

dear editor, The group of reticulate pigmentary disorders includes the rare autosomal dominant Dowling–Degos disease (DDD) and Galli–Galli disease (GGD; OMIM 179850, 615327 and 615696).1 In light of substantial clinical, histological and mutational overlap between GGD and DDD, they are considered to belong to the same entity.2, 3 Mutations in KRT5 (encoding keratin 5) have been associated with GGD/DDD since 2006.2, 3, 4, 5 With the development of whole‐exome sequencing, mutations in POFUT1 (encoding protein O‐fucosyltransferase 1)6, 7 and POGLUT1 (encoding protein O‐glucosyltransferase 1)8 have been shown to underlie some cases of GGD/DDD. We report mutations in POGLUT1 in three families of European ancestry.
In family 1 the mother presented, aged 75 years, with a 40‐year history of reticulate hyperpigmented scaling plaques on the neck, lateral proximal arms, proximal legs and popliteal fossae (Fig. 1a, b). Light microscopy revealed epidermal acanthosis, hypergranulosis and hyperkeratosis with parakeratosis and suprabasilar acantholysis (Fig. 1c). Direct DNA sequencing of KRT5 was negative for mutations. A whole‐exome sequencing approach was taken using DNA from the affected mother (Appendix S1 and Table S1; see Supporting Information). During this time mutations were reported in POFUT1 and POGLUT1. A previously reported heterozygous nonsense mutation was identified in POGLUT1, p.Arg218*; c.652C>T, and confirmed by Sanger sequencing. The mutation was identified in her affected son but not in two unaffected daughters.
Figure 1.
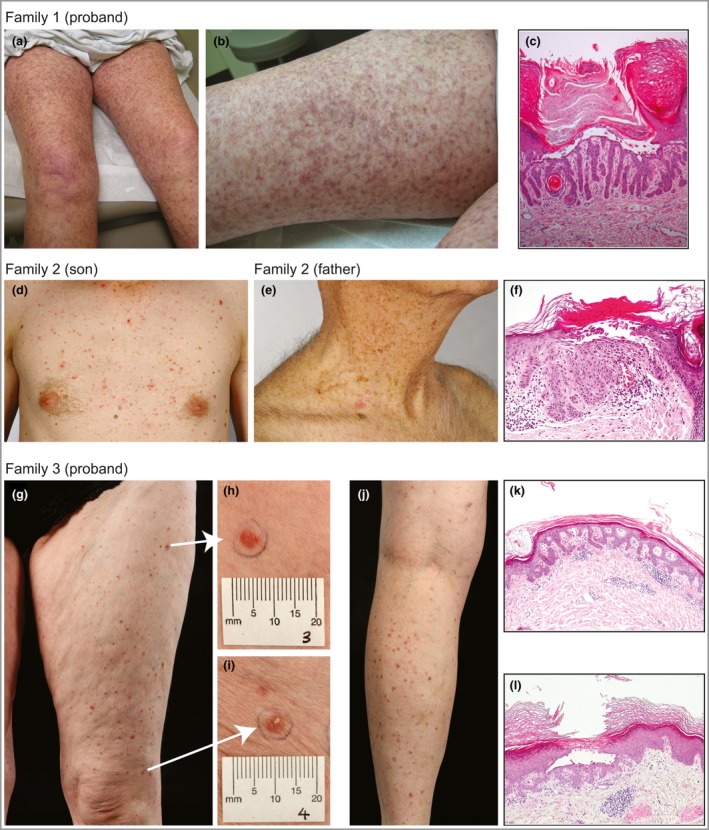
Clinical findings. (a, b) Reticulate hyperpigmented scaling plaques on the proximal legs of the proband of family 1. (c) Light microscopy of three punch biopsies from the proband of family 1 revealed similar findings of filiform digitate downgrowth of the epidermal rete ridges, presence of small horn cysts, epidermal acanthosis, hypergranulosis, and hyperkeratosis with parakeratosis and suprabasilar acantholysis. (d) Small red‐brown macules and crusted papules widely distributed over the trunk, neck, back, abdomen and limbs of the proband of family 2. The large body folds, hands and feet, and face were spared. (e) The father of the proband of family 2 had similar clinical features, limited to the neck region without involvement of the large flexures. (f) Light microscopy shows some apical acantholysis and parakeratosis with a moderate dermal inflammatory infiltrate. (g–j) The proband of family 3 with multiple small (approximately 5‐mm‐diameter) red‐brown scaly papules, present predominantly on the legs but also on the forearms with some postinflammatory change from previous lesions. (k) Light microscopy from the proband of family 3, a medium‐power view showing elongation of the rete ridge and modest inflammatory infiltrate. (l) A medium‐power view of a second biopsy from the right anterior thigh. There is an intraepidermal vesicle, as a result of acantholysis of the keratinocytes, some of which show dyskeratosis. Note the elongation of the rete ridges immediately below the vesicle.
