

Abstract
A comparison of P4 activations mediated by low‐valent β‐diketiminato (L) cobalt complexes is presented. The formal Co0 source [K2(L3Co)2(μ2:η1,η1‐N2)] (1) reacts with P4 to form a mixture of the monoanionic complexes [K(thf)6][(L3Co)2(μ2:η4,η4‐P4)] (2) and [K(thf)6][(L3Co)2(μ2:η3,η3‐P3)] (3). The analogue CoI precursor [L3Co(tol)] (4 a), however, selectively yields the corresponding neutral derivative [(L3Co)2(μ2:η4,η4‐P4)] (5 a). Compound 5 a undergoes thermal P atom loss to form the unprecedented complex [(L3Co)2(μ2:η3,η3‐P3)] (6). The products 2 and 3 can be obtained selectively by an one‐electron reduction of their neutral precursors 5 a and 6, respectively. The electrochemical behaviour of 2, 3, 5 a, and 6 is monitored by cyclic voltammetry and their magnetism is examined by SQUID measurements and the Evans method. The initial CoI‐mediated P4 activation is not influenced by applying the structurally different ligands L1 and L2, which is proven by the formation of the isostructural products [(LCo)2(μ2:η4,η4‐P4)] [L=L3 (5 a), L1 (5 b), L2 (5 c)].
Keywords: cyclic voltammetry, paramagnetic NMR, small molecule activation, white phosphorus, β-diketiminates
Introduction
The activation of white phosphorus (P4) with transition metal (TM) complexes with the objective of generating organophosphorus compounds has been an ongoing research topic.1 For this purpose, an understanding of the P4 transformation pathway in the coordination sphere of transition metals is necessary. Thus, a variety of Pn ligand moieties were stabilized to give insight into the stepwise P4 degradation and aggregation processes using well‐established ligand systems such as the CpR family (Cp=cyclopentadienyl).1 However, over the last years, β‐diketiminato (nacnac=L) ligand systems have gained increasing attention in mild P4 activations using MI precursors: The initial P4 fixation step of an intact P4 tetrahedron at a metal center was achieved at an electron‐rich CuI nacnac compound.2 In reactions with transition metal complexes of Groups 53 and 8–10,4 products with modified [P2]2−, [P4]0, [P4]2− and [P8]4− ligands, respectively, were obtained. So far, for nacnac systems, a [P3]3− ligand was found solely in compound [(L3VR)2(cyclo‐P3)]n− (R=N(tolyl)2, n=0,1; R=O(dipp), n=0) and [{L3V(N(tolyl)2)}2(μ2:η3,η2‐cyclo‐P3)] (A, Scheme 1).3c
Scheme 1.
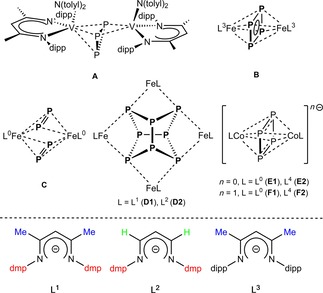
Selected examples of Pn‐TM complexes containing ligands L0–L4 (L0=CH[CHN(2,6‐iPr2C6H3)]2, L1=CH[C(Me)N(2,6‐Me2C6H3)]2, L2=CH[CHN(2,6‐Me2C6H3)]2, L3=CH[C(Me)N(2,6‐iPr2C6H3)]2, L4=CH[C(Me)N(2,6‐Et2C6H3)]2). Bottom: Comparison of the nacnac ligands used in this study, L1, L2 and L3.5
However, cyclo‐P3 complexes of the type [LM(μ2:η3,η3‐P3)M′L′]±n have been structurally characterized using neutral, tridentate triphos (1,1,1‐tris(diphenylphosphinomethyl)ethane) and etriphos (1,1,1‐tris(diethylphosphinomethyl)ethane) ligands in different combinations with 3d metals (Fe, Co, Ni) and 4d metals (Rh, Pd).6 The influence of the ligand substituents in FeI‐mediated P4 transformation has recently been illustrated by a comparative study using a set of ligands L1‐L3 (Scheme 1).7 Despite the application of the same reaction conditions, different products were obtained, which are sensitively dependent on small changes of the ligand substituents. 