

ABSTRACT
Adenosine (A) to inosine (I) RNA editing is important for life in metazoan organisms. Dysregulation or mutations that compromise the efficacy of A to I editing results in neurological disorders and a shorten life span. These reactions are catalyzed by adenosine deaminases acting on RNA (ADARs), which hydrolytically deaminate adenosines in regions of duplex RNA. Because inosine mimics guanosine in hydrogen bonding, this prolific RNA editing alters the sequence and structural information in the RNA landscape. Aicardi-Goutières syndrome (AGS) is a severe childhood autoimmune disease that is one of a broader set of inherited disorders characterized by constitutive upregulation of type I interferon (IFN) referred to as type I interferonopathies. AGS is caused by mutations in multiple genes whose protein products, including ADAR1, are all involved in nucleic acid metabolism or sensing. The recent crystal structures of human ADAR2 deaminase domain complexed with duplex RNA substrates enabled modeling of how AGS causing mutations may influence RNA binding and catalysis. The mutations can be broadly characterized into three groups; mutations on RNA-binding loops that directly affect RNA binding, “second-layer” mutations that can alter the disposition of RNA-binding loops, and mutations that can alter the position of an α-helix bearing an essential catalytic residue.
KEYWORDS: ADAR; Aicardi-Goutieres Syndrome; base-flipping; A to I; inosine; RNA editing
Introduction
RNA is frequently edited after being transcribed by RNA polymerase. These enzyme-catalyzed transformations include inserting, deleting, converting or modifying ribonucleotides.1,2 One of the most abundant RNA modifications found in metazoans is the deamination of adenosine to inosine (A-to-I). This reaction is catalyzed by the class of enzymes called adenosine deaminases acting on RNA (ADAR).3 Like the nucleoside cytidine and adenosine deaminases, ADARs require a zinc-activated water to hydrate the nucleobase followed by a loss of ammonia to yield the hydrolytic deamination product. However, ADARs also require the binding of inositol hexakisphosphate (IHP) in the protein core to maintain stability and activity.4 Another difference with the nucleoside deaminases is that ADARs only deaminate nucleotides in regions of duplex secondary structure.
In the ADAR deamination reaction, the resulting inosine nucleotide product behaves more like guanosine in hydrogen-bonding patterns, consequently A-to-I edits can significantly alter the RNA function. Variations in function include changes in codon meaning (i.e., recoding) in mRNA, altering splice sites, and changing RNA secondary structure. Over three million RNA A-to-I edits have been documented in healthy human tissues.5 A to I editing is critical for normal cellular function and dysregulated ADAR activity can result in a variety of neurological disorders such as epilepsy and Prader Willi Syndrome, depression, schizophrenia, amyotrophic lateral sclerosis, as well as cancer.6-10
There are three ADARs in humans; ADAR1, ADAR2, and ADAR3. Interestingly, while ADAR1 and ADAR2 are both catalytically active and expressed in almost all tissues, the enigmatic ADAR3 is expressed mainly in the brain and has no detectable catalytic activity. Genetic knockouts of ADAR1 and ADAR2 in mice result in embryonic or neonatal death respectively11-13 establishing the significance of ADARs and RNA A-to-I editing in organismal health.
ADAR protein structures are modular, with all ADARs possessing a homologous catalytic or deamination domain comprising roughly the C-terminal 400 amino acids and N-terminal nucleic acid binding domains14,15 (Fig. 1A). ADAR1 is 1226 residues in length, and contains two Z-DNA binding domains (Z-DBD) and three double-stranded RNA binding domains (dsRBDs) within its N-terminal ∼800 residues, while ADAR2 is 701 residues and contains only two dsRBDs and no Z-DBD. ADAR3 also contains two dsRBDs plus and an N-terminal Arginine-rich segment, in addition to a non-functional “catalytic domain.”
Figure 1.

Primary structure of ADAR. (A) Schematic representation of the modular domains of the three human ADARs. Catalytic deaminase domain is represented in yellow. (B) Sequence alignment of ADAR deaminase domains from diverse organisms. The secondary structural elements observed in the human ADAR2 X-ray structure are represented above the sequence alignment. The seven AGS-causing missense mutations are marked with cyan-colored triangles below the sequence. The catalytic Glutamate is marked by a magenta square, and the ADAR-specific RNA-binding loop is highlighted by a green line.
