotide substitutions, five insertions, and seven deletions. The majority of the variants reside in the noncoding regions of the CYP2D6 gene: 18 (30%) were upstream, 20 (33%) intronic, and five (8%) downstream gene variants. Supp. Figure S1 shows the distribution of these variants across the CYP2D6 locus, alongside the variants on the Roche AmpliChip CYP450 test and those included in PharmGKB. Eighteen variants in the coding regions represent 10 missense mutations, two frameshift mutations, one in‐frame deletion, and one splice‐acceptor variant, with the remaining four being synonymous variants. Of the 61 unique variants, nine have not previously been described in dbSNP. Of these, seven are associated with a long T‐homopolymer stretch, leaving one novel deletion (22:g.42126133_42126135delTGT), which was present in 17 of the 24 individuals in this study in the downstream region of CYP2D6 gene region, and one SNV (22:g.42131610G>C) present in one sample in the upstream gene region. Sanger sequencing confirmed both novel variants (Supp. Fig. S2).
Phenotype predictions for these individuals for the separate haplogroup sequences were all in agreement with those obtained from the AmpliChip (Table 1). Moreover, using the PacBio data, we were able to more specifically type a subset of the haplogroup calls. One of the CYP2D6*1 haplogroups could now be called specifically as CYP2D6*1B based on the presence of the 22:g.42126963C>T variant. Several samples were called to have the CYP2D6*4ABDJK haplogroup by the Roche AmpliChip CYP450 test based on the 1846G>A (rs3892097) variant, whereas the PacBio was able to refine the call to CYP2D6*4A, based on all seven variants for this haplogroup. Similarly, CYP2D6*2ABD haplotypes, based on two variants, could now be matched to CYP2D6*2A; based on 14 variants, CYP2D6*35 haplogroups could more precisely be defined as CYP2D6*35A and CYP2D6*6 as CYP2D6*6A.
In total, 19 variants were not present in PharmGKB, meaning data linking these to specific haplogroups is currently lacking. Nine of these variants were found in more than one haplogroup, and 10 variants were unique to specific haplotypes. The downstream gene variant 22:g.42126079C > T was only present in the CYP2D6*17 haplogroup, the intronic 22:g.42129545G > A variant only in CYP2D6*6B, and the two upstream gene variants, 22:g.42131610G > C and 22:g.42131631A > T, were both present in one of the CYP2D6*9 haplotypes. Also, two intronic and one upstream gene variant were detected in CYP2D6*1 haplotypes only, two of which, 22:g.42129623C > T and 22:g.42130522G > A, had previously also been identified by Twist et al. (2016). One of the CYP2D6*35 haplogroup samples contained the 22:g.42126944C > T missense variant, but PolyPhen [Adzhubei et al., 2010], SIFT [Kumar et al., 2009], and Condel [González‐Pérez and López‐Bigas, 2011] scored this as “tolerated,” “benign,” and “neutral,” respectively. Two of the variants not listed in PharmGKB were found in CYP2D6*2A haplotypes, with one located in the upstream gene region (22:g.42131114A > G) and one missense variant (22:g.42127611C > T), with “deleterious” SIFT and Condel scores and “benign” according to PolyPhen.
The PacBio data were able to identify CNVs for samples that carry at least two distinctive CYP2D6 haplotype sequences. For samples 4, 21, and 22, an unequal subread coverage for the distinct haplogroups was observed, indicating a duplication of one of the haplogroups. For instance, in sample 4, the ratio of reads for the CYP2D6 * 2A and CYP2D6*1B diplotype was 2.3 to 1, indicating a duplication of the *2A haplogroup sequence for this individual, which was consistent with the results derived from the AmpliChip CYP450 test as well as the Triplex PCR fragment to detect CYP2D6 gene duplication events (Supp. Fig. S3). For sample 20, a single CYP2D6*2A haplotype was observed, and the Triplex PCR indicated a gene duplication, supporting a duplication of the CYP2D6*2A haplogroup in combination with a single CYP2D6*2A haplogroup on the other chromosome, indicating a CYP2D6*2A/*2AXN diplotype.
