

ABSTRACT
MicroRNAs (miRNAs) regulate the expression of mRNA through sequence-specific binding of the 3′ untranslated region (UTR). The seed sequence of miRNAs is the key determinant for target site recognition. Paralogous miRNAs, which share the same seed sequences but differ in their 3′ regions, are known to regulate largely overlapping groups of mRNAs. However, no study has analyzed functional differences between paralogous miRNAs with proper experimental methods. In this study, we compared the targets of paralogous miRNAs, miR-221 and miR-222. Using a nuclease-mediated genome engineering technique, we established knockout cell lines for these miRNAs, and precisely analyzed differences in target regulation. We found that miR-221 and miR-222 suppress the previously identified targets, CDKN1B and CDKN1C, differentially. Whereas both miRNAs suppressed CDKN1B, only miR-221 suppressed CDKN1C. From transcriptome analyses, we found that several different target mRNAs were regulated by each of miR-221 and miR-222 independently, although a large number of mRNAs responded commonly to miR-221 and miR-222. This is the first study to compare the mRNA regulations by paralogous miRNAs and illustrate that paralogous miRNAs with the same seed sequence also have difference in target regulation.
KEYWORDS: Knockout, microRNA, miR-221, miR-222, TALEN
Introduction
MicroRNAs (miRNAs) are small regulatory RNAs involved in diverse physiologic processes. Their inaccurate expression leads to various diseases.1,2 miRNA genes are transcribed by RNA polymerase II as long primary transcripts, called primary miRNAs (pri-miRNAs), and are cleaved by the complex of DROSHA and DGCR8 proteins.3 The resultant hairpin RNAs, called precursor miRNAs (pre-miRNAs), are exported into the cytoplasm by Exportin 5 protein. In the cytoplasm, pre-miRNAs are further cleaved into miRNA duplexes by DICER, and loaded into ARGONAUTE (AGO) protein complex. Between the miRNA duplexes, only one strand, which is called guide strand, remains in the AGO protein. The other strand called the passenger strand is discarded. This strand selection is determined by the thermodynamic stability of nucleotides at each end of miRNA duplexes. Depending on their origin between the 5′ and 3′ arms in the pre-miRNAs, the final single stranded miRNAs, also called mature miRNAs, are designated as 5p and 3p miRNAs, respectively.
In the complex with AGO proteins, miRNAs bind to mRNAs in a sequence-specific manner. The primary binding of nucleotides occurs in the 2nd to 8th position from the 5′ end of the miRNA, which is called ‘seed’ sequence. This is critical because miRNAs with highly similar sequences, but with a single nucleotide difference in the seed sequence region, were shown to suppress non-overlapping groups of mRNAs.4 The structure of AGO with guide DNA and target RNA duplexes also suggests the importance of seed regions for miRNA function.5 Because of the importance of seed binding, diverse algorithms were devised to predict the targets of miRNAs based on the strict requirement of a seed match.6
However, several studies also suggested an important role for regions other than seed sequences. In an early study, it was shown that compensation by strong binding of the 3′ region of miRNA to mRNA overcame weak base-pairing at the seed region.7 Moreover, another study reported other types of miRNA-targeting mechanisms wherein the central region of miRNAs has a major role in binding to target sites.8 In a recent transcriptome-wide study, it was also shown that approximately 40% of miRNA-dependent AGO binding did not occur at the seed match-containing region.9 These reports suggested that it is important to consider the sequences outside the seed region for the prediction of miRNA targets. However, this functional importance has not been fully established compared with that of seed sequences.
Because of the importance of the seed region for miRNA function, its sequences are highly conserved, at the species level, for diverse miRNAs. Moreover, between paralogous miRNAs, which have originated from a common ancestor, seed sequences are identical even though the 3′ regions differ. However, for each paralogous miRNA, sequences at the 3′ region are usually similar among different species.10 This suggests that miRNA sequences in the 3′ region may also have important functional roles. Nevertheless, few studies have identified functional differences between paralogous miRNAs.
In this study, we compared the function of paralogous miRNAs, specifically miR-221 and miR-222, through a genome engineering technique. We ablated each miRNA, and measured functional impacts based on target reporters and transcriptome-wide analyses. From these analyses, we found that although they share the common seed sequence and have overlapping functions, miR-221 and miR-222 also have different target mRNAs and functions.
Results
Nuclease-mediated knockout of miR-221 and miR-222
To understand the functional differences between paralogous miRNAs with the same seed sequence, we ablated miRNA-coding sequences through a genome engineering technique. For knockout, we selected miR-221 and miR-222, which reside in close proximity on the X chromosome, and constitute a miR-222∼221 cluster (tilde indicates a transcript spanning the indicated gene cluster) (Fig. 1A). From the miR-221 and miR-222 loci, 3p strand miRNAs are mainly incorporated into AGO, and accordingly, the function of miR-221–3p and miR-222–3p have been extensively studied. They share the same seed sequence, and differ by four nucleotides at their middle and 3′ regions. In the miRBase database, they were registered from diverse vertebrate species.10 To analyze their sequence conservation, we collected the sequences of the hairpin regions of miR-221 and miR-222 from different species with available genome sequences. Strikingly, the sequences were determined to be identical at the entire 3p region for both miR-221 and miR-222 (Fig. S1). This implies that in addition to the seed sequences, those sequences of middle and 3′ region are also important for the function of these miRNAs.
Figure 1.
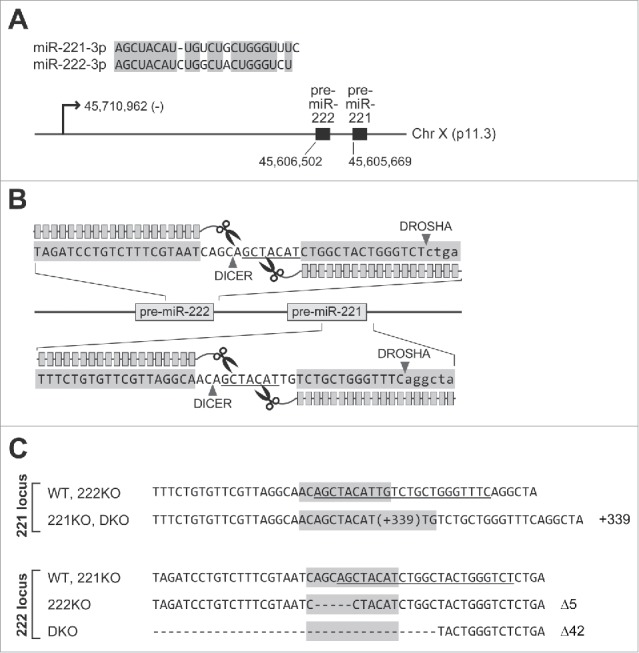
TALEN-mediated knockout of miR-221 and miR-222. (A) Sequence alignment of miR-221–3p and miR-222–3p (upper panel), and genomic information of pri-miR-222∼221 cluster (lower panel) are shown. The transcriptional start site of pri-miR-222∼221 (GRCh37/hg19) was identified in our recent study.30 The 5′ ends of the pre-miRNAs were annotated by using the sequencing data from diverse cell lines.4,24 (B) Depiction of TALEN constructs used for the knockout of miR-221 and miR-222. Two TALEN constructs were used as a pair for the knockout of each miRNA. The consecutive boxes designate TAL-effector DNA binding domains while the scissor indicates the nuclease domain. The seed sequences (2nd to 8th nucleotides from 5′ end of the miRNA) are underlined. Note that the TALEN constructs were designed to cleave the DNA region corresponding to seed sequences of miRNAs. (C) Confirmation of DNA mutation by sequencing. For wild type and knockout cell lines, the DNA sequences adjacent to targeted regions are shown. On the right, the numbers of nucleotides inserted or deleted are shown. The underlined nucleotides indicate the sequences of miR-221–3p and miR-222–3p. The DNA region designated for TALEN-mediated cleavage is shaded with gray color.
To introduce mutations into the miRNAs, we used the previously established transcription activator-like effector nucleases (TALENs).4 The TALEN constructs were designed to cleave the seed sequence regions, resulting in a functional blockade even with a small mutation (Fig. 1B). For knockout, we selected the SNU-638 gastric cancer cell line, which has a near diploid genome.11 Because this cell line is derived from a male patient, and has a normal X chromosome karyotype, a single allele for the miR-222∼221 cluster exists in these cells. Thus, mutation of just one allele enabled us to obtain knockout cell lines. After transfection of TALEN constructs, targeting miR-221 and miR-222, into SNU-638 cells, we selected single cell clones. By sequencing the genomic DNA encompassing the targeted region, we confirmed the mutation for miR-221 (221KO) and miR-222 (222KO) (Fig. 1C). To make double knockout (DKO) cells for both miR-221 and miR-222, we transfected TALEN constructs for miR-222 into the 221KO clone. Finally, a cell line with mutations in both miRNAs was obtained (DKO).
Verification of miR-221 and miR-222 knockout cell lines
Based on genomic DNA sequences, we predicted the secondary structures of miR-221 and miR-222 from wild type and knockout cells (Fig. 2A). Because of the large insertion of DNA into the sequence of miR-221, miRNAs are expected to be not expressed in 221KO and DKO clones. To confirm the defect in miRNA expression, we extracted total RNAs and performed Northern blot analysis. As expected, miR-221 was not produced in these cell lines (Fig. 2B).
Figure 2.
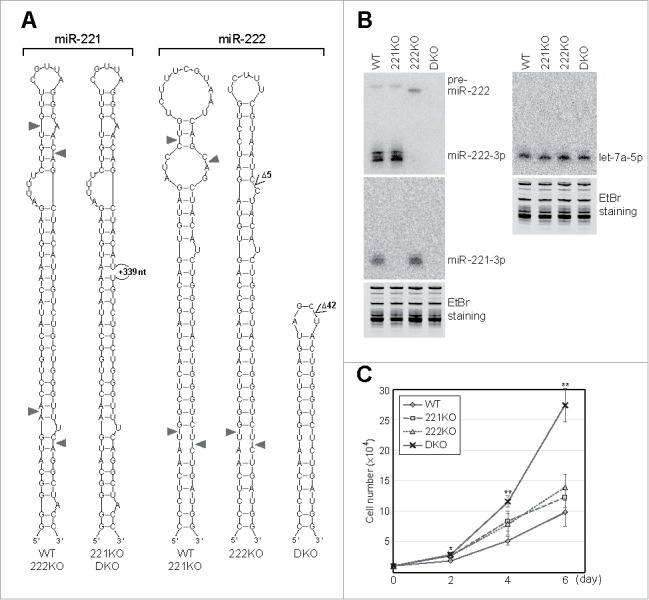
Verification of miR-221 and miR-222 knockout cell lines. (A) The predicted secondary structures of miR-221 and miR-222 from the DNA sequences of wild type and knockout cell lines. The cleavage sites of DROSHA and DICER are indicated with triangles. (B) The expression of miR-221 and miR-222 was measured by Northern blot analysis. The level of representative miRNA, let-7a, was also measured as a control. The image of the gel stained with ethidium bromide (EtBr) is also shown as a loading control. (C) The proliferation rates of wild type and knockout cells were measured. Ten thousand cells were plated at day 0, and the cell numbers were counted two, four, and six days later. Error bars show standard errors from six independent biologic replicates (n = 6). P values were calculated using two-tailed t-test (*: P < 0.01, **: P < 0.001). Note that only the proliferation of DKO cells shows significant increase in all time points.
The predicted secondary structure of miR-222 in the 222KO clone showed a stable double-stranded stem region and smaller terminal and internal loops compared with that of miR-222 in wild type cells. However, miR-222 was not detected in the 222KO clone (Fig. 2B). Because of observed accumulation of pre-miR-222, the DICER processing step was expected to be obstructed in the 222KO clone. In the case of miR-222 in the DKO clone, DROSHA cleavage was expected to be blocked because pre-miR-222 was not observed (Fig. 2B). This was expected as the hairpin of pre-miR-222 became too small for cleavage by DROSHA in the DKO clone (Fig. 2A).
The expression level of let-7a, which was selected as a control miRNA, was similar among wild type and knockout cell lines. Moreover, the knockout of miR-221 did not affect the expression of miR-222, and vice versa (Fig. 2B). These data suggest that TALEN constructs that we used were specific.
Previous studies reported both oncogenic and tumor-suppressive functions of miR-221 and miR-222 in different types of human cancer cells.12 To test the effect of miRNA ablation on the SNU-638 cells that we used, cell proliferation rate was monitored by cell counting (Fig. 2C). The knockout of either miR-221 or miR-222 slightly increased the cell proliferation rate compared with the proliferation of wild type cells. In comparison to this, the proliferation was significantly increased when both miRNAs were deleted. This result shows that miR-221 and miR-222 together exert an anti-proliferative effect in the SNU-638 cells.
Differences in target suppression between miR-221 and miR-222
To analyze the functional effects of miR-221 and miR-222 knockout on target regulation, we used reporter constructs for known targets of these miRNAs, specifically CDKN1B and CDKN1C (Fig. 3A). The 3′ untranslated region (UTR) of CDKN1B has two binding sites for miR-221–3p and miR-222–3p, whereas that of CDKN1C has one site. In previous studies, ectopic expression of synthetic miRNA mimics for both miR-221 and miR-222 suppressed the expression of both targets in the cells.13-18 In addition, the expression of these targets was increased by treatment with inhibitors targeting both miRNAs. Accordingly, it was concluded that CDKN1B and CDKN1C are targets of both miR-221 and miR-222.13-18
Figure 3.
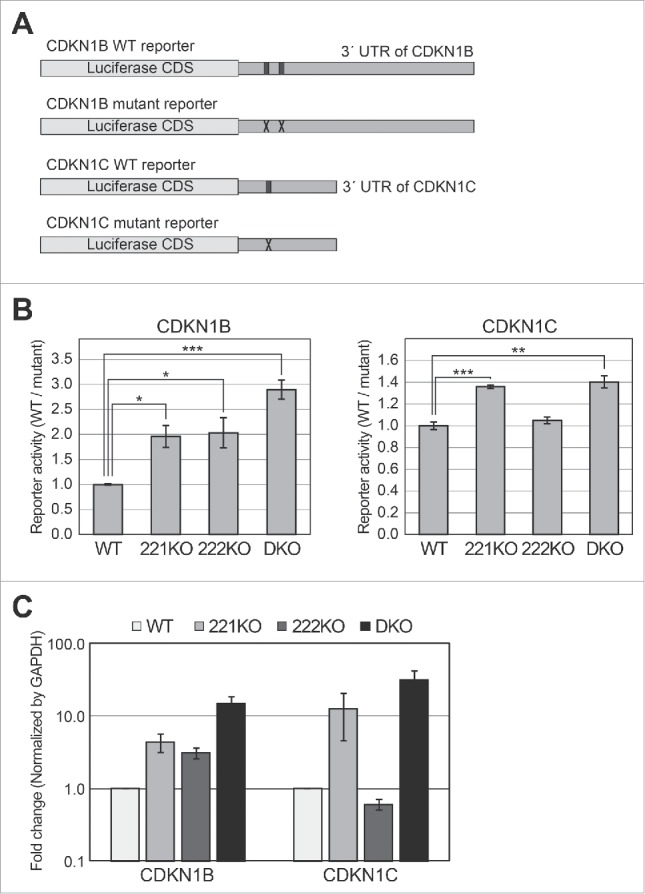
Reporter assays using targets of miR-221 and miR-222 in knockout cells. (A) Schematic representation of reporter constructs used in this experiment. The black boxes in the 3′ UTR of the wild type (WT) reporters indicate the binding sites of miR-221–3p and miR-222–3p. The ‘X’ marks in mutant reporters indicate the mutation introduced to block base-pairing between miRNAs and their target sites (Fig. S2). (B) Luciferase assays were performed using the CDKN1B and CDKN1C reporters. After the transfection of WT or mutant reporters into the cells, luciferase activity was measured. After normalizing the activity of the WT reporter by that of the mutant reporter, we compared the relative activities among wild type and knockout cells. Error bars show standard errors from four independent biologic replicates (n = 4). P values were calculated using a one-tailed t-test (*: P < 0.01, **: P < 0.001, ***: P < 0.0001). (C) Quantitative PCR experiments were performed to measure the mRNA levels of CDKN1B and CDKN1C in wild type and knockout cells, respectively. Error bars show standard errors from five independent biologic replicates (n = 5).
We transfected a luciferase construct harboring the 3′ UTR sequence of CDKN1B or CDKN1C into wild type or knockout cells. For the normalization control, we used the same constructs but with mutant binding sites for miR-221 and miR-222 (Fig. 3A).18 We found that the activity of CDKN1B reporter was increased by knockout of both miR-221 (221KO) and miR-222 (222KO) (Fig. 3B). In addition, the increase in reporter activity was more prominent in the DKO cells compared with that of individual knockouts. When we measured the mRNA level of CDKN1B in the knockout cells, the same pattern with luciferase activity was observed (Fig. 3C). This suggests that these two miRNAs suppress CDKN1B cooperatively.
In the case of CDKN1C, the reporter activity was increased in 221KO and DKO cells (lacking expression of miR-221) (Fig. 3B). However, activity was unchanged, compared with control, when the luciferase construct was introduced into knockout cells of miR-222 (222KO). Moreover, the degree of de-repression was nearly identical between 221KO and DKO cells, although the expression level of miR-222 was very different between these two cell lines (Fig. 3B). We confirmed that the mRNA level of CDKN1C also increased only in the 221KO and DKO cells (Fig. 3C). These data suggest that even though miR-221 and miR-222 have the same seed sequence, only miR-221 has a major effect on the suppression of CDKN1C, which is contrary to previous reports.13-18 The knockout of endogenous miRNA genes in this study has fewer side effects compared with ectopic expression of miRNA mimics or inhibitors in previous studies (see Discussion). Our results clearly show differential regulation of target genes with paralogous miRNAs, in the cell lines that we tested.
The difference between the responses of CDKN1B and CDKN1C to miR-221 and miR-222 shows that the sequences outside of the seed region are also important for specific suppression of target mRNAs. Because there is no critical difference in the number of base-paired nucleotides between miRNA and target mRNA (Fig. S2), we still do not understand the reason for differential regulation of CDKN1C between miR-221 and miR-222.
Global analysis of mRNA profile in the knockout cells
To analyze differences in global mRNA regulation between miR-221 and miR-222, we performed RNA sequencing (RNA-seq) analysis on RNAs from wild type and knockout cells. After mRNA enrichment, we applied Illumina deep sequencing. Based on RNA-seq data, we filtered out mRNAs with low expression, removed those with deviation between biologic duplicates greater than 2-fold. To increase the reliability of mRNA candidates for target analysis, we collected the data made by crosslinking and immunoprecipitation followed by high-throughput sequencing (CLIP-seq) for AGO proteins,19 and selected only the mRNAs with AGO binding signals (see Materials and Methods). Finally, 5,605 mRNAs were obtained for the sequence analyses.
We analyzed the 3′ UTR sequences of these mRNAs, and found that 461 contained a fully complimentary 7-mer motif required for binding the seed sequence of miR-221 and miR-222. When we calculated the cumulative fraction of mRNAs based on their fold changes, mRNAs with complimentary 7-mer sequences were enriched slightly but significantly in all the knockout cells (Figs. 4A-C). These results suggest that knockout of either miR-221 or miR-222 is sufficient to increase the expression of seed sequence-containing target mRNAs in a global level. Moreover, the fold changes of mRNAs between knockout cells showed high correlation values (Figs. 4D-F). Thus, miR-221 and miR-222 share a large proportion of common targets. Our data also suggests that sum of the amount of these two miRNAs in the cells is important for target suppression, and depletion of either miRNA could de-repress the expression of target mRNAs.
Figure 4.
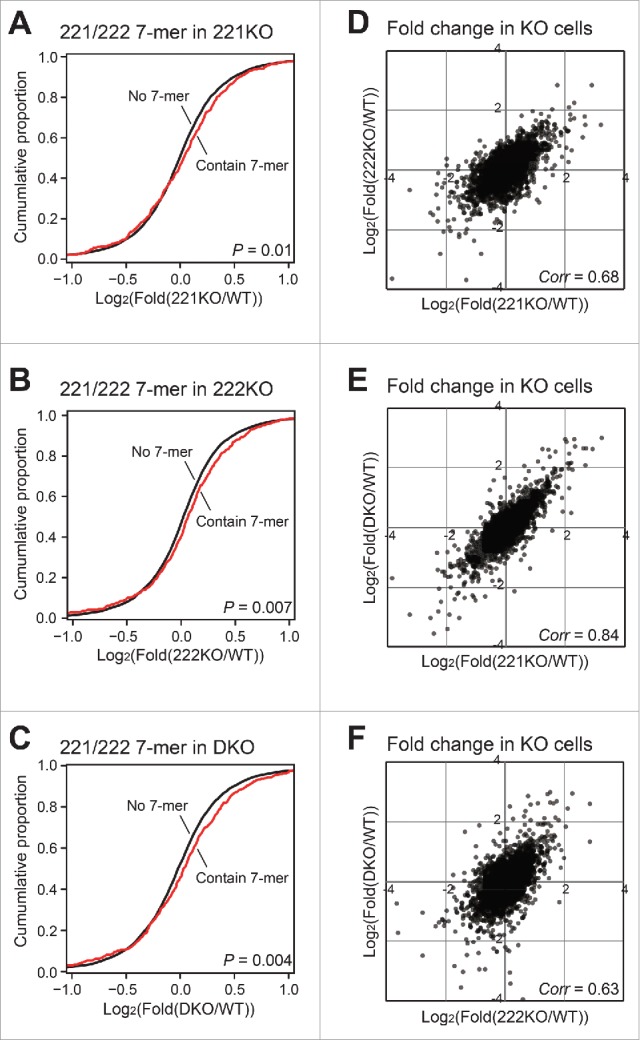
Transcriptome analyses of miR-221 and miR-222 knockout cells. (A-C) Based on the RNA-seq data from wild type and knockout cells, we selected 5,605 mRNAs (see Materials and Methods). Among these mRNAs, we identified those with 3′ UTR containing the 7-mer motif common to miR-221–3p and miR-222–3p. The fold changes between 7-mer containing mRNAs and the other mRNAs were compared by cumulative plots. The levels of mRNAs from (A) 221KO, (B) 222KO, and (C) DKO cells were compared with those of wild type cells for the calculation of fold changes. The P values were calculated by the Kolmogorov–Smirnov test. (D-F) The fold changes of mRNAs between knockout cells were compared. The correlation values for each comparison were shown.
Analysis of the targets of miR-221 and miR-222
To analyze how many targets overlap between miR-221 and miR-222, we compared the elevated mRNAs among the knockout cells. For this, only the mRNAs with expression change between knockout and wild type (KO/WT) greater than 1.3-fold in at least one knockout library were chosen among the mRNAs selected in Fig. 4. This filtering resulted in 136 mRNAs with AGO-CLIP signal and 7-mer motif at their 3′ UTR (Fig. 5A and Table S1).
Figure 5.
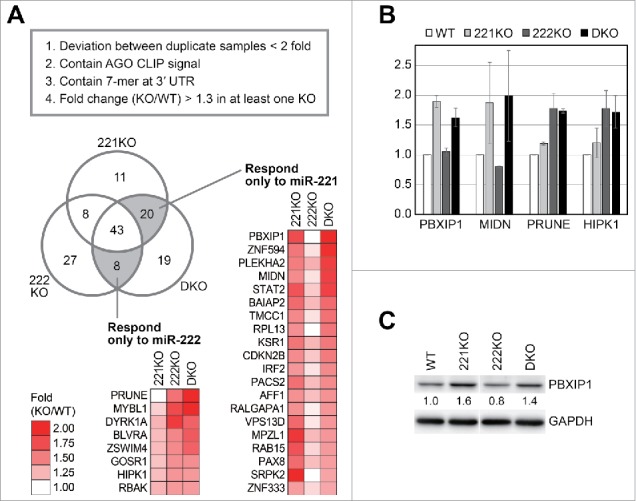
Target analyses of miR-221 and miR-222. (A) From the RNA-seq data, we removed the mRNAs with high deviation in fold changes between biologic duplicates, and selected those with AGO-CLIP signal and 7-mer motif at their 3′ UTR. Then, we selected only the mRNAs increased more than 1.3-fold in any knockout cells compared with wild type cells. To collect mRNAs that responded only to miR-221 or miR-222, the fold changes of mRNAs between knockout cell lines were compared, and the number of mRNAs in each population was shown in a Venn diagram. We selected mRNAs that were commonly increased in 221KO and DKO but not in 222KO, as those responding only to miR-221. The mRNAs responding only to miR-222 were also selected by the similar way. The gene names and fold changes of mRNAs in these groups were shown with heat maps. (B and C) We selected several genes, and confirmed their expression change in the knockout cells through (B) quantitative PCR and (C) Western blotting. Error bars in (B) show standard errors from two or three independent biologic replicates (n = 2 or 3). The gap between the lanes in (C) indicates a discontinuous lane from a single gel.
When increased mRNAs were compared among the knockout libraries, the largest fraction of mRNAs was included in the group commonly overlapped among all libraries, which accounts for about one third of mRNAs (43/136) (Fig. 5A and Table S1). Thus, the mRNAs suppressed by one of the miRNAs between miR-221 and miR-222 have a tendency to be suppressed also by the other. However, there were still several mRNAs which responded only to either miR-221 or miR-222. Twenty mRNAs responded only to miR-221 ablation (commonly upregulated in both 221KO and DKO, but not in 222KO), and eight mRNAs were increased only by miR-222 knockout (commonly upregulated in both 222KO and DKO, but not in 221KO) (Fig. 5A and Table S1). We selected several genes, and confirmed their expression change in the knockout cells (Figs. 5B and C). In summary, our data show that in addition to common targets of miR-221 and miR-222, there are also specific targets for each of these miRNAs although these two miRNAs have the same seed sequence.
Discussion
Functional studies of miRNAs generally use synthetic oligonucleotides such as miRNA mimics and inhibitors. Because the ectopic expression of small RNA mimics perturbs the endogenous population of AGO-associated miRNAs, these studies have the potential to lead to wrong interpretation.20 Conversely, miRNA inhibitors cannot discriminate between miRNAs with similar sequences.21 Therefore, these experimental methods are not suitable to study the functional differences between paralogous miRNAs with highly similar sequences. In this study, we used knockout experiments to study functional differences between paralogous miRNAs. Recent studies reported the successful knockout of miRNA genes using TALEN and Cas9 nucleases.4,22,23 We used a TALEN-based knockout technique to specifically delete miR-221 and miR-222 (paralogous miRNAs), which did not noticeably affect expression of the other miRNA (Fig. 2B). Because more than half of miRNAs in humans have paralogous counterparts,10 knockout techniques will be useful for the functional dissection of these miRNAs.
It is plausible that the miRNAs produced from 5p arms of miR-221 and miR-222 hairpins (miR-221–5p and miR-222–5p) have functional effects in cells. When we analyzed the expression of these 5p miRNAs using public data, we found that their expression levels are very low among diverse cancer cell lines (Table S2).24 We also could not detect a signal for miR-221–5p or miR-222–5p using Northern blot analysis. Although we could not rule out a possible role for these 5p miRNA species entirely, it is probable that they have a limited role in the cells.
One unexpected finding from this study was the similar CDKN1C reporter activity between wild type and 222KO cells. In previous studies, we and other researchers showed suppression of CDKN1C through ectopic expression of a miR-222 mimic, and activation by a miRNA inhibitor.13-18 However, our knockout study suggests that CDKN1C is not suppressed by miR-222 despite having a seed match at its 3′ UTR. In contrast, this target was suppressed by miR-221, a paralogous miRNA. Thus, each paralogous miRNA could exert its suppressive effect by differentially acting on the same target mRNAs. This also suggests that the existence of seed binding sequences does not guarantee the suppression of mRNAs by corresponding miRNAs. This notion was further verified by analyzing global mRNAs expression profiles in the knockout cells (Fig. 5). We found that several mRNAs were regulated exclusively to each either miR-221 or miR-222. We could not identify any rule determining this specific regulation. In a recent study, it was reported that the 3′ region of miRNA contributes to the specific binding of miRNAs from the same family into target mRNAs.25 However, this finding does not apply to our results, because the number of base-pairs between 3′ region of miRNA and target mRNA was similar between the binding of miR-221 and miR-222 into CDKN1C (Fig. S2). Thus, there maybe is another rule governing the interaction between these two miRNA-target pairs. Further studies are required to establish the rules for target site recognition by miRNAs.
Materials and methods
TALEN-mediated knockout of miRNAs
The design of TALEN constructs was reported in our previous study.4 The reporter construct for the enrichment of knockout cells was also described in a previous paper.26 The TALEN plasmids for miR-221 and miR-222 knockout and the reporter construct were transfected into SNU-638 cells using Lipofectamine 2000 (Life Technologies) or 4D-nucleofector (Amaxa). Two days after transfection, H-2Kk positive cells were separated using a magnetic activated cell sorting system (MACS, Miltenyi Biotec) according to the manufacturer's protocol. After isolation of single cells, individual clones were analyzed using a mismatch-sensitive T7 endonuclease I (T7E1) assay. The mutant clones were further checked by genotyping using fluorescent PCR, and genomic DNA sequences were confirmed by Sanger sequencing.
Northern blot analysis
Total RNA was prepared using TRIzol reagent (Life Technologies). For Northern blot analysis, 10 ug of total RNA was separated on 15% denaturing polyacrylamide gel, and transferred onto a Hybond-NX membrane (Amersham). The membrane was hybridized with a radioactively labeled oligonucleotide probe complementary to each miRNA.
Luciferase assay
WT and mutant reporters of CDKN1B and CDKN1C were created previously.18 Wild type and miRNA knockout SNU-638 cells were seeded onto 24-well plates (100,000 cells per well). The following day, 0.36 ug of each luciferase reporter was co-transfected with 0.04 ug of pRL-CMV vector, which expresses Renilla luciferase, the control for transfection efficiency. After 24 h of transfection, cell lysates were prepared and luciferase assay was performed using a Dual luciferase reporter assay kit (Promega) according to the manufacturer's protocol. The activity from the WT reporter was normalized to that of the mutant reporter.
RNA-seq analysis
From total RNA of wild type and knockout cells, mRNAs with poly(A) tails were enriched using oligo(dT) Dynabeads (Life Technologies). Sequencing libraries were made using TruSeq Stranded Total RNA Library Prep Kit (Illumina). The quality of the libraries was verified using an Agilent 2100 Bioanalyzer (Agilent). For the sequencing of libraries, HiSeq 2500 (Illumina) was used with 50 sequencing cycles. The analyzed sequences were aligned into the human reference genome (GRCh37/hg19) using the STAR algorithm,27 and assembled using Cufflinks.28 We only selected transcripts with an average value of FPKM (Fragments Per Kilobase of transcript per Million mapped reads) greater than 1, and removed the transcripts whose FPKM value is 0 in any library. Those transcripts with deviation in fold changes (KO/WT) between biologic duplicates greater than 2-fold, were also removed. In addition, only the mRNAs with AGO binding signals from CLIP-seq experiments were chosen.19 Finally, 5,605 transcripts passed these criteria and were used for the analyses in this study. For analysis of genes containing the 7-mer motif (common to miR-221 and miR-222 seed sequences), we downloaded 3′ UTR sequences from the University of California Santa Cruz (UCSC) Table Browser (http://genome.ucsc.edu/cgi-bin/hgText).29
Data deposition
The RNA-seq data in this manuscript have been deposited in the Gene Expression Omnibus (GEO) under the accession number GSE80064.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the members of the Department of Biochemistry, Chonnam National University Medical School, for their technical help and discussion.
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (NRF-2015R1C1A1A02036313, NRF-2016R1A4A1009895), and by Chonnam National University (2015–3036).
References
- 1.Kim YK. Extracellular microRNAs as Biomarkers in Human Disease. Chonnam Med J 2015; 51:51-7; PMID:26306299; http://dx.doi.org/ 10.4068/cmj.2015.51.2.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farazi TA, Hoell JI, Morozov P, Tuschl T. MicroRNAs in human cancer. Adv Exp Med Biol 2013; 774:1-20; PMID:23377965; http://dx.doi.org/ 10.1007/978-94-007-5590-1_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15:509-24; PMID:25027649; http://dx.doi.org/ 10.1038/nrm3838 [DOI] [PubMed] [Google Scholar]
- 4.Kim YK, Wee G, Park J, Kim J, Baek D, Kim JS, Kim VN. TALEN-based knockout library for human microRNAs. Nat Struct Mol Biol 2013; 20:1458-64; PMID:24213537; http://dx.doi.org/ 10.1038/nsmb.2701 [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Juranek S, Li H, Sheng G, Tuschl T, Patel DJ. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 2008; 456:921-6; PMID:19092929; http://dx.doi.org/ 10.1038/nature07666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215-33; PMID:19167326; http://dx.doi.org/ 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol 2005; 3:e85; PMID:15723116; http://dx.doi.org/ 10.1371/journal.pbio.0030085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell 2010; 38:789-802; PMID:20620952; http://dx.doi.org/ 10.1016/j.molcel.2010.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, Rudensky AY. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell 2012; 48:760-70; PMID:23142080; http://dx.doi.org/ 10.1016/j.molcel.2012.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014; 42:D68-73; PMID:24275495; http://dx.doi.org/ 10.1093/nar/gkt1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chun YH, Kil JI, Suh YS, Kim SH, Kim H, Park SH. Characterization of chromosomal aberrations in human gastric carcinoma cell lines using chromosome painting. Cancer Genet Cytogenet 2000; 119:18-25; PMID:10812166; http://dx.doi.org/ 10.1016/S0165-4608(99)00217-4 [DOI] [PubMed] [Google Scholar]
- 12.Garofalo M, Quintavalle C, Romano G, Croce CM, Condorelli G. miR221/222 in cancer: their role in tumor progression and response to therapy. Curr Mol Med 2012; 12:27-33; PMID:22082479; http://dx.doi.org/ 10.2174/156652412798376170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galardi S, Mercatelli N, Giorda E, Massalini S, Frajese GV, Ciafre SA, Farace MG. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J Biol Chem 2007; 282:23716-24; PMID:17569667; http://dx.doi.org/ 10.1074/jbc.M701805200 [DOI] [PubMed] [Google Scholar]
- 14.Gillies JK, Lorimer IA. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 2007; 6:2005-9; PMID:17721077; http://dx.doi.org/ 10.4161/cc.6.16.4526 [DOI] [PubMed] [Google Scholar]
- 15.le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafrè SA, et al.. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J 2007; 26:3699-708; PMID:17627278; http://dx.doi.org/ 10.1038/sj.emboj.7601790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visone R, Russo L, Pallante P, De Martino I, Ferraro A, Leone V, Borbone E, Petrocca F, Alder H, Croce CM, et al.. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr Relat Cancer 2007; 14:791-8; PMID:17914108; http://dx.doi.org/ 10.1677/ERC-07-0129 [DOI] [PubMed] [Google Scholar]
- 17.Medina R, Zaidi SK, Liu CG, Stein JL, van Wijnen AJ, Croce CM, Stein GS. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res 2008; 68:2773-80; PMID:18413744; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-6754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim YK, Yu J, Han TS, Park SY, Namkoong B, Kim DH, Hur K, Yoo MW, Lee HJ, Yang HK, et al.. Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res 2009; 37:1672-81; PMID:19153141; http://dx.doi.org/ 10.1093/nar/gkp002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang YC, Di C, Hu B, Zhou M, Liu Y, Song N, Li Y, Umetsu J, Lu ZJ. CLIPdb: a CLIP-seq database for protein-RNA interactions. BMC Genomics 2015; 16:51; PMID:25652745; http://dx.doi.org/ 10.1186/s12864-015-1273-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan AA, Betel D, Miller ML, Sander C, Leslie CS, Marks DS. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat Biotechnol 2009; 27:549-55; PMID:19465925; http://dx.doi.org/ 10.1038/nbt0709-671a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012; 3:1; PMID:22230293; http://dx.doi.org/ 10.1186/1758-907X-3-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang Q, Meng X, Meng L, Chang N, Xiong J, Cao H, Liang Z. Small indels induced by CRISPR/Cas9 in the 5′ region of microRNA lead to its depletion and Drosha processing retardance. RNA Biol 2014; 11:1243-9; PMID:25590615; http://dx.doi.org/ 10.1080/15476286.2014.996067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uhde-Stone C, Sarkar N, Antes T, Otoc N, Kim Y, Jiang YJ, Lu B. A TALEN-based strategy for efficient bi-allelic miRNA ablation in human cells. RNA 2014; 20:948-55; PMID:24717974; http://dx.doi.org/ 10.1261/rna.042010.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009; 138:673-84; PMID:19703394; http://dx.doi.org/ 10.1016/j.cell.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broughton JP, Lovci MT, Huang JL, Yeo GW, Pasquinelli AE. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol Cell 2016; 64:320-33; PMID:27720646; http://dx.doi.org/ 10.1016/j.molcel.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim H, Kim MS, Wee G, Lee CI, Kim H, Kim JS. Magnetic separation and antibiotics selection enable enrichment of cells with ZFN/TALEN-induced mutations. PLoS One 2013; 8:e56476; PMID:23441197; http://dx.doi.org/ 10.1371/journal.pone.0056476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29:15-21; PMID:23104886; http://dx.doi.org/ 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511-5; PMID:20436464; http://dx.doi.org/ 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 2004; 32:D493-6; PMID:14681465; http://dx.doi.org/ 10.1093/nar/gkh103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeong G, Lim YH, Kim YK. Precise mapping of the transcription start sites of human microRNAs using DROSHA knockout cells. BMC Genomics 2016; 17:908; PMID:27835943; http://dx.doi.org/ 10.1186/s12864-016-3252-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.