2,6‐Diisopropylphenyl (dipp) substituents as the ligands’ aromatic flanking groups support the formation of the dinuclear complexes [(L3Fe)2(μ2:η4,η4‐P4)] (B)7 and [(L0Fe)2(μ2:η2,η2‐P2)2] (C).4c The latter was synthesized by the Driess group.4c The ligands L0 and L3 only differ in their backbone substituents. However, for sterically less demanding 2,6‐dimethylphenyl (dmp) substituents, the formation of the tetranuclear complexes [(LFe)4(μ4:η2,η2,η2,η2‐P8)] [L=L1 (D1), L2 (D2)] with dimerized P4 units was observed.7 These results demonstrate that the product formation is affected by both the aromatic flanking groups and the ligand backbone substituents. Simultaneously, we investigated the [L3Co]‐mediated transformations of white phosphorus, which resulted in novel P4‐ and P3‐containing complexes (vide infra). In the meantime, Driess and co‐workers reported P4 activation by [LCo] fragments leading to the neutral complexes [(LCo)2(μ2:η4,η4‐P4)] [L=L0 (E1), L4 (E2)] (Scheme 1).4b One‐electron reduction led to the monoanionic products [K(dme)4][(LCo)2(μ2:η4,η4‐P4)] [L=L0 (F1), L4 (F2)] and transformed the [P4]0 middle deck into a [P4]2− ligand. Recently, Wolf and co‐workers have reported on [(BIAN)Co]−‐mediated P4 activations with a nacnac‐related bidentate redox non‐innocent BIAN (1,2‐bis(2,6‐diisopropylphenylimino)acenaphthene) ligand system yielding compounds containing [P4]4− moieties.8
Motivated by our first results with [L3Co] compounds, we speculated that the P4 activation outcome should be sensitive to the oxidation state of the precursor (Co0 versus CoI). Additionally, we wanted to address the question of the ligands’ influence (L1–L3) in CoI‐mediated P4 activations and we were intrigued by the observed P‐atom extrusion from the initially obtained P4 middle deck to form P3 compounds. The latter ones are still quite rare in comparison to P4 ligand complexes.
Here, we report on the P4 activation by a formal Co0 precursor yielding the monoanionic [K(thf)6][(L3Co)2(μ2:η4,η4‐P4)] (2) and [K(thf)6][(L3Co)2(μ2:η3,η3‐P3)] (3). Through a CoI‐mediated P4 transformation at room temperature or under thermolytic conditions, the corresponding neutral relatives are obtained, which generate 2 and 3 selectively after subsequent one‐electron reduction. The redox chemistry of the products was investigated by cyclic voltammetry (CV), and their magnetic behavior was examined both in solution (Evans method) and in the solid state (SQUID).
Results and Discussion
The formal Co0 precursor [K2(L3Co)2(μ2:η1,η1‐N2)] (1) was synthesized by a one‐pot reaction and was isolated as two different solvomorphs, 1⋅solv (solv=n‐hexane9 or OEt2).10 The X‐ray structures of 1⋅solv consist of two [L3Co] fragments bridged by a N2 unit. Two potassium atoms cover the N2 moiety and are coordinated in the phenyl pockets of the dipp substituents.11 The N−N distance in 1⋅n‐hexane/OEt2 is 1.215(3) and 1.220(4) Å, respectively, which is in line with the that (1.220(2) Å) of the previously reported [K2(L5Co)2(μ2:η1,η1‐N2)] [L5=CH[C(tBu)N(2,6‐iPr2C6H3)]2].12 The presence of a [N2]2− moiety in 1⋅n‐hexane is supported by Raman spectroscopy (νNN=1568 cm−1).9 The reaction of 1 with P4 proceeds by N2 evolution, showing that the formal [N2]2− species is re‐oxidized and revealing 1 as a formal dicobalt(0) starting material.
Conducting the reaction in 1:1 stoichiometry leads to the complete consumption of P4 and the formation of a mixture of the monoanionic complexes [K(thf)6][(L3Co)2(μ2:η4,η4‐P4)] (2) and [K(thf)6][(L3Co)2(μ2:η3,η3‐P3)] (3), which were detected by 1H NMR spectroscopy.13 The appearance of a cyclo‐P3 moiety in product 3 indicates that an extrusion of one P atom from the cyclo‐P4 moiety in 2 is possible. However, if the reaction is conducted with two equivalents of P4, compound 2 is the only product found in the 1H NMR spectrum.