Four other samples (12, 14, 15, and 19) resulted in a single CYP2D6 haplogroup sequence. The Duplex PCR assay detected CYP2D6*5 gene deletions for 12, 14, and 19 (Supp. Fig. S4), indicating the PacBio sequence represents the gene copy of one chromosome only. For sample 12, this gene deletion was not detected with the AmpliChip CYP450 test, which reported a CYP2D6*4/*4 diplotype instead of CYP2D6*4/*5, making this the only discordant call between the two assays. The negative call for a CYP2D6*5 gene deletion for sample 15 implies it carries two identical CYP2D6*10D haplogroup sequences.
These data indicate that the two‐step barcoding scheme can successfully be used for sample multiplex barcoding and sequencing of multiple long‐range PCR amplicons on a single SMRT cell, generating high‐quality phased variant data that can successfully be used for reliable CYP2D6 genotyping.
Discussion
In this paper, we present a reliable method for full‐length sequencing of the 6.6‐kb CYP2D6 gene, an important pharmacogenetic gene. We describe a two‐step sample barcoding scheme for long‐amplicon sequencing on the PacBio RSII to determine the haplogroup sequences and diplotype. Sample‐specific barcodes are introduced via M13 forward and reverse sequences, which are widely used in both research and clinical genetics laboratories and have previously been used in NGS for adding sample‐specific barcodes [De Leeneer et al., 2011]. Therefore, use of the M13 sequences will facilitate the implementation of PacBio for (long) amplicon sequencing in place of standard Sanger sequencing. In contrast to the direct barcoding with fusion primers, our setup with generic M13 barcodes is scalable and flexible and can easily be adapted to profiling other genomic targets without the need for significant additional investments. For example, extending the existing setup by two target loci for 12 individuals can be achieved by designing just four additional target‐specific M13 tailed, standard desalted, primers. To achieve the same for the direct barcoding method would require an additional 48, expensive, HPLC‐purified fusion primers. The generic use of the barcode primers makes the setup cost‐efficient, flexible, and easy to implement for a wide range of targets with minimal optimization time. Compared with the RocheAmpliChip CYP450 test and the Pharmgenomics Genochip CYP2D6, the PacBio setup for CYP2D6 genotyping results in an approximately 90% and 20% cost reduction per sample, respectively (reagents only, excluding laboratory utilities, equipment depreciation, and labor for data analysis and wet‐lab). The cost per sample will decrease even more with the higher output of the PacBio Sequel system, making (long)amplicon sequencing on the PacBio a suitable alternative to Sanger and targeted short‐read approaches.
PacBio long‐amplicon sequencing can overcome several of the shortcomings of existing CYP2D6 genotyping methodologies. The ability to handle and sequence long DNA fragments makes it possible to move from a preselected set of variants or only the exonic regions to the entire 6.6‐kb CYP2D6 locus, including the promoter region, all introns, and the downstream gene region. Long reads also aid in discriminating the locus of interest from potential off‐target sequences such as the CYP2D7 pseudogene. This can be achieved at the level of primer design during experiment setup by the larger pool of unique primer sequences in a larger sequence space, as well as during data analysis based on sequence identity. For the CYP2D6 experiments, no high‐identity off‐target sequence alignments were observed, indicating pseudogene contamination was not present in our data. In addition, the long‐sequence reads provide the opportunity for high‐quality variant phasing, that is, to identify the exact distribution of multiple heterozygous variants on the two separate CYP2D6 haplogroups across multi‐kb regions that is especially of importance for low‐polymorphic samples. Our data showed clear CYP2D6 haplogroup sequence separation based on a range of 3–30 heterozygous variants allowing us to make genotype calls on the separate individual haplogroup sequences.
