

Abstract
The outer membrane vesicle (OMV) derived from Porphyromonas gingivalis plays an essential role in causing inflammation which, in turn, plays an important part in the pathogenesis of cardiovascular diseases such as atherosclerosis and thromboembolism. However, the contribution of oral bacteria to vascular calcification is yet to be determined. Here, we evaluated the effect of OMV on vascular smooth muscle cell (VSMC) calcification both in vitro and ex vivo. We established a reproducible P. gingivalis OMV‐induced differentiation and calcification model of VSMCs in vitro. The results indicate that OMV promotes VSMC calcification in a concentration‐dependent manner, modulating the expression of bone markers and SMC markers both on genes and proteins that are important for osteoblastic differentiation and mineralization of VSMCs. We also showed that the key osteogenic transcription factor, runt‐related transcription factor 2 (Runx2), which is affected by upstream extracellular‐regulated kinase (ERK) signaling, is a key regulator of OMV‐induced VSMC differentiation and calcification. Taken together, our research demonstrates that Runx2 is a crucial component of OMV‐induced calcification of VSMCs, and ERK signaling plays a vital role in mediating Runx2 up‐regulation and VSMC calcification.
Keywords: extracellular‐regulated kinase, outer membrane vesicle, Porphyromonas gingivalis, runt‐related transcription factor 2, vascular calcification
Abbreviations
- ALP
alkaline phosphatase
- ColIA1
collagen typeIalpha 1
- ERK
extracellular‐regulated kinase
- OC
osteocalcin
- OMV
outer membrane vesicle
- Runx2
runt‐related transcription factor 2
- SMA
smooth muscle‐specific α‐actin
- VSMC
vascular smooth muscle cell
Porphyromonas gingivalis induces a local chronic host inflammatory response that ultimately leads to inflammatory alveolar bone resorption and tooth loss, which are the key features of periodontal disease 1. Evidence suggests that P. gingivalis‐mediated periodontal disease is the risk factor of cardiovascular disease because periodontal pathogenic bacteria have been detected in heart valve lesions, thrombus, and atheromatous plaque 2, 3, 4.
The outer membrane vesicle (OMV), released by P. gingivalis as extracellular membrane vesicles, retains major virulence including proteins, muramic acid, lipopolysaccharide (LPS), gingipain, fimbriae, and capsule 5, 6. OMV could therefore be internalized into host cells via a lipid raft‐dependent endocytic pathway 7, resulting in the activation of inflammatory pathways. OMV can be detected in the peripheral blood and cerebrospinal fluid in patients with severe bacterial infections 8, 9, which may be closely related to cardiovascular disease.
Vascular calcification, the accumulation of calcium deposits in the blood vessel, is a hallmark of atherosclerosis, causing angiosclerosis and reduced elasticity of the vessel 10. A variety of vascular cells, including vascular smooth muscle cells (VSMCs), myofibroblasts, vascular mesenchymal progenitors, and endothelial cells, could be involved in vascular calcification 11, 12. Many studies have now supported the theory that vascular calcification is an active cell‐driven process associated with osteoblast differentiation of vascular cells 13, 14, 15, which is manifested as the expression of numerous bone‐related molecules including alkaline phosphatase (ALP), collagen type I alpha 1 (ColIA1), osteocalcin (OC), and mineralized bone‐like structures 16, 17, 18. Runt‐related transcription factor 2 (Runx2) is a key transcription factor that regulates osteoblast 19 and chondrocyte 20 differentiation. Runx2 induces ALP activity and expression of bone‐related matrix proteins, including ColIA, OC, and osteopontin (OPN) 21, 22, 23, 24. Increased expression of Runx2 is associated with VSMC calcification in vitro 24, 25, 26, 27, revealing a potential role of Runx2 in vascular calcification. Pathogen‐induced inflammatory factors play a pivotal role in atherosclerotic arteries, potentially enhancing the differentiation and mineralization of vascular cells. Furthermore, extracellular‐regulated kinase (ERK) signaling is an important pathway to identify promising targets for therapeutic intervention in inflammatory diseases 28. ERK activation increases Runx2 transcriptional activity 29. The ERK pathway is an important connection between the cell surface and nucleus to control diverse cellular functions, including proliferation, differentiation, migration, and survival 30. The level of Runx2 is a decisive factor in the occurrence of osteoblastic differentiation 31. Our previous study demonstrated that OMV could induce oxidative stress activation through ERK1/2 9. However, the contribution of inflammatory factors derived from oral pathogenic bacteria to vascular calcification remains to be determined.
In the current report, we hypothesized that P. gingivalis‐derived OMV would affect VSMC differentiation and mineralization. Using an in vitro and ex vivo approach, we find that activation of Runx2 is necessary for OMV‐induced VSMC calcification. These studies demonstrate for the first time that periodontal pathogens may play a pivotal role in vascular calcification.
Materials and methods
Materials
Cell culture media were purchased from Invitrogen Technologies (Carlsbad, CA, USA). Real‐time PCR primers were purchased from Sangon Biotech (Shanghai, China). The miScript SYBR Green PCR kit was purchased from QIAGEN (Hilden, Germany). The Alkaline Phosphatase Colorimetric Assay Kit was purchased from Biovision (Milpitas, CA, USA). The Calcio Arsenazo III Proced was purchased from STANBIO laboratory (Boerne, TX, USA). The Pierce ECL Western Blotting KIT was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Specific antibodies against Runx2, SM22α, α‐SMA, and OC were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against phosphorylated ERK1/2 and total ERK were obtained from Santa Cruz Technology (Santa Cruz, CA, USA). Specific β‐actin antibody was purchased from Sigma‐Aldrich. All secondary antibodies were purchased from Thermo Scientific (Waltham, MA, USA).