A second family with GGD/DDD, present in two generations, was also negative for KRT5 mutation. The proband was a 40‐year‐old man who progressively developed, starting from adolescence, small red‐brown macular and papular crusted lesions (Fig. 1d). His father had similar clinical features (Fig. 1e). Light microscopy showed filiform digitate downgrowth of epidermal rete ridges, presence of small horn cysts, apical acantholysis and parakeratosis with a moderate dermal inflammatory infiltrate (Fig. 1f). POFUT1 and POGLUT1 were screened by Sanger sequencing (Appendix S1). A previously unreported heterozygous missense mutation was identified in POGLUT1, p.Gly170Glu; c.509G>A, in the affected father and affected son, but not in the unaffected mother or unaffected brother (Fig. 2a, b). p.Gly170 is highly conserved in the encoded enzyme across species. In silico prediction tools, MutationTaster (http://www.mutationtaster.org/) and PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), predict this to be a disease‐causing variant. It is not in the dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), NHLBI Exome Variant Server (http://evs.gs.washington.edu/EVS/) or ExAC (http://exac.broadinstitute.org/) databases. No mutations were identified in POFUT1.
Figure 2.
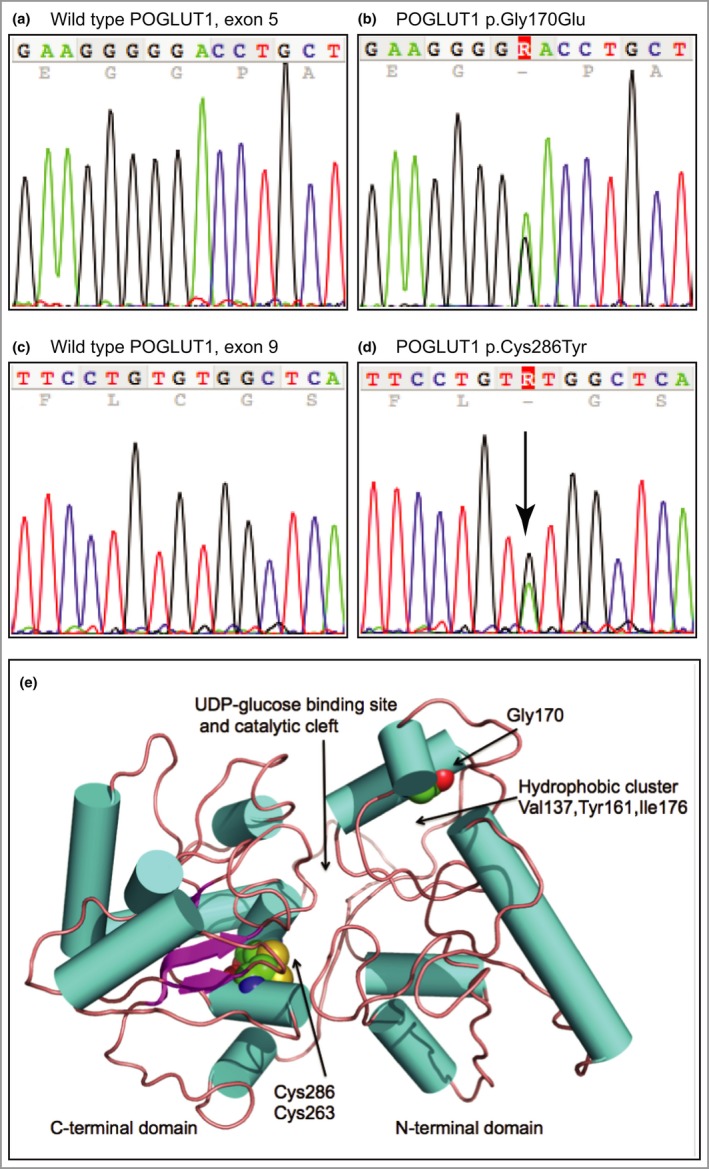
Mutation analysis and protein modelling. (a) Wild‐type POGLUT1 sequence of exon 5 showing nucleotides c.502–516. (b) Equivalent region as in (a) from the proband of family 2, showing the heterozygous mutation c.509G>A leading to missense mutation p.Gly170Glu. (c) Wild‐type POGLUT1 sequence showing nucleotides c.850–864 of exon 9. (d) Equivalent region as in (c) from the proband of family 3, showing the heterozygous mutation c.857G>A resulting in missense mutation p.Cys286Tyr. (e) The protein model of human POGLUT1. The enzyme structure is shown in cartoon format with cylinders (cyan) to represent α‐helices, and arrows (purple) β‐strands that are linked by coils (brown). The residues Gly170, Cys263 and Cys286 are depicted in CPK representation coloured N blue, S yellow, O red and C green. The N‐terminal domain consists of residues 1–180. The modelling predicts that each missense mutation, p.Gly170Glu and p.Cys286Tyr, would compromise enzyme activity due to a reduction in stability of the protein fold near the active site.