Recently, two papers have described the use of long sequence read NGS platforms for CYP2D6 genotyping. Using the MinION platform, Ammar et al. (2015) sequenced a 5‐kb PCR amplicon without any sample multiplexing options. The high‐error rate of the MinION system, leading to high number of false‐positive variants, is the main limitation of this approach. Qiao et al. (2016) first performed separate amplification of a “downstream” and a “upstream” fragment, with the latter being used for assessing CYP2D6 CNVs by gel analysis. Both amplicons were used in a nested PCR and barcoding PCR reaction prior to PacBio RSII sequencing, bringing the total number of PCRs to three. Although successful in identifying the CYP2D6 diplotypes, the study was also limited to a ∼5‐kb sequence fragment covering only the coding sequences of the CYP2D6 gene, potentially missing variants affecting regulatory sequence features located in the upstream or downstream gene regions, for example, the promoter region. In our study, we had identified and confirmed the existence of two novel variants in these regions, indicating the added value of including the upstream and downstream gene regions. A main difference between the PacBio studies is that Qiao et al. (2016) corrected PacBio errors based on alignments to a predefined reference, whereas we used the PacBio Long Amplicon Analysis tool, which is an assumption‐free approach, that is, it is independent of any predefined reference sequence. The Long Amplicon analysis is therefore more appropriate for analyzing complex structural rearrangements that would be difficult to align to an existing predefined reference.
Translating a CYP2D6 genotype to a CYP2D6 phenotype is notoriously difficult, and there is currently no standardized process [Hicks et al., 2014]. Although all PacBio‐derived genotypes were in agreement with those obtained from the RocheAmpliChip CYP450 test, many additional variants were detected in the PacBio data, only a subset (n = 42) of which had previously been associated with specific genotypes as described in the PharmGKB [Whirl‐Carrillo et al., 2012] (www.pharmgkb.org; version February 4, 2016). This leaves 19 variants for which the contribution to the CYP2D6 phenotype is currently unresolved. Some of these variants may potentially help to explain the large interindividual variation in CYP2D6 metabolic capacity typically observed within CYP2D6 phenotype groups [Schenk et al., 2007]. However, the majority of these variants resided in intronic or either the upstream and downstream gene regions or were missense variants without consequence according to SIFT, PolyPhen, and Condel scores. One missense variant in a CYP2D6*2A haplogroup (22:g.42127611C>T; 3180G>A; rs78209835; NP_000097.3:p.Asp337Asn) had SIFT and Condel scores indicating a deleterious effect on protein function, whereas its PolyPhen scored a “benign” effect. Unfortunately, we do not have CYP2D6 protein activity data for this individual, making it unclear whether the variant indeed affects protein function. Further research is needed to determine the exact contribution of these variants.
In addition to these challenges in predicting a CYP2D6 phenotype from a CYP2D6 genotype, an additional level of complexity lies at the in silico prediction of the variant effects. Different tools are available; however, depending on the settings of these analyses, concordance between these tools may be low [McCarthy et al., 2014]. Also, these prediction tools are based on different sets of assumptions and therefore may produce conflicting predictions for the same variant. In addition, the choice of the reference sequence may affect the prediction results. Prediction tools based on the GRCh19 genome release, representing a CYP2D6*2 haplogroup, may have a different sequence context of immediate adjacent bases for a specific variant compared with the GRCh38 release, representing a CYP2D6*1 sequence, potentially leading to erroneous conclusions on the effect of a variant. Finally, most tools predict the effect of individual variants, ignoring potential cumulative effects of multiple phased variants across the entire gene locus of the separate haplotypes.
Although the PacBio approach for CYP2D6 variant phasing and haplotyping was successful, we are aware that a relatively low number of samples were profiled and that these were selected from a mainly Caucasian collection, potentially excluding relevant haplotypes from non‐European descent. Also, no samples with a CYP2D6‐7 fusion gene were included in this study. An in‐depth analysis of the variants' effects on CYP2D6 mRNA splicing are not included as these require extensive confirmation of the in silico predictions at the RNA level.