ADARs recognize their RNA substrates partially through the dsRBDs, which characteristically span two minor grooves, but because they interact with 2′-hydroxyls and phosphodiester backbones, dsRBDs typically bind non-specifically to dsRNA. Recent evidence suggests the sequence specificity that dictates which adenosines get deaminated by ADARs is partially mediated through the C-terminal deaminase domain.16,17 This suggests dsRBDs draw the ADARs to the RNA substrate and the catalytic domain plays an important role in determining which adenosines get edited to inosine.
While many diseases mentioned above are associated with dysregulated ADAR activity, two disorders, Dyschromatosis Symmetrica Hereditaria (DSH) and Aicardi-Goutières Syndrome (AGS), can be attributed to specific mutations mapped to the ADAR1 gene. Additionally, most of the mutations of these disorders map to the catalytic domain18-20 corroborating the important role the catalytic domain has on identifying RNA targets and efficiently editing these dsRNA sites. Patients with DSH, commonly found in East Asian countries, display a rare pigmentary genodermatosis, but are otherwise asymptomatic healthy individuals.21
AGS is a severe childhood autoimmune disease that has a genetic basis but also clinical and biochemical similarities to congenital infection.22,23 It is one of a broader set of inherited disorders characterized by constitutive upregulation of type I interferon (IFN) (i.e., type I interferonopathies).23,24 AGS is caused by mutations in multiple genes whose protein products, including ADAR1, are all involved in nucleic acid metabolism or sensing, suggesting a variety of nucleic acid ligands trigger the AGS-associated IFN response.24 Sensing nucleic acids that are of viral origin and distinguishing these from host nucleic acids is a key aspect of the human immune response. Recent studies suggest the presence of increased levels of cytosolic double stranded RNA arising from defects in ADAR1 activity caused by AGS mutations leads to interferon induction.25,26 Normal ADAR1 activity would create multiple IU mismatches in these RNAs, preventing them from binding to the cytosolic dsRNA sensor IFIH1 (also known as MDA5).26,27 While AGS has also been linked to mutations in the TREX1, RNASEH2A, RNASEH2B RNASEH2C, SAMHD1 and IFIH genes,22 this review will focus on AGS-causing mutations mapped to the ADAR1 deaminase domain and the likely consequences of these mutations on RNA recognition by the deaminase domain. There are currently no structures of ADAR1 available. However, the 400 residue ADAR1 and ADAR2 deaminase domains are 39% identical and 59% similar in amino acid sequence (Fig. 1B), indicating their overall structures will be very similar. In addition, our recently determined crystal structure of human ADAR2 deaminase domain complexed with dsRNA revealed key RNA recognition elements on the deaminase domain surface and provides a useful structural model for predicting the effect of AGS mutations found in the ADAR1 deaminase domain.28 Throughout this paper we will utilize the single-letter amino acid code to designate ADAR1 numbering and the three-letter code for ADAR2 numbering.
Structure of ADAR2 catalytic domain-RNA complex
The structure of human ADAR2 catalytic domain was first determined in 2005 in the absence of any RNA substrate.4 This structure determined the overall fold, the active site zinc ion, and the unexpected presence of inositol hexakisphosphate (IHP) buried in the protein core that likely helps stabilize the active fold. We recently reported four crystal structures of the human ADAR2 catalytic domain bound to 23 bp RNA duplexes of different sequences each containing the nucleoside analog 8-azanebularine (8-azaN) in place of adenosine at the edited site.28 Upon bonding in the ADAR active site, 8-azanebularine becomes hydrated mimicking the proposed intermediate thus forming a tight-binding dsRNA-ADAR complex amenable for structural studies.29,30
The dsRNA-ADAR structure revealed approximately 20 bp of the dsRNA interacts with amino acid residues found entirely on loops between secondary structural elements of ADAR (Fig. 2). Both strands of RNA interact with ADAR with the majority of the contacts mediated through the phosphodiester-ribose backbone near the editing site. The overall RNA duplex bends very little upon binding ADAR, however the width of the major groove widens opposite the edited site, but is compensated by the narrowing of the adjacent minor groove.