The solid state structure of 2⋅2 thf reveals a salt consisting of two [K(thf)6]+ cations and two crystallographically distinguishable [(L3Co)2(μ2:η4,η4‐P4)]− monoanions (Figure 1).9 Each anion is a centrosymmetric dicobalt complex that consists of two [L3Co] fragments bridged by a planar cyclo‐P4 ligand. The P−P distances amount to 2.1913(10)–2.1951(10) Å in anion 1 and 2.1897(10)–2.2004(10) Å in anion 2, respectively. These values correspond well with the cyclo‐[P4]2− moiety (2.178(1) and 2.207(1) Å) of the reported compound [(L3Fe)2(μ2:η4,η4‐P4)] (B).7 The central P4 ligands in 2 are almost square planar with interior angles of 86.07(3) and 93.92(3)° in anion 1 and 86.38(3) and 93.62(3)° in anion 2. The Co−P distances range from 2.3362(7)–2.4149(7) Å in anion 1 and 2.3441(7)–2.4190(7) Å in anion 2. Selected atomic distances of compound 2 are summarized in Table 1. Minor deviations within the atomic parameters of compound 2⋅2 thf and the related compounds [K(dme)4][(LCo)2(μ2:η4,η4‐P4)] [L=L0 (F1), L4 (F2)] can be explained by small changes in the organic environment of the counter ion and the nacnac ligands of the complex monoanions. They may affect the Co⋅⋅⋅Co′ distances and their coordination geometry (torsion angle Θ between the Co⋅⋅⋅Co′ axis and the plane formed by the nitrogen atoms and the methine carbon in the ligand backbone; Figure 1 for graphical presentation of Θ).14
Figure 1.
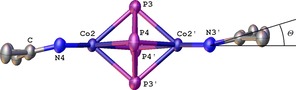
Anionic part of the molecular structure of 2. Hydrogen atoms and aromatic flanking groups are omitted for clarity; thermal ellipsoids are drawn at 50 % probability level. The torsion angle Θ is depicted spanning between the Co⋅⋅⋅Co′ axis and the plane formed by the nitrogen atoms and the methine carbon in the ligand backbone.
Table 1.
Comparison of P−P and Co⋅⋅⋅Co′ atomic distances and torsion angles Θ in anions of [K(solv)x][(LCo)2(μ2:η4,η4‐P4)] (L=L3 (2)9, L0 (F1)4b, L4 (F2)4b).
| Complex | 2:anion 1 | 2:anion 2 | F1 4b | F2 4b,4a |
|---|---|---|---|---|
| d(P−P) [Å] | 2.1913(10) | 2.1897(10) | 2.1739(7) | 2.154(1)[a] |
| 2.1951(10) | 2.2004(10) | 2.1976(7) | 2.163(1)[a] | |
| 2.225(1)[a] | ||||
| 2.230(1)[a] | ||||
| d(Co⋅⋅⋅Co′) [Å] | 3.603 | 3.625 | 3.626 | 3.603 |
| Θ [°] | 13.87(6) | 15.87(6) | 15.33(4) | 6.60(8)[a] |
| 14.97(7) [a] |
[a] Anion of F2 is not centrosymmetric. Therefore, four individual d(P−P) and two Θ values are given.
The monoanionic [(L3Co)2(μ2:η3,η3‐P3)]− was obtained in two different solvomorphs [K(dme)4][(L3Co)2(μ2:η3,η3‐P3)]⋅dme (3 a) and [K(thf)6][(L3Co)2(μ2:η3,η3‐P3)]⋅2 thf (3 b). Both compounds are ionic and consist of solvent (DME or THF) molecules, one solvent‐saturated potassium counter ion, and the [(L3Co)2(μ2:η3,η3‐P3)]− monoanion (Figures 2 and 3). In both X‐ray structures, the complex anions are built from two [L3Co] fragments bridged by a P3 triangle. In 3 a, the L3 ligand planes are almost parallel to each other with an dihedral angle of 2.00(7)° (N1‐N2 versus N3‐N4). However, in 3 b, the ligand planes are in a twisted conformation with a dihedral angel of 74.2(4)° (N1‐N2 versus N3‐N4, Figure 2).
Figure 2.

Comparison of the anions in the molecular structures of 3 a (left) and 3 b (right). Hydrogen atoms and aromatic flanking groups are omitted for clarity; thermal ellipsoids of Co and P atoms are drawn at 50 % probability level; major component of disordered cyclo‐P3 ligand is drawn in 3 a.
Figure 3.
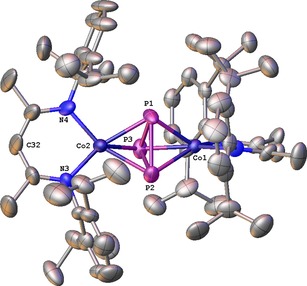
Anionic part of the molecular structure of 3 b. Hydrogen atoms are omitted for clarity; thermal ellipsoids are drawn at 50 % probability level.