Preparation of P. gingivalis OMV
Preparation of OMV was performed from culture supernatants of P. gingivalis ATCC 33277 as previously described 6, 32, 33. In brief, the supernatant of a 2‐day culture of standard strain 33277 was collected by centrifugation at 4000 g for 20 min at 4 °C, then filtered through a 0.22‐μm polyvinylidene difluoride (PVDF) filter and ultracentrifuged at 150 000 g for 3 h at 4 °C in a 40 Ti rotor. The resulting OMV pellet was resuspended and the protein concentration was tested by the Bradford assay 34 using BSA as a criterion. Purified OMV was washed twice in 0.22‐μm filtered 0.01M PBS at 150 000 g for 2 h at 4 °C. OMV was isolated multiple times following the same protocol throughout the whole experiment. To avoid protein degradation, OMV was stored at −20 °C for short‐term storage (< 7 days) and −80 °C for long‐term storage (< 30 days). Transmission electron microscopy (TEM) was performed using a FEI Philips CM200EFG instrument (FEI Company, Eindhoven, The Netherlands) as previously described 35.
VSMC isolation and cell culture
Primary VSMCs were isolated from the aortas of C57BL/6 mice (Sichuan University, Chengdu, China) as described previously 36, 37 and confirmed to express smooth muscle‐specific β‐actin antibody (β‐SMA) (Sigma‐Aldrich) by flow cytometry. All experiments were performed using VSMCs at passages 3–5.
Animal welfare and experimental procedures were implemented in line with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006). The Animal Ethics Committee of Beijing Institute of Translational Medicine authorized the study.
In vitro VSMC calcification
Vascular smooth muscle cell calcification was induced by the addition of OMVs (0.1, 1 or 10 ng·mL−1) in osteogenic base media comprising 0.25 mm L‐ascorbic acid, 10 mm β‐glycerophosphate and 10−8 m dexamethasone (Gibco, Grand Island, NY, USA) for 3 weeks. Media changes were carried out every 3 days. Commercially available ultrapure P. gingivalis lipopolysaccharide (LPS) (Invivogene, San Diego, CA, USA) was used at a concentration of 10 ng·mL−1 as a comparison. Mineralization was determined by Alizarin Red staining (Sigma‐Aldrich).
Analysis of cell viability
The 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay was used for determining cell viability and proliferation. In experiments, 2 × 104 cells were added to each well on 24‐well gelatin‐coated tissue culture plates and stimulated with different concentrations of P. gingivalis OMV or LPS. After incubation for 4, 24, 48, and 72 h at 37 °C, 0.5 mg·mL−1 MTT (Sigma) in PBS was added to each well. The plates were incubated for 4 h at 37 °C, then the culture medium was removed and 500 μL of DMSO was added to each well followed by 5 min of incubation on a shaker. Optical density (OD) of each well was detected at 450 nm using a UV‐1200 spectrophotometer (Mapada, Shanghai, China). Cell viability data were expressed as percentages of the control.
Total calcium measurement
Cells were dissolved in 0.5 mol·L−1 hydrochloric acid by shaking overnight at 4 °C. Total calcium in the cell lysates was determined by the Arsenazo III method according to the manufacturer's instructions.
Real‐time polymerase chain reaction analysis
The effect of OMV on the expression of VSMC markers was determined by real‐time PCR. Total RNAs were isolated from VSMCs using Trizol reagent and reverse‐transcribed into cDNA. SYBR Green‐based real‐time PCR reactions were performed using specific primers for murine ALP 38, ColIA1 16, OC 17, Runx2 20, SM22α 21, α‐SMA 21,ERK1/2 29, and β‐actin as the control 38. Quantitative real‐time PCR was performed with the miScript SYBR Green PCR Kit on an iCycler Thermal Cycler (Bio‐Rad, Hercules, CA, USA) according to the manufacturer's instructions.
Western blot analysis
Cells were treated with 10 ng·mL−1 OMV for 3 weeks and suspended in radioimmunoprecipitation (RIPA) lysis buffer. Proteins were separated and the preparation was transferred. Protein concentration was measured according to the protocol 37, 39. PVDF membranes were incubated overnight with primary antibodies for Runx2, ALP, ColIA1, OC (1/1000 dilution), SM22α,α‐SMA (1/2000), phosphorylated ERK1/2, total ERK (1/1000),β‐actin (1/6000). Secondary antibodies (1/10 000 for target proteins and 1/20 000 for β‐actin) were added for 1 h at room temperature. Protein bands were quantified using enhanced chemiluminescence ECL reagent (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Detection of Runx2 Transactivity
To determine the transcriptional activity of the Runx2 protein, VSMCs were transfected with p6xRunxLuc luciferase reporter plasmid (0.5 μg) and SV containing a cDNA for Renilla reniformis luciferase (0.5 μg) using FuGENE 6. A minimal SV40 promoter was used to encode the Renilla luciferase gene downstream. VSMCs were washed after 24 h and treated with 10 ng·mL−1 OMV for another 48 h. The transfection efficiency of the luciferase reporter was normalized by Renilla Luciferase Activity System (Promega, Madison, WI, USA).
Aortic ring culture and calcification
Under anesthesia, the aortas were instantly removed from 2‐month‐old C57BL/6 mice (25–30 g) prior to euthanasia. The vasculatures were placed into sterile dishes containing PBS, 1 μg·mL−1 penicillin, and 100 μg·mL−1 streptomycin (Pen‐Strep, Thermo Scientific), and refrigerated at 4 °C for 1 h. Under aseptic conditions, the aortas were dissected under a microscope and washed five times in PBS: Pen‐Strep (the same concentration as above) to remove all visible residuals. The aortas were cut into rings of about 3 ± 1 mm. Individual rings were placed into wells of sterile 12‐well plates (Costar, Sigma‐Aldrich) containing 1 mL Dulbecco's Modified Eagle's Medium (DMEM) with 1 mm sodium pyruvate/phenol red, Pen‐Strep, 2 mm glutamine, and 10% fetal calf serum (FCS). All aortic samples were placed at room temperature in an atmosphere of 5% CO2. Vessels were equilibrated at least 1 h before replacing the medium with fresh medium and the addition of stimulants. Vessels were incubated with PBS (control), OMV (10 ng·mL−1), or OMV cotreated with ERK inhibitor (20 μm) for 3 weeks with media/stimulator changes every 3 days. At 3 weeks, the vessels were embedded in paraffin and cut into 8‐μm serial sections. Frozen aortic sections were stained using silver nitrate by Von Kossa (Sigma‐Aldrich) to detect calcium deposits. Stained specimens were examined under a light microscope (DM3000, Lecia, Wetzlar, Germany).
Statistical analysis
Quantitative data are expressed as means ± SD of at least three separate experiments. Statistical significance was determined using one‐way ANOVA and significance was established at a P value < 0.05.
Results
Porphyromonas gingivalis OMV induces VSMC calcification in a concentration‐dependent manner
Outer membrane vesicle purity and structure were determined using TME. The purified OMV presented with small spherical structures in the absence of periplasmic components such as cytoplasmic, inner membrane proteins, and cellular fragments. Porphyromonas gingivalis OMV was found to be spherical in shape and have a size distribution of 80–150 nm, as determined by TEM (Fig. 1A). We then examined the direct effect of OMV or LPS on VSMC calcification in vitro. We found that OMV at concentration above 50 ng·mL−1 led to cell death within 17 days (data not shown). OMV at nontoxic concentrations of 0.1–10 ng·mL−1 induced VSMC calcification in a concentration‐dependent manner, presented as an MMT assay histogram (Fig. 1B), Alizarin Red staining (Fig. 1C) and total calcium levels (Fig. 1D). No significant change in cell viability was observed after treatment with the lower concentrations of OMV (0.1–10 ng·mL−1) even after 3 days. In addition, the presence of 10 ng·mL−1 LPS caused a mild effect toward calcifying VSMCs. OMV‐induced calcium levels were much higher than those of LPS activation at the same concentration. Taken together, we established a reproducible P. gingivalis OMV‐induced differentiation and calcification model of VSMCs in vitro.
Figure 1.
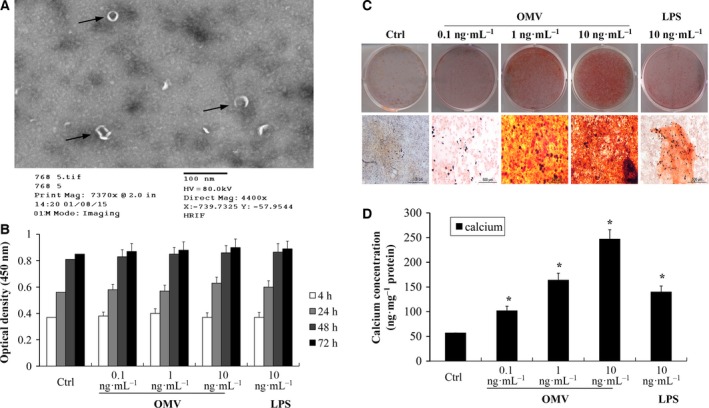
Porphyromonas gingivalis OMV induces VSMC calcification. (A) Image of purified OMV identification through TEM. (B) The cell viability was measured after 4, 24, 48, and 72 h using the MTT assay. The y‐axis represents mean ± SD values of optical densities measured at 450 nm from four wells of one representative experiment. (C) VSMCs were exposed to 0.1, 1, and 10 ng·mL−1 OMV and 10 ng·mL−1 LPS in osteogenic media for 3 weeks with changing media every 3 days. VSMC calcification was determined by Alizarin Red staining. Representative images of stained dishes (upper) and microscopic views (×40, lower) are shown. (D) Total calcium levels were determined by the Arsenazo III method according to the manufacturer's instructions. The results from three independent experiments performed in duplicate are shown (*P < 0.001, compared with control).
OMV induces the expression of bone markers and down‐regulates SMC markers
Accumulating evidence indicates that vascular calcification resembles the process of osteogenesis 40, 41. We therefore evaluated the effect of OMV on osteogenic differentiation of VSMCs and measured the expression of bone and SMC markers by real‐time PCR and western blot. Expression of the bone markers ALP, ColIA1, and OC were increased in 10 ng·mL−1 OMV‐treated osteogenic media at 2 weeks (Fig. 2A,C) both at RNA and protein level. The Alkaline Phosphatase Colorimetric Assay showed increased expression of ALP activity following exposure to 10 ng·mL−1 OMV for 15 days (Fig. 2B). In contrast, there was a dramatic decline in SM22α and α‐SMA expression during VSMV osteogenic differentiation (Fig. 2D–F). These data demonstrate that induction of bone markers and suppression of SMC‐specific phenotype are associated with OMV‐induced VSMC calcification.
Figure 2.
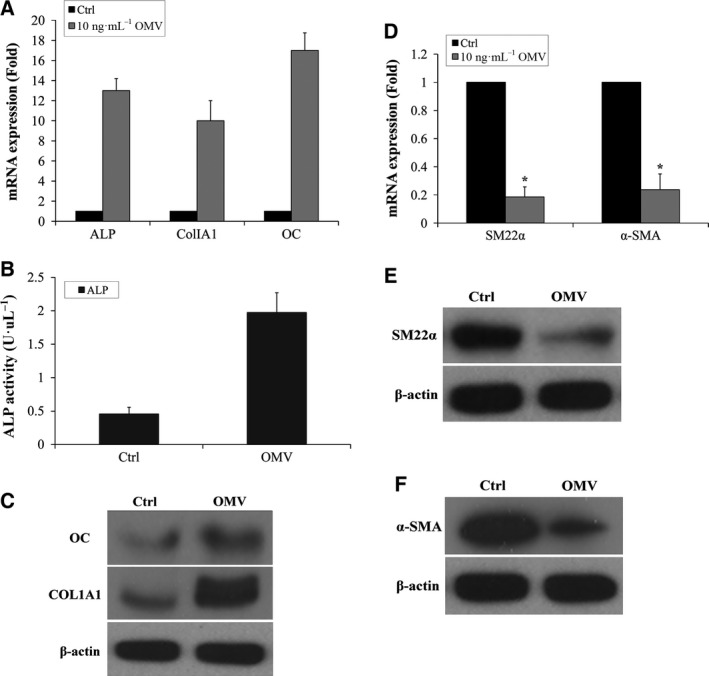
OMV induces the expression of bone markers and down‐regulates SMC Markers. (A) Expression of bone markers ALP, ColIA1, and OC was determined by RT‐PCR during VSMC calcification. VSMCs were exposed to 10 ng·mL−1 OMV in osteogenic media for 2 weeks. (B) The Alkaline Phosphatase Colorimetric Assay was performed in VSMCs exposed to 10 ng·mL−1 OMV for 15 days. (C) Expression of ColIA1 and OC protein was determined by western blot analysis. (D) Expression of SMC markers SM22α and α‐SMA was determined by RT‐PCR in VSMCs exposed to 10 ng·mL−1 OMV in osteogenic media for 2 weeks. (E) Expression of SMC marker SM22α protein. (F) Expression of SMC marker α‐SMA protein. Results from three independent experiments performed in duplicate are shown (*P < 0.001, compared with control).
OMV induces Runx2 expression and transactivity in VSMCs
Runx2 is a key transcription factor that regulates osteoblast differentiation and has been shown to induce ALP activity and regulate expression of many bone martric markers 23. We therefore determined the potential effect of OMV on the expression and transactivity of Runx2 in VSMCs. Quantitative real‐time PCR confirmed an increase in Runx2 mRNA when VSMCs were treated with 10 ng·mL−1 OMV for 3–21 days (Fig. 3A). Furthermore, western blot analysis confirmed that the OMV‐induced increase in Runx2 mRNA translated to protein expression (Fig. 3B). Runx2 protein expression was increased in VSMCs exposed to OMV in a time‐dependent manner. To determine the transcriptional activity of the Runx2 protein, a luciferase receptor assay was performed. A multimeric Runx2 promotor receptor was transfected into VSMCs and then the cells were induced by OMV (Fig. 3C). The data suggest that OMV induces Runx2 expression and transactivity in VSMCs, which may contribute to cellular calcification.
Figure 3.
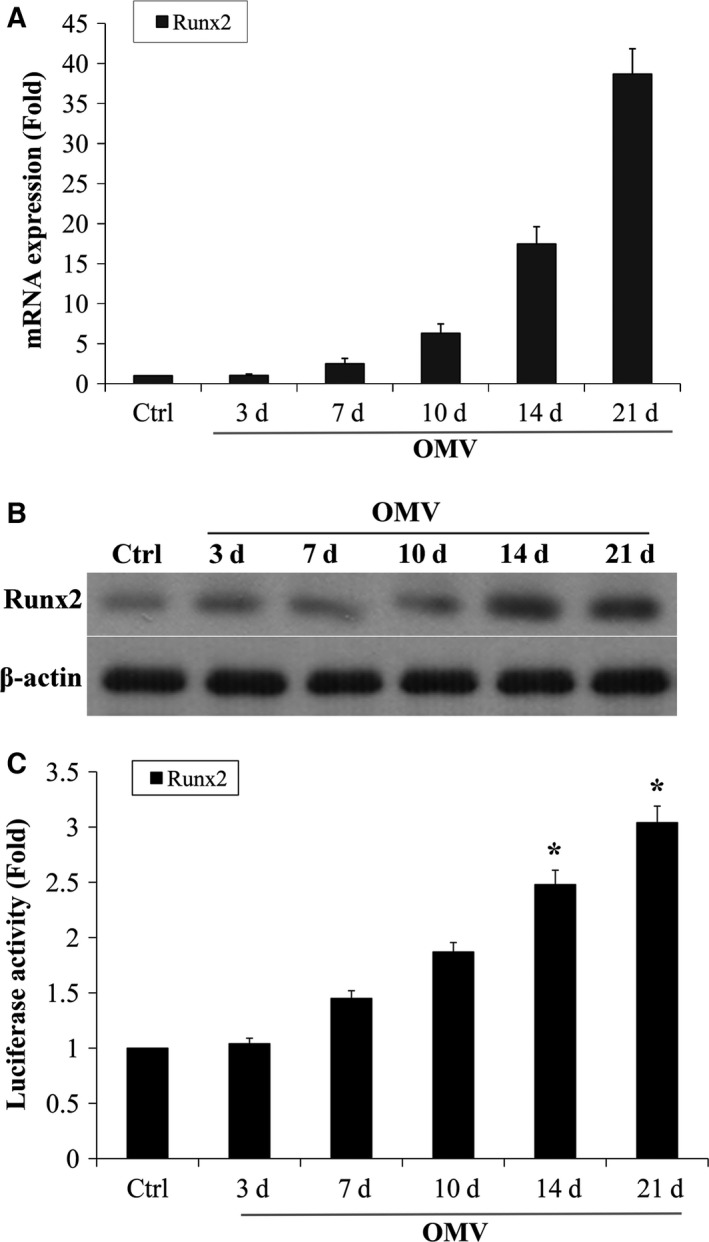
OMV induces Runx2 expression and transactivity in VSMCs. (A) Expression of Runx2 in VSMCs exposed to 10 ng·mL−1 OMV in osteogenic media for 3–21 days was determined by RT‐PCR. Results from three independent experiments performed in duplicate are shown (*P < 0.001, compared with control). (B) Expression of Runx2 protein was determined by western blot analysis. (C) Runx2 transactivity in VSMCs was determined with the use of p6xRunxLuc and a Renilla luciferase expression plasmid. The transfection efficiency of the luciferase receptor was normalized by Renilla Luciferase Activity System. Results from three independent experiments performed in duplicate are shown (*P < 0.001, compared with control).
ERK signaling mediates Runx2 activation and VSMC calcification
Extracellular‐regulated kinase is an important member of the mitogen‐activated protein kinase (MAPK) family and is activated by oxidative stimulation with H2O2 or OMV in a variety of cells 16, 42, 43. We therefore investigated whether activation of phosphorylated ERK1/2 by OMV was required for VSMC calcification. We determined that OMV induced rapid activation of ERK1/2, an important mediator for intracellular calcium transduction (Fig. 4A). Moreover, pharmacological inhibition of ERK1/2 by PD98059 (20 μm) prevented OMV‐induced VSMC calcification (Fig. 4B). Furthermore, the increased expression of Runx2, ALP, ColIA1, and OC was significantly interdicted by MAPK inhibition, while OMV‐induced down‐regulation of SMA22α was not restored (Fig. 4C). Our data indicate that ERK1/2 is able to participate in the up‐regulation of Runx2 and VSMC calcification.
Figure 4.
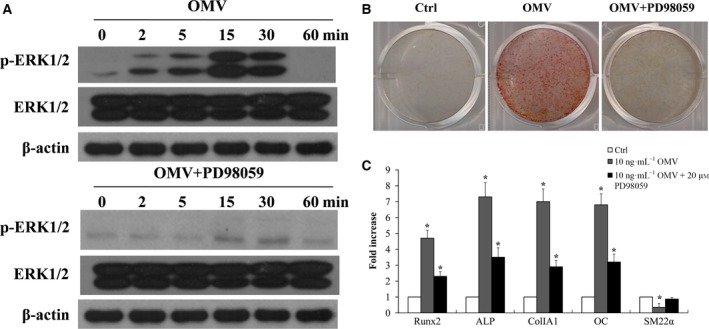
ERK signaling mediates Runx2 activation and VSMC calcification. (A) Expression of phosphorylation ERK1/2 was determined in VSMCs exposed to 10 ng·mL−1 OMV in osteogenic media by western blot analysis. Representative results from three independent experiments in duplicate are shown (upper). VSMCs were pretreated with ERK1/2 inhibitor (PD98059) 20 μm for 30 min and then exposed to 10 ng·mL−1 OMV in osteogenic media. The treatment was repeated every 3 days, and cells were treated for 2 weeks. Expression of p‐ERK1/2 was determined in VSMCs exposed to 10 ng·mL−1 OMV cotreated with PD98059 by western blot analysis (lower). (B) VSMC calcification was determined by Alizarin Red staining. (C) Effect of MAPK inhibitor on the expression of Runx2, ALP, ColIA1, OC, and SM22α was determined by RT‐PCR. VSMCs were pretreated with PD98059 (20 μm) for 30 min and then exposed to 10 ng·mL−1 OMV for 2 weeks in osteogenic media. The results are presented as the fold increase relative to the control group (mean ± SD, n = 3). Representative results from five independent experiments in duplicate are shown (*P < 0.01, compared with control).
Representative immunohistochemistry image of VSMC calcification in mice aortas
For further study into the effect of ERK1/2 on OMV‐induced Runx2 activation and VSMC calcification, ex vivo experimentation was performed. An aorta ring culture model was used to detect calcium deposits in VSMCs following Von Kossa staining. Aorta rings were cotreated with ERK1/2 inhibitor and OMV for 3 weeks. The results showed that OMV stimulated vascular calcification compared with the control, and this effect was blocked when PD98059 was added (Fig. 5A–C). This result further confirmed the activation role of ERK1/2 on the OMV‐induced expression of Runx2 up‐regulation and VSMC calcification. This data demonstrates that Runx2 is an essential component of OMV‐induced calcification of VSMCs, and ERK signaling plays a vital role in mediating Runx2 up‐regulation and VSMC calcification.
Figure 5.
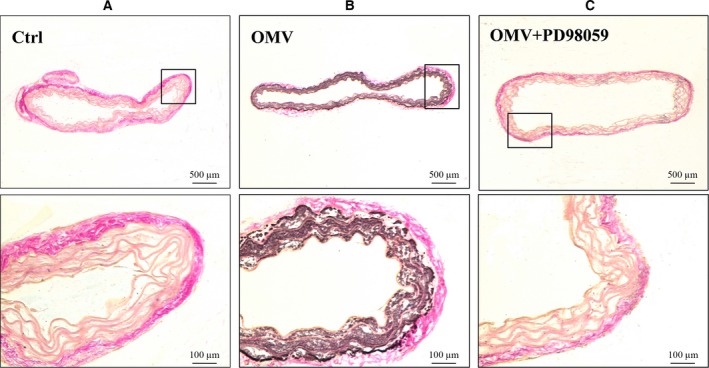
Immuohistochemistry for OMV‐induced VSMC calcification in mouse aorta. (A) After 3 weeks of aorta ring culture with PBS, aortic sections were stained using silver nitrate to detect calcium deposits. (B) Aorta rings cultured in the stimulation of 10 ng·mL−1 OMV for 3 weeks. (C) Aorta rings were cotreated with 10 ng·mL−1 OMV and ERK inhibitor. Representative images from three independent experiments are performed in duplicate. Higher magnification images of the boxed areas are shown under each image.
Discussion
Epidemiological links between periodontitis and angiocardiopathy predict that a direct correlation exists between these two diseases. One of the rationales for this mechanistic link is that periodontitis patients have frequent bacteremic episodes, with LPS, gingipain, and other virulence factors of periodontal disease often detected in their circulation. The presence of serum lipids (and their modification), engagement of receptors on inflammatory cytokines and cells, invasion of endothelial cells, and diffusion of atheromatous lesions could therefore be attributed to bacteria and their components that could further promote inflammatory responses, thus contributing to atherosclerotic plaque formation. OMV retains virulence and can be internalized by host cells or released into the circulation. In the sera of periodontitis patients, significantly stronger reactivity was observed on the OMV‐induced wild‐type strain than the isogenic OMV‐depleted strain 5. One possible hypothesis is that OMV may exert its function by serving as a transport vehicle for toxin delivery.
To the best of our knowledge, there are no published data illustrating the causal association between periodontitis and vascular calcification. First, we thus evaluated the effects of OMV or LPS on VSMC calcification, which indicated that OMV induced osteogenic differentiation and calcification of VSMCs in a dose‐dependent manner at a non‐toxic concentration (0.1–10 ng·mL−1). This result was further confirmed by measuring total production of calcium, the deposition of which in the vessel wall is a characteristic of advanced atherosclerosis, with reduced elasticity and compliance of the vessel wall 1. Our result identified a positive connection between P. gingivalis‐derived OMV and VSMC calcification. Many studies indicate that P. gingivalis LPS plays a critical role in the pathogenesis of cardiovascular disease. Interestingly, LPS‐only induction plays a relatively minor role in stimulating vascular cellular calcification, indicating that other virulence components of OMV might be involved in the calcification process. Here, we focused on the impact of OMV on VSMC calcification and found that OMV‐induced VSMC calcification was relevant to down‐regulation of VSMC‐specific markers and increased expression of bone markers, indicating a phenotypic change of VSMCs into osteoblast‐like cells.
More importantly, we demonstrated that VSMC differentiation following stimulation with OMV is mediated by Runx2, a key transcription factor for osteoblast and chondrocyte differentiation 19, 20, 27, 39, 44. ERK1/2 plays a pivotal role in Runx2 activation and subsequently accelerates VSMC calcification 16, 42, 45. Porphyromonas gingivalis‐derived OMV has the ability to activate the MAPK pathway 9, which may lead to activation of Runx2. We found that OMV‐activated ERK signaling was required for OMV‐induced calcification of VMSCs. In contrast, pharmacological inhibition of MAPK by PD98059 46 reduced the expression and transactivity of Runx2, as well as the expression of bone markers during VSMC calcification. These results are consistent with activated intracellular signaling increases in gene and transcriptional expression of Runx2 during osteoblast and chondrocyte differentiation 47. Our data provide further support that Runx2 is an essential component of OMV‐induced calcification of VSMCs and suggests that ERK1/2 is able to participate in the up‐regulation of Runx2 and VSMC calcification.
In summary, our results reveal an activation role of P. gingivalis‐derived OMV in mediating VSMC calcification both in vitro and ex vivo. Runx2, which is affected by upstream of ERK signaling, exerts a great influence on OMV‐induced VSMC differentiation and calcification. Our findings provide vital molecular insight into the effect of ERK1/2‐Runx2 on vascular calcification and a possible causal relationship between periodontitis and vascular calcification.
Author contributions
YJ designed the experiment. WWY and BG completed the project and acquired the data. WYJ helped with the animal experiment. YJ and WWY interpreted the data and wrote the paper. WWY and BG contributed equally to this work and are joint first authors.
Acknowledgements
We thank Prof. Wenbiao Wang (Aviation General Hospital of China Medical University [AVIC]), Associate Prof. Baorong Zhang (Department of Stomatology, AVIC), and Prof. Yi Ding (West China College of Stomatology, Sichuan University) for helpful discussion and technical guidance. This study was supported by Science and Technology Project of Beijing 2014–2016 (2014JY0330) and Healthy Care Program of the People's Liberation Army 2015–2017 (14BJZ10).
References
- 1. Graves DT, Li J and Cochran DL (2011) Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res 90, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bullon P, Cordero MD, Quiles JL, Morillo JM, del Carmen Ramirez‐Tortosa M and Battino M (2011) Mitochondrial dysfunction promoted by Porphyromonas gingivalis lipopolysaccharide as a possible link between cardiovascular disease and periodontitis. Free Radic Biol Med 50, 1336–1343. [DOI] [PubMed] [Google Scholar]
- 3. Deshpande RG, Khan M and Genco CA (1998) Invasion strategies of the oral pathogen Porphyromonas gingivalis: implications for cardiovascular disease. Invasion Metastasis 18, 57–69. [DOI] [PubMed] [Google Scholar]
- 4. Iwai T, Inoue Y, Umeda M, Huang Y, Kurihara N, Koike M and Ishikawa I (2005) Oral bacteria in the occluded arteries of patients with Buerger disease. J Vasc Surg 42, 107–115. [DOI] [PubMed] [Google Scholar]
- 5. Nakao R, Takashiba S, Kosono S, Yoshida M, Watanabe H, Ohnishi M and Senpuku H (2014) Effect of Porphyromonas gingivalis outer membrane vesicles on gingipain‐mediated detachment of cultured oral epithelial cells and immune responses. Microbes Infect 16, 6–16. [DOI] [PubMed] [Google Scholar]
- 6. Cecil JD, O'Brien‐Simpson NM, Lenzo JC, Holden JA, Chen YY, Singleton W and Reynolds EC (2016) Differential responses of pattern recognition receptors to outer membrane vesicles of three periodontal pathogens. PLoS One 11, e0151967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Farge E, Ojcius DM, Subtil A and Dautry‐Varsat A (1999) Enhancement of endocytosis due to aminophospholipid transport across the plasma membrane of living cells. Am J Physiol 276, C725–C733. [DOI] [PubMed] [Google Scholar]
- 8. Bai D, Nakao R, Ito A, Uematsu H and Senpuku H (2015) Immunoreactive antigens recognized in serum samples from mice intranasally immunized with Porphyromonas gingivalis outer membrane vesicles. Pathog Dis 73, 1–16. [DOI] [PubMed] [Google Scholar]
- 9. Jia Y, Guo B, Yang W, Zhao Q, Jia W and Wu Y (2015) Rho kinase mediates Porphyromonas gingivalis outer membrane vesicle‐induced suppression of endothelial nitric oxide synthase through ERK1/2 and p38 MAPK. Arch Oral Biol 60, 488–495. [DOI] [PubMed] [Google Scholar]
- 10. Trion A and van der Laarse A (2004) Vascular smooth muscle cells and calcification in atherosclerosis. Am Heart J 147, 808–814. [DOI] [PubMed] [Google Scholar]
- 11. Hayashi K, Nakamura S, Nishida W and Sobue K (2006) Bone morphogenetic protein‐induced MSX1 and MSX2 inhibit myocardin‐dependent smooth muscle gene transcription. Mol Cell Biol 26, 9456–9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Witztum JL (1994) The oxidation hypothesis of atherosclerosis. Lancet 344, 793–795. [DOI] [PubMed] [Google Scholar]
- 13. Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J and Stem D (1995) Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule‐1 (VCAM‐1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 96, 1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scheidegger KJ, Butler S and Witztum JL (1997) Angiotensin II increases macrophage‐mediated modification of low density lipoprotein via a lipoxygenase‐dependent pathway. J Biol Chem 272, 21609–21615. [DOI] [PubMed] [Google Scholar]
- 15. Zoccali C, Mallamaci F and Tripepi G (2004) Novel cardiovascular risk factors in end‐stage renal disease. J Am Soc Nephrol 15 (Suppl 1), S77–S80. [DOI] [PubMed] [Google Scholar]
- 16. Cai H (2005) Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res 68, 26–36. [DOI] [PubMed] [Google Scholar]
- 17. Gutierrez J, Ballinger SW, Darley‐Usmar VM and Landar A (2006) Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res 99, 924–932. [DOI] [PubMed] [Google Scholar]
- 18. Cseko C, Bagi Z and Koller A (2004) Biphasic effect of hydrogen peroxide on skeletal muscle arteriolar tone via activation of endothelial and smooth muscle signaling pathways. J Appl Physiol (1985) 97, 1130–1137. [DOI] [PubMed] [Google Scholar]
- 19. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley‐Usmar VM, McDonald JM and Chen Y (2008) Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem 283, 15319–15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pagani F, Francucci CM and Moro L (2005) Markers of bone turnover: biochemical and clinical perspectives. J Endocrinol Invest 28, 8–13. [PubMed] [Google Scholar]
- 21. Gallop PM, Lian JB and Hauschka PV (1980) Carboxylated calcium‐binding proteins and vitamin K. N Engl J Med 302, 1460–1466. [DOI] [PubMed] [Google Scholar]
- 22. Collin‐Osdoby P (2004) Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res 95, 1046–1057. [DOI] [PubMed] [Google Scholar]
- 23. Ducy P, Zhang R, Geoffroy V, Ridall AL and Karsenty G (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89, 747–754. [DOI] [PubMed] [Google Scholar]
- 24. Lian JB, Javed A, Zaidi SK, Lengner C, Montecino M, van Wijnen AJ, Stein JL and Stein GS (2004) Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit Rev Eukaryot Gene Expr 14, 1–41. [PubMed] [Google Scholar]
- 25. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G and Giachelli CM (2001) Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res 89, 1147–1154. [DOI] [PubMed] [Google Scholar]
- 26. Mori K, Shioi A, Jono S, Nishizawa Y and Morii H (1999) Dexamethasone enhances in vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 19, 2112–2118. [DOI] [PubMed] [Google Scholar]
- 27. Chen NX, Duan D, O'Neill KD, Wolisi GO, Koczman JJ, Laclair R and Moe SM (2006) The mechanisms of uremic serum‐induced expression of bone matrix proteins in bovine vascular smooth muscle cells. Kidney Int 70, 1046–1053. [DOI] [PubMed] [Google Scholar]
- 28. Procaccia S, Kraus S and Seger R (2010) Determination of ERK activity: anti‐phospho‐ERK antibodies and in vitro phosphorylation. Methods Mol Biol 661, 39–58. [DOI] [PubMed] [Google Scholar]
- 29. Jun JH, Yoon WJ, Seo SB, Woo KM, Kim GS, Ryoo HM and Baek JH (2010) BMP2‐activated Erk/MAP kinase stabilizes Runx2 by increasing p300 levels and histone acetyltransferase activity. J Biol Chem 285, 36410–36419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shaul YD and Seger R (2007) The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 1773, 1213–1226. [DOI] [PubMed] [Google Scholar]
- 31. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW and Franklin RA (2007) Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773, 1263–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imamura T, Potempa J, Pike RN, Moore JN, Barton MH and Travis J (1995) Effect of free and vesicle‐bound cysteine proteinases of Porphyromonas gingivalis on plasma clot formation: implications for bleeding tendency at periodontitis sites. Infect Immun 63, 4877–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee EY, Bang JY, Park GW, Choi DS, Kang JS, Kim HJ, Park KS and Gho YS (2007) Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli . Proteomics 7, 3143–3153. [DOI] [PubMed] [Google Scholar]
- 34. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 35. Chen YY, Peng B, Yang Q, Glew MD, Veith PD, Cross KJ, Glodie KN and Reynolds EC (2011) The outer membrane protein LptO is essential for the O‐deacylation of LPS and the co‐ordinated secretion and attachment of A‐LPS and CTD proteins in Porphyromonas gingivalis . Mol Microbiol 79, 1380–1401. [DOI] [PubMed] [Google Scholar]
- 36. Chen Y, Kelm RJ Jr, Budd RC, Sobel BE and Schneider DJ (2004) Inhibition of apoptosis and caspase‐3 in vascular smooth muscle cells by plasminogen activator inhibitor type‐1. J Cell Biochem 92, 178–188. [DOI] [PubMed] [Google Scholar]
- 37. Chen Y, Budd RC, Kelm RJ Jr, Sobel BE and Schneider DJ (2006) Augmentation of proliferation of vascular smooth muscle cells by plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol 26, 1777–1783. [DOI] [PubMed] [Google Scholar]
- 38. Qu Q, Perala‐Heape M, Kapanen A, Dahllund J, Salo J, Vaananen HK and Harkonen P (1998) Estrogen enhances differentiation of osteoblasts in mouse bone marrow culture. Bone 22, 201–209. [DOI] [PubMed] [Google Scholar]
- 39. Sun Y, Byon CH, Yuan K, Chen J, Mao X, Anderson PG and Chen Y (2012) Smooth muscle cell‐specific runx2 deficiency inhibits vascular calcification. Circ Res 111, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Towler DA, Shao JS, Cheng SL, Pingsterhaus JM and Loewy AP (2006) Osteogenic regulation of vascular calcification. Ann N Y Acad Sci 1068, 327–333. [DOI] [PubMed] [Google Scholar]
- 41. Bostrom K, Watson KE, Stanford WP and Demer LL (1995) Atherosclerotic calcification: relation to developmental osteogenesis. Am J Cardiol 75, 88B–91B. [DOI] [PubMed] [Google Scholar]
- 42. Martindale JL and Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192, 1–15. [DOI] [PubMed] [Google Scholar]
- 43. Liu H, Li X, Qin F and Huang K (2014) Selenium suppresses oxidative‐stress‐enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J Biol Inorg Chem 19, 375–388. [DOI] [PubMed] [Google Scholar]
- 44. Engelse MA, Neele JM, Bronckers AL, Pannekoek H and de Vries CJ (2001) Vascular calcification: expression patterns of the osteoblast‐specific gene core binding factor alpha‐1 and the protective factor matrix gla protein in human atherogenesis. Cardiovasc Res 52, 281–289. [DOI] [PubMed] [Google Scholar]
- 45. Jersmann HP, Hii CS, Ferrante JV and Ferrante A (2001) Bacterial lipopolysaccharide and tumor necrosis factor alpha synergistically increase expression of human endothelial adhesion molecules through activation of NF‐kappaB and p38 mitogen‐activated protein kinase signaling pathways. Infect Immun 69, 1273–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kuper C, Beck FX and Neuhofer W (2012) Toll‐like receptor 4 activates NF‐kappaB and MAP kinase pathways to regulate expression of proinflammatory COX‐2 in renal medullary collecting duct cells. Am J Physiol Renal Physiol 302, F38–F46. [DOI] [PubMed] [Google Scholar]
- 47. Fujita T, Azuma Y, Fukuyama R, Hattori Y, Yoshida C, Koida M, Ogita K and Komori T (2004) Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K‐Akt signaling. J Cell Biol 166, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]