Figure 2.
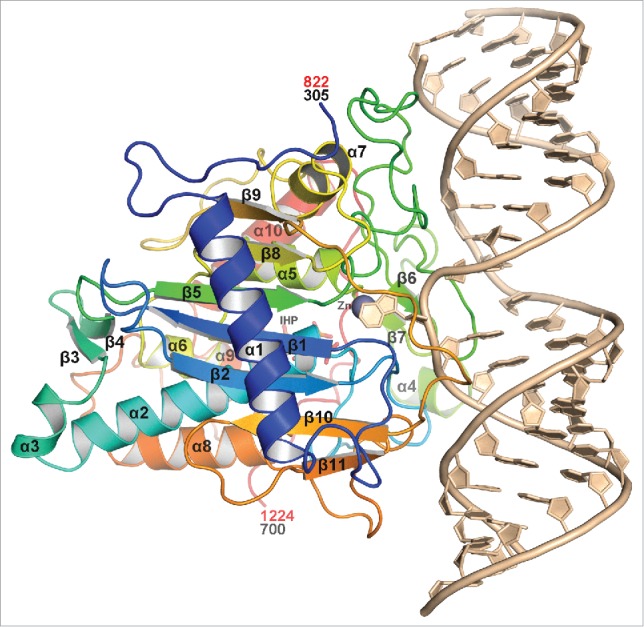
Overall ribbon view of hADAR2d complexed with dsRNA substrate. The protein is colored in the rainbow spectrum starting with blue at the N-terminus and ending with red at the C-terminus. The secondary structure elements are labeled. RNA is colored in sand with the 8-azaN flipped out interacting with the Zn2+ ion, gray colored sphere. IHP, which is mostly obscured in this view, is drawn as sticks. Numbers in red indicate equivalent hADAR1 numbering.
Strictly conserved Glu488 (ADAR2 numbering) pushes out the edited base flipping it ∼180° out of the RNA duplex into the ADAR active site, where it interacts with the zinc ion and the invariant active site residue Glu396. Glu488 is flanked by conserved Glycine residues Gly487 and Gly489, allowing the deep penetration of Glu488 into the RNA duplex minor groove, forcing out the edited adenine and occupying the vacated space, where it hydrogen-bonds to the orphan base on the opposite strand (Fig. 3).
Figure 3.
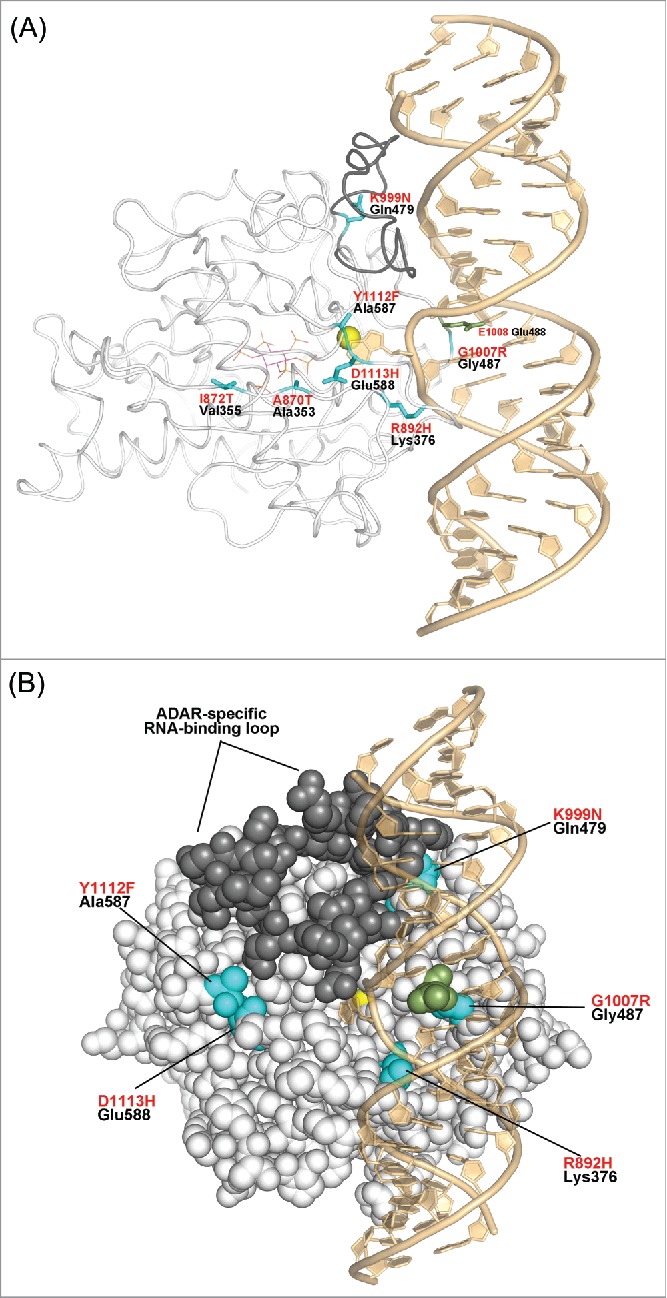
hADAR1 AGS-causing mutants mapped onto the hADAR2-dsRNA complex structure. (A) The hADAR2 structure drawn as white-colored tube, with the ADAR-specific RNA-binding loop colored in darker gray, which was disordered in the hADAR2d RNA-free structure.4 The hADAR2 residues equivalent to the hADAR1 AGS-causing mutations are show in cyan with black labels. hADAR1 AGS-causing mutations are labeled in red text, mapped onto the structure. Glu488, which pushed out the edited base is drawn in green sticks, Zn2+ ion in yellow sphere, and IHP is dawn in sticks with magenta-colored carbons. (B) Space-filling representation rotated slightly with respect to (a).