The different complex anion conformations may originate from packing effects directed by the unequally shaped counter cations. The cyclo‐P3 middle deck is disordered over two positions in 3 a (occupancy 81:19).15 The middle deck in 3 b, however, is localized at one distinct position. As can be seen in Table 2, the P−P distances in 3 a are similar to the ones in the nacnac containing compound [{L3V(N(tolyl)2)}2(μ2:η3,η2‐cyclo‐P3)] (A),3c displaying a cyclo‐[P3]3− moiety. In 3 b, they compare better with the ones in [(triphos)Co(μ2:η3,η3‐cyclo‐P3)Fe(etriphos)](PF6)2 (G).6b Overall, they are in line with P−P single bonds [for comparison: P−P single bond in white phosphorus determined by X‐ray diffraction: 2.209(5) Å,16 electron diffraction: 2.1994(3) Å,17 Raman spectroscopy: 2.2228(5) Å,18 and DFT calculations: 2.1994(3) Å17]. The Co−P distances in 3 a are between 2.2046(17) and 2.3684(8) Å and for 3 b in the range of 2.248(3) and 2.277(3) Å. The Co⋅⋅⋅Co′ distance in 3 a is 3.7359(5) and amounts to 3.724(2) Å in 3 b, which is slightly elongated compared to 2 (3.603 and 3.625 Å, Table 1).
Table 2.
Comparison of selected atomic distances and angles in the [(L3Co)2(μ2:η3,η3‐P3)]− anion in 3 a (major component) and 3 b, [{L3V(N(tolyl)2)}2(μ2:η3,η2‐cyclo‐P3)] (A)3c and the dication in [(triphos)Co(μ2:η3,η3‐cyclo‐P3)Fe(etriphos)](PF6)2 (G).6b
| Complex | 3 a | 3 b | A 3c | G 6b |
|---|---|---|---|---|
| d(P−P) [Å] | 2.1674(13) | 2.217(4) | 2.1658(10) | 2.226(8) |
| 2.1790(16) | 2.224(4) | 2.1804(9) | 2.229(8) | |
| 2.3303(17) | 2.237(4) | 2.2155(9) | 2.234(8) | |
| ∢(P‐P‐P) [°] | 57.34(5) | 59.59(13) | 59.03(3) | – |
| 57.82(5) | 59.93(14) | 59.68(3) | ||
| 64.84(5) | 60.48(13) | 61.29(3) | ||
| d(M⋅⋅⋅M′) [Å] | 3.7359(5) | 3.724(2) | 4.460 | 3.80 |
| Θ [°] | 9.43(7) 12.22(7) | 8.7(3) 13.5(6) | – | – |
The 1H NMR spectra in [D8]THF display signals between 11.42 and −35.29 ppm for 2 9 and 8.15 and −12.85 ppm for 3, respectively. Except for the THF and DME signals, respectively, the 1H NMR spectra of 3 a, b do not deviate from each other. No signals are detected in the 31P{1H} NMR spectra for 2 and 3 due to their paramagnetic nature. Their magnetic moment (μeff) in [D8]THF solution (RT) was determined by the Evans method: 3.90 μB (2) and 3.51 μB (3). In the solid state, these values are confirmed by SQUID measurements displaying a gradual decrease of the magnetic moment in the temperature range from 300 to 2 K of 3.80 to 3.30 μB in 2 and 3.58 to 1.70 μB in 3 a. Therefore, the electronic structure of 2 is best described as containing a [P4]2− moiety bridging mixed valence CoI and CoII centers. This is in agreement with the previously reported compounds F1 and F2.4b Compound 3, however, contains a [P3]3− ligand, which is bridged by two CoII metal centers.
As mentioned above, starting from the formal Co0 precursor 1, we obtained the compounds 2 and 3 as a mixture of products, the ratio of which is sensitively dependent on stoichiometry and reaction conditions. To discover an alternative approach, we targeted the use of the CoI starting material [L3Co(tol)] (4 a), which was speculated to yield the neutral analogues of 2 and 3. After their one‐electron reduction, the compounds 2 and 3 should be accessible.
Therefore, the CoI compound [L3Co(tol)] (4 a) was reacted with P4, and [(L3Co)2(μ2:η4,η4‐P4)] (5 a) was selectively formed. Metric parameters and the characterization of compound 5 a are discussed in detail below.