Sequencing Software Generated Variant Calls in Reference to Genome Build GRCh38
To match the variant calls with CYP2D6 haplotypes, we checked the Website of the Human Cytochrome P450 (CYP) Allele Nomenclature Committee [Sim and Ingelman‐Sundberg, 2010, 2013] (http://www.cypalleles.ki.se). Unfortunately, variants reported in the CYP2D6 haplotype table do not follow existing HGVS standards [Den Dunnen and Antonarakis, 2000] (www.HGVS.org/mutnomen). Helpful but indirect links for some variants are given to dbSNP, but overall the variants cannot be used easily. Similarly, the haplotype tables provided by PharmGKB [Whirl‐Carrillo et al., 2012] (www.pharmgkb.org) and the SuperCYP Cytochrome P450 database [Preissner et al., 2010] (http://bioinformatics.charite.de/supercyp/) suffer from the same problem, where nonstandard variant description is used. We therefore decided to add all CYP2D6 reference haplotypes, using standardized variant descriptions, to the LOVD‐powered CYP2D6 gene variant database (www.LOVD.nl/CYP2D6). In addition, in collaboration with an international workgroup [Kalman et al., 2016], we generated an upgraded CYP2D6 haplotype table reporting all variants in relation to all commonly used CYP2D6 reference sequences (including genome builds GRCh19 and GRCh38, RefSeqGene record NG_008376.3, LRG_303, and reference transcript NM_000106.4). Finally, we submitted all CYP2D6 data reported here to the CYP2D6database. As we sequenced the entire genomic gene segment, including introns and direct gene flanking regions, our data extended several alleles with variants hitherto un‐reported as being part of these alleles.
In summary, using the two‐step barcoding approach, we show that multiplex sequencing of 12 samples for full‐length CYP2D6 generated reliable sequence information. This approach is cost‐efficient and could represent an improved alternative for existing CYP2D6 diplotyping technologies for both clinical‐level genotyping and research purposes.
Disclosure statement: The authors have no conflict of interest to declare.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Communicated by Graham R. Taylor
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammar R, Paton TA, Torti D, Shlien A, Bader GD. 2015. Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Res 4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank PCD, Swen JJ, Guchelaar H‐J, van der Straaten T. 2015. GenoChip CYP2D6 macroarray as a method to genotype for CYP2D6 variants: results of a validation study in a Caucasian population. Pharmacogenomics 16:681–687. [DOI] [PubMed] [Google Scholar]
- Carneiro MO, Russ C, Ross MG, Gabriel SB, Nusbaum C, DePristo MA. 2012. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics 13:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Dunnen JT, Antonarakis SE. 2000. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15:7–12. [DOI] [PubMed] [Google Scholar]
- De Leeneer K, Hellemans J, De Schrijver J, Baetens M, Poppe B, van Criekinge W, de Paepe A, Coucke P, Claes K. 2011. Massive parallel amplicon sequencing of the breast cancer genes BRCA1 and BRCA2: opportunities, challenges, and limitations. Hum Mutat 32:335–344. [DOI] [PubMed] [Google Scholar]
- Dezentjé VO, Opdam FL, Gelderblom H, Hartigh den J, van der Straaten T, Vree R, Maartense E, Smorenburg CH, Putter H, Dieudonné AS, Neven P, van de Velde CJH, et al. 2015. CYP2D6 genotype‐ and endoxifen‐guided tamoxifen dose escalation increases endoxifen serum concentrations without increasing side effects. Breast Cancer Res Treat 153:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drögemöller BI, Wright GEB, Niehaus DJH, Emsley R, Warnich L. 2013. Next‐generation sequencing of pharmacogenes: a critical analysis focusing on schizophrenia treatment. Pharmacogenet Genomics 23:666–674. [DOI] [PubMed] [Google Scholar]
- Eichelbaum M, Spannbrucker N, Steincke B, Dengler HJ. 1979. Defective N‐oxidation of sparteine in man: a new pharmacogenetic defect. Eur J Clin Pharmacol 16:183–187. [DOI] [PubMed] [Google Scholar]
- Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT. 2011. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 32:557–563. [DOI] [PubMed] [Google Scholar]
- Gaedigk A. 2013. Complexities of CYP2D6 gene analysis and interpretation. Int Rev Psychiatry 25:534–553. [DOI] [PubMed] [Google Scholar]
- Gaedigk A, Coetsee C. 2008. The CYP2D6 gene locus in South African Coloureds: unique allele distributions, novel alleles and gene arrangements. Eur J Clin Pharmacol 64:465–475. [DOI] [PubMed] [Google Scholar]
