

ABSTRACT
Contribution of autophagy and regulation of related proteins to the degeneration of retinal pigment epithelium (RPE) in age-related macular degeneration (AMD) remain unknown. We report that upregulation of KRT8 (keratin 8) as well as its phosphorylation are accompanied with autophagy and attenuated with the inhibition of autophagy in RPE cells under oxidative stress. KRT8 appears to have a dual role in RPE pathophysiology. While increased expression of KRT8 following autophagy provides a cytoprotective role in RPE, phosphorylation of KRT8 induces pathologic epithelial-mesenchymal transition (EMT) of RPE cells under oxidative stress, which is mediated by MAPK1/ERK2 (mitogen-activated protein kinase 1) and MAPK3/ERK1. Inhibition of autophagy further promotes EMT, which can be reversed by inhibition of MAPK. Thus, regulated enhancement of autophagy with concurrent increased expression of KRT8 and the inhibition of KRT8 phosphorylation serve to inhibit oxidative stress-induced EMT of RPE cells as well as to prevent cell death, suggesting that pharmacological manipulation of KRT8 upregulation through autophagy with combined inhibition of the MAPK1/3 pathway may be attractive therapeutic strategies for the treatment of AMD.
KEYWORDS: age-related macular degeneration (AMD), apoptosis, autophagy, cell survival, epithelial-mesenchymal transition (EMT), KRT8 (keratin 8), mitogen-activated protein kinases 1 and 3 (MAPK1/3)
Introduction
Age-related macular degeneration (AMD) is the progressive degeneration of the retinal pigment epithelium (RPE), retina, and choriocapillaris among the elderly, and is one of the leading causes of blindness in the world. AMD is mainly divided into dry (80% to 90%) and wet forms (10% to 20%), and choroidal neovascularization (CNV) is a clinical hallmark of wet (neovascular) AMD.1 CNV with the subsequent development of subretinal fluid accumulation, hemorrhage, exudation, and scarring could lead to the loss of vision. Currently, the main treatment target for CNV is vascular endothelial growth factor (VEGF), and thus, anti-VEGF agents have been used as a standard treatment of CNV.2 However, new therapeutic targets are being studied continuously to increase both the safety and efficacy of the treatment of this multifactorial disease and prevent vision loss.3 Moreover, there is currently no proven effective treatment of dry AMD.
Although extensive research has focused on understanding the basic and clinical aspects of AMD, including studies of AMD risk genotypes,4-6 the pathogenesis of AMD remains unknown. To date, oxidative stress, hypoxia, chronic inflammation, and accumulation of drusen have been suggested as the underlying cellular pathological causes.7
The eye requires high levels of oxygen, especially when it is continuously exposed to light, because biochemical reactions of light-electrical energy conversion require increased metabolic rate in the RPE and retina. Generation of reactive oxygen species (ROS) during these processes in the outer retina is involved in light-induced retinal degeneration.8,9 The RPE is also exposed to an oxidative environment, which is caused by high oxygen tensions in the epithelium and accumulation of lipofuscin.10 Particularly, the nondegradable components in lipofuscin generate ROS in the presence of light, which is toxic to the RPE.11 Prolonged exposure to light causes RPE cells to consume a large amount of oxygen to perform visual cycle processes, nutrient transport, and phagocytosis of photoreceptor outer segments. These processes significantly increase the oxygen demand and production of ROS in the RPE.
Accumulation of oxidized proteins in aged cells and in age-related disorders is caused partly as the autophagic pathways become less efficient with age.12 The levels of proteins related to autophagy are increased in older non-AMD donor RPE and retinas, whereas those from late AMD donors have lower autophagy protein levels.13 During aging, oxidative stress and protein misfolding in RPE cells produce functional abnormalities in RPE.14 Well-controlled autophagy could facilitate the clearance of toxic protein aggregates or misfolded proteins in RPE, thus increasing the ability of RPE to tolerate oxidative stress13,15,16 Zhao et al. demonstrate that activation of MTOR (mechanistic target of rapamycin) and the epithelial-mesenchymal transition (EMT) including RPE transdifferentiation are the general RPE stress responses, resulting in the loss of RPE characteristics.17 However, the role of autophagy in AMD pathology, the changes in proteins related to autophagy that protect the RPE against oxidative stress as well as the relationship between autophagy and EMT in the RPE remain unknown.
Failure of or impaired RPE adaptive mechanisms against oxidative stress could be the major cause of pathogenesis of AMD. In our previous study, we profiled and characterized the whole proteome of the aqueous humor (AH) from patients with AMD as well as that of ARPE-19 cells under oxidative stress to investigate the molecular and proteomic changes in the RPE during aging or under oxidative stress conditions.18 Some of the secretory proteins from the RPE, retina, and CNV were identified in the AH, which are selectively upregulated in patients with AMD as compared to controls. Interestingly, among them, KRT8 (keratin 8), which is a well-known epithelial marker protein, was found to be elevated more than 2-fold in AMD patients as compared to controls. Only a few studies have reported a role of KRT8 in the eye other than as a marker of mature RPE cells.19 In addition, CTSD (cathepsin D), which is one of the principal lysosomal proteases in the RPE and known to accumulate in cells following the activation of autophagy, was found to be elevated more than 1.5-fold in AMD patients compared to controls.20 This upregulation of CTSD might be required for the breakdown of toxic aggregated materials in autolysosomes in the RPE when under oxidative stress; thus, autophagic activity might increase in some AMD patients as a survival mechanism in response to the pathologic conditions of AMD.
Herein, we investigated the changes in novel proteins related to putative endogenous adaptive and protective mechanisms operating under oxidative stress, which prevent apoptosis and EMT in RPE cells. Changes in KRT8 and autophagy marker proteins were scrutinized as potential endogenous cytoprotective mechanisms triggered by AMD or oxidative stress, which is a characteristic feature of an aging RPE. We found that the expression of KRT8 as well as autophagy markers increases under oxidative stress conditions, possibly contributing to cytoprotection in RPE cells. Based on the results, we suggest that activated autophagy and upregulation of KRT8 along with its phosphorylation suppresses apoptotic cell death. However, under prolonged oxidative stress, the same processes invoke the dedifferentiation of RPE cells through EMT. These findings will improve our understanding of the molecular pathology of AMD in the RPE under oxidative stress.
Results
Superoxide radicals induce autophagy in retinal pigment epithelial cells
We first examined generation of oxygen radicals in RPE cell lines (ARPE-19, hTERT-RPE), and primary RPE cells exposed to paraquat or H2O2 for 12 or 24 h by monitoring the increase in fluorescence in cells (Fig. 1A, and Fig. S1A, B). The addition of paraquat or peroxide elevated the concentration of oxygen radicals in the RPE cells. Since oxidative conditions or formation of ROS is known to induce autophagy,12 we next investigated whether the oxidative stress generated by paraquat induces autophagy in cultured ARPE-19 cells. Prolonged exposure to oxidative stress after 24 h of paraquat treatment resulted in accumulation of microtubule-associated protein light chain 3B/MAP1LC3B (LC3B) puncta, which is a widely used marker for autophagosomes (Fig. 1B). Counting of GFP-LC3B puncta showed that ROS generated by paraquat induces autophagy in the ARPE-19 cells with a 4- to 5-fold increase in the number GFP-LC3B puncta, compared to untreated cells. However, treatment with the autophagy inhibitor 3-methyladenine (3-MA) inhibits autophagosome formation, as indicated by the absence of obvious puncta in the ARPE-19 cells under oxidative stress (Fig. 1B). We also investigated the formation of autophagic vacuoles under ROS stress using a cationic amphiphilic tracer dye.21 Formation of paraquat-induced punctate structures, representing autophagic vacuoles, was inhibited by autophagy inhibitors such as 3-MA and bafilomycin A1 (Baf A1) in both primary RPE cells and ARPE-19 cells (Fig. 1C, and Fig. S1C).
Figure 1.
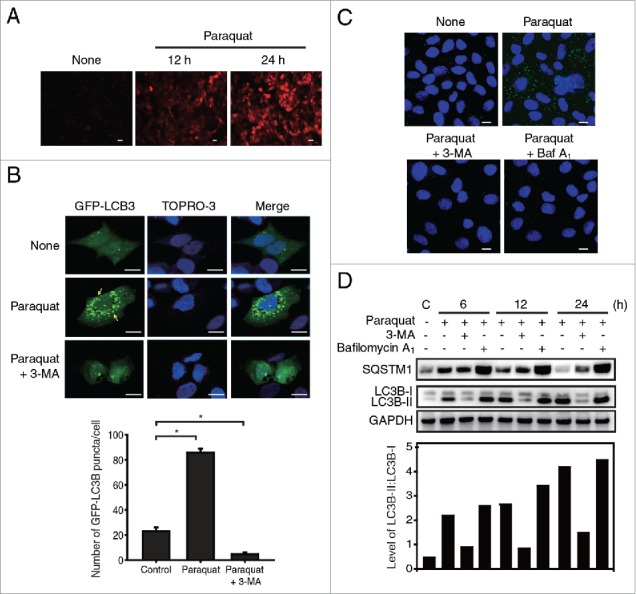
Oxidative stress induces autophagy in RPE cells. (A) Fluorescence microscopic images of DCFDA-stained ARPE-19 cells exposed to paraquat (400 μM) for 12 and 24 h; scale bar: 20 µm. (B) The localization of GFP-LC3B puncta in ARPE-19 cells treated with paraquat (400 μM) in the absence or presence of 3-MA (10 mM) for 24 h; scale bar: 10 µm. Nuclei were fluorescently stained with TOPRO-3 and are represented with blue fluorescence. Bar graph indicates the average number of GFP-LC3B puncta per cell. The data are presented as the mean ± SD, n = 3. *P < 0.05 vs. control. (C) Cells were treated with 400 μM paraquat in the absence or presence of either 10 mM 3-MA or 50 nM Baf A1 for 24 h. Fluorescence microscopic images of autophagic vacuoles formed in human primary RPE cells, which are stained as green puncta. Nuclei are indicated by blue fluorescence (TOPRO-3 staining). Scale bar: 5 µm. (D) Western blot analysis of SQSTM1 and LC3B-II/I in ARPE-19 cells. ARPE-19 cells were treated with paraquat (400 μM) in the absence or presence of 3-MA (10 mM) or Baf A1 (50 nM) for 6, 12, and 24 h. Bar graph indicates the ratio of LC3B-II to LC3B-I in the western blot analysis images.
Subsequently, we investigated the progression of autophagy in ARPE-19 cells by monitoring LC3B conversion and SQSTM1 (sequestosome 1) degradation (Fig. 1D). RPE cells treated with paraquat exhibited the conversion of LC3B-I (cytosolic form) to LC3B-II (membrane-bound lipidated form), which is an early event of autophagy during autophagosome formation. In addition, prolonged exposure to paraquat resulted in gradual degradation of SQSTM1, suggesting that autophagosomes were readily fused with lysosomes, and their contents were subsequently degraded in the autolysome.22 In contrast, 3-MA treatment suppressed the conversion of LC3B-I to LC3B-II and degradation of SQSTM1 in RPE cells under oxidative stress. Alternative autophagy inhibitor, Baf A1, which blocks the late steps of autophagy by inhibiting fusion between autophagosomes and lysosomes via inhibiting the vacuolar-type H+-ATPase (V-ATPase),23 did not affect LC3B conversion but inhibited degradation of SQSTM1 in paraquat-treated cells. Together, these results indicate that RPE cells under oxidative stress undergo autophagy processes, which can be blocked by 3-MA and Baf A1 in the early and late phases of autophagy, respectively.
Activation of the AKT-MTOR pathway and autophagy induction protects RPE cells from apoptotic cell death under oxidative stress
To address whether ROS-induced autophagy can protect RPE cells under oxidative stress from apoptotic cell death, we monitored apoptotic cell death using flow cytometry analysis (Fig. 2A, B and C). A 24 h paraquat treatment did not affect the fate of ARPE-19 cells, whereas addition of autophagy inhibitors such as 3-MA or Baf A1 significantly increased the population of apoptotic cells by over 30% under oxidative stress conditions (Fig. 2A). The apoptotic cell death of human primary RPE cells was also observed after autophagy inhibitor treatment under oxidative stress (Fig. 2B). Under the ROS stress condition, both ARPE-19 and primary RPE cells, which were treated with autophagy inhibitors, showed a comparable and significant increment in apoptotic cell death (Fig. 2C). To confirm apoptosis in ARPE-19 cells, the cells under oxidative stress with or without autophagy inhibitors were analyzed by monitoring the activities of apoptotic enzymes, CASP3 (caspase 3) and CASP7. Paraquat treatment did not induce the activity of CASP3 and CASP7, while inhibition of autophagy with 3-MA or Baf A1 significantly enhanced CASP activity by approximately 2.5-fold (Fig. 2D).
Figure 2.
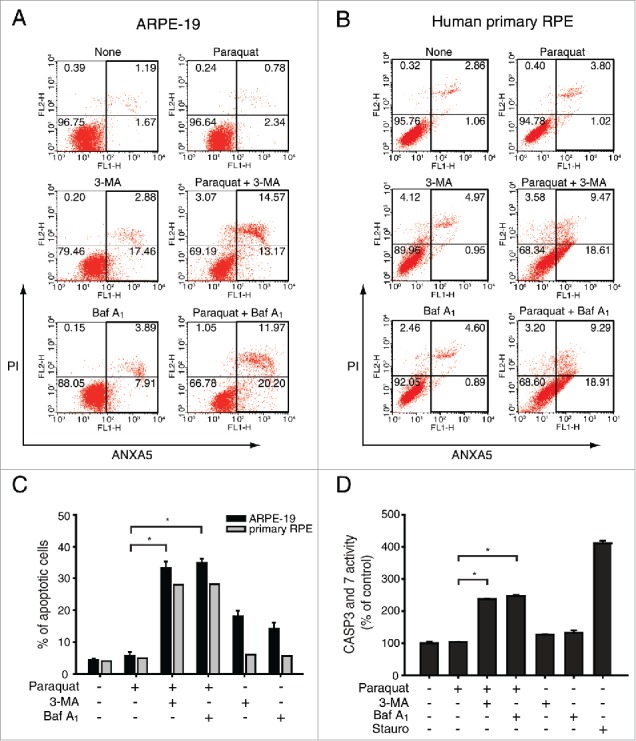
Inhibition of autophagy increases apoptotic cell death in RPE cells under oxidative stress. ARPE-19 and human primary RPE cells were treated with 3-MA (10 mM) or Baf A1 (50 nM) in the absence or presence of paraquat (400 μM) for 24 h. (A) ARPE-19 and (B) human primary RPE cells were stained with FITC-conjugated ANXA5 and propidium iodide (PI) and analyzed by FACS. (C) The graph represents the mean percentage of apoptotic cells (n = 3). *P < 0.01 vs. paraquat. (D) Apoptotic enzyme CASP3 and CASP7 activity was measured after ARPE-19 cells were treated with 400 μM paraquat in the absence or presence of either 10 mM 3-MA or 50 nM Baf A1 for 24 h. Cells were also treated with staurosporine (1 μM) to serve as positive controls for apoptotic cell death. The data are presented as the mean ± SD, n = 3. *P < 0.01 vs. paraquat.
It has been previously reported that PPP2/PP2A (protein phosphatase 2) is downregulated in ARPE-19 cells exposed to oxidative stress,24 possibly leading to the stimulation of autophagy.25,26 In APRE-19 cells exposed to paraquat, significant downregulation of PPP2 was observed as early as 60 min, but the PPP2 level was restored to that of the control at 24 h (Fig. S2). Inactivation of PPP2 has been known to induce phosphorylation of AKT, a survival or antiapoptotic protein.27 Based on the result that RPE cells under oxidative stress evaded apoptotic cell death (Fig. 2), we hypothesized that the amount of phosphorylated AKT would increase due to PPP2 inactivation.
It has been recently reported that administration of oxidants to mice caused the activation of the AKT-MTOR (mechanistic target of rapamycin) pathway, resulting in dedifferentiation of the retinal pigment epithelium and accompanying photoreceptor degeneration.17 To address how the downregulation of PPP2 affects phosphorylation of AKT and MTOR during the early stages of ROS induction in the ARPE-19 cells, we monitored the progression of AKT and MTOR phosphorylation in ARPE-19 cells treated with paraquat (Fig. S2). Phosphorylation of AKT (Ser473) and MTOR (Ser2481) was augmented during the downregulation of PPP2, and the autophagy process was delayed due to reduced conversion of LC3B in response to the activation of the AKT-MTOR pathway. This suggests that downregulation of PPP2 induces the activation of the AKT-MTOR pathway before ROS-mediated autophagy in ARPE-19 cells under oxidative stress. Therefore, decreased levels of early-elevated p-AKT and p-MTOR and the subsequent onset of autophagy played a positive role in the survival of RPE cells under oxidative stress conditions in the present study.
Oxidative stress induces upregulation of KRT8 during autophagy
In our previous proteomics analysis of the exosomes present in the AH of neovascular AMD patients, KRT8 (keratin 8) and CTSD were found to be increased significantly as compared to the controls.18 Because exosomes derived from the AH contained various molecular constituents of the RPE, we supposed that exosomes from the AH of neovascular AMD patients might represent a physiologic condition in AMD patients. Thus, we selected KRT8 as a potential marker protein associated with the autophagy induced by oxidative stress.
We first examined the effect of oxidative stress on KRT8 expression in ARPE-19 cells. Expression of KRT8 as well as its phosphorylated form (p-KRT8) was enhanced in a manner that was dependent on both paraquat concentration and exposure time (Fig. 3A). Enhancement of KRT8 expression was concomitant with autophagy progression, as indicated by SQSTM1 degradation and LC3B conversion. We next examined whether inhibition of autophagy progression affects the expression of KRT8 and its phosphorylated form (Fig. 3B). When the cells were treated with paraquat (400 μM) for 24 h, expression of both KRT8 and its phosphorylated form was enhanced without altering the p-KRT8:total KRT8 ratio. However, these cells showed decreased expression of KRT8 after cotreatment with an autophagy inhibitor, 3-MA. On the other hand, the cells under oxidative stress did not significantly attenuate the upregulation of KRT8 and p-KRT8 when treated with Baf A1, an autophagy inhibitor working in the late stages. Regulation of KRT8 phosphorylation is likely not directly involved in autophagy because the p-KRT8:total KRT8 ratio did not significantly change under oxidative stress.
Figure 3.
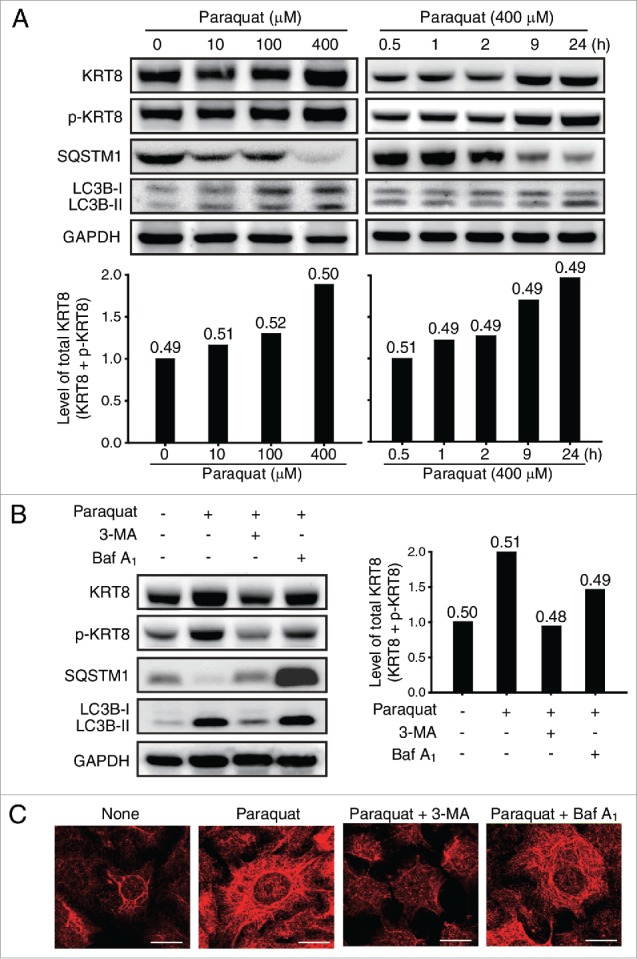
Oxidative stress upregulates KRT8 and its phosphorylated form. (A) Western blot analysis of KRT8 and its phosphorylated form (p-KRT8), and autophagy markers such as SQSTM1 and LC3B-II/I in ARPE-19 cells. Cells were exposed to various concentrations of paraquat (0 to 400 μM) for 24 h (left panel). The same western blot analysis was performed for the cells that were treated with paraquat (400 μM) for various times (0.5 to 24 h) (right panel). Bar graphs indicate the level of total KRT8 expression (KRT8 + p-KRT8). The protein band intensities were normalized to GAPDH. The p-KRT8:total KRT8 ratio was determined from the western blot analysis, and it is shown on the top of each bar graph as a number. (B) Western blot analysis of KRT8, p-KRT8, SQSTM1, and LC3B-II/-I present in ARPE-19 cells treated with paraquat (400 μM) in the absence or presence of either 10 mM 3-MA or 50 nM Baf A1 for 24 h. Bar graphs indicate the total KRT8 expression level (KRT8 + p-KRT8) normalized to GAPDH, and the p-KRT8:total KRT8 ratio is shown as a number on the top of each bar graph. (C) Fluorescence microscopy images of ARPE-19 cells immunostained for KRT8 (red fluorescence). Cells were exposed to paraquat (400 μM) in the absence or presence of either 3-MA (10 mM) or Baf A1 (50 nM) for 24 h. Scale bar: 10 µm.
To further examine KRT8 expression in the RPE under oxidative stress, we performed immunocytochemistry analysis using fluorescent antibodies specific to KRT8 (Fig. 3C). The result showed that KRT8 expression increased throughout the cells, from the circumference of the nucleus to the cell periphery, upon exposure to paraquat for 24 h. However, cotreatment with paraquat and 3-MA markedly decreased expression of KRT8, especially in the cell periphery. In contrast, cotreatment with Baf A1 did not diminish the expression level of KRT8, which was elevated due to oxidative stress. These immunocytochemistry results were consistent with the KRT8 expression pattern analyzed from the western blot assay (Fig. 3B). These results suggest that the oxidative stress induces upregulation of both KRT8 and its phosphorylated form in RPE cells. More importantly, the enhanced expression of KRT8 was attenuated when RPE cells under oxidative stress were treated with 3-MA, indicating that KRT8 upregulation was related to an early stage of autophagy progression. This result was further corroborated by the observation that the KRT8 upregulation was not diminished with Baf A1 treatment, which blocks the late steps of autophagy.
KRT8 facilitates autophagosome-lysosome fusion during autophagy in RPE cells
Since KRT8 expression was observed to be related with the early stage of autophagy in RPE cells under the oxidative stress, we examined whether inactivation of autophagy initiation by knockdown of ATG5 (autophagy-related 5) diminishes KRT8 expression under oxidative stress. ATG5 is required for autophagosome elongation due to its ubiquitin ligase activity.28 ATG5 knockdown with ATG5-specific siRNA resulted in decreased expression of KRT8 and its phosphorylated form, as compared with untreated RPE cells under oxidative stress (Fig. 4A). Thus, expression of the intermediate filament KRT8 is enhanced during the initial stages of autophagy induced by oxidative stress in RPE cells.
Figure 4.
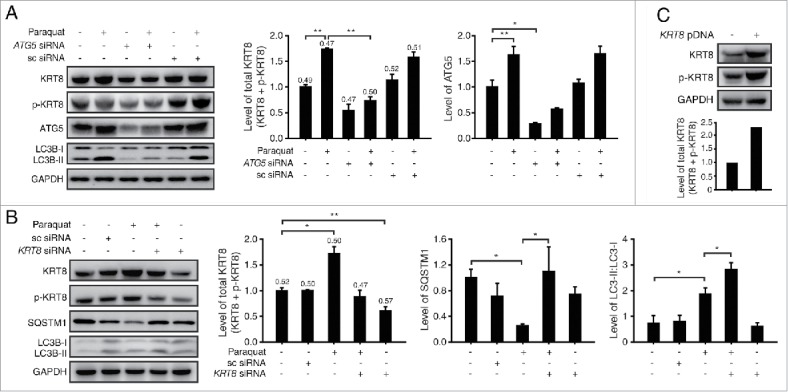
KRT8 expression is related to autophagy progression in RPE cells under oxidative stress. Western blot analysis of ARPE-19 cells after paraquat (400 μM) treatment of 24 h in the absence or presence of either (A) anti-ATG5 siRNA or (B) anti-KRT8 siRNA. Bar graphs indicate each protein expression level or the total KRT8 expression level (KRT8 + p-KRT8), and the numbers on the top of the quantitative bar graph represent the p-KRT8:total KRT8 ratio. Each protein band intensity was normalized to GAPDH. (C) Western blot analysis of KRT8 and its phosphorylated form (p-KRT8) in ARPE-19 cells overexpressing ARPE-19 and KRT8. Bar graphs show KRT8 and p-KRT8 expression levels normalized to GAPDH in western blot analysis.
Next, we investigated the relationship between KRT8 and autophagy through the knockdown of KRT8 (Fig. 4B). Under oxidative stress, KRT8 knockdown using anti-KRT8 siRNA resulted in accumulation of LC3B-II and decreased degradation of SQSTM1. Because LC3B-I to LC3B-II conversion and SQSTM1 degradation occurs at early and late stage of autophagy, respectively, knockdown of KRT8 is likely to affect the later stages of autophagy such as the fusion between autophagosomes and lysosomes. Thus, we tested the potential relationship between the late stage of autophagy (i.e., autophagosome-lysosome fusion) and KRT8 expression using paraquat-treated RPE cells. We induced oxidative stress in RPE cells with different expression levels of KRT8, using paraquat, KRT8-overexpressing cells (Fig. 4C), and KRT8-specific siRNA treated cells (i.e., KRT8 knockdown cells). To observe the autophagosome-lysosome fusion in ARPE-19 cells at different KRT8 expression levels, autophagosomes were traced with GFP-LC3B and lysosomes were stained with lysosome-specific fluorescent dye (LysoTracker Red DND-99), respectively. The ARPE-19 cells under oxidative stress and a control group showed accumulation of GFP-LC3B puncta and lysosomal organelles with partial colocalization (Fig. 5A). Autophagosomes traced with GFP-LC3B puncta and lysosomes mostly colocalized in the KRT8-overexpressing cells, as visualized with orange-colored fluorescence in the merged image. On the contrary, GFP-LC3B puncta did not overlap with lysosomes in ARPE-19 cells with KRT8 knockdown. Thus, it is possible that enhanced expression of KRT8 during the paraquat-induced autophagic process facilitates autophagosome-lysosome fusion in ARPE-19 cells under oxidative stress.
Figure 5.
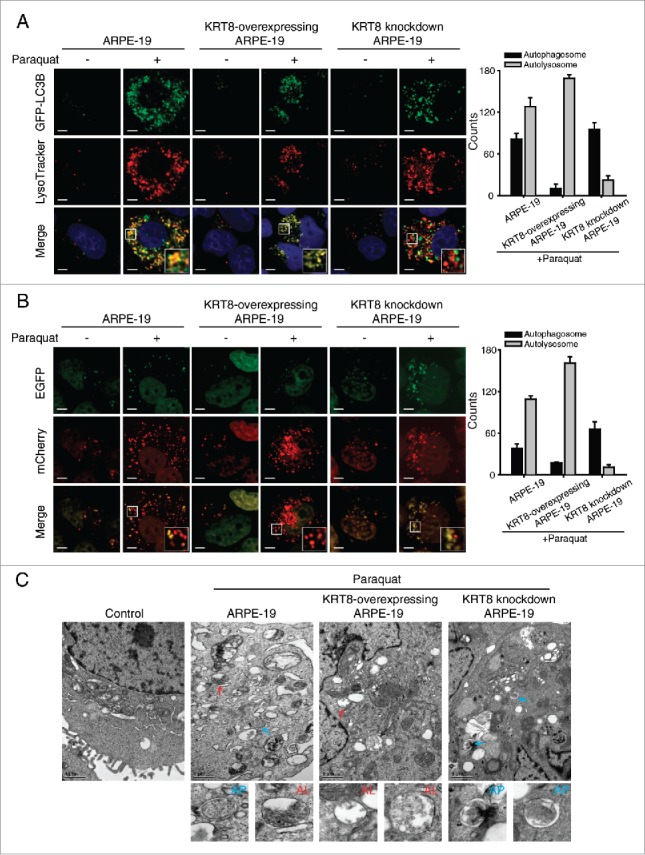
KRT8 facilitates autophagosome-lysosome fusion in RPE cells under oxidative stress. ARPE-19 cells, which were differently prepared for KRT8 expression, were transfected with (A) GFP-LC3B or (B) mCherry-EGFP-LC3B. The cells were then incubated in the absence or presence of paraquat (400 μM) for 24 h. (A) Fluorescence microscopy images of the cells were obtained by monitoring GFP-LC3B puncta (green fluorescence) and the lysosomes were stained with LysoTracker Red DND-99 (red fluorescence). Insets show an enlarged image of the merged field of interest. Scale bar: 3 µm. The fluorescence levels of GFP-LC3B puncta and lysosomes were quantified and presented as a graph; counts are the mean ± SD of 3 individual experiments. (B) The autophagosomes (yellow puncta) and autolysosomes (red puncta) in the mCherry-EGFP-LC3B-transfected cells were visualized with a confocal fluorescence microscope. Insets show an enlarged image of the merged field of interest. Scale bar: 3 µm. Bar graph indicates the counts of autophagosomes and autolysosomes in each cell. (C) Transmission electron microscopy images showing autophagic vesicles in the cells, which were prepared differently than for KRT8 expression. Blue-colored arrows indicate double-membrane autophagosomes (AP), and red arrows indicate single-membrane autolysosomes (AL). The enlarged images show each autophagic vacuole that is indicated by an arrow.
To further determine if the loss of KRT8 negatively affects the fusion between autophagosomes and lysosomes, we transfected cells with mCherry-EGFP-LC3B to label autophagic vacuoles (yellow) and performed cell imaging analysis after inducing autophagy (Fig. 5B). The autophagosomes are stained as yellow fluorescence puncta due to the FRET of mCherry (red) and EGFP (green) fluorescence. After autophagosome-lysosome fusion, EGFP is released from mCherry-EGFP-LC3B and degraded in lysosomes. Thus, autolysosome formation will lead to a fluorescence change from yellow to red. Similar to the above result (Fig. 5A), the fusion to form autolysosomes, as indicated by an increase in red puncta, was observed in the KRT8-overexpressing cells treated with paraquat. However, the KRT8 knockdown cells did not show red fluorescence and maintained yellow fluorescence puncta, indicating that autophagosomes did not readily fuse with lysosomes. These results indicate that KRT8 enhances autophagy clearance by facilitating autophagosome-lysosome fusion.
We next examined the role of KRT8 in autophagosome blockage and clearance in ARPE-19 cells under oxidative stress conditions using transmission electron microscopy (Fig. 5C). Autophagic flux was observed in ARPE-19 cells when under oxidative stress, as indicated by the presence of autophagosomes with a double membrane and autolysosomes with degraded autophagic cargo inside. Autolysosome formation was facilitated in the KRT8-overexpressing cells treated with paraquat, as many autolysosomes reflecting autophagy clearance appeared in the cytosol. In contrast, the KRT8 knockdown ARPE-19 cells showed only the double membranous vacuole form of autophagosomes, indicating diminished autolysosome formation due to autophagosome blockage. Together with the fusion assay data on LC3B fluorescence puncta, the transmission electron microscopy results indicate that KRT8 facilitated autophagosome clearance by enhancing the fusion process between autophagosomes and lysosomes.
KRT8 protects RPE cells from apoptotic cell death
KRT8, an intermediate filament, is known to maintain and support the shape and compartmentalization of organelles such as the mitochondria.29 In addition to its structural role, KRT8 has been reported to play a cytoprotective role against apoptosis through the modulation of mitochondria shape and function.30 To determine whether KRT8 protects RPE cells from apoptotic cell death under oxidative stress, we monitored apoptotic cell death in RPE cells with different expression levels of KRT8 (Fig. 6A). When the KRT8-overexpressing cells were treated with paraquat, the apoptotic cell count did not alter significantly, compared to the control group (ARPE-19). Interestingly, overexpression of KRT8 in RPE cells significantly decreased apoptosis to normal levels, as compared with RPE cells cotreated with paraquat and 3-MA or Baf A1 (Fig. 6A and B). Thus, apoptotic cell death caused by autophagy inhibitors in RPE cells under oxidative stress (see also Fig. 2) was attenuated with KRT8 expression. We analyzed the population of apoptotic cells using KRT8 knockdown cells, which were prepared with KRT8-specific siRNA, under oxidative stress. When these cells were subjected to paraquat treatment, the apoptotic cell count significantly increased by approximately 9-fold as compared with the control cells. The apoptotic cell population also increased than the paraquat-treated control cells (ARPE-19), when cellular autophagy was inhibited with 3-MA or Baf A1 under oxidative stress conditions.
Figure 6.
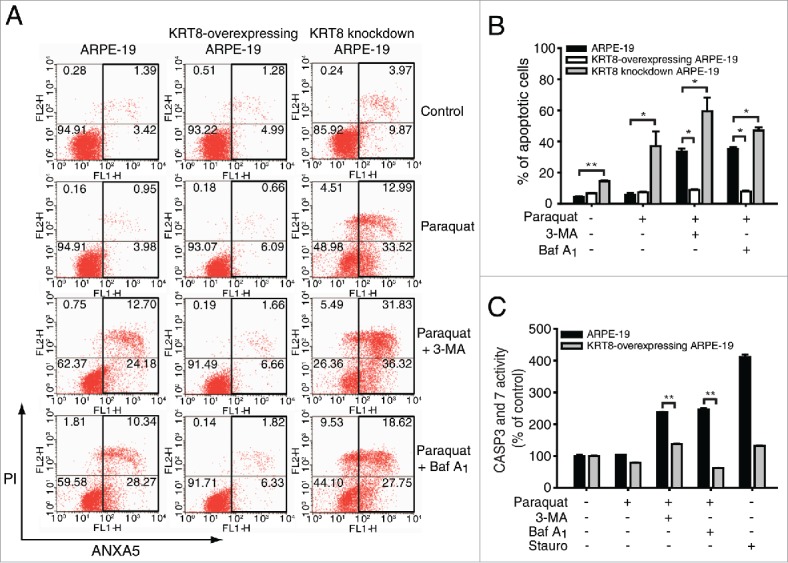
KRT8 protects RPE cells from apoptosis. ARPE-19 cells, which were differently prepared for KRT8 expression, were incubated with 400 μM paraquat in the absence or presence of either 10 mM 3-MA or 50 nM Baf A1 for 24 h. (A) A population of apoptotic cells was analyzed by FACS. (B) The bar graphs represent the mean percentage of apoptotic cells (n = 3). *P < 0.05 vs. control group, **P < 0.01 vs. control group. (C) Apoptotic enzyme CASP3 and CASP7 activity was measured for ARPE-19 and KRT8-overexpressing ARPE-19 cells. Staurosporine (1 μM) treatment was used as a positive control for apoptotic cell death. The data are presented as the mean ± SD, n = 3. **P < 0.01 vs. control group.
Further, to confirm the cytoprotective role of KRT8 against apoptotic cell death in ARPE-19 cells under oxidative stress, the KRT8-overexpressing RPE cells under oxidative stress with or without autophagy inhibitors were analyzed by monitoring CASP3 and CASP7 activity (Fig. 6C). Treatment with autophagy inhibitors such as 3-MA or Baf A1, significantly enhanced the CASP activity in RPE cells under oxidative stress (Fig. 2D). However, the elevated CASP3 and CASP7 activity was diminished in KRT8-overexpressing cells, which is consistent with the results obtained using flow cytometry. Thus, KRT8 expression in ARPE-19 cells under oxidative stress protects the cells from apoptotic cell death, playing a positive role in cell survival of RPE cells under oxidative stress conditions. This effect is likely brought about by KRT8 compensating for the blocking of autophagy by 3-MA or Baf A1 to cope with oxidative damage in the cells.
Oxidative stress induces MAPK1-mediated KRT8 phosphorylation and perinuclear reorganization in RPE cells
The structures of intermediate filaments including KRT8 respond dynamically to various cellular conditions such as stress and mitosis. Particularly, assembly and reorganization of KRT8 often requires its phosphorylation.31 KRT8 is phosphorylated by the mitogen-activated protein kinase (MAPK) family at Ser73.32 First, we determined whether KRT8 is phosphorylated by the MAPK kinase family in ARPE-19 cells under oxidative stress. During oxidative stress, phosphorylation of KRT8 was inhibited by a MAP2K1 inhibitor (PD98059), but not by a MAPK8/9/10 inhibitor (SP600125) in RPE cells (Fig. 7A). This result indicates that MAP2K1 is responsible for KRT8 phosphorylation at Ser73 during oxidative stress. When the ARPE-19 cells were treated with paraquat for 24 h, the level of p-MAPK1, an active form of MAPK1, increased and was accompanied by the upregulation of p-KRT8 (Fig. 7B). However, PD98059 suppressed paraquat-induced MAPK1 activation and KRT8 phosphorylation in RPE cells under oxidative stress, in which PD98059 attenuated MAPK1 through inhibiting MAP2K1, the upstream kinase of MAPK1. When the RPE cells were treated with paraquat, the MAP2K1 inhibitor PD98059 caused neither apoptotic cell death nor inhibition of autophagosome-lysosome fusion during autophagy (Fig. S5).
Figure 7.
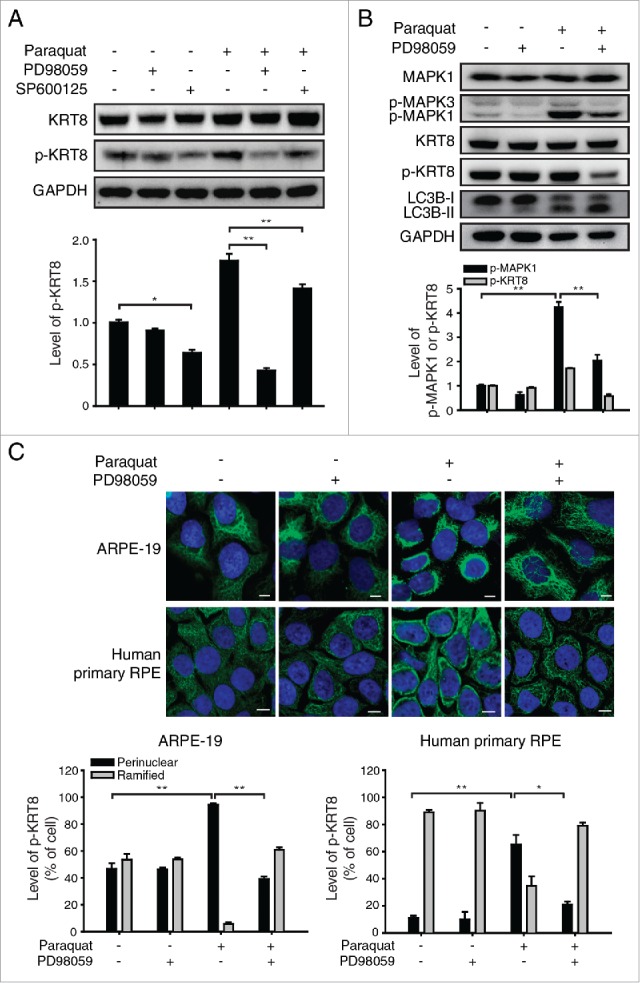
Oxidative stress induces MAPK1 mediated-KRT8 phosphorylation and its reorganization in RPE cells. (A) Western blot analysis of KRT8 and its phosphorylated form (p-KRT8) in ARPE-19 cells. ARPE-19 cells were treated with MAPK8/9/10 inhibitor; SP600125 (10 μM) or MAP2K1 inhibitor; PD98059 (20 μM) in the absence or presence of paraquat (400 μM) for 24 h. Bar graph indicates the expression level of p-KRT8 normalized to GAPDH in the western blot analysis. Data are presented as the mean ± SD, n = 3 (*P < 0.05, **P < 0.01). (B) ARPE-19 cells were exposed to paraquat (400 μM) for 24 h in the absence or presence of PD98059 (20 μM). Western blot analysis for expression levels of MAPK1, phosphorylated MAPK1/3 (p-MAPK1/3), KRT8, p-KRT8, and LC3B-II/I. Bar graph shows p-MAPK1 and p-KRT8 expression level in western blot analysis images. The protein band intensities were normalized to GAPDH, expressed relative to control. Data are presented as the mean ± SD, n = 3. **P < 0.01. (C) Both ARPE-19 (upper images) and human primary RPE cells (bottom images) were treated with paraquat (400 μM) in the absence or presence of PD98059 (20 μM) for 36 h. The cells were immunostained for p-KRT8 (green fluorescence). Nuclei are represented with blue fluorescence (TOPRO-3 staining). Scale bar: 3 µm. Bar graphs indicate the percentage of p-KRT8 distribution as either a ramified presence or a perinuclear presence in cells. Data are presented as the mean ± SD, n = 3. *P < 0.05; **P < 0.01.
Next, we determined the presence of p-KRT8 in both ARPE-19 and human primary RPE cells under oxidative stress. It has been reported that KRT8 and p-KRT8 are usually abundant in the cytoplasm of epithelial cells under normal conditions. 31 RPE cells under normal conditions showed widespread p-KRT8 in the cytoplasm, while paraquat treatment, which upregulates p-KRT8 resulted in the perinuclear presence of p-KRT8 (Fig. 7C). The reorganization of p-KRT8 was suppressed by PD98059 treatment in RPE cells under oxidative stress. This result suggests that KRT8 phosphorylation and its perinuclear reorganization are mediated by MAPK1 activation.
KRT8 phosphorylation elicits epithelial-mesenchymal transition and migration in RPE cells under oxidative stress
Besides increased intraocular level of VEGF, loss of epithelial features19 and junction integrity in the RPE33 are responsible for the development and unhampered growth of CNV, subsequent hemorrhage, and disciform scar formation that eventually result in the loss of vision when left untreated. When RPE cells are exposed to stress for a long-term, cells collapse their secure barrier to invade new blood vessels easily into the retina.34 Based on this pathology, we investigated the epithelial to mesenchymal transition (EMT) that causes loss of cell-cell tight junctions in epithelial cells and their ability to migrate.35
To monitor EMT in RPE cells under the oxidative stress, we examined the expression of CDH1/E-cadherin and VIM (vimentin), which are markers for epithelial and mesenchymal stages, respectively, when RPE cells were exposed to paraquat under various conditions for 48 h (Fig. 8A). Normal RPE cells maintain the epithelial morphology and show high expression of CDH1 in cell-cell junction with little expression of VIM in the cytoplasm. However, cells under continuous oxidative stress underwent EMT and exhibited downregulation of CDH1 and overexpression of VIM. Importantly, when the RPE cells were treated with the MAP2K1 inhibitor (PD98059) and paraquat, paraquat-induced EMT was significantly suppressed through the maintenance of CDH1 expression and inhibition of VIM expression.
Figure 8.
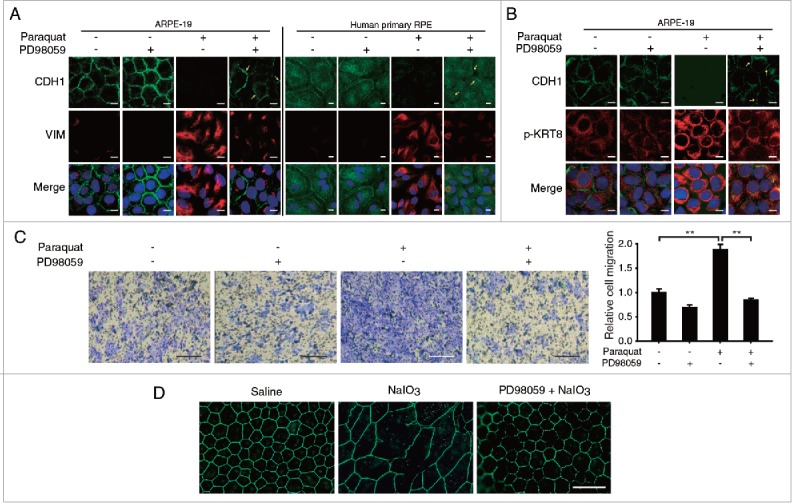
Oxidative stress induces epithelial-mesenchymal transition as well as RPE degeneration in vitro and in vivo. Both ARPE-19 and human primary RPE cells were treated with paraquat (400 μM) in the absence or presence of PD98059 (20 μM) for 48 h. The cells were immunostained for CDH1 (green fluorescence) and (A) VIM or (B) p-KRT8 (red fluorescence). The nuclei were visualized with TOPRO-3 staining (pseudo-colored blue). Scale bar: 10 µm. (C) ARPE-19 cells after paraquat (400 μM) treatment of 36 h in the absence or presence of PD98059 (20 μM) were assayed for cell migration using a transwell. Images show the presence of migrated cells, which were stained with methylene blue. Scale bar: 0.5 mm. Bar graph represents the relative number of migrated cells, which was quantified by measuring the blue-colored area. The data are presented as the mean ± SD, n = 3. **P < 0.01. (D) Immunohistochemical analysis of mice retinas at 7 to 9 wk of age using antibodies against the tight junction marker TJP1/ZO-1 (green fluorescence). Saline treated group received a single intravenous injection of 0.9% NaCl and served as controls. Mice were intravenously injected with NaIO3 (20 mg/kg) in the presence or absence of PD98059 (10 mg/kg). Animals were killed 2 wk after a single intravenous injection. Fluorescent images were obtained using a confocal microscope. Scale bar: 50 µm.
Next, we monitored distribution and expression level of CDH1 and p-KRT8 in ARPE-19 cells under various conditions to investigate the relationship between the perinuclear reorganization of phosphorylated KRT8 and progression of EMT under oxidative stress conditions (Fig. 8B). In ARPE-19 cells under normal conditions, CDH1 was expressed in cell-cell junctions, and p-KRT8 was widely distributed in a ramified form in the cytoplasm. When the cells were treated with paraquat, expression of p-KRT8 significantly increased, and p-KRT8 was distributed in the perinuclear region, accompanied by decreased CDH1 expression. When the MAP2K1 inhibitor PD98059 was added to the cells under oxidative stress, the reorganization and expression of p-KRT8 were inhibited, and intact CDH1 was present in tight junctions. Thus, these results indicate that the phosphorylation of KRT8 and its subsequent perinuclear reorganization are closely related to EMT progression in ARPE-19 cells under oxidative stress. Furthermore, we examined the migratory properties of stressed ARPE-19 cells by performing the transwell migration assay, in which the migrated cells were traced with methylene blue staining. The ARPE-19 cells under oxidative stress showed an enhanced transwell migratory effect as compared with the control group (Fig. 8C). This migratory property of the paraquat-treated RPE cells was significantly reversed by cotreatment with PD98059, indicating that KRT8 phosphorylation via MAPK1 was responsible for EMT and subsequent cellular migration.
To confirm the effect of KRT8 phosphorylation that results in loss of RPE junction integrity, oxidative stress was induced using iodate in mice; C57BL/6 mice were treated with saline (n = 6) or NaIO3 (20 mg/kg, n = 5). As shown in Fig. 8D, homogenous hexagonal staining of TJP1/ZO-1 was clearly seen in the controls (saline, left), whereas there was a highly disorganized pattern of staining in NaIO3 injected mice (NaIO3, center). To study the effect of PD98059 treatment on NaIO3-induced RPE degeneration in vivo, mice were immediately administrated with PD98059 (10 mg/kg, n = 4) after tail vein injection of NaIO3. MAP2K1 inhibitor PD98059 (10 mg/kg) markedly reversed the damaging effect of NaIO3 in the RPE (PD98059+NaIO3, right), suggesting that RPE degeneration induced by oxidative stress could be prevented by MAP2K1 inhibition. Together with in vivo results, the results suggest that prolonged oxidative stress leads to EMT in RPE cells and subsequent loss of cell junction integrity via KRT8 phosphorylation, which can be attenuated by inhibiting MAPK1 activity and preventing the phosphorylation of KRT8.
Discussion
In this study, we found that a short-term challenge by reactive oxygen species generated with paraquat (oxidative stress) in ARPE-19 or human primary RPE cells did not result in cell death due to induction of AKT phosphorylation via inactivation of PPP2 in the early phase and subsequent activation of autophagy (Fig. S2, Figs. 1 and 2). However, inhibition of autophagy under similar conditions led to increased cell death (Fig. 2). Thus, we speculated that RPE cells in vivo could maintain their functional and anatomic integrity in their young or healthy state, when their autophagy processes work efficiently despite continuous exposure to oxidative stress. However, in aged cells or during pathological conditions, autophagy is impaired, leading to RPE cell death, and development of degenerative diseases such as AMD.34 Previous studies have shown that a low level of autophagy is necessary to degrade and recycle long lived or toxic proteins and damaged organelles to maintain cell viability and cellular homeostasis.36-38
Cells can respond to a variety of insults, including oxidative stress with increased autophagic activity,39 and mutations that compromise the autophagy system result in increased stress sensitivity.40 Autophagy, a key cellular adaptive mechanism for maintaining cellular homeostasis involving the efficient clearance of harmful protein aggregates, is decreased in aged cells or in cells exposed to various stress condition including AMD in the eye.13,37,41-44 Although autophagy could be both protective and harmful in the setting of oxidative stress or an environment causing AMD, it remains to be determined whether increasing the level autophagy could be used as a preventive mechanism in RPE, and several recent studies have demonstrated that enhanced autophagy protects the RPE from oxidative damage and cell death.13,15,16,36,37 Studies also have reported that enhanced autophagy rescues cellular function in various diseases other than AMD such as Huntington disease45 or Mallory-Denk body formation in liver diseases.46 Thus, based on results of our studies along with others,47,48 treatments that enhance autophagy (i.e., rapamycin) would be expected to increase resistance to oxidative stress.
In this and in our previous studies,18 we observed that KRT8 was significantly upregulated in patients with neovascular AMD as well as in the RPE under oxidative stress. Our data indicated that the upregulated KRT8 after the early stage of autophagy under oxidative stress in RPE cells expedites autophagosome-lysosome fusion, the late stage of autophagy (Fig. 3, Fig. 4 and 5). Furthermore, overexpression of KRT8 was shown to protect RPE cells under oxidative stress from apoptotic cell death (Fig. 6). In epithelial cells, the predominant intermediate filaments are the keratins, which constitute the largest and most complex class of intermediate filaments.49 They are expressed in epithelial cells throughout the body, where they form structural networks to span the cell cytoplasm linking the plasma membrane, nucleus, and other cytoskeletal components.50 Keratins are divided into type I (K9-K20) and type II (K1-K8) and organized into bundles, composed of heterodimers; one type I and one type II keratin monomer.31 Each type I keratin has a specific type II keratin partner, depending on a tissue- or differentiation type; KRT8, a major component of intermediate filament in epithelial cells, has been known to pair with KRT18 of type II.31 Disruption of keratins can weaken the mechanical integrity of the cell. Besides providing structural support for the cells, accumulating evidence has shown that KRT8, with its filament partner KRT18, plays an important role in modulating cellular response to apoptotic stimuli.29,30 KRT8-KRT18 is reported to be responsible for cell resistance to tumor necrosis factor-induced cytotoxicity51,52 or Fas-mediated apoptosis.53 Thus, we suggest that elevated levels of KRT8 contribute to the enhanced survival of the RPE under pathological conditions.
We also demonstrated that activated MAPK1 mediates phosphorylation of KRT8 under oxidative stress (Fig. 7A). Phosphorylation of KRT8-KRT18 is required for the regulation of KRT8-KRT18 filament organization, turnover, and interaction with other proteins.54-56 KRT8 phosphorylation is caused by cell cycle progression, exposure to various growth factors, or stress-activated kinases including MAPK kinase family.57,58 We observed that oxidative stress leads to phosphorylation of KRT8 and its reorganization to a perinuclear place in the RPE cells; this can be reduced by the MAP2K1 inhibitor, PD98059 (Fig. 7). According to several studies, phosphorylation of keratin, which results in its reorganization, has been known to increase cell migration in epithelial cells.59-61 We found that oxidative stress induces EMT, and cell migration in RPE is mediated by phosphorylated KRT8 reorganization (Figs. 7 and 8). EMT, whose loss of epithelial and gain of mesenchymal character, is one of the key events in tumor progression, metastasis, hypoxia, fibrosis, and aging.62 Because maintenance of the epithelial characteristics of RPE is essential for the health of the overlying photoreceptors as well as the underlying choriocapillaris, stress-induced failure of RPE functions, well before overt RPE cell loss or death, may contribute to the pathogenesis and progression of AMD.17,63 Thus, maintaining RPE integrity by inhibiting KRT8 phosphorylation should prove to be clinically useful. According to our findings, the MAP2K1 inhibitor diminishes KRT8 phosphorylation and reorganization, and suppresses oxidative stress-induced cell degeneration both in vitro and in vivo (Fig. 8).
As summarized in Fig. 9, our results demonstrate that short-term exposure to oxidative stress does not trigger RPE cell death. Instead, this oxidative stress triggers autophagy, an antiapoptosis and cytoprotective process of the RPE. In addition, we suggest that upregulation of KRT8 with the inhibition of its phosphorylation is an essential cytoprotective mechanism and is required for preserving RPE integrity. We provided evidence to show that autophagy could have an essential role in the coping mechanisms of RPE cells under oxidative stress, and upregulation of KRT8 is a key component of this response. Elevated KRT8 in stressed RPE cells may be responsible for resistance to apoptosis. Besides, phosphorylated KRT8 induces EMT with enhanced cell migration via perinuclear reorganization. Because the loss of the epithelial features of RPE might be an early sign of the development of degenerative eye diseases such as AMD, our results indicate that KRT8 might be a new target for oxidative stress in RPE. Assuming that oxidative stress and aging are the major underlying causes of AMD, the upregulation of KRT8 and downregulation of phosphorylated KRT8 may have therapeutic applications in AMD, as these changes can promote resistance to oxidative stress and prevent RPE degeneration. Our previous study18 demonstrates that levels of KRT8 and proteins related to autophagy in AMD patients are higher than those in control subjects, indicating that these biomarker proteins could be novel targets for the treatment of AMD. Further studies will be thus needed to substantiate that targeting autophagy and KRT8 can be a therapeutic approach to stimulate endogenous protective mechanisms as well as to enhance cell survival while suppressing EMT, thus subsequently preventing the deterioration of neighboring photoreceptors and visual loss.
Figure 9.
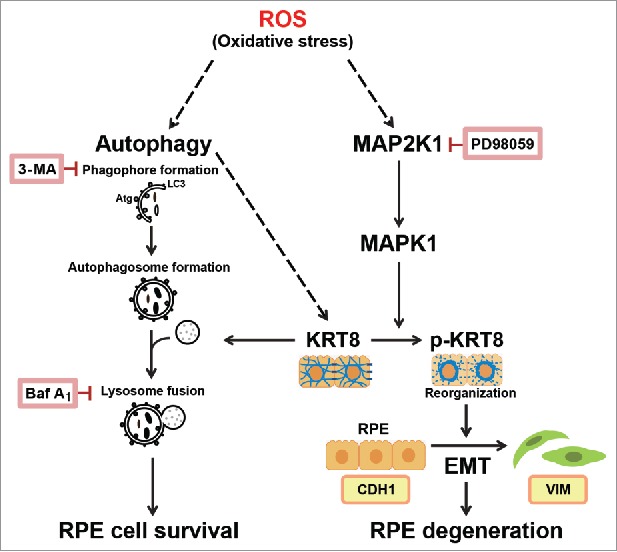
Proposed mechanism of the cellular fate of the RPE under oxidative stress via autophagy and the epithelial-mesenchymal transition (EMT).
Materials and methods
Reagents and antibodies
Methyl viologen dichloride hydrate (paraquat; 856177), 3-methyladenine (M9281), bafilomycin A1 (B1793), PD98059 (P215), SP600125 (S5567), and 2′,7′-dichlorofluorescin diacetate (D6883) were purchased from Sigma-Aldrich (numbers in parentheses indicate catalog numbers). Dulbecco's modified Eagle's medium: nutrient mixture F-12 (DMEM-F12; 11330032) and Opti-MEM (31985070) were supplied by GIBCO®. Plasmid DNAs (pSELECT-GFP-LC3B [setz-gfplc3b] and pUNO1-hKRT8, coding for the sequence of Homo sapiens (Hs)/human KRT8 [uno1-hkrt8]) were purchased from InvivoGen. The pBABE-puro mCherry-EGFP-LC3B plasmid was obtained from Addgene (22418), which was originally deposited by Jayanta Debnath (University of California at San Francisco, USA). Cyto-ID® autophagy detection kit was purchased from Enzo Life Sciences (ENZ-51031). The siRNA of KRT8 (Santa Cruz Biotechnology, sc-35156) and ATG5 (Cell Signaling Technology, 6345S) were supplied as indicated. The negative control siRNA (SN-1001) was purchased from Bioneer. TOPRO-3 (T3605) was purchased from Invitrogen Life Technologies. The following antibodies were used: anti-PPP2/PP2A was from BD Biosciences (610556), anti-LC3B, AKT, p-AKT, MTOR, p-MTOR, CDH1/E-cadherin, ATG5 (for detection of ATG12-ATG5), and PathScan® EMT Duplex IF Antibody Kit were purchased from Cell Signaling Technology (3868s, 9272, 9271, 2972s, 2974s, 3195, 2630, and 7771); anti-GAPDH, KRT8, and p-KRT8 were from Abcam (ab9484, ab9023, and ab32579); anti-SQSTM1, MAPK1, p-MAPK1/3, and horseradish peroxidase-conjugated anti-rabbit and mouse immunoglobulin were from Santa Cruz Biotechnology (sc-28359, sc-154, and sc-7383); anti-TJP1/ZO-1 was purchased from Life Technologies (61-7300); fluorescence (Alexa Fluor 488 and 555)-conjugated anti-mouse and rabbit immunoglobulin were purchased from Invitrogen Life Technologies (A11008, A11001, and A21428) (Table 1).
Cell culture and transfection
ARPE-19 cells (retinal pigment epithelial cell line; purchased from American Type Culture Collection, ATCC CRL-2302) were maintained in DMEM-F12 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Human primary RPE cells purchased from Lonza Biologics (00194987) were maintained in Retinal Pigment Epithelial Cell Basal Medium (Lonza Biologics, 195409) containing supplements (l-glutamine, GA-1000, and bFGF). ARPE-19 and human primary RPE cells were cultured at 37°C in 5% CO2. To prepare the ARPE-19 cells overexpressing KRT8, cells were seeded in a 60 mm dish and incubated with Opti-MEM medium at 37°C for 2 h. Cells were then transfected with 5 μg of the KRT8 overexpression plasmid (pUNO1-hKRT8) using Lipofectamine 2000 (Invitrogen Life Technologies, 11668019). After 6 h, cells were washed with phosphate-buffered saline (PBS; Welgene, LB 201-02), and maintained in complete DMEM-F12 medium with 20 μg/mL blasticidin (InvivoGen, ant-bl-1). To knockdown KRT8 expression in ARPE-19 cells, cells were transfected with anti-KRT8 siRNA. Cells were seeded (∼2.5 × 105) in a 6-well plate and incubated with Opti-MEM medium at 37°C for 2 h. Cells were then transfected with 50 nM of KRT8 siRNA or negative control siRNA using Lipofectamine 2000. After incubation at 37°C for 6 h, cells were washed with PBS and complete DMEM-F12 (supplemented with 10% FBS, 1% penicillin/streptomycin) was added.
Reactive oxygen species detection
Paraquat-induced ROS formation in ARPE-19 cells was detected by fluorescence microscopy. ARPE-19 cells (∼80% confluent) in a 6-well plate were incubated with 400 μM paraquat at 37°C for 12 or 24 h. Cells were then loaded with 10 μM 2′,7′-dichlorofluorescin diacetate (DCFDA) for 10 min at 37°C in the dark. At the end of incubation, the medium containing DCFDA was aspirated. Cells were then washed once with serum free DMEM-F12 medium, and then supplemented with complete DMEM-F12 medium. Cells were examined under a fluorescence microscope (AxioVert 200, Carl Zeiss, Oberkochen, Germany) with excitation and emission set at 525 and 550 nm, respectively. Fluorescence of oxidized DCFDA in cells was captured with AxioCam using AxioVision software (Carl Zeiss).
Autophagy assay; GFP-LC3B analysis
ARPE-19 cells were seeded in a 60-mm dish and incubated with Opti-MEM medium at 37°C for 2 h. Next, cells were transfected with 4 μg of pSELECT-GFP-LC3B using Lipofectamine 2000. After 6 h, cells were washed with PBS and complete DMEM-F12 medium with 400 μM paraquat, 10 mM 3-MA and/or 50 nM of Baf A1 for 24 h. Cells were fixed in 4% paraformaldehyde at room temperature for 1 h. Cells were then washed twice with PBS and stained with TOPRO-3 (1:1000), and observed under a confocal fluorescence microscope (Olympus FV-1000 spectral, Tokyo, Japan) with excitation and emission set at 525 and 550 nm, respectively.
Autophagosome fusion assay; mCherry-EGFP-LC3B analysis
ARPE-19 cells were seeded in a 12-well plate and incubated with Opti-MEM medium at 37°C for 2 h. Next, cells were transfected with 1 μg of pBABE-puro mCherry-EGFP-LC3B using Lipofectamine 2000. After transfection, cells were treated with 400 μM paraquat for 24 h. Cells were fixed in 4% paraformaldehyde at room temperature for 1 h. Cells were then washed twice with PBS and observed for EGFP fluorescence and mCherry fluorescence with excitation laser at 488 and 543 nm, respectively, under a confocal fluorescence microscope (Olympus FV-1000 spectral). The fluorescence images were captured at 495 to 530 nm (EGFP) and at 590 to 650 nm (mCherry) and emission signals were quantified with FV10-ASW 2.0 (Olympus software).
Transmission electron microscopy
ARPE-19 cells were seeded in a 100-mm dish and incubated with 400 μM paraquat for 24 h. Cells were fixed with 2% paraformaldehyde and 2% glutaraldehyde in 0.05 M sodium cacodylate buffer (pH 7.2) at 4°C for 2 h. Cells were washed 3 times with 0.05 M sodium cacodylate buffer (pH 7.2) and postfixed with 1% osmium tetroxide and 0.05 M cacodylate buffer at 4°C for 2 h. After postfixation, cells were washed twice with distilled water and en bloc stained in 0.5% uranyl acetate at 4°C overnight. The cells were dehydrated with various concentrations of ethanol and transitioned into 100% propylene oxide. The dehydrated cells were infiltrated with Spurr resin (Electron Microscopy Sciences, 14300) in propylene oxide and polymerized in Spurr resin at 70°C overnight. The cells were sequentially stained with 2% uranyl acetate and Reynolds' lead citrate after ultramicrotome sectioning (MTX, RMC, Tucson, AZ, USA). The prepared cells were observed by transmission electron microscopy (LIBRA 120, Carl Zeiss, Oberkochen, Germany).
Autophagy assay; autophagy fluorescence detection
Cells were washed using the Cyto-ID® Autophagy detection kit (Enzo Life Sciences, ENZ-51031) according to the manufacturer's protocol. ARPE-19 and human RPE cells were exposed to 400 μM paraquat in the absence or presence either 10 mM 3-MA or 50 nM Baf A1 for 24 h. Cells were washed with 1X assay buffer provided with the detection kit and incubated in a culture medium containing 2 μL/mL of Cyto-ID® Green Detection Reagent and 1 μL/mL of TOPRO-3 for 30 min at 37°C. Cells were washed with 1X Assay buffer and fixed with 4% paraformaldehyde. The fluorescence was measured with a confocal microscope (Olympus FV-1000 spectral).
Western blot analysis
Cells were first lysed in RIPA buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 0.1% SDS [Bio Basic Inc., SB0485], 0.5% sodium deoxycholate [Sigma-Aldrich, D6750], 1.0% Tergitol® [Sigma-Aldrich, NP40], 1 mM phenylmethanesulfonylfluoride, 10 mM 2-mercaptoethanol). The protein concentration was determined using the Bradford method (Bio-Rad, 5000006). A total of 30 μg of protein was separated by gradient SDS polyacrylamide gel electrophoresis (4-12% Tris-glycine gradient gel; Invitrogen, NP0322). Proteins were then transferred to an Immobilon-P PVDF membrane (Millipore, IPVH00010), and the membranes were blocked with 5% skim milk in TBST buffer (25 mM Tris, 150 mM NaCl, 2 mM KCl, 0.1% Tween 20 [Sigma-Aldrich, P5927], pH 7.4) for 1 h at room temperature, followed by overnight incubation in primary antibodies (1:500 to 1:2000) at 4°C. The membrane was subsequently incubated with horseradish peroxidase-conjugated anti-rabbit and mouse immunoglobulin, and signals were detected with an enhanced chemiluminescence system (Millipore, Bedford, MA, USA) by using G:BOX Chemi XL (Syngene, Ozyme, Saint-Quentin-en-Yvelines). The protein band intensities were measured by densitometry (GeneTools software included in G:BOX Chemi XL) and normalized to each corresponding loading control GAPDH.
Flow cytometric analysis
After the cells were treated with paraquat plus each inhibitor (3-MA, Baf A1, and PD98059) and/or anti-KRT8 siRNA for 24 h, they were washed with cold PBS and resuspended in staining buffer containing fluorescein-5-isothiocyanate, ANXA5, and propidium iodide, provided in the apoptosis detection kit (BD Biosciences, 556547), according to the manufacturer's instructions. Flow cytometric data were obtained using a FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA, USA) equipped with BD CellQuest Pro software (BD Biosciences). Caspase-3 and 7 activity in apoptotic cells was measured using the Caspase-Glo 3/7 reagent (Promega, G8091) according to the manufacturer's instructions, and a VICTOR X3 Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA) was used to measure the luminescence emitted from the cleaved substrates.
Cell migration assay
Cellular migration assays were performed using ARPE-19 cells in a 24-well plate with transwell inserts (Costar®, 6.5 mm, 5.0-μm pore size, Corning Costar; CLS3421). Approximately 1×105 cells were seeded into transwell inserts with serum free DMEM-F12 medium. The transwell inserts, which contained the cells, were transferred into the lower chamber with DMEM-F12 medium supplemented with 10% FBS, 1% penicillin/streptomycin and 400 μM paraquat and/or 20 μM of PD98059. After incubation for 36 h at 37°C in a 5% CO2 atmosphere, cells were fixed with 4% paraformaldehyde for 1 h at room temperature. Cells were then permeabilized with 100% methanol for 20 min at room temperature. The cells were stained with methylene blue, and nonmigrated cells were removed using cotton tips. The migrated cells were observed under a light microscope (Eclipse TS100; Nikon, Tokyo, Japan).
Experimental animals and immunostaining
Mice were maintained in accordance with the policies of the Konkuk University Institutional Animal Care and Use Committee (IACUC). These mice were housed in a controlled barrier facility within the Konkuk University Laboratory Animal Research Center. All animals were handled in accordance with the policies set forth in the ARVO Statement for the Use of Animals in Ophthalmic and Visual Research. C57BL/6 male mice at an age of 7 to 9 wk were purchased from the Orient Bio (Seongnam, Korea) and were allowed one wk to acclimate to the facility before the following experiments. All mice were anesthetized with CO2 gas deeply and their eyes enucleated immediately. Anterior eye cups were dissected on cold PBS. After removal of the retina carefully, RPE/choroids were fixed 4% paraformaldehyde. RPE/choroids flat mounts were incubated in blocking solution including 1% BSA (Sigma-Aldrich, A7906) and 0.2% Triton X-100 for 30 min at room temperature. Tissues were incubated with TJP1/ZO-1 antibody for overnight at 4°C. Flat mounts were rinsed in PBS containing 0.5% BSA and then incubated with a secondary antibody 1 h at room temperature and mounted. All stained samples were captured using a CCD camera (Axio Cam MRc, Carl Zeiss, Oberkochen, Germany).
For cellular immunostaining, ARPE-19 and primary RPE cells were cultured in 24-well plate with auto-coverglass (12 mm ∅). Cells were fixed with 4% paraformaldehyde for 1 h at room temperature. Cells were then blocked with 3% BSA for 1 h after permeabilized with 0.2% Triton X-100 for 15 min. Cells were incubated overnight at 4°C with the primary antibodies (1:200; KRT8 and p-KRT8 or 1:100; PathScan® EMT Duplex IF Kit Primary Antibody Cocktail) and treated with the fluorescence-conjugated secondary antibodies (1:1000; Alexa Fluor 488 and 555) for 2 h at room temperature in the dark. The cells were washed 3 times with PBS for 10 min each after every step, and the nuclei were stained with TOPRO-3 (1:500 diluted). Cells were mounted on the coverslip with ProLong Gold antifade reagent (Invitrogen Life Technologies, P36934) and observed with a confocal microscope (Olympus FV-1000 spectral). To quantify the fluorescence intensity distribution of p-KRT8, 3 linear region of interests (ROIs) of equal length were placed in the perinuclear and cytoplasmic regions of each cell (software included in Olympus FV-1000). The normalized intensity profile for all ROI was obtained and used as an indicator of p-KRT8 distribution under different conditions.
Statistical analysis
Replicate data are expressed as means ± standard error of mean (s.e.m.). Paired data were evaluated by the Student t test, and a one-way analysis of variance (ANOVA) was used for multiple comparisons. A value of P < 0.05 was considered statistically significant.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- AH
aqueous humor
- AMD
age-related macular degeneration
- ATG5
autophagy-related 5
- Baf A1
bafilomycin A1
- CASP
caspase
- CDH1/E-cadherin
cadherin 1
- CNV
choroidal neovascularization
- CTSD
cathepsin D; DCFDA, 2′,7′-dichlorofluorescin diacetate
- EMT
epithelial-mesenchymal transition
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- KRT8
keratin 8
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3
- MAPK1/ERK2
mitogen-activated protein kinase 1
- MAPK3/ERK1
mitogen-activated protein kinase 3
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- PPP2/PP2A
protein phosphatase 2
- ROI
region of interest
- ROS
reactive oxygen species
- RPE
retinal pigment epithelium
- SQSTM1
sequestosome 1
- TJP1/ZO-1
tight junction protein 1
- VEGF
vascular endothelial growth factor
- VIM
vimentin
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (NRF-2012M3A9B2028336 and NRF-2015R1A2A2A01005591 to D.-E. K. and H. C., respectively.).
References
- [1].Bressler NM, Bressler SB, Congdon NG, Ferris FL 3rd, Friedman DS, Klein R, Lindblad AS, Milton RC, Seddon JM. Potential public health impact of age-related eye disease study results: AREDS report no. 11. Arch Ophthalmol 2003; 121:1621-4; http://dx.doi.org/ 10.1001/archopht.121.11.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology 2013; 120:2292-9; PMID:23642856; http://dx.doi.org/ 10.1016/j.ophtha.2013.03.046 [DOI] [PubMed] [Google Scholar]
- [3].Jaffe GJ, Eliott D, Wells JA, Prenner JL, Papp A, Patel S. A Phase 1 Study of Intravitreous E10030 in combination with ranibizumab in neovascular age-related macular degeneration. Ophthalmology 2016; 123:78-85; PMID:26499921; http://dx.doi.org/ 10.1016/j.ophtha.2015.09.004 [DOI] [PubMed] [Google Scholar]
- [4].Churchill AJ, Carter JG, Lovell HC, Ramsden C, Turner SJ, Yeung A, Escardo J, Atan D. VEGF polymorphisms are associated with neovascular age-related macular degeneration. Hum Mol Genet 2006; 15:2955-61; PMID:16940309; http://dx.doi.org/ 10.1093/hmg/ddl238 [DOI] [PubMed] [Google Scholar]
- [5].Brantley MA Jr., Fang AM, King JM, Tewari A, Kymes SM, Shiels A. Association of complement factor H and LOC387715 genotypes with response of exudative age-related macular degeneration to intravitreal bevacizumab. Ophthalmology 2007; 114:2168-73; PMID:18054635 [DOI] [PubMed] [Google Scholar]
- [6].Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, et al.. Seven new loci associated with age-related macular degeneration. Nat Genet 2013; 45:433-9, 39e1-2; PMID:23455636; http://dx.doi.org/ 10.1038/ng.2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arjamaa O, Nikinmaa M, Salminen A, Kaarniranta K. Regulatory role of HIF-1alpha in the pathogenesis of age-related macular degeneration (AMD). Ageing Res Rev 2009; 8:349-58; PMID:19589398; http://dx.doi.org/ 10.1016/j.arr.2009.06.002 [DOI] [PubMed] [Google Scholar]
- [8].Ranchon I, LaVail MM, Kotake Y, Anderson RE. Free radical trap phenyl-N-tert-butylnitrone protects against light damage but does not rescue P23H and S334ter rhodopsin transgenic rats from inherited retinal degeneration. J Neurosci 2003; 23:6050-7; PMID:12853423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yamashita H, Horie K, Yamamoto T, Nagano T, Hirano T. Light-induced retinal damage in mice. Hydrogen peroxide production and superoxide dismutase activity in retina. Retina 1992; 12:59-66; PMID:1565873 [PubMed] [Google Scholar]
- [10].Schmitz-Valckenberg S, Fleckenstein M, Scholl HP, Holz FG. Fundus autofluorescence and progression of age-related macular degeneration. Surv Ophthalmol 2009; 54:96-117; PMID:19171212 [DOI] [PubMed] [Google Scholar]
- [11].Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis 1999; 5:32; PMID:10562656 [PMC free article] [PubMed] [Google Scholar]
- [12].Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749-60; PMID:17347651; http://dx.doi.org/ 10.1038/sj.emboj.7601623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, Lewin A, Grant M, Dunn W Jr, Ding J, et al.. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014; 10:1989-2005; PMID:25484094; http://dx.doi.org/ 10.4161/auto.36184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kaarniranta K, Salminen A, Eskelinen EL, Kopitz J. Heat shock proteins as gatekeepers of proteolytic pathways-Implications for age-related macular degeneration (AMD). Ageing Res Rev 2009; 8:128-39; PMID:19274853 [DOI] [PubMed] [Google Scholar]
- [15].Johansson I, Monsen VT, Pettersen K, Mildenberger J, Misund K, Kaarniranta K, Schonberg S, Bjorkoy G. The marine n-3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy 2015; 11:1636-51; PMID:26237736; http://dx.doi.org/ 10.1080/15548627.2015.1061170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang L, Cano M, Handa JT. p62 provides dual cytoprotection against oxidative stress in the retinal pigment epithelium. Biochim Biophys Acta 2014; 1843:1248-58; PMID:24667411; http://dx.doi.org/ 10.1016/j.bbamcr.2014.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhao C, Yasumura D, Li X, Matthes M, Lloyd M, Nielsen G, Ahern K, Snyder M, Bok D, Dunaief JL, et al.. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest 2011; 121:369-83; PMID:21135502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kang GY, Bang JY, Choi AJ, Yoon J, Lee WC, Choi S, Yoon S, Kim HC, Baek JH, Park HS, et al.. Exosomal proteins in the aqueous humor as novel biomarkers in patients with neovascular age-related macular degeneration. J Proteome Res 2014; 13:581-95; PMID:24400796; http://dx.doi.org/ 10.1021/pr400751k [DOI] [PubMed] [Google Scholar]
- [19].Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci 1996; 37:855-68; PMID:8603870 [PubMed] [Google Scholar]
- [20].Wang Z, Zhang J, Wang Y, Xing R, Yi C, Zhu H, Chen X, Guo J, Guo W, Li W, et al.. Matrine, a novel autophagy inhibitor, blocks trafficking and the proteolytic activation of lysosomal proteases. Carcinogenesis 2013; 34:128-38; PMID:23002236; http://dx.doi.org/ 10.1093/carcin/bgs295 [DOI] [PubMed] [Google Scholar]
- [21].Guo S, Liang Y, Murphy SF, Huang A, Shen H, Kelly DF, Sobrado P, Sheng Z. A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy 2015; 11:560-72; PMID:25714620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of P62/Sqstm1. Method Enzymol 2009; 452:181-97; http://dx.doi.org/ 10.1016/S0076-6879(08)03612-4 [DOI] [PubMed] [Google Scholar]
- [23].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Antony R, Lukiw WJ, Bazan NG. Neuroprotectin D1 induces dephosphorylation of Bcl-xL in a PP2A-dependent manner during oxidative stress and promotes retinal pigment epithelial cell survival. J Biol Chem 2010; 285:18301-8; PMID:20363734; http://dx.doi.org/ 10.1074/jbc.M109.095232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yorimitsu T, He C, Wang K, Klionsky DJ. Tap42-associated protein phosphatase type 2A negatively regulates induction of autophagy. Autophagy 2009; 5:616-24; PMID:19223769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Qi Z, Yang W, Liu Y, Cui T, Gao H, Duan C, Lu L, Zhao C, Zhao H, Yang H. Loss of PINK1 function decreases PP2A activity and promotes autophagy in dopaminergic cells and a murine model. Neurochem Int 2011; 59:572-81; PMID:21672589; http://dx.doi.org/ 10.1016/j.neuint.2011.03.023 [DOI] [PubMed] [Google Scholar]
- [27].Leopoldino AM, Squarize CH, Garcia CB, Almeida LO, Pestana CR, Polizello AC, Uyemura SA, Tajara EH, Gutkind JS, Curti C. Accumulation of the SET protein in HEK293T cells and mild oxidative stress: cell survival or death signaling. Mol Cell Biochem 2012; 363:65-74; PMID:22143534; http://dx.doi.org/ 10.1007/s11010-011-1158-x [DOI] [PubMed] [Google Scholar]
- [28].Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657-68; PMID:11266458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta 2006; 1757:692-99; PMID:16729962 [DOI] [PubMed] [Google Scholar]
- [30].Tao GZ, Looi KS, Toivola DM, Strnad P, Zhou Q, Liao J, Wei YQ, Habtezion A, Omary MB. Keratins modulate the shape and function of hepatocyte mitochondria: a mechanism for protection from apoptosis. J Cell Sci 2009; 122:3851-55; PMID:19825937; http://dx.doi.org/ 10.1242/jcs.051862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Snider NT, Omary MB. Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat Rev Mol Cell Biol 2014; 15:163-77; PMID:24556839; http://dx.doi.org/ 10.1038/nrm3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liao J, Ku NO, Omary MB. Stress, apoptosis, and mitosis induce phosphorylation of human keratin 8 at Ser-73 in tissues and cultured cells. J Biol Chem 1997; 272:17565-73; PMID:9211903 [DOI] [PubMed] [Google Scholar]
- [33].Bailey TA, Kanuga N, Romero IA, Greenwood J, Luthert PJ, Cheetham ME. Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 2004; 45:675-84; PMID:14744914 [DOI] [PubMed] [Google Scholar]
- [34].Ambati J, Fowler BJ. Mechanisms of age-related macular degeneration. Neuron 2012; 75:26-39; PMID:22794258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tamiya S, Liu LH, Kaplan HJ. Epithelial-Mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Invest Ophthalmol Vis Sci 2010; 51:2755-63; PMID:20042656; http://dx.doi.org/ 10.1167/iovs.09-4725 [DOI] [PubMed] [Google Scholar]
- [36].Toops KA, Tan LX, Jiang Z, Radu RA, Lakkaraju A. Cholesterol-mediated activation of acid sphingomyelinase disrupts autophagy in the retinal pigment epithelium. Mol Biol Cell 2015; 26:1-14; PMID:25378587; http://dx.doi.org/ 10.1091/mbc.E14-05-1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Valapala M, Wilson C, Hose S, Bhutto IA, Grebe R, Dong A, Greenbaum S, Gu L, Sengupta S, Cano M, et al.. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/betaA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014; 10:480-96; PMID:24468901; http://dx.doi.org/ 10.4161/auto.27292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115:2679-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Girardot F, Monnier V, Tricoire H. Genome wide analysis of common and specific stress responses in adult drosophila melanogaster. BMC Genomics 2004; 5:74; PMID:15458575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wu H, Wang MC, Bohmann D. JNK protects Drosophila from oxidative stress by trancriptionally activating autophagy. Mech Dev 2009; 126:624-37; PMID:19540338; http://dx.doi.org/ 10.1016/j.mod.2009.06.1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ryhanen T, Hyttinen JM, Kopitz J, Rilla K, Kuusisto E, Mannermaa E, Viiri J, Holmberg CI, Immonen I, Meri S, et al.. Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy-mediated proteolysis in human retinal pigment epithelial cells. J Cell Mol Med 2009; 13:3616-31; PMID:19017362; http://dx.doi.org/ 10.1111/j.1582-4934.2008.00577.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One 2009; 4:e4160; PMID:19129916; http://dx.doi.org/ 10.1371/journal.pone.0004160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205 [DOI] [PubMed] [Google Scholar]
- [44].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204 [DOI] [PubMed] [Google Scholar]
- [45].Yoon SY, Choi JE, Kweon HS, Choe H, Kim SW, Hwang O, Lee H, Lee JY, Kim DH. Okadaic acid increases autophagosomes in rat neurons: implications for Alzheimer's disease. J Neurosci Res 2008; 86:3230-9; PMID:18615642; http://dx.doi.org/ 10.1002/jnr.21760 [DOI] [PubMed] [Google Scholar]
- [46].Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology 2008; 47:2026-35; PMID:18454506; http://dx.doi.org/ 10.1002/hep.22294 [DOI] [PubMed] [Google Scholar]
- [47].Nussenblatt RB, Byrnes G, Sen HN, Yeh S, Faia L, Meyerle C, Wroblewski K, Li Z, Liu B, Chew E, et al.. A randomized pilot study of systemic immunosuppression in the treatment of age-related macular degeneration with choroidal neovascularization. Retina 2010; 30:1579-87; PMID:20847709; http://dx.doi.org/ 10.1097/IAE.0b013e3181e7978e [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Stahl A, Paschek L, Martin G, Gross NJ, Feltgen N, Hansen LL, Agostini HT. Rapamycin reduces VEGF expression in retinal pigment epithelium (RPE) and inhibits RPE-induced sprouting angiogenesis in vitro. FEBS Lett 2008; 582:3097-102; PMID:18703055; http://dx.doi.org/ 10.1016/j.febslet.2008.08.005 [DOI] [PubMed] [Google Scholar]
- [49].Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem 1994; 63:345-82; PMID:7979242 [DOI] [PubMed] [Google Scholar]
- [50].Lau AT, Chiu JF. The possible role of cytokeratin 8 in cadmium-induced adaptation and carcinogenesis. Cancer Res 2007; 67:2107-13; PMID:17332340 [DOI] [PubMed] [Google Scholar]
- [51].Caulin C, Ware CF, Magin TM, Oshima RG. Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J Cell Biol 2000; 149:17-22; PMID:10747083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Inada H, Izawa I, Nishizawa M, Fujita E, Kiyono T, Takahashi T, Momoi T, Inagaki M. Keratin attenuates tumor necrosis factor-induced cytotoxicity through association with TRADD. J Cell Biol 2001; 155:415-26; PMID:11684708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gilbert S, Loranger A, Daigle N, Marceau N. Simple epithelium keratins 8 and 18 provide resistance to Fas-mediated apoptosis. The protection occurs through a receptor-targeting modulation. J Cell Biol 2001; 154:763-73; PMID:11514590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Coulombe PA, Omary MB. ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr Opin Cell Biol 2002; 14:110-22; PMID:11792552 [DOI] [PubMed] [Google Scholar]
- [55].Ku NO, Omary MB. Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J Cell Biol 2000; 149:547-52; PMID:10791969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ku NO, Liao J, Omary MB. Phosphorylation of human keratin 18 serine 33 regulates binding to 14-3-3 proteins. EMBO J 1998; 17:1892-906; PMID:9524113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].He T, Stepulak A, Holmstrom TH, Omary MB, Eriksson JE. The intermediate filament protein keratin 8 is a novel cytoplasmic substrate for c-Jun N-terminal kinase. J Biol Chem 2002; 277:10767-74; PMID:11781324 [DOI] [PubMed] [Google Scholar]
- [58].Feng L, Zhou X, Liao J, Omary MB. Pervanadate-mediated tyrosine phosphorylation of keratins 8 and 19 via a p38 mitogen-activated protein kinase-dependent pathway. J Cell Sci 1999; 112(Pt 13):2081-90; PMID:10362538 [DOI] [PubMed] [Google Scholar]
- [59].Busch T, Armacki M, Eiseler T, Joodi G, Temme C, Jansen J, von Wichert G, Omary MB, Spatz J, Seufferlein T. Keratin 8 phosphorylation regulates keratin reorganization and migration of epithelial tumor cells. J Cell Sci 2012; 125:2148-59; PMID:22344252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chung BM, Rotty JD, Coulombe PA. Networking galore: intermediate filaments and cell migration. Curr Opin Cell Biol 2013; 25:600-12; PMID:23886476; http://dx.doi.org/ 10.1016/j.ceb.2013.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Park MK, Lee HJ, Shin J, Noh M, Kim SY, Lee CH. Novel participation of transglutaminase-2 through c-Jun N-terminal kinase activation in sphingosylphosphorylcholine-induced keratin reorganization of PANC-1 cells. Biochim Biophys Acta 2011; 1811:1021-9; PMID:21840417; http://dx.doi.org/ 10.1016/j.bbalip.2011.07.007 [DOI] [PubMed] [Google Scholar]
- [62].Sabbah M, Emami S, Redeuilh G, Julien S, Prevost G, Zimber A, Ouelaa R, Bracke M, De Wever O, Gespach C. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat 2008; 11:123-51; PMID:18718806; http://dx.doi.org/ 10.1016/j.drup.2008.07.001 [DOI] [PubMed] [Google Scholar]
- [63].Zanzottera EC, Messinger JD, Ach T, Smith RT, Curcio CA. Subducted and melanotic cells in advanced age-related macular degeneration are derived from retinal pigment epithelium. Invest Ophthalmol Vis Sci 2015; 56:3269-78; PMID:26024109; http://dx.doi.org/ 10.1167/iovs.15-16432 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.