A 52‐year‐old woman, in family 3, presented at age 50 years with a 10‐year history of multiple, sometimes itchy, small red‐brown scaly lesions. These started on her legs then spread onto her forearms with a few lesions on her trunk (Fig. 1g–j). Her mother had similar lesions. Light microscopy of different skin biopsies variably showed focal hyperkeratosis, focal parakeratosis, acantholysis and elongation of rete ridges with a mild patchy chronic inflammatory infiltrate in the upper dermis (Fig. 1k). One biopsy showed acantholysis, (Fig. 1l). Screening of KRT5 and POFUT1 did not yield any mutations. A heterozygous missense mutation was identified in POGLUT1, p.Cys286Tyr; c.857G>A, in both affected individuals (Fig. 2c, d). PolyPhen‐2 and MutationTaster predict this previously unreported variant as disease causing; it is not in the dbSNP, NHLBI Exome Variant Server or ExAC databases. p.Cys286 is highly conserved across a number of species.
Mutations in POGLUT1 were reported by Basmanav et al.8 in 13 unrelated individuals. The majority were nonsense mutations, but there were also a splice‐site and a missense mutation. We identified one previously reported nonsense mutation, p.Arg218*, and two previously unreported missense mutations, p.Gly170Glu and p.Cys286Tyr. The p.Arg218* mutation results in a truncated form of POGLUT1 that lacks part of the substrate‐binding site required for high‐affinity binding of UDP‐glucose.8
Using homology modelling, we investigated the effects of the two missense mutations (Fig. 2e). Gly170 is on a short helical segment with C‐alpha hydrogens directed into a hydrophobic pocket that likely stabilizes a subdomain, part of the N‐terminal domain. This pocket is about 15 Å distant from the site of catalysis. A mutation to glutamate places a polar and acidic side chain into this hydrophobic environment. The result would be potentially destabilizing electrostatic interactions and steric clashes with Val137, Tyr161 and Ile176. Cys286 is in the C‐terminal domain, close to Cys263. The model strongly suggests that a disulfide bond would be formed by these two residues. Mutation of Cys286 to tyrosine would remove the highly stabilizing disulfide bond that holds together distinct elements of secondary structure, and then, depending on which side‐chain rotamers were adopted, to clash with Phe278, His282, Leu283, Val260 and Ser288, which form part of the hydrophobic core of the C‐terminal domain. Again, the consequence would be to destabilize the protein fold at a position about 15 Å from the site of catalysis. These models predict that each missense mutation, p.Gly170Glu and p.Cys286Tyr, would compromise enzyme activity due to a reduction in stability of the protein fold near the active site.