- Gaedigk A, Ndjountché L, Divakaran K, DiAnne Bradford L, Zineh I, Oberlander TF, Brousseau DC, McCarver DG, Johnson JA, Alander SW, Wayne Riggs K, Steven Leeder J. 2007. Cytochrome P4502D6 (CYP2D6) gene locus heterogeneity: characterization of gene duplication events. Clin Pharmacol Ther 81:242–251. [DOI] [PubMed] [Google Scholar]
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. 2008. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther 83:234–242. [DOI] [PubMed] [Google Scholar]
- González‐Pérez A, López‐Bigas N. 2011. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks JK, Swen JJ, Gaedigk A. 2014. Challenges in CYP2D6 phenotype assignment from genotype data: a critical assessment and call for standardization. Curr Drug Metab 15:218–232. [DOI] [PubMed] [Google Scholar]
- Kalman LV, Agúndez J, Appell ML, Black JL, Bell GC, Boukouvala S, Bruckner C, Bruford E, Caudle K, Coulthard SA, Daly AK, del Tredici A, et al. 2016. Pharmacogenetic allele nomenclature: international workgroup recommendations for test result reporting. Clin Pharmacol Ther 99:172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup 1000 Genome Project Data Processing . 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahgoub A, Dring LG, Idle JR, Lancaster R, Smith RL. 1977. Polymorphic hydroxylation of debrisoquine in man. Lancet 310:584–586. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17:10–12. [Google Scholar]
- McCarthy DJ, Humburg P, Kanapin A, Rivas MA, Gaulton K, Cazier J‐B, Donnelly P. 2014. Choice of transcripts and software has a large effect on variant annotation. Genome Med 6:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen RP, Sangkuhl K, Klein TE, Altman RB. 2009. Cytochrome P450 2D6. Pharmacogenet Genomics 19:559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissner S, Kroll K, Dunkel M, Senger C, Goldsobel G, Kuzman D, Guenther S, Winnenburg R, Schroeder M, Preissner R. 2010. SuperCYP: a comprehensive database on cytochrome P450 enzymes including a tool for analysis of CYP‐drug interactions. Nucleic Acids Res 38:D237–D243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao W, Yang Y, Sebra R, Mendiratta G, Gaedigk A, Desnick RJ, Scott SA. 2016. Long‐read single molecule real‐time full gene sequencing of cytochrome P450‐2D6. Hum Mutat 37:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen MC, Desmeules J, Daali Y, Chiappe A, Diemand A, Rey C, Chabert J, Dayer P, Hochstrasser D, Rossier MF. 2008. The AmpliChip CYP450 test: cytochrome P450 2D6 genotype assessment and phenotype prediction. Pharmacogenomics J 9:34–41. [DOI] [PubMed] [Google Scholar]
- Schenk PW, van Fessem MAC, Verploegh‐Van Rij S, Mathot RAA, van Gelder T, Vulto AG, van Vliet M, Lindemans J, Bruijn JA, van Schaik RHN. 2007. Association of graded allele‐specific changes in CYP2D6 function with imipramine dose requirement in a large group of depressed patients. Mol Psychiatry 13:597–605. [DOI] [PubMed] [Google Scholar]
- Sim SC, Ingelman‐Sundberg M. 2010. The Human Cytochrome P450 (CYP) Allele Nomenclature website: a peer‐reviewed database of CYP variants and their associated effects. Hum Genomics 4:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim SC, Ingelman‐Sundberg M. 2013. Update on allele nomenclature for human cytochromes P450 and the Human Cytochrome P450 Allele (CYP‐allele) Nomenclature Database. Methods Mol Biol 987:251–259. [DOI] [PubMed] [Google Scholar]
- The Netherlands Trial Register 1509 . Accessed at: http://www.trialregister.nl/trialreg/admin/rctview.asp?TC=1509.
- Travers KJ, Chin C‐S, Rank DR, Eid JS, Turner SW. 2010. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res 38:e159–e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twist GP, Gaedigk A, Miller NA, Farrow EG, Willig LK, Dinwiddie DL, Petrikin JE, Soden SE, Herd S, Gibson M, Cakici JA, Riffel AK, et al. 2016. Constellation: a tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole‐genome sequences. NPJ Genomic Med 1:15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairavan R. 2004. AutoGenomics, Inc. Pharmacogenomics 5:585–588. [DOI] [PubMed] [Google Scholar]
- Whirl‐Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, Thorn CF, Altman RB, Klein TE. 2012. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther 92:414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PEM. 2008. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat 29:6–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information