Refluxing 5 a for three hours (110 °C, toluene) gives rise to the loss of one phosphorus atom and the formation of [(L3Co)2(μ2:η3,η3‐P3)] (6),19 which was clearly characterized by mass spectrometry20 and 1H NMR spectroscopy.21 The dinuclear compound contains two [L3Co] fragments, and the bridging middle deck exhibits a savage disorder within its cyclo‐P3 moiety. We emphasize that the P−P distances cannot be precisely described. However, the initially localized electron density unambiguously displays triangle‐shaped cyclo‐P3 constitution and enables an estimation of the P−P distances in 6 (approx. d(P−P): 2.147(3), 2.223(2), 2.235(2) Å). These values are comparable with the ones found in 3 a (2.1674(13), 2.1790(16), 2.3303(17) Å) and 3 b (2.217(4), 2.224(4) and 2.237(4) Å) and are elongated compared to the ones in A (2.1658(10), 2.1804(9) and 2.2155(9) Å).3c The Co⋅⋅⋅Co′ distance in 6 is 3.747 Å and therefore comparable to the ones in 3 a (3.7359(5) Å) and 3 b (3.724(2) Å), but elongated compared to its precursor complex 5 a (3.610 Å, vide infra).
The 1H NMR spectrum of 6 reveals signals between 20.06 and −12.68 ppm. No signal is detected in the 31P{1H} NMR spectrum. The magnetic moment (μeff) of 6 in C6D6 solution is 2.97 μB at room temperature (Evans method).22 This value is confirmed in the solid state by a SQUID measurement. A successive decrease from 2.7 to 2.0 μB was measured in the temperature range from 300 to 2 K (see the Supporting Information). The values are in agreement with antiferromagnetically coupled CoII and CoIII metal centers.
Electrochemistry
The electrochemical properties of the complexes 5 a and 6 were probed by cyclic voltammetry (CV) in THF solution containing Bu4NPF6 electrolyte (0.1 mol L−1, 295 K, see Supporting Information for further details).20 An irreversible oxidation was detected at E 1/2=−0.34 V for 5 a and E 1/2=−0.11 V for 6 (vs. Cp2Fe/Cp2Fe+). The compounds 5 a and 6 each reveal one reversible reduction at E 1/2=−1.62 V (vs. Cp2Fe/Cp2Fe+). The complexes 2 9 and 3 confirm these values by the corresponding electrochemical behavior. For 3, an additional reduction event was monitored at −2.52 V (vs. Cp2Fe/Cp2Fe+).
We experimentally performed the reduction of 5 a and 6, respectively, with one equivalent of potassium graphite in THF at room temperature. The corresponding anionic compounds [K(thf)6][(L3Co)2(μ2:η4,η4‐P4)] (2) and [K(thf)6][(L3Co)2(μ2:η3,η3‐P3)] (3), respectively, are selectively and quantitatively formed, which was proven by 1H NMR spectroscopy of the crude reaction solution. On a preparative scale, the isolated yields obtained as single crystals are 41 % for 2 and 62 % for 3. Consequently, regarding selectivity, this synthetic route is superior to the Co0‐mediated P4 activation, which, in contrast, yielded a mixture of products.
Impact of ligand design
Three β‐diketimines (L1H, L2H, L3H) were synthesized to provide a comparable hybrid ligand set L1–L3 with backbone (R=H, Me) and aromatic (Ph*=dmp or dipp) substituents (Scheme 1 and Scheme 2), and to investigate the influence of the ligand design on the CoI‐mediated P4 transformation. The [LCo(tol)] [L=L3 (4 a), L1 (4 b), L2 (4 c)] starting materials were prepared in one‐pot reactions (see the Supporting Information). All conducted P4 activation reactions were performed under the same conditions ([LCo(tol)]:P4=2:1, toluene, 2–3 h, RT) and yielded similar isolated products [(LCo)2(μ2:η4,η4‐P4)] [L=L3 (5 a), L1 (5 b), L2 (5 c)]. The crystals of all the new compounds 5 a–c were grown from saturated toluene solutions, and single crystal X‐ray diffraction was performed. The molecular structures of 5 a–c are shown in Figure S5 in the Supporting Information. As a representative, compound 5 a is presented in Figure 4 a. Its P4 moiety is rectangularly shaped, consequently spanned by two shorter and two longer P−P atom distances. Together with two coordinating Co atoms, the [P4Co2] complex core builds a distorted octahedron. In 5 a–c, the shorter P−P atom distances are between 2.1256(6) and 2.1301(7) Å and the longer P−P distances are between 2.2513(10) and 2.2980(7) Å. Compared with a phosphorus single bond in the tetrahedral P4, the planar rectangular‐shaped P4 moieties in 5 a–c contain a pair of shorter and a pair of elongated P−P bonds. The Co⋅⋅⋅Co′ distances in 5 a–c are between 3.502 and 3.610 Å and, therefore, any bonding interaction can be ruled out. Due to the centrosymmetric molecular structure (P21/n in 5 a–c), the ligands are parallel to each other. In 5 a–c, the torsion angels Θ (between the Co⋅⋅⋅Co′ axis and the plane formed by the nitrogen atoms and the methine carbon in the ligand backbone) are between 3.40(6) and 12.32(6)°. In 5 b, c (and E1, 2 4b), the P−P edges of the cyclo‐P4 unit are nearly parallel or rectangular, respectively, compared to the N−N axis of coordinating nitrogen atoms (compare ∢(NN‐PPshort)=ω 1 and ∢(NN‐PPlong)=ω 2, see Figure 4 b). The structural parameters of 5 a–c and E1, 2 are summarized in Table 3.