Comparing the ADAR2 RNA-bound structure to RNA-free structure revealed the structures are very similar with a root-mean-squared deviation (rmsd) of 0.296Å for 296 equivalent residues. The largest deviations occur in RNA-binding loops, and a surface loop involved in a crystal lattice contact. More importantly, the dsRNA-ADAR2 structure revealed the conformation of a 17-residue loop (residues 458–474) that was disordered in the RNA-free crystal structure (Fig. 3). Upon binding dsRNA substrate, this loop becomes ordered because it partially enters the major groove 5′ of the edited site, and interacts with both strands of the RNA phosphodiester backbone. These residues are part of a larger loop (454–477), which we call the ADAR-specific RNA-binding loop because, while conserved in each ADAR family, the sequence varies between ADAR1, ADAR2 and ADAR3 suggesting it may help dictate RNA substrate specificity.
Mapping Aicardi-Goutières syndrome (AGS) mutations on the ADAR2-RNA complex structure
Of the eight AGS missense mutations identified in human ADAR1 gene, seven of them map to the catalytic domain.19 Remarkably only three of the seven mutations map to locations in the ADAR structure directly involved in RNA binding and include mutants; R892H, K999N, and G1007R (Fig. 3). The R892H mutant was identified as part of a double mutant with P193A, which is located in the N-terminal Z-DNA/Z-RNA–binding domain. Therefore, it is unknown if the R892H mutation alone will confer AGS disease. R892 corresponds to Lys376 in ADAR2 that interacts with phosphates of nucleotides G13 and A14 adjacent to the edited base A12. Therefore, the conservative substitution of R892H is expected to alter RNA orientation and positioning of the edited base, thereby possibly reducing its catalytic efficiency. This mutation could also conceivably change ADAR1 substrate specificity because subtle differences in sequence-influenced RNA structure may affect binding affinity at this site.
K999N AGS mutant of ADAR1 maps to Gln479 in the ADAR2 structure. This residue lies at the C-terminal end of the ADAR-specific RNA-binding loop, much of which is structurally disordered in the absence of RNA.4 In ADAR2 the side chain of Gln479, which points away from the RNA, is near the phosphate of U15 of the non-edited strand (i.e.,6 Å away). In ADAR1, it is likely K999 will interact with the RNA phosphate backbone at this location. Therefore, the K999N substitution found in the AGS mutant is expected to alter RNA binding affinity and affect target RNA deamination efficiency.
G1007R is the most radical substitution found of all the AGS causing ADAR1 mutants. ADAR1 G1007 maps to Gly487 of ADAR2, which immediately precedes the base-flipping residue of Glu488. These residues are part of the invariant GEG triplet sequence where the flanking glycines allow for the close approach and intercalation of Glu488 in the minor groove of RNA substrate and flipping out of the edited adenine base. Consequently, mutating either glycine next to Glu488 will drastically alter the enzymes ability to promote base flipping. Indeed, the base-flipped conformation revealed in our ADAR-RNA crystal structures would not be possible with the G1007R mutation. This is consistent with activity studies indicating that the G1007R mutation in ADAR1 blocks editing activity.19,25 It is interesting to note that all the AGS patients carrying the G1007R mutation are heterozygous,19 likely because of the deleterious consequence of this mutation, while AGS patients with the other mutations are homozygous. Indeed, complete loss of ADAR1 protein activity is embryonic lethal, so it is expected that patients carrying highly inhibitory mutations would not be homozygous.
Two of the AGS causing mutations that do not map to the RNA interface are A870T and I872T. Like the R892H mutant, both A870T and I872T mutants were identified as part of a double mutant also with P193A,19 so it is unknown how large of an effect the P193A mutant may have on AGS. Nevertheless, both A870T and I872T lie on strand β1 and point toward and interact with helix α2 (Figs. 3A, and 4). A870 of ADAR1 is equivalent to Ala353 of ADAR2 and makes van der Waals contact (3.5–4.0Å) with the aliphatic portion of the essential catalytic residue, Glu396 (E912 of ADAR1) (Fig. 4). Therefore, the A870T mutation will change the disposition of the catalytic glutamate residue altering catalytic deamination efficiency. The I872T mutant lies further away from the active site, but I872 of ADAR1 can form a hydrophobic interaction with F919 and the aliphatic portion of R916 on helix α2 (Fig. 4). ADAR1 active site residue E912, along with the zinc ligand H910, lie at the N-terminal end of helix α2, so the I872T mutant may also shift the helix axis, altering catalytic activity. It is interesting to note that conserved residues R916 and R917, which lie on the opposite side of helix α2 near I872, interact with IHP suggesting that IHP may help position this helix for catalytic efficiency (Fig. 4).
Figure 4.
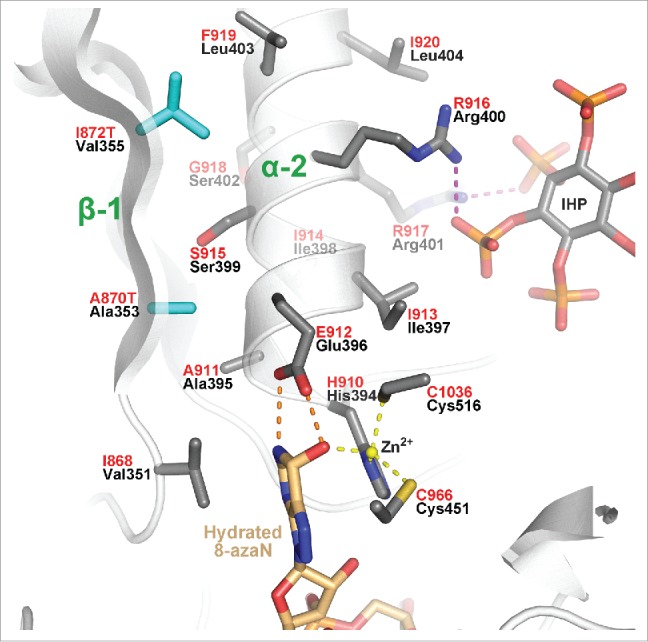
Close-up view of active site revealing the location of the two buried ADAR1 AGS-causing mutations A870T and I872T (cyan) mapped on the hADAR2 structure. Invariant catalytic residue Glu396/E912 hydrogen-bonds (orange dashed lines) with flipped-out edited base analog; 8-azaN. Zinc coordination is illustrated with yellow dashed lines, and conserved Arginines of helix α2 that interact with IHP phosphates are shown with magenta dashed lines.
The final two AGS-causing mutations, Y1112F and D1113H do not map near the RNA interface or the active site. However, these residues can be categorized as “second layer” mutations, because they lie adjacent to RNA-interacting loops. These mutations map to Ala587 and Glu588 of ADAR2 respectively (Fig. 5) and lie at the N-terminal side of the β9-β10 loop, which is a long loop that extends across much of the protein on the side facing the RNA (Fig. 2). Lys594 of ADAR2 (K1120 in ADAR1) lies at the C-terminal end of this loop and protrudes in the major groove of the RNA substrate where it interacts with phosphate of G5 of the complementary unedited strand (Fig. 5). ADAR1 residue D1113 corresponds to Glu588 of ADAR2, which forms a salt link with highly conserved Arg349 (R866 ADAR1) in the α1-β1 loop (Fig. 5). Arg349 is adjacent to Arg348, which like Lys594, also interacts with the phosphate of G5 of the unedited strand. Interestingly, Arg348 is conserved in the ADAR2 family, but is actually a conserved Glycine in the ADAR1 family (G865). However, on the other side of highly conserved Arg349 (R866 ADAR1) is Lys350 (K867 ADAR1), which is conserved in all ADAR families. While this Lys350 does not directly interact with RNA substrate in the ADAR2-RNA structure, it points toward the major grove and lies close to the phosphate adjacent to the flipped-out 8-azaN. The D1113H mutation in ADAR1 would disrupt the ion pair with R866 and distort both the loop that contains K867 as well the loop that contains D1113 and conserved R1116 that is directed to the major groove of RNA (Fig. 5).
Figure 5.
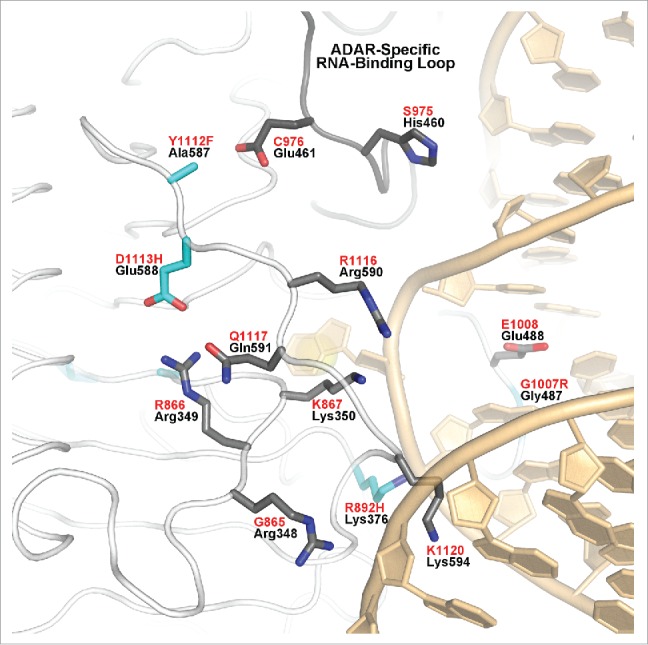
“Second-layer” AGS mutations of hADAR1 Y1112F and D1113H (cyan) mapped on hADAR2 structure. Ala587, equivalent to ADAR1 Y1112F, points toward Glu461, C976 in ADAR1, which is part of the ADAR-specific RNA-binding loop (shown with gray-colored main chain tube). hADAR1 and hADAR2 numbering are in red and black, respectively.
Lying next to D1113H in ADAR1 is another AGS-causing mutation, Y1112F (Fig. 5). Y1112F maps to Ala587 in the ADAR2 structure. In ADAR2, the Ala587 side chain points toward Glu461 in the ADAR-specific RNA-binding loop (gray-colored main chain Fig. 5). These two residues in ADAR1 are Y1112 and C976, which may contact each other either through hydrophobic interactions or hydrogen bonding. The AGS-causing mutation of Y1112F could disrupt this interaction triggering a shift or rearrangement of the ADAR-specific RNA-binding loop (Fig. 5). Thus, the Y1112F mutation may alter RNA interactions resulting in less efficient deamination of normally targeted adenosines, or deamination of new adenosines from a distorted ADAR-specific RNA-binding loop mediating new RNA contacts.
Conclusions
Recently reported crystal structures of human ADAR2 deaminase domain bound to duplex RNA and the high degree of similarity between the deaminase domains of ADAR1 and ADAR2 allows us to postulate consequences of AGS mutations found in the ADAR1 deaminase domain. Importantly, besides G1007R, which will prevent base flipping and block editing entirely, other AGS mutations are expected to have more subtle effects on editing activity by creating conservative changes immediately at the protein-RNA interface (R892H, K999N), by altering the position of an α-helix bearing an essential catalytic residue (I872T, A870T) or by changing the positioning of RNA-binding loops on the deaminase domain surface (Y1112F, D1113H). This analysis is consistent with the observation that some level of editing activity is maintained for all mutants except G1007R.19,25 Further structural studies of human ADAR1 in complex with RNA are needed to test these predictions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Grosjean H. Fine-tuning of RNA functions by modification and editing. Berlin; New York: Springer, 2005. [Google Scholar]
- 2.Zipeto MA, Jiang Q, Melese E, Jamieson CH. RNA rewriting, recoding, and rewiring in human disease. Trends Mol Med 2015; 21:549-59; PMID:26259769; http://dx.doi.org/ 10.1016/j.molmed.2015.07.001 [DOI] [PubMed] [Google Scholar]
- 3.Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988; 55:1089-98; PMID:3203381; http://dx.doi.org/ 10.1016/0092-8674(88)90253-X [DOI] [PubMed] [Google Scholar]
- 4.Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 2005; 309:1534-9; PMID:16141067; http://dx.doi.org/ 10.1126/science.1113150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Picardi E, Manzari C, Mastropasqua F, Aiello I, D'Erchia AM, Pesole G. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep 2015; 5:14941; PMID:26449202; http://dx.doi.org/ 10.1038/srep14941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallo A, Locatelli F. ADARs: allies or enemies? The importance of A-to-I RNA editing in human disease: from cancer to HIV-1. Biol Rev Camb Philos Soc 2012; 87:95-110; PMID:21682836; http://dx.doi.org/ 10.1111/j.1469-185X.2011.00186.x [DOI] [PubMed] [Google Scholar]
- 7.Silberberg G, Lundin D, Navon R, Ohman M. Deregulation of the A-to-I RNA editing mechanism in psychiatric disorders. Hum Mol Genet 2012; 21:311-21; PMID:21984433; http://dx.doi.org/ 10.1093/hmg/ddr461 [DOI] [PubMed] [Google Scholar]
- 8.Maas S, Kawahara Y, Tamburro KM, Nishikura K. A-to-I RNA editing and human disease. RNA Biol 2006; 3:1-9; PMID:17114938; http://dx.doi.org/ 10.4161/rna.3.1.2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slotkin W, Nishikura K. Adenosine-to-inosine RNA editing and human disease. Genome Med 2013; 5:105; PMID:24289319; http://dx.doi.org/ 10.1186/gm508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morabito MV, Abbas AI, Hood JL, Kesterson RA, Jacobs MM, Kump DS, Hachey DL, Roth BL, Emeson RB. Mice with altered serotonin 2C receptor RNA editing display characteristics of Prader-Willi syndrome. Neurobiol Dis 2010; 39:169-80; PMID:20394819; http://dx.doi.org/ 10.1016/j.nbd.2010.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000; 406:78-81; PMID:10894545; http://dx.doi.org/ 10.1038/35017558 [DOI] [PubMed] [Google Scholar]
- 12.Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 2004; 279:4894-902; PMID:14615479; http://dx.doi.org/ 10.1074/jbc.M311347200 [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 2004; 279:4952-61; PMID:14613934; http://dx.doi.org/ 10.1074/jbc.M310162200 [DOI] [PubMed] [Google Scholar]
- 14.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci U S A 1994; 91:11457-61; PMID:7972084; http://dx.doi.org/ 10.1073/pnas.91.24.11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature 1996; 379:460-4; PMID:8559253; http://dx.doi.org/ 10.1038/379460a0 [DOI] [PubMed] [Google Scholar]
- 16.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2011; 2:319; PMID:21587236; http://dx.doi.org/ 10.1038/ncomms1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eifler T, Pokharel S, Beal PA. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry 2013; 52:7857-69; PMID:24124932; http://dx.doi.org/ 10.1021/bi4006539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou Y, Chen J, Gao M, Zhou F, Du W, Shen Y, Yang S, Zhang XJ. Five novel mutations of RNA-specific adenosine deaminase gene with dyschromatosis symmetrica hereditaria. Acta dermato-venereologica 2007; 87:18-21; PMID:17225010; http://dx.doi.org/ 10.2340/00015555-0168 [DOI] [PubMed] [Google Scholar]
- 19.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al.. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 2012; 44:1243-8; PMID:23001123; http://dx.doi.org/ 10.1038/ng.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing Q, Wang M, Chen X, Qian X, Qin W, Gao J, Wu S, Gao R, Feng G, He L. Identification of a novel ADAR mutation in a Chinese family with dyschromatosis symmetrica hereditaria (DSH). Arch Dermatol Res 2005; 297:139-42; PMID:16133458; http://dx.doi.org/ 10.1007/s00403-005-0589-1 [DOI] [PubMed] [Google Scholar]
- 21.Hayashi M, Suzuki T. Dyschromatosis symmetrica hereditaria. J Dermatol 2013; 40:336-43; PMID:22974014; http://dx.doi.org/ 10.1111/j.1346-8138.2012.01661.x [DOI] [PubMed] [Google Scholar]
- 22.Crow YJ, Livingston JH. Aicardi-Goutieres syndrome: an important Mendelian mimic of congenital infection. Dev Med Child Neurol 2008; 50:410-6; PMID:18422679; http://dx.doi.org/ 10.1111/j.1469-8749.2008.02062.x [DOI] [PubMed] [Google Scholar]
- 23.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 2015; 15:429-40; PMID:26052098; http://dx.doi.org/ 10.1038/nri3850 [DOI] [PubMed] [Google Scholar]
- 24.Livingston JH, Crow YJ. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutieres Syndrome and Beyond. Neuropediatrics 2016; 47:355-60; PMID:27643693; http://dx.doi.org/ 10.1055/s-0036-1592307 [DOI] [PubMed] [Google Scholar]
- 25.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al.. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 2014; 9:1482-94; PMID:25456137; http://dx.doi.org/ 10.1016/j.celrep.2014.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA EDITING RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015; 349:1115-20; PMID:26275108; http://dx.doi.org/ 10.1126/science.aac7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liddicoat BJ, Chalk AM, Walkley CR. ADAR1, inosine and the immune sensing system: distinguishing self from non-self. Wiley Interdiscip Rev RNA 2016; 7:157-72; PMID:26692549; http://dx.doi.org/ 10.1002/wrna.1322 [DOI] [PubMed] [Google Scholar]
- 28.Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, Beal PA. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol 2016; 23:426-33; PMID:27065196; http://dx.doi.org/ 10.1038/nsmb.3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haudenschild BL, Maydanovych O, Veliz EA, Macbeth MR, Bass BL, Beal PA. A transition state analogue for an RNA-editing reaction. J Am Chem Soc 2004; 126:11213-9; PMID:15355102; http://dx.doi.org/ 10.1021/ja0472073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phelps KJ, Tran K, Eifler T, Erickson AI, Fisher AJ, Beal PA. Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res 2015; 43:1123-32; PMID:25564529; http://dx.doi.org/ 10.1093/nar/gku1345 [DOI] [PMC free article] [PubMed] [Google Scholar]