POGLUT1 adds O‐linked glucose to serine residues in epidermal growth factor‐like repeats of Notch receptors, and is an essential regulator of Notch signalling. Loss of Notch1 and Notch2 in mice results in abnormal pigmentation; these and other studies suggest that mutations in POGLUT1 resulting in aberrations in Notch signalling would lead to abnormal pigmentation and keratinocyte morphology.9, 10
Differences were noted between our cases. In family 2, lesions were widely scattered over the entire trunk and limbs (son) and on the neck and upper trunk region (father), rather than in a reticulated pattern. Similarly, in family 3 there was no reticulate rash compared with that of family 1. With only a small number of cases reported it is difficult to draw any genotype–phenotype correlation.
These findings expand the spectrum of mutations in POGLUT1 and confirm POGLUT1 as the third candidate gene, along with KRT5 and POFUT1, to consider in diagnosis of GGD/DDD.
Supporting information
Appendix S1. Supplementary methods.
Table S1. POGLUT1 primers for polymerase chain reaction and sequencing.
Acknowledgments
We thank all of the patients and families involved in this study.
Funding sources: F.J.D.S. and N.J.W. were supported by a grant from the Pachyonychia Congenita Project (to F.J.D.S.; www.pachyonychia.org). The Centre for Dermatology and Genetic Medicine at the University of Dundee is supported by a Wellcome Trust Strategic Award (098439/Z/12/Z to W.H.I.M.).
Conflicts of interest: none declared.
References
- 1. Muller CS, Tremezaygues L, Pföhler C, Vogt T. The spectrum of reticulate pigment disorders of the skin revisited. Eur J Dermatol 2012; 22:596–604. [DOI] [PubMed] [Google Scholar]
- 2. Hanneken S, Rütten A, Pasternack SM et al Systematic mutation screening of KRT5 supports the hypothesis that Galli‐Galli disease is a variant of Dowling‐Degos disease. Br J Dermatol 2010; 163:197–200. [DOI] [PubMed] [Google Scholar]
- 3. Schmieder A, Pasternack SM, Krahl D et al Galli‐Galli disease is an acantholytic variant of Dowling‐Degos disease: additional genetic evidence in a German family. J Am Acad Dermatol 2012; 66:e250–1. [DOI] [PubMed] [Google Scholar]
- 4. Betz RC, Planko L, Eigelshoven S et al Loss‐of‐function mutations in the keratin 5 gene lead to Dowling‐Degos disease. Am J Hum Genet 2006; 78:510–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reisenauer AK, Wordingham SV, York J et al Heterozygous frameshift mutation in keratin 5 in a family with Galli‐Galli disease. Br J Dermatol 2014; 170:1362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Basmanav FB, Fritz G, Lestringant GG et al Pathogenicity of POFUT1 in Dowling‐Degos disease: additional mutations and clinical overlap with reticulate acropigmentation of Kitamura. J Invest Dermatol 2015; 135:615–18. [DOI] [PubMed] [Google Scholar]
- 7. Li M, Cheng R, Liang J et al Mutations in POFUT1, encoding protein O‐fucosyltransferase 1, cause generalized Dowling‐Degos disease. Am J Hum Genet 2013; 92:895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Basmanav FB, Oprisoreanu AM, Pasternack SM et al Mutations in POGLUT1, encoding protein O‐glucosyltransferase 1, cause autosomal‐dominant Dowling‐Degos disease. Am J Hum Genet 2014; 94:135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aubin‐Houzelstein G, Djian‐Zaouche J, Bernex F et al Melanoblasts' proper location and timed differentiation depend on Notch/RBP‐J signaling in postnatal hair follicles. J Invest Dermatol 2008; 128:2686–95. [DOI] [PubMed] [Google Scholar]
- 10. Kumano K, Masuda S, Sata M et al Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res 2008; 21:70–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary methods.
Table S1. POGLUT1 primers for polymerase chain reaction and sequencing.