Scheme 2.
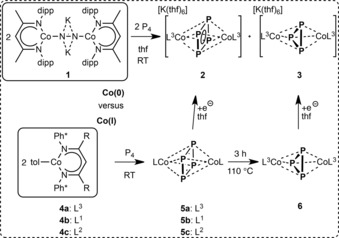
Performed Co0‐ and CoI‐mediated P4 transformations.
Figure 4.
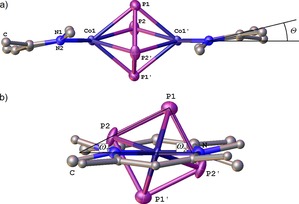
a) Molecular structure of the compound 5 a. Hydrogen atoms and aromatic flanking groups are omitted for clarity; thermal ellipsoids are drawn at 50 % probability level. The torsion angle Θ is depicted, which spans between the Co⋅⋅⋅Co′ axis and the plane formed by the nitrogen atoms and the methine carbon in the ligand backbone; b) view along Co1⋅⋅⋅Co1′ axis, revealing the angles ω 1 and ω 2, which span between the N−N axis of coordinating nitrogen atoms and the edges of the cyclo‐P4 unit.
Table 3.
Comparison of P−P and Co⋅⋅⋅Co′ atomic distances in neutral [(LCo)2(μ2:η4,η4‐P4)] [L=L3 (5 a), L1 (5 b), L2 (5 c), L0 (E1), L4 (E2)].
| Complex | 5 a | 5 b | 5 c | E1 4b | E2 4b |
|---|---|---|---|---|---|
| d(P−P) short [Å] | 2.1295(10) | 2.1256(6) | 2.1301(7) | 2.1237(13) | 2.1298(14) |
| d(P−P) long [Å] | 2.2513(10) | 2.2972(6) | 2.2980(7) | 2.2984(13) | 2.2889(15) |
| d(Co⋅⋅⋅Co′) [Å] | 3.610 | 3.502 | 3.503 | 3.491 | 3.533 |
| Θ [°] | 12.22(8) | 12.32(6) | 3.40(6) | 14.88(9) | 7.0(1) |
| ω 1 [°] | 26.34(6) | 2.18(4) | 1.97(4) | 1.96(7) | 2.58(8) |
| ω 2 [°] | 62.36(6) | 87.87(4) | 87.95(4) | 88.02(7) | 87.43(8) |
The 1H NMR spectra of the compounds 5 a–c display signals between 11.99 and −28.61 ppm and reveal their paramagnetic nature in solution. Therefore, no signals are detected in their 31P{1H} NMR spectra. Their magnetic moment (μeff) in solution (RT) was determined by the Evans method: 3.02 μB (5 a 22 in C6D6), 2.42 μB (5 b in C6D6), 1.84 μB (5 c in [D8]THF). In the solid state, however, the SQUID measurements of 5 a and 5 b display diamagnetic behavior in the temperature range of 2–300 K. Their electronic structure in the solid state is best described as two antiferromagnetically coupled CoI centers bridged by a [P4]0 ligand similar to the previously reported compounds E1, 2.4b In solution, exclusively one signal set for the ligand is observed in the 1H NMR spectrum of 5 a–c, respectively, suggesting the integrity of each dinuclear compound in solution on the NMR time scale (Figure S15).
Conclusion
We reported different [LCoI]‐mediated P4 activations yielding neutral complexes [(LCo)2(μ2:η4,η4‐P4)] (L=L1, L2, L3), each containing a similar rectangular‐shaped [P4]0 moiety. In contrast to the P4 activation by LFeI compounds, for the Co system, the ligand substituents (L1–L3) do not alter the reaction outcome. For the ligand system L3, we demonstrate that one P atom can be extruded thermolytically to generate an unprecedented neutral cyclo‐[P3]3−‐containing compound [(L3Co)2(μ2:η3,η3‐P3)]. As a novel approach, we present the P4 transformation with a formal [L3Co0] precursor, which generates corresponding monoanions with cyclo‐[P4]2− and cyclo‐[P3]3− ligands as a mixture of products. Each product was selectively accessed through the one‐electron reduction of its neutral precursor.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was initially supported by the Deutsche Forschungsgemeinschaft. The European Research Council (ERC) is acknowledged for the support in the SELFPHOS AdG‐339072 project. We are thankful to Dr. M. Bodensteiner for his valuable support regarding the solution of X‐ray structures. We extend our thanks to Dr. F. Dielmann for the initial X‐ray measurement of 5 a and to Dr. W. Patterson for the Raman measurement of 1⋅n‐hexane.
F. Spitzer, C. Graßl, G. Balázs, E. Mädl, M. Keilwerth, E. M. Zolnhofer, K. Meyer, M. Scheer, Chem. Eur. J. 2017, 23, 2716.
Contributor Information
Fabian Spitzer, http://www.uni‐regensburg.de/chemie‐pharmazie/anorganische‐chemie‐scheer/.
Prof. Dr. Manfred Scheer, Email: manfred.scheer@chemie.uni-regensburg.de.
References
- 1.
- 1a. Tofan D., Cossairt B. M., Cummins C. C., Inorg. Chem. 2011, 50, 12349–12358; [DOI] [PubMed] [Google Scholar]
- 1b. Caporali M., Gonsalvi L., Rossin A., Peruzzini M., Chem. Rev. 2010, 110, 4178–4235; [DOI] [PubMed] [Google Scholar]
- 1c. Scheer M., Balázs G., Seitz A., Chem. Rev. 2010, 110, 4236–4256; [DOI] [PubMed] [Google Scholar]
- 1d. Khan S., Sen S. S., Roesky H. W., Chem. Commun. 2012, 48, 2169–2179; [DOI] [PubMed] [Google Scholar]
- 1e. Giffin N. A., Masuda J. D., Coord. Chem. Rev. 2011, 255, 1342–1359. [Google Scholar]
- 2. Spitzer F., Sierka M., Latronico M., Mastrorilli P., Virovets A. V., Scheer M., Angew. Chem. Int. Ed. 2015, 54, 4392–4396; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4467–4472. [Google Scholar]
- 3.
- 3a. Tran B. L., Singhal M., Park H., Lam O. P., Pink M., Krzystek J., Ozarowski A., Telser J., Meyer K., Mindiola D. J., Angew. Chem. Int. Ed. 2010, 49, 9871–9875; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10067–10071; [Google Scholar]
- 3b. Camp C., Maron L., Bergman R. G., Arnold J., J. Am. Chem. Soc. 2014, 136, 17652–17661; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Pinter B., Smith K. T., Kamitani M., Zolnhofer E. M., Tran B. L., Fortier S., Pink M., Wu G., Manor B. C., Meyer K., Baik M.-H., Mindiola D. J., J. Am. Chem. Soc. 2015, 137, 15247–15261. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Yao S., Xiong Y., Milsmann C., Bill E., Pfirrmann S., Limberg C., Driess M., Chem. Eur. J. 2010, 16, 436–439; [DOI] [PubMed] [Google Scholar]
- 4b. Yao S., Lindenmaier N., Xiong Y., Inoue S., Szilvási T., Adelhardt M., Sutter J., Meyer K., Driess M., Angew. Chem. 2015, 127, 1266–1270; [DOI] [PubMed] [Google Scholar]
- 4c. Yao S., Szilvasi T., Lindenmaier N., Xiong Y., Inoue S., Adelhardt M., Sutter J., Meyer K., Driess M., Chem. Commun. 2015, 51, 6153–6156. [DOI] [PubMed] [Google Scholar]
- 5.The ligands L0–L4 are called “nacnac” in this report due to their structural similarities. More precisely, L0 and L2 can be regarded as vinylogous amidines and therefore are part of the “vinamidine” ligand family. See: Lloyd D., McNab H., Angew. Chem. Int. Ed. Engl. 1976, 15, 459–468; [Google Scholar]; Angew. Chem. 1976, 88, 496–504. [Google Scholar]
- 6.
- 6a.For (triphos)Co,Ni(triphos) and (triphos)Ni,Ni(triphos), see: Di Vaira M., Midollini S., Sacconi L., J. Am. Chem. Soc. 1979, 101, 1757–1763; [Google Scholar]
- 6b.for (triphos)Fe,Co(etriphos) and (triphos)Co,Co(triphos) (in footnote): Bianchini C., Di Vaira M. D., Meli A., Sacconi L., Inorg. Chem. 1981, 20, 1169–1173; [Google Scholar]
- 6c.for (triphos)Co,Rh(triphos) and (triphos)Ni,Rh(triphos): Bianchini C., Di Vaira M., Meli A., Sacconi L., J. Am. Chem. Soc. 1981, 103, 1448–1452; [Google Scholar]
- 6d.for (triphos)Pd,Pd(triphos): Dapporto P., Sacconi L., Stoppioni P., Zanobini F., Inorg. Chem. 1981, 20, 3834–3839; [Google Scholar]
- 6e.for a review, see: Di Vaira M., Sacconi L., Angew. Chem. Int. Ed. Engl. 1982, 21, 330–342; [Google Scholar]; Angew. Chem. 1982, 94, 338–351; for additional examples: [Google Scholar]
- 6f. Scherer O. J., Werner B., Heckmann G., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1991, 30, 553–555; [Google Scholar]; Angew. Chem. 1991, 103, 562–563; [Google Scholar]
- 6g. Mädl E., Balázs G., Peresypkina E. V., Scheer M., Angew. Chem. Int. Ed. 2016, 55, 7702–7707; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7833–7838; [Google Scholar]
- 6h. Nakanishi Y., Ishida Y., Kawaguchi H., Inorg. Chem. 2016, 55, 3967–3973. [DOI] [PubMed] [Google Scholar]
- 7. Spitzer F., Graßl C., Balázs G., Zolnhofer E. M., Meyer K., Scheer M., Angew. Chem. Int. Ed. 2016, 55, 4340–4344; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4412–4416. [Google Scholar]
- 8.S. Pelties, T. Maier, D. Herrmann, B. de Bruin, C. Rebreyend, S. Gärtner, I. G. Shenderovich, R. Wolf, Chem. Eur. J DOI: 10.1002/chem.201603296. [DOI] [PubMed]
- 9.F. Spitzer, Master thesis, University of Regensburg 2013.
- 10.After lithiation of L3H and subsequent transmetalation with CoBr2, its reduction was performed with an excess of potassium graphite under N2 atmosphere.
- 11.In 1⋅n-hexane the NCCCN ligand planes are orientated parallel to each other. However, in 1⋅OEt2, they are twisted (dihedral angel of coordinating N atoms: 32.38(9)°). Further analytical data (X-ray, 1H NMR, Evans method, EA, Raman spectrum) and pictures of 1⋅solv are presented in the Supporting Information.
- 12. Ding K., Pierpont A. W., Brennessel W. W., Lukat-Rodgers G., Rodgers K. R., Cundari T. R., Bill E., Holland P. L., J. Am. Chem. Soc. 2009, 131, 9471–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For initial investigations see reference [9]. The identification of products 2 and 3 in the 1H NMR spectra was enabled after their selective preparation.
- 14.The dependency of torsion angle Θ and Fe⋅⋅⋅Fe′ distance was already recognized in citation [7].
- 15.Due to the disorder of the cyclo-P3 moiety, only the geometric parameters of the major component are discussed. Furthermore, it has to be noted that the positions of the disordered P3 and P3A atoms are close to each other and hence, the involved bond lengths might not be determined with high accuracy.
- 16.In β-P4: Simon A., Borrmann H., Craubner H., Phosphorous and Sulfur 1987, 30, 507–510. [Google Scholar]
- 17. Cossairt B. M., Cummins C. C., Head A. R., Lichtenberger D. L., Berger R. J. F., Hayes S. A., Mitzel N. W., Wu G., J. Am. Chem. Soc. 2010, 132, 8459–8465. [DOI] [PubMed] [Google Scholar]
- 18. Brassington N. J., Edwards H. G. M., Long D. A., J. Raman Spectrosc. 1981, 11, 346–348. [Google Scholar]
- 19.The crystal structures of 6 and 5 a were initially reported in reference [20]. However, to present improved structural values, crystals of 6 and 5 a were re-measured.
- 20.C. Grassl, PhD thesis, University of Regensburg, 2013.
- 21.During the thermolysis, a black, pyrophoric precipitate is formed. The CHN analysis of the washed and dried precipitate suggests only some organic content (C%: ≈35, H%: ≈5, N%: ≈3). Exclusively, compound 6 and L3H were detected in the 1H NMR spectrum (reaction control) as the only soluble products formed during the thermolysis reaction.
- 22.For initial investigations see reference [20]. For further information see the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary