

ABSTRACT
PRKAA (protein kinase, AMP-activated, α catalytic subunit) regulates mitochondrial biogenesis, function, and turnover. However, the molecular mechanisms by which PRKAA regulates mitochondrial dynamics remain poorly characterized. Here, we report that PRKAA regulated mitochondrial fission via the autophagy-dependent degradation of DNM1L (dynamin 1-like). Deletion of Prkaa1/AMPKα1 or Prkaa2/AMPKα2 resulted in defective autophagy, DNM1L accumulation, and aberrant mitochondrial fragmentation in the mouse aortic endothelium. Furthermore, autophagy inhibition by chloroquine treatment or ATG7 small interfering RNA (siRNA) transfection, upregulated DNM1L expression and triggered DNM1L-mediated mitochondrial fragmentation. In contrast, autophagy activation by overexpression of ATG7 or chronic administration of rapamycin, the MTOR inhibitor, promoted DNM1L degradation and attenuated mitochondrial fragmentation in Prkaa2-deficient (prkaa2−/−) mice, suggesting that defective autophagy contributes to enhanced DNM1L expression and mitochondrial fragmentation. Additionally, the autophagic receptor protein SQSTM1/p62, which bound to DNM1L and led to its translocation into the autophagosome, was involved in DNM1L degradation by the autophagy-lysosome pathway. Gene silencing of SQSTM1 markedly reduced the association between SQSTM1 and DNM1L, impaired the degradation of DNM1L, and enhanced mitochondrial fragmentation in PRKAA-deficient endothelial cells. Finally, the genetic (DNM1L siRNA) or pharmacological (mdivi-1) inhibition of DNMA1L ablated mitochondrial fragmentation in the mouse aortic endothelium and prevented the acetylcholine-induced relaxation of isolated mouse aortas. This suggests that aberrant DNM1L is responsible for enhanced mitochondrial fragmentation and endothelial dysfunction in prkaa knockout mice. Overall, our results show that PRKAA deletion promoted mitochondrial fragmentation in vascular endothelial cells by inhibiting the autophagy-dependent degradation of DNM1L.
KEYWORDS: AMPK, autophagy, DNM1L, endothelial dysfunction, mitochondrial fission, PRKAA/AMPK catalytic subunit α
Introduction
Mitochondria are dynamic, energy-producing organelles that frequently undergo cycles of fusion and fission to generate networks and to form smaller, individual mitochondria, respectively.1,2 Mitochondrial dynamics is an important process that maintains normal mitochondrial function. A perturbation of this process is associated with mitochondrial dysfunction in various diseases, including neurodegenerative diseases and diabetes.3,4 In addition, inhibition of DNM1L (dynamin 1-like), a mitochondrial fission protein, restores NOS3/eNOS (endothelial nitric oxide synthase 3) phosphorylation and cyclic guanosine monophosphate production, suggesting that increased mitochondrial fission contributes to endothelial dysfunction.5 Endothelial dysfunction is an initial step in the development of cardiovascular diseases. Mitochondrial fusion is controlled by fusion proteins, including MFN1 (mitofusin 1), MFN2, and OPA1 (optic atrophy 1). Several mitochondrial outer membrane proteins, including FIS1 (fission, mitochondrial 1), MFF (mitochondrial fission factor), and MIEF1/2 (mitochondrial elongation factor 1/2), have been proposed to promote mitochondrial fission by recruiting DNM1L.6 DNM1L is a primarily cytoplasmic protein that translocates to the mitochondria upon activation and initiates mitochondrial fragmentation.7-9 DNM1L activity and cellular localization are highly regulated by post-translational modifications, including phosphorylation, ubiquitination, sumoylation, and N-linked nitrosylation.10 A recent study using pharmacological and genetic approaches further suggests that DNM1L expression may be regulated by autophagy.11 However, the molecular mechanisms controlling DNM1L protein turnover remain elusive.
AMPK (protein kinase, AMP-activated) is a heterotrimer that is composed of α, β, and γ subunits. Each subunit has at least 2 isoforms.12,13 Although the major isoform of PRKAA in endothelial cells is PRKAA1,14 both PRKAA1 and PRKAA2 may be equally important in maintaining endothelial function. Activation of either PRKAA1/AMPKα115 or PRKAA2/AMPKα216 can improve endothelial function by phosphorylating NOS3/eNOS at both Ser1177 and Ser635 to increase nitric oxide release. AMPK activation promotes mitochondrial biogenesis through upregulation of the PPARGC1A/PGC-1α (peroxisome proliferative activated receptor, gamma, coactivator 1 α), which regulates the transcription of many genes that are involved in mitochondrial energy metabolism, mitochondrial physiology, and the oxidation of glucose and fatty acids.17-19 Activation of AMPK also reduces the production of mitochondrial reactive oxygen species (ROS) and increases cellular antioxidant potential.20-22 Pharmacological activation of AMPK with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) markedly increases mitochondrial uncoupling protein-2 expression and reduces superoxide production and prostacyclin synthase nitration in diabetic mice.23 Similarly, metformin-activated AMPK normalizes high glucose-induced, mitochondria-derived ROS production via the induction of magnesium-superoxide dismutases and the activation of the AMPK-PPARGC1A pathway.24 Furthermore, AMPK activation increases mitochondrial autophagy by phosphorylating ULK1 (unc51-like kinase 1) and prevents apoptosis upon nutrient deprivation.25,26 Recent studies suggest that AMPK may have different effects on mitochondrial dynamics in different cell types. Mahendra et al27 report that C-peptide treatment activates AMPK and prevents mitochondrial fission in endothelial cells from streptozotocin-injected diabetic mice. Jia et al28 report that AICAR treatment decreases palmitate-induced mitochondrial fission in aortic endothelial cells; this effect is partially attributed to the enhancement of DNM1L phosphorylation. In addition, AMPK activation attenuates mitochondrial fragmentation in fatty acid-treated INS-1E β-cells29 and in cisplatin-treated human renal proximal tubular epithelial cells.30 However, Toyama et al31 identify that AMPK activation triggers mitochondrial fission by phosphorylating MFF in human osteosarcoma U2OS cells. Thus, the contribution of PRKAA to mitochondrial dynamics in different cell types needs to be clarified.
In the present study, we examined mitochondria fission, autophagy, DNM1L expression, and endothelial function in both global and endothelial cell-specific prkaa knockout mice. Genetic deletion of Prkaa1 or Prkaa2 in endothelial cells suppressed autophagy, leading to DNM1L accumulation and increasing DNM1L-dependent mitochondrial fragmentation. Consistently, gene silencing of DNM1L or administration of mdivi-1 attenuated PRKAA deficiency-induced mitochondrial fragmentation and endothelial dysfunction. Our results suggest that PRKAA is essential for maintaining the balance between mitochondrial fusion and fission.
Results
Deletion of Prkaa1 or Prkaa2 induces mitochondrial fragmentation in the aortic endothelium and in isolated endothelial cells
To determine the role of PRKAA in regulating mitochondrial dynamics, we analyzed mitochondrial morphology in wild-type (WT), Prkaa1-deficient (prkaa1−/−), and Prkaa2-deficient (prkaa2−/−) mouse aortas. Western blot analysis confirmed that deletion of either Prkaa1 or Prkaa2 gene significantly reduced PRKAA1/AMPKα1 and PRKAA2/AMPKα2 expression, and inhibited the phosphorylation of PRKAA and its downstream substrate, ACACA/ACC (acetyl-coenzyme A carboxylase α) (Fig. 1A and Fig. S1A and S1B). In WT aortic endothelial cells, mitochondria appeared as a typical filamentous morphology with integrated double membranes and tight cristae. In contrast, mitochondria became smaller and punctate with significantly short in length (Fig. 1B), and mitochondrial number was increased in prkaa1−/− or prkaa2−/− mice aortic endothelial cells (Fig. S2A), which were suggestive of mitochondrial fragmentation.
Figure 1.
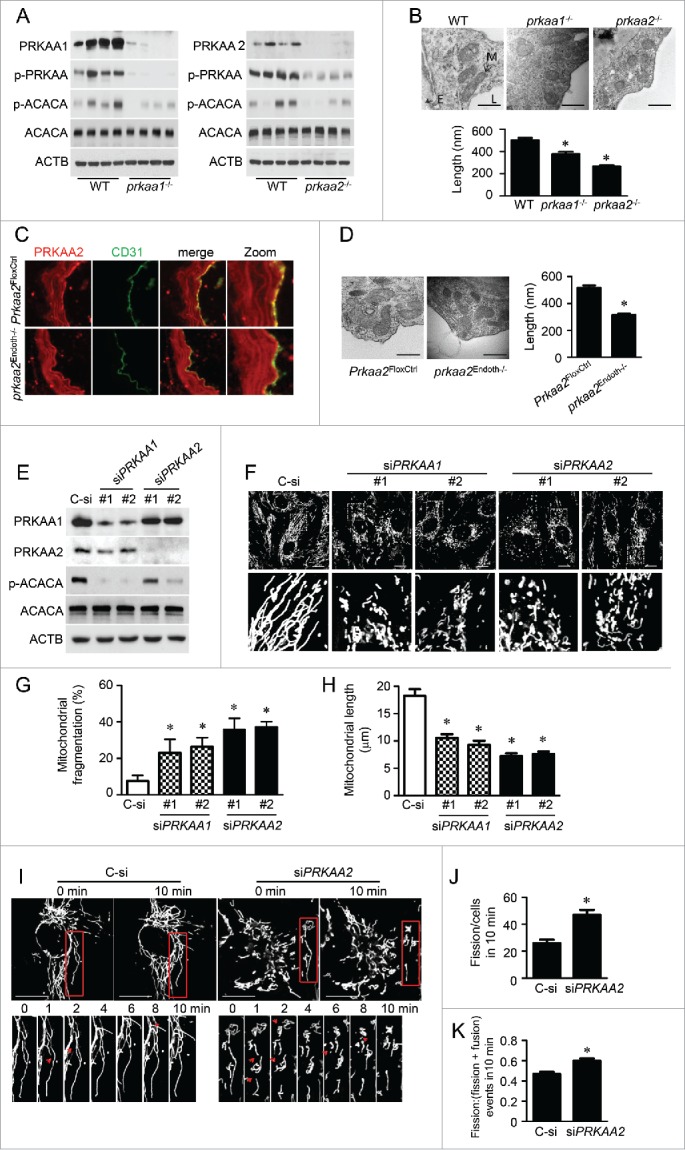
PRKAA deletion induces mitochondrial fragmentation in aortic endothelial cells. (A) Protein levels of PRKAA1/AMPKα1, PRKAA2/AMPKα2, ACACA, phosphorylation of PRKAA at Thr172 and ACACA at Ser79 in aortas isolated from wild-type (WT), prkaa1−/−, and prkaa2−/− mice were analyzed by western blotting. (B) Representative transmission electron micrographs of mitochondria and quantification of mitochondrial length in the aortic endothelium of WT, prkaa1−/−, and prkaa2−/− mice. Scale bars: 500 nm; L, lumen; M, mitochondria; E, endothelium. n = 5 mice, * P < 0.05 vs. WT. (C) Mouse aortas isolated from Prkaa2flox/flox (Prkaa2FloxCtrl) and endothelial-specific prkaa2 knockout (prkaa2Endoth−/−) mice were stained with antibodies against PRKAA2/AMPKα2 (Red) and PECAM1 (Green) and observed using fluorescence microscopy. (D) Representative electron micrographs of mitochondria and quantification of mitochondrial length in the aortic endothelium of Prkaa2FloxCtrl and prkaa2Endoth−/− mice. Scale bars: 500 nm. n = 5 mice, * P < 0.05 vs. Prkaa2FloxCtrl. (E and F) Human umbilical vein endothelial cells (HUVECs) were transfected with control small interfering RNA (siRNA; control, C-si), PRKAA1 siRNA (siPRKAA1), or PRKAA2 siRNA (siPRKAA2) for 48 h. (E) Protein levels of PRKAA1/AMPKα1, PRKAA2/AMPKα2, ACACA, and phosphorylation of ACACA at Ser79 were analyzed by western blotting. (F) Representative confocal images of the mitochondrial morphology in HUVECs stained with MitoTracker® Deep Red FM. Scale bars: 10 µm. Quantification of mitochondrial fragmentation of the cells (G) and average mitochondrial length (H). n ≥ 100, *P < 0.05 vs. C-si. (I to K) Mitochondrial morphology in live HUVECs stained with MitoTracker® Deep Red FM was captured using time-lapse confocal microscopy. Images were collected at 1 min intervals for 10 min. (I) Representative time-lapse confocal images. Scale bars: 10 µm. Quantification of mitochondrial fission (J) and the ratio of fission and fission-plus-fusion events (K) in live HUVECs. n = 15, *P < 0.05 vs. C-si.
We further verified that deletion of Prkaa2 induced mitochondrial fission in endothelial-specific Prkaa2 knockout (prkaa2Endoth−/−) mice. Immunofluorescence staining indicated that PRKAA2/AMPKα2 protein levels in the aortic endothelium were significantly reduced in prkaa2Endoth−/− mice (Fig. 1C). Similar to our findings in prkaa1−/− and prkaa2−/− mice with global deletion of the Prkaa isoforms, mitochondria became smaller, with shorter length (Fig. 1D), and increased number (Fig. S2B) in aortic endothelial cells from prkaa2Endoth−/− mice.
Next, we used a small interfering RNA (siRNA) approach to demonstrate the effect of PRKAA on mitochondrial dynamics in vitro using human umbilical vein endothelial cells (HUVECs). To minimize off-target effects of siRNA, we used 2 pairs of siRNAs, which target different PRKAA1 or PRKAA2 mRNA sequences. These siRNA significantly reduced PRKAA1/AMPKα1 or PRKAA2/AMPKα2 protein levels and inhibited ACACA phosphorylation (Fig. 1E and Fig. S1C). In control siRNA-transfected HUVECs, mitochondria exhibited elongated tubules and were highly interconnected. However, in PRKAA1 siRNA- or PRKAA2 siRNA-transfected HUVECs, mitochondria became shorter and more spherical (Fig. 1F to 1H) and mitochondrial number increased (Fig. S3), which were indicative of mitochondrial fragmentation.
To establish the role of PRKAA in regulating mitochondrial dynamics, we used time-lapse confocal imaging to measure mitochondrial fusion and fission in live HUVECs transfected with control siRNA or PRKAA2 siRNA. Mitochondrial fission occurred at a higher frequency in the cells transfected with PRKAA2 siRNA than in the cells transfected with control siRNA (47 ± 4 vs. 26 ± 2 events/cell in 10 min, Fig. 1I and 1J). The proportion of fission events (assay as the ratio of fission/(fission + fusion) events) also increased in HUVECs transfected with PRKAA2 siRNA (Fig. 1K). These results suggest that PRKAA inhibition induces mitochondrial fragmentation.
Prkaa deletion increases mitochondrial fragmentation by upregulating DNM1L and reducing DNM1L Ser637 phosphorylation
To gain insight into the mechanism by which PRKAA regulates mitochondrial dynamics, we examined the expression of mitochondrial dynamics-related proteins, including DNM1L, FIS1, MFN2, and OPA1,32 in WT, prkaa1−/−, and prkaa2−/− mice. Compared with WT mouse aortas, prkaa1−/− and prkaa2−/− mouse aortas exhibited higher DNM1L protein levels. Conversely, the deletion of either Prkaa1 or Prkaa2 did not alter the expression of FIS1, MFN2, and OPA1 (Fig. 2 A). Immunohistochemical staining further confirmed that in comparison to WT aortic endothelial cells, DNM1L expression was significantly higher in aortic endothelial cells from prkaa1−/−, prkaa2−/−,or prkaa2Endoth−/− mice (Fig. 2 B and C).
Figure 2.
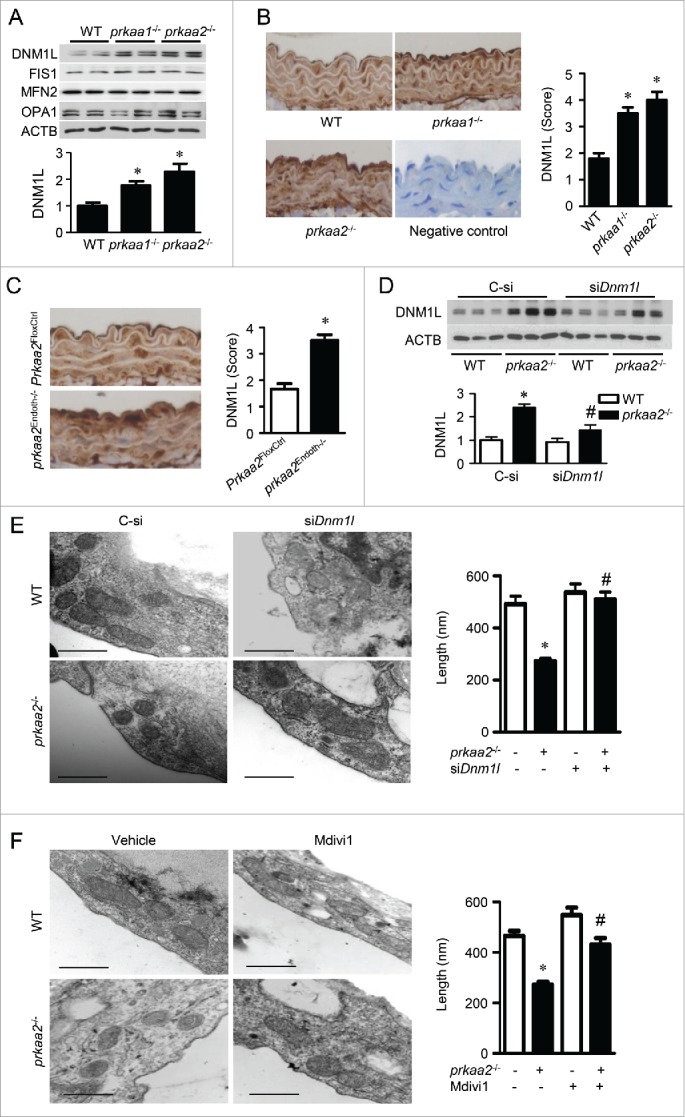
Prkaa2 deletion increases DNM1L expression and DNM1L-dependent mitochondrial fission in aortic endothelial cells. (A) Western blot analysis of mitochondrial dynamics-related proteins, including DNM1L, FIS1 (fission, mitochondrial 1), MFN2 (mitofusin 2), and OPA1 (optic atrophy 1), in WT, prkaa1−/− and prkaa2−/− mouse aortas. n = 5 mice, *P < 0.05 vs. WT. (B) Representative images of immunohistochemical staining and quantification of positive staining for DNM1L in aortas from WT, prkaa1−/−, and prkaa2−/− mice. n = 5, *P < 0.05 vs. WT. (C) Immunohistochemical staining for DNM1L in aortas from Prkaa2FloxCtrl and prkaa2Endoth−/− mice. Positive staining for DNM1L was quantified, as described in Materials and Methods. n = 5 mice, *P < 0.05 vs. Prkaa2FloxCtrl. (D and E) WT and prkaa2−/− mice were retro-orbitally injected with control (C-si or Dnm1l siRNA (siDnm1l, 5mg/kg). (D) DNM1L protein levels in the aorta were analyzed by western blotting. (E) Representative transmission electron micrographs of mitochondria and quantification of mitochondrial length in mouse aortas. Scale bars: 500 nm. n = 6 mice, *P < 0.05 vs. WT, #P < 0.05 vs. C-si. (F) WT and prkaa2−/− mice were treated with mdivi-1 (1.2 mg/kg/d, osmotic pump) or vehicle (dimethyl sulfoxide [DMSO]) for 14 d. Representative transmission electron micrographs of mitochondria and quantification of mitochondrial length in mouse aortas. n = 6, *P < 0.05 vs. WT; #P < 0.05 vs. vehicle.
Given that DNM1L upregulation is associated with increased mitochondrial fission in the PRKAA-deficient endothelium, we examined whether DNM1L mediates mitochondrial fission in PRKAA-deficient endothelial cells in vivo and in cultured endothelial cells. To examine whether DNM1L accumulation is required for PRKAA deficiency-induced mitochondrial fission, we retro-orbitally injected prkaa2−/− mice with Dnm1l siRNA (5 mg/kg) to prevent the accumulation of DNM1L in mouse aortas.33 Western blot analysis verified that transfection of Dnm1l siRNA significantly reduced DNM1L expression in prkaa2−/− mice (Fig. 2D). Consistently, mitochondrial fragmentation in prkaa2−/− mice was attenuated (Fig. 2E and Fig. S4A). Moreover, administration of mdivi-1 (1.2 mg/kg/d, 2 wk), a selective DNM1L inhibitor,34 also attenuated mitochondrial fragmentation in prkaa2−/− mice (Fig. 2F and Fig. S4B).
In cultured HUVECs, gene silencing of either PRKAA1 or PRKAA2 significantly increased DNM1L protein levels (Fig. 3A and B). In contrast, transfection with either PRKAA1 or PRKAA2 siRNA had no effect on the expression of FIS1, MFN2, and OPA1 (Fig. 3 A), all of which are important for mitochondrial dynamics. Moreover, DNM1L protein expression is higher in prkaa1−/− or prkaa2−/− mouse embryonic fibroblasts (MEFs) (Fig. S5). To determine whether MFF plays a role in regulating mitochondrial dynamics in endothelial cells, we detected its expression in endothelial cells and U2OS cells. Large amount of MFF was expressed in U2OS cells, but it was rarely detected in HAECs and HUVECs (Fig. S6).
Figure 3.
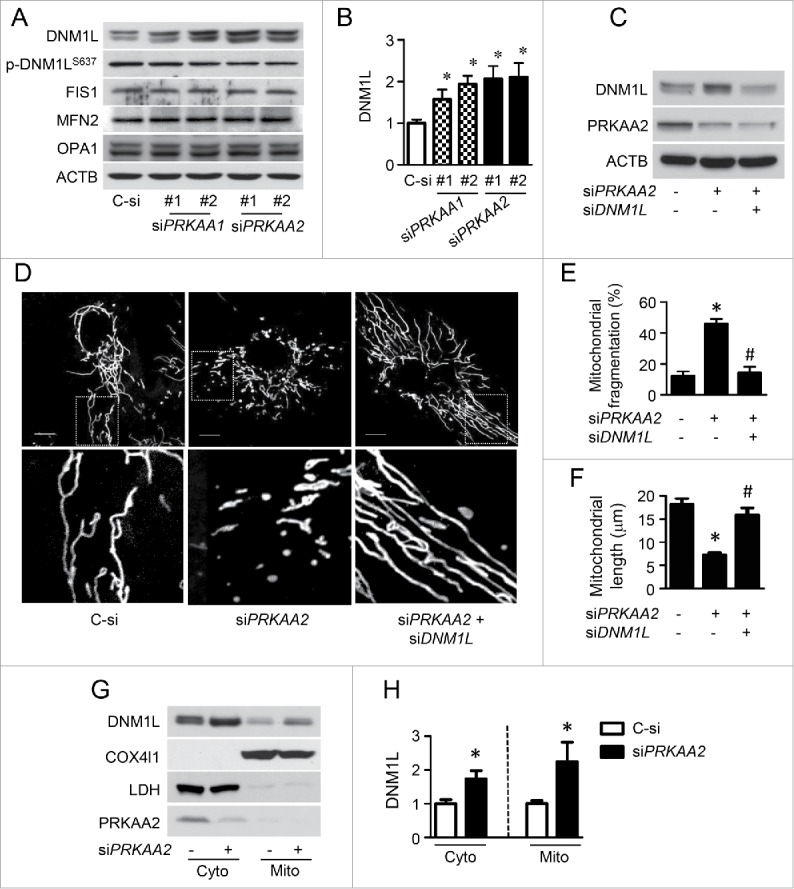
PRKAA deletion increases DNM1L-dependent mitochondrial fission via upregulation of DNM1L expression and translocation into mitochondria. (A and B) HUVECs were transfected with control (C-si), siPRKAA1, or siPRKAA2 for 48 h. Mitochondrial dynamics-related proteins were then analyzed by western blotting. n = 4 independent experiments, *P < 0.05 vs. C-si (C to F) HUVECs were transfected with C-si, siPRKAA2, or siPRKAA2 plus siDNM1L for 48 h. (C) Levels of PRKAA2/AMPKα2 and DNM1L were measured by western blotting. (D) Representative images of mitochondrial morphology. Scale bars: 10 µm. Quantification of mitochondrial fragmentation of the cells (E) and average mitochondrial length (F). n ≥ 100, *P < 0.05 vs. C-si, #P < 0.05 vs. siPrkaa2. (sG and H) Representative western blot (G) and quantification of DNM1L (H) in cytoplasmic and mitochondrial fractions from HUVECs transfection with Prkaa2 siRNA. COX4I1 and LDH were used as mitochondrial and cytosolic markers, respectively.
To investigate whether DNM1L is required for mitochondrial fission in PRKAA2/AMPKα2-deficient endothelial cells, we transfected HUVECs with control, PRKAA2, or PRKAA2 plus DNM1L siRNA (Fig. 3C). In control siRNA-transfected cells, mitochondria had an elongated, tubular-like appearance. After gene silencing of PRKAA2, mitochondria became shorter and smaller, indicating an increase in mitochondrial fission. Cotransfection of cells with PRKAA2 and DNM1L siRNA significantly decreased mitochondrial fission and reduced the number of cells containing fragmented mitochondria (Fig. 3D to F). We also examined the effects of PRKAA on DNM1L subcellular distribution. We found that PRKAA2 siRNA increased DNM1L protein levels in cytosolic and mitochondrial fractions by 1.7- and 2.2-fold, respectively (Fig. 3G and H). Knockdown of either PRKAA1 or PRKAA2 reduced DNM1L phosphorylation at S637 (Fig. 3A), which inhibits DNM1L activity and prevents its translocation to mitochondrial fission sites.29 Therefore, DNM1L accumulation and inactivation was required for mitochondrial fission in PRKAA2/AMPKα2-deficient endothelial cells.
DNM1L inhibition attenuates endothelial dysfunction in prkaa2−/− mice
Next, we tested whether DNM1L upregulation contributes to oxidative stress and endothelial dysfunction in prkaa2−/− mouse. Reduction of DNM1L was associated with a significant decrease in superoxide production in carotid artery from Dnm1l siRNA-injected prkaa2−/− mice (Fig. 4A and B). In parallel with the increase in mitochondrial fragmentation and superoxide production, aortic rings from prkaa2−/− mice showed impaired acetylcholine (Ach)-induced, endothelium-dependent vasorelaxation, compared with aortic rings from WT mice (Fig. 4C). Similarly endothelium-dependent vasorelaxation was also impaired in prkaa2Endoth−/− mouse aortas (Fig. S7). The impairment of Ach-induced, endothelium-dependent relaxation in aortic rings from prkaa2−/− mice was prevented by administration of Dnm1l siRNA (Fig. 4C), indicating that mitochondrial fission contributes to endothelial dysfunction in prkaa2−/− mice. However, Dnm1l siRNA had no effect on endothelium-independent vasorelaxation, which was induced by a NO donor, sodium nitroprusside (SNP) (Fig. 4D).
Figure 4.
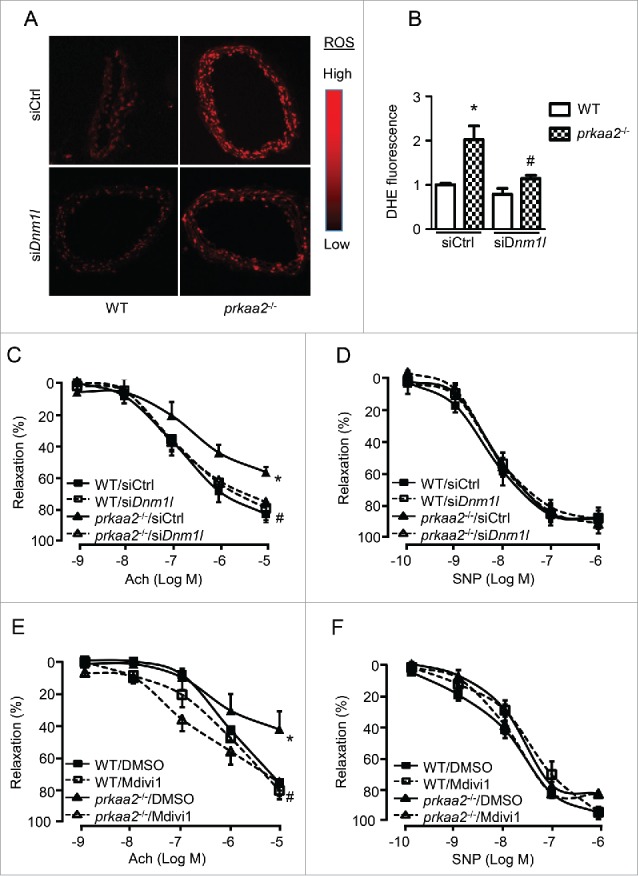
Inhibition of mitochondria fission attenuates oxidative stress and endothelial dysfunction in prkaa2−/− mice. (A) WT and prkaa2−/− mice were injected with control (siCtrl) or siDnm1l (5 mg/kg). Four d after treatment, frozen sections of carotid artery were incubated with dihydroethidium (DHE; 5 µM) for 30 min. Representative fluorescence microscopic images of DHE-stained artery (A) and quantification of intensity grade showing with color lookup table (B). n = 5; *P < 0.05 vs. WT; #P < 0.05 vs. siCtrl. (C) Aortas were contracted with U46619 (30 nM). Endothelium-dependent vasodilator responses were measured in the presence of acetylcholine (Ach, 10−9 to 10−5 M). (D) Endothelium-independent vasodilator responses were measured in the presence of sodium nitroprusside (SNP, 10−10 to 10−6 M). n = 6 to 8; *P < 0.05 vs. WT; #P < 0.05 vs. siCtrl. (E and F) WT and prkaa2−/− mice were treated with mdivi-1 (1.2 mg/kg/d) or vehicle (DMSO) for 14 d. (E) Aortas were contracted with U46619. Endothelium-dependent vasodilator responses were measured in the presence of Ach (10−9 to 10−4 M). (F) Endothelium-independent vasodilator responses were measured in the presence of SNP (10−10 to 10−6 M). n = 6 to 8; *P < 0.05 vs. WT; #P < 0.05 vs. vehicle.
We further determined the role of mitochondrial fission in PRKAA deletion-induced endothelial dysfunction by chronically administering prkaa2−/− mice with mdivi-1. Administration of mdivi-1 did not alter Ach-induced relaxation in aortas from WT mice but increased Ach-induced relaxation in aortas from prkaa2−/− mice (Fig. 4E). However, there was no significant difference in endothelium-independent relaxation among these groups (Fig. 4F), thus indicating that mitochondrial fission contributes to endothelial dysfunction in prkaa2−/− mice.
Inhibition of mitochondrial fusion impairs endothelium-dependent vasorelaxation
Next, we determined whether inhibition of mitochondrial fusion impairs NO bioactivity and endothelial function. MFN2 is a mitochondrial protein that promotes mitochondrial fusion. Efficient knockdown of Mfn2 by siRNA (Fig. S8A) triggered mitochondrial fragmentation (Fig. 5A and B) and superoxide production in HUVECs (Fig. S8B). Furthermore, transfection with Mfn2 siRNA attenuated calcium ionophore A23187-induced NOS3 phosphorylation and NO production (Fig. 5C and D).
Figure 5.
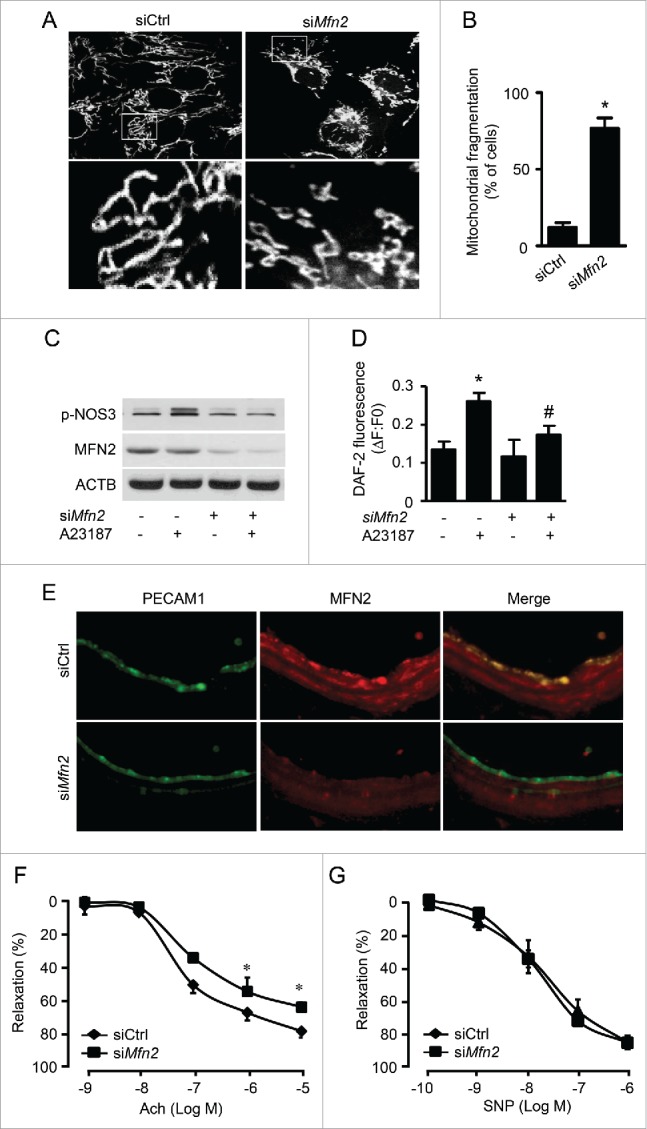
Inhibited mitochondrial fusion impairs endothelial function. (A) HUVECs were transfected with control (siCtrl) or MFN2 siRNA (siMFN2) for 48 h, and mitochondria were labeled with MitoTracker® Deep Red FM. Mitochondrial morphology was analyzed using fluorescence microscopy. Scale bars: 5 µm. (B) Mitochondrial fragmentation was determined, as described in Materials and Methods. n ≥ 100; *P < 0.05 vs. siCtrl. (C) HUVECs were transfected with siCtrl or siMFN2 for 48 h and then treated with A23187 or DMSO for 30 min. Phosphorylation of NOS-3 was measured by western blotting. (D) Nitric oxide concentration in the cells was assessed using the DAF-2 fluorescence probe. *P < 0.05 vs. DMSO; #P < 0.05 vs. siCtrl. (E) WT mouse carotid arteries were transfected with siCtrl or siMfn2 using electroporation for 48 h, stained with antibodies against MFN2 (Red) and PECAM1 (Green), and observed using fluorescence microscopy. (F) Endothelium-dependent vasodilator responses were measured in the presence of Ach (10−9 to 10−5 M). (G) Endothelium-independent vasodilator responses were measured in the presence of SNP (10−10 to 10−6 M). n = 4, * P < 0.05 vs. siCtrl.
To determine whether inhibition of mitochondrial fusion impairs endothelial function ex vivo, we transfected carotid arteries with Mfn2 siRNA by electroporation, as described by Isakson.35 Immunofluorescence staining revealed that MFN2 protein expression dramatically decreased in the endothelium and smooth muscle cells of Mfn2 siRNA-transfected arteries (Fig. 5E). Mfn2 siRNA-transfected arteries also exhibited impaired Ach-induced vessel relaxation. The maximal Ach-induced relaxation was less in Mfn2 siRNA-transfected arteries than in control siRNA-transfected arteries. The endothelium-independent vasorelaxation evoked by SNP did not differ between Mfn2 siRNA-transfected arteries and control siRNA-transfected arteries (Fig. 5F and G), indicating that the inhibition of mitochondrial fusion impairs endothelial dysfunction.
PRKAA2/AMPKα2 deletion suppresses autophagy in endothelial cells
Previous studies demonstrated that PRKAA inactivation suppresses autophagy,36,37 and Dnm1L is degraded by autophagy in neurons.11 Therefore, we investigated whether PRKAA ablation leads to DNM1L accumulation by inhibiting the autophagy-dependent degradation of DNM1L. In prkaa2−/− mouse aortas, the conversion of cytoplasmic LC3B-I to autophagosomal membrane-bound LC3B-II was dramatically reduced. Protein levels of SQSTM1/p62, a marker for inhibited or defective autophagy, were significantly increased (Fig. 6A). Immunohistochemical staining indicated that SQSTM1 mainly accumulated in the aortic endothelium (Fig. 6B). In addition, electron micrographic analysis of the aortic endothelium was performed to quantify double membrane-bounded autophagic vacuoles, which contained a variety of undigested cytoplasmic components and organelles such as mitochondria. Significantly fewer autophagosomes were detected in aortas of prkaa2−/− mice than those of WT mice, which further confirmed the reduction of autophagy in PRKAA-deficient aortas (Fig. 6C).
Figure 6.
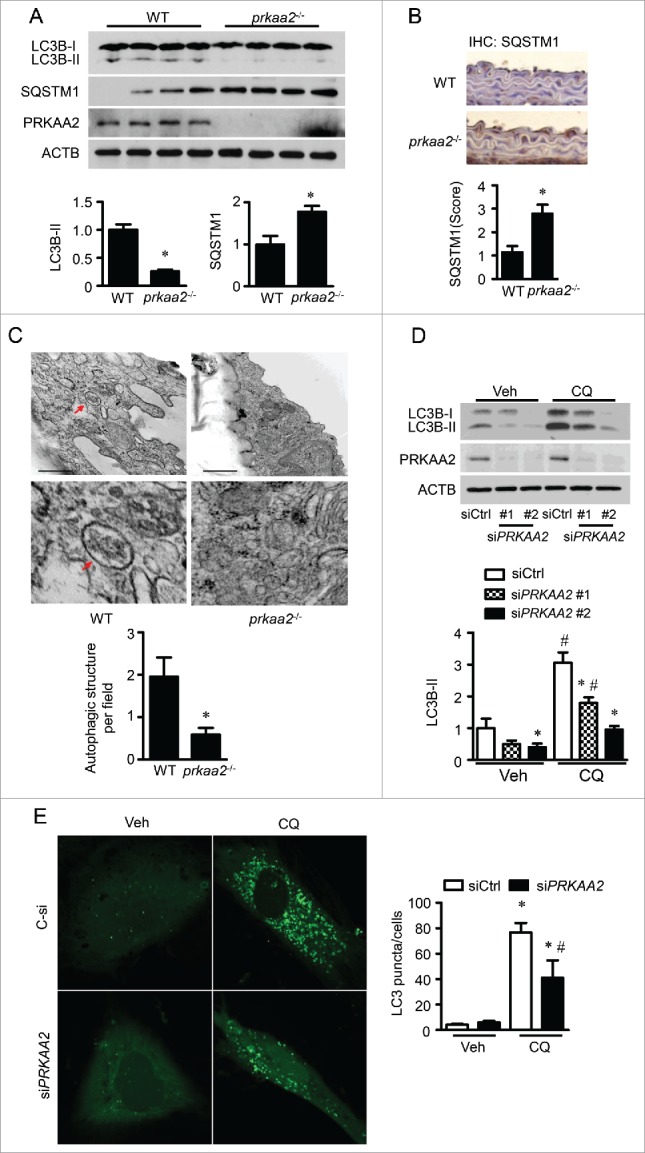
PRKAA deletion inhibits autophagic flux. (A) Levels of LC3B and SQSTM1 in WT and prkaa2−/− mouse aortas were determined by western blotting. n = 7 mice, *P < 0.05 vs. WT. (B) Immunohistochemical staining for SQSTM1 in WT and prkaa2−/− mouse aortas and quantification of positive staining for SQSTM1 in the aortas. n = 5, *P < 0.05 vs. WT. (C) Electron microscopic analysis of autophagic vacuoles (AV) and quantification of AV in WT and prkaa2−/− mouse aortas. n = 6 mice, *P < 0.05 vs. WT. (D) HUVECs were transfected with control (siCtrl) or siPRKAA2 for 24 h and then treated with bafilomycin A1 (BafA, 5 nM) for 24 h. Protein levels of LC3B and PRKAA2/AMPKα2 were measured by western blotting. n = 4. *P < 0.05 vs. siCtrl, #P < 0.05 vs. Veh. (E) HUVECs were cotransfected with siCtrl or siPRKAA2 and GFP-LC3B adenovirus for 24 h, and then treated with CQ for 24 h. Quantification of GFP-LC3B puncta formation in HUVECs was analyzed using fluorescence microscopy. n ≥ 50, *P < 0.05 vs. no CQ treatment; #P < 0.05 vs. siCtrl.
To analyze autophagy flux, chloroquine (CQ) was used to inhibit lysosomal activity38 in PRKAA2 siRNA-transfected HUVECs. PRKAA2 siRNA transfection reduced LC3B-II protein levels in HUVECs treated with or without CQ (Fig. 6D). To determine the effect of PRKAA on autophagosome formation, HUVECs were cotransfected with PRKAA2 siRNA and GFP-LC3 adenovirus by electroporation for 48 h. CQ treatment increased autophagosome formation, as indicated by punctate GFP-LC3, in control siRNA-transfected cells. However, transfection with PRKAA2 siRNA attenuated CQ-enhanced autophagosome formation (Fig. 6E). Therefore, inactivation of PRKAA led to an impairment of autophagosome formation.
PRKAA2 deletion inhibits the autophagy-dependent degradation of DNM1L
Next, we examined whether suppression of autophagy increases DNM1L protein levels and mitochondrial fission. Treatment of HUVECs with CQ increased the expression of DNM1L at both 8 and 24 h. To ensure that CQ treatment inhibited autophagy, LC3B levels were also analyzed. LC3B-II levels increased after CQ treatment (Fig. 7A), thus confirming inhibition of autophagosomal-lysosomal fusion. To investigate whether the proteasome is involved in DNM1L degradation, we treated HUVECs with proteasome inhibitors, MG132 and lactacystin. MG132 and lactacystin treatment did not dramatically change DNM1L level (Fig. S9A and B). These data suggest that DNM1L can be degraded through the autophagy-lysosome pathway.
Figure 7.

PRKAA deficiency-inhibited autophagy results in DNM1L accumulation. (A) HUVECs were treated with 3 µM CQ for 8 or 24 h. Protein levels of DNM1L and LC3B were analyzed by western blotting. n = 4. *P < 0.05 vs. no CQ. (B and D) HUVECs were transfected with control (siCtrl) or ATG7 siRNA (siATG7) for 48 h. (B) Protein levels of DNM1L, ATG7, and LC3B were analyzed by western blotting. n = 4. *P < 0.05 vs. siCtrl. (C) Mitochondrial morphology was analyzed by fluorescence microscopy. Scale bars: 5 µm. (D) Mitochondrial fission was determined, as described in Materials and Methods. n ≥ 100; *P < 0.05 vs. siCtrl. (E) HUVECs were incubated with cycloheximide (CHX, 1 µg/ml) for 4 to 24 h. DNM1L protein levels were analyzed by western blotting. n = 4. *P < 0.05 vs. no CHX. (F) HUVECs were transfected with C-si or siPRKAA2 for 24 h and then incubated with CHX for 8 or 24 h. After treatment, DNM1L protein levels were analyzed by western blotting. n = 4. *P < 0.05 vs. no CHX, #P < 0.05 vs. siCtrl. (G to I) HUVECs were transfected with siCtrl or siPRKAA2 for 24 h, after which they were transfected with adenoviruses encoding GFP (Ad-GFP) or ATG7 (Ad-ATG7) for 24 h. (G and H) Protein levels of DNM1L, p-ACACA, ATG7, LC3B, and SQSTM1 were measured by western blotting. n = 4. *P < 0.05 vs. siCtrl, #P < 0.05 vs. GFP (I) Mitochondrial morphology was analyzed using fluorescence microscopy. Scale bars: 10 µm. (J) Mitochondrial fission was determined, as described in Materials and Methods. n ≥ 100; *P < 0.05 vs. siCtrl, #P < 0.05 vs. Ad-GFP. (K) HUVECs were transfected with siCtrl, 2 independent siPRKAA1, or 2 independent siPRKAA2 for 48 h. Mitochondrial proteins, including VDAC, TOMM20 and CYCS/cytochrome C, were analyzed by western blotting.
To verify that the increased expression of DNM1L was autophagy mediated, we used the gene silencing approach to examine the effect of autophagy on DNM1L expression. Gene silencing of ATG7 (autophagy-related 7), an essential protein for autophagosome formation, decreased ATG7 protein expression and reduced the conversion of LC3B-1 to LC3B-II in association with increased DNM1L protein expression (Fig. 7B). Similar results were also observed in atg7−/− and atg5−/− MEFs (Fig. S10A). Additionally, cytosolic and mitochondrial DNM1L was increased in atg7−/− MEFs (Fig. S10B). We further examined whether autophagy inhibition increases mitochondrial fission. In control siRNA-transfected cells, mitochondria had an elongated tubular-like appearance. However, ATG7 siRNA-transfected HUVECs showed a significant increase in mitochondrial fragmentation, and the mitochondria became shorter and smaller (Fig. 7C and D). This suggests that autophagy suppression induces DNM1L-dependent mitochondrial fission.
To establish that defective autophagy mediates DNM1L accumulation in PRKAA-deficient cells, we examined whether activated autophagy reduces DNM1L accumulation in PRKAA-deficient cells. First, we examined DNM1L protein stability in HUVECs that were treated with cycloheximide (CHX) to inhibit protein synthesis. DNM1L protein levels gradually decreased in control siRNA-transfected cells. After 24 h of CHX treatment, DNM1L was almost completely undetected. However, DNM1L protein levels displayed slightly reduced by CHX treatment in PRKAA2 siRNA-transfected cells (Fig. 7E and F). We further examined whether ATG7 overexpression can prevent DNM1L accumulation in PRKAA-deficient HUVECs. Transfection of HUVECs with the ATG7 adenovirus (Ad-ATG7) significantly increased ATG7 protein expression. Gene silencing of PRKAA2 inhibited autophagy activity, as evidenced by a reduction in the conversion of LC3B-I to LC3B-II. Therefore, the inhibition of autophagy resulted in cytosolic DNM1L accumulation. Overexpression of ATG7 prevented autophagy deficiency and markedly reduced DNM1L accumulation in HUVECs (Fig. 7G and H. We also examined whether the increase in autophagy can inhibit mitochondrial fission in PRKAA2 siRNA-transfected cells. In Ad-GFP-transfected cells, PRKAA2/AMPKα2 inhibition increased mitochondrial fission, as indicated by disrupted mitochondrial networks. Overexpression of ATG7 also prevented the loss of mitochondrial networks in PRKAA2 siRNA-transfected cells (Fig. 7I and J). To clarify whether the DNM1L accumulation results from increased numbers of mitochondria in PRKAA-deficient cells, we determined mitochondrial mass by measuring mitochondrial markers, VDAC, CYCS/cytochrome C, and TOMM20/Tom20 in HUVECs transfected with PRKAA1 or PRKAA2 siRNA. The western blot analysis indicated that PRKAA deficiency had no effect on the expression of VDAC, CYCS, and TOMM20 (Fig. 7K). Therefore, our findings suggest that autophagy deficiency in PRKAA-deficient cells is a major mechanism for cytosolic DNM1L accumulation and mitochondrial fragmentation.
Chronic administration of rapamycin attenuates DNM1L accumulation and mitochondrial fission in prkaa2−/− mouse aortas
To understand the role of autophagy in PRKAA deletion-induced, DNML-dependent mitochondrial fission in vivo, we assessed the impact of rapamycin, an autophagy inducer,39 on DNM1L expression and mitochondrial fission in prkaa2−/− mice. Chronic administration of rapamycin inhibited MTOR phosphorylation at Ser2481, which was suggestive of MTOR signaling inhibition (Fig. 8A). Inhibition of MTOR was also associated with the activation of autophagy, as evidenced by higher LC3B-II protein levels and lower SQSTM1 levels in rapamycin-treated mouse aortas than in vehicle-treated mouse aortas (Fig. 8A to C). At the same time, rapamycin treatment reduced DNM1L accumulation (Fig. 8D to F) and attenuated mitochondrial fragmentation in prkaa2−/− mouse aortas (Fig. 8G and H). In PRKAA siRNA-transfected HUVECs, rapamycin treatment also reduced DNM1L accumulation (Fig. 8I).
Figure 8.
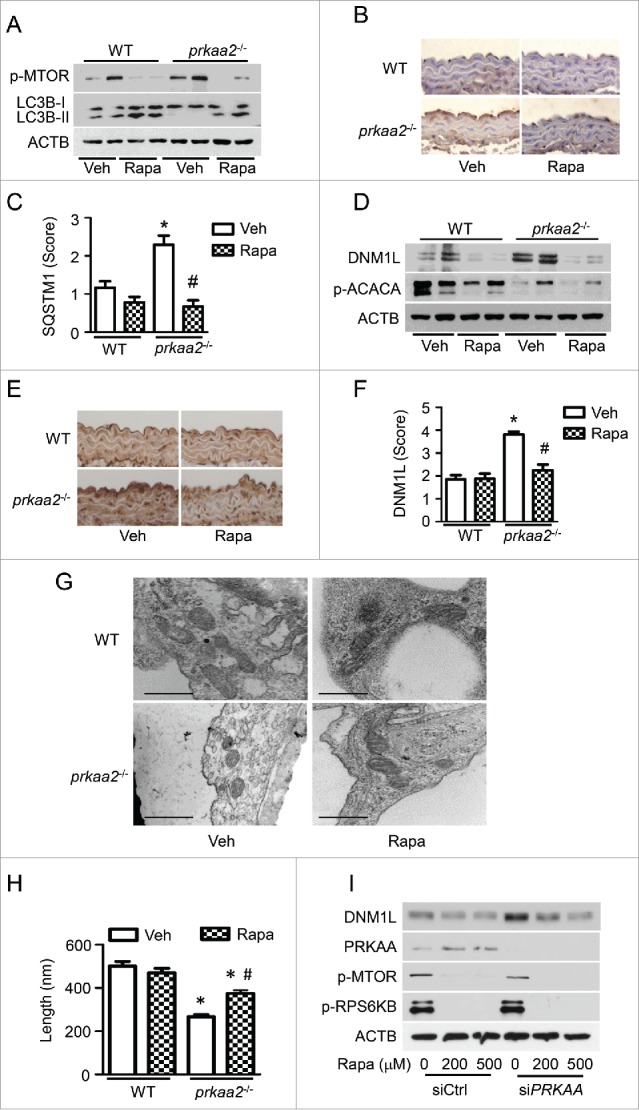
Activation of autophagy reduces DNM1L expression and mitochondrial fission in prkaa2−/− mice. WT and prkaa2−/− mice were orally administrated with rapamycin (Rapa, 14 mg/kg in diet) for 1 week. (A) Protein levels of phosphorylated MTOR at Ser2481 (p-MTOR) and LC3B in aortas were measured by western blotting. (B and C) Immunohistochemical staining for SQSTM1 in aortas from rapamycin-treated WT and prkaa2−/− mice, and quantification of positive staining for SQSTM1 in the aortas. *P < 0.05 vs. WT; #P < 0.05 vs. Vehicle. (D) Western blot analysis of DNM1L and p-ACACA protein expression in mouse aortas. (E and F) Immunohistochemical staining for DNM1L in aortas from rapamycin-treated WT and prkaa2−/− mice. Positive staining for DNM1L was quantified, as described in Materials and Methods. *P < 0.05 vs. WT; #P < 0.05 vs. Vehicle. (G) Representative electron micrographs of aortic endothelial mitochondria. Scale bars: 500 nm. (H) Mitochondrial length was quantified, as described in Materials and Methods. n = 6 mice, *P < 0.05 vs. WT, #P < 0.05 vs. Vehicle. (I) HUVECs were transfected with control (C-si) or siPRKAA for 24 h, after which they were treated with different concentrations of rapamycin for 24 h. Cell lysates were subjected to western blot analysis for DNM1L, PRKAA, p-MTOR, and phosphorylated RPS6KB at Thr389 (p-RPS6KB).
The interaction between SQSTM1 and DNM1L regulates DNM1L degradation and mitochondrial dynamics
SQSTM1 is one factor that targets specific cargos for autophagy. To examine whether SQSTM1 mediates DNM1L degradation in PRKAA-deficient cells, we performed coimmunoprecipitation using SQSTM1 and DNM1L antibodies. In control siRNA-transfected HUVECs, endogenous SQSTM1 associated with DNM1L. This association was significantly increased in PRKAA2 siRNA-transfected cells (Fig. 9A and B). Immunofluorescence staining confirmed the interaction between DNM1L and SQSTM1 (Fig. 9C). Given that PRKAA inactivation leads to defective autophagy, we examined whether inhibition of autophagy can increase the interaction between DNM1L and SQSTM1. When autophagy was inhibited by bafilomycin A1 (BafA), the association of DNM1L with SQSTM1 significantly increased (Fig. 9D and E). Similar results were also observed in atg7−/− MEFs (Fig. 9F and G), in which autophagy was significantly inhibited.
Figure 9.
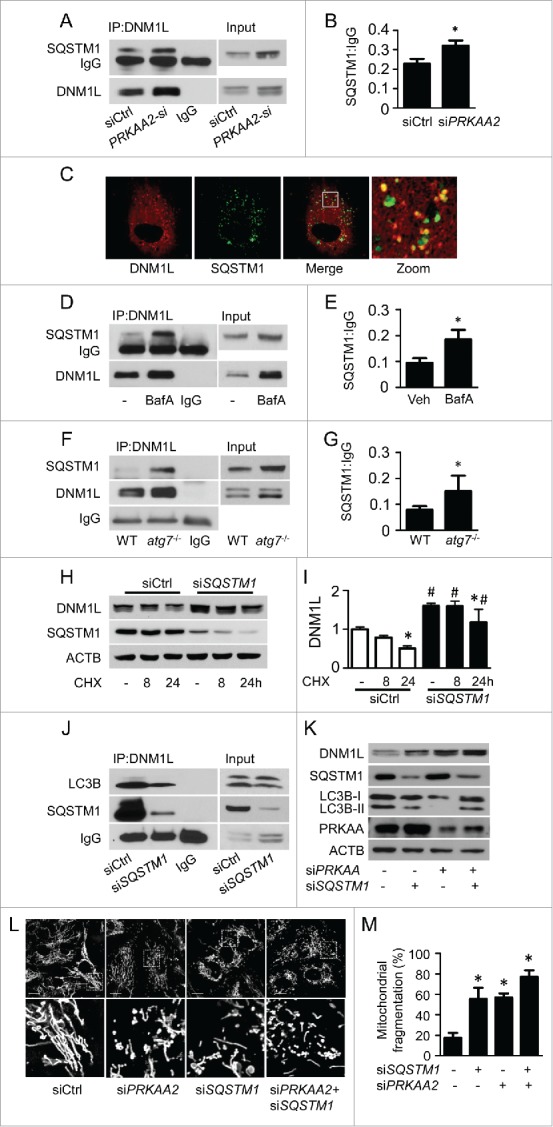
SQSTM1 is required for the autophagy-dependent degradation of DNM1L. (A and B) HUVECs were transfected with control (siCtrl) or siPRKAA2 for 48 h, and the interaction between DNM1L and SQSTM1 was determined by immunoprecipitation and western blotting. (C) The colocalization of DNM1L and SQSTM1 was assessed in HUVEC transfection with plasmid coding for cherry-DNM1L and GFP-SQSTM1. Scale bars: 5 µm. (D and E) HUVECs were transfected with or without BafA for 24 h, and the interaction between DNM1L and SQSTM1 was examined by immunoprecipitation and western blotting. (sF and G) The interaction between DNM1L and SQSTM1 in WT and atg7−/− MEFs was measured by immunoprecipitation and western blotting. (H and I) HUVECs were transfected with C-si and SQSTM1 siRNA (siSQSTM1) for 24 h, after which they were incubated with CHX for 8 to 24 h. DNM1L protein levels were analyzed by western blotting. (J) HUVECs were transfected with siCtrl and siSQSTM1 for 48 h. The interactions among SQSTM1, DNM1L, and LC3B were determined using immunoprecipitation and western blotting. (K to M) HUVECs were cotransfected with siPRKAA2 and siSQSTM1 for 48 h. (K) Protein levels of DNM1L, PRKAA, LC3B, and SQSTM1 were measured by western blotting. (L) Mitochondrial morphology was analyzed using fluorescence microscopy. Scale bars: 10 µm. (M) Mitochondrial fission was determined, as described in Materials and Methods. n = 100; *P < 0.05 vs. siCtrl.
Furthermore, SQSTM1 siRNA attenuated DNM1L degradation (Fig. 9H and I) and markedly reduced the association between LC3B and DNM1L (Fig. 9J), suggesting that SQSTM1 delivers DNM1L to the autophagosome, where it is subsequently degraded. To establish that SQSTM1 mediates DNM1L degradation in PRKAA-deficient cells, we examined DNM1L degradation in PRKAA-deficient cells that were transfected with control or SQSTM1 siRNA. Notably, knockdown of SQSTM1 impaired the degradation of DNM1L in PRKAA-deficient HUVECs (Fig. 9K). In addition, knockdown of SQSTM1 significantly induced mitochondrial fragmentation in PRKAA-deficient cells (Fig. 9L and M).
Discussion
In the present study, we demonstrate that PRKAA deletion suppressed autophagy, leading to DNM1L accumulation and mitochondrial fragmentation. Inhibition of DNM1L prevented mitochondrial fragmentation and endothelial dysfunction, suggesting that PRKAA plays a critical role in regulating mitochondrial dynamics in the development of endothelial dysfunction. Mechanistically, we found that inactivation of PRKAA promoted the interaction between SQSTM1 and DNM1L, and increased the autophagy-dependent degradation of DNM1L. Our data indicate that PRKAA controls mitochondrial dynamics via the SQSTM1-mediated autophagic degradation of DNM1L (Fig. S11).
PRKAA is an important protein in the maintenance of mitochondrial homeostasis and metabolic function. It increases mitochondrial biogenesis and promotes the autophagy-dependent clearance of damaged mitochondria.40 AMPK activation increases mitochondrial mass and improves mitochondrial function via PPARGC1A-mediated mitochondrial biogenesis in mammalian cells. Recently, we have demonstrated that PRKAA1/AMPKα1 deficiency inhibits autophagy-dependent mitochondrial clearance, resulting in the accumulation of damaged mitochondria and hemolytic destruction of erythrocytes.26 In this study, we show that PRKAA plays an essential and new role in regulating mitochondrial dynamics. In the prkaa1−/− and prkaa2−/− mice aortic endothelium, disruption of mitochondrial fusion-fission balance resulted in aberrant mitochondrial fragmentation. Similar findings were observed in PRKAA1 siRNA- or PRKAA2 siRNA-transfected cells. Consistent with our findings, AMPK activation by pharmacological means has been associated with mitochondrial elongation in endothelial cells,,27,28 pancreatic cells29 and kidney.30 In contrast, AMPK activation by electron transport chain inhibitors trigger mitochondrial fragmentation in human osteosarcoma U2OS cells.31 Mitochondrial morphology and function in cancer cells are dramatically different from those of original, nontransformed cell types.41 For instance, mitochondria in cancer cells exhibit fragmentation and high rates of glycolysis.42 Moreover, cancer cells are particularly sensitive to AMPK signaling. AMPK pharmaceutical activators, metformin and AICAR, exhibit inhibitory effects on cellular metabolism, proliferation, and growth in pancreatic43 prostate,44 and breast cancer cells.45 Therefore, the effect of PRKAA on mitochondrial dynamics might be different between cancer cells and endothelial cells. Numerous molecules have been discovered to participate in the regulation of mitochondrial dynamics. AMPK inactivation significantly increased DNM1L protein levels but had no effects on the expression of FIS1, MFN2, and OPA1. DNM1L siRNA transfection significantly decreased mitochondrial fission and reduced the number of cells containing fragmented mitochondria in PRKAA-deficient HUVECs, suggesting that PRKAA deficiency triggers DNM1L-mediated mitochondrial fission in endothelial cells. However, in human U2OS osteosarcoma cells, AMPK activation induces mitochondrial fission by phosphorylating MFF at Ser155 and Ser172 and recruiting DNM1L to mitochondria.31 Although MFF is expressed as multiple splice variants across various cell types, its expression is variable in different human tissues and cell types.46,47 The Human Protein Atlas Program reports that MFF protein is undetectable in endothelial cells in colon and cerebral cortex tissue (http://www.proteinatlas.org/ENSG00000168958-MFF/tissue/primary+data). Consistently, we found that a large amount of MFF is expressed in U2OS cells, but it is rarely detected in HAECs and HVUECs. These results suggest that the AMPK-MFF-mitochondrial fragmentation mechanism might be absent in endothelial cells.
DNM1L is subjected to regulation at multiple levels, including transcription, proteasomal degradation, sumoylation, S-nitrosylation, O-glycosylation, and reversible phosphorylation. Previous studies have shown that AMPK directly phosphorylates DNM1L at Ser637, thus preventing DNM1L translocation to mitochondria and inhibiting mitochondrial fragmentation.29,48 In this study, we identified a new mechanism by which PRKAA regulates DNM1L protein expression. PRKAA2/AMPKα2 deficiency markedly increased DNM1L protein levels but did not affect its mRNA expression. Inhibition of autophagy resulted in DNM1L accumulation. In contrast, overexpression of ATG7, an important molecule regulating autophagosome formation, can increase autophagy in the cells transfected with control siRNA and prevented the autophagy suppression in the cells transfected with PRKAA2 siRNA. Furthermore, ATG7 overexpression markedly reduced DNM1L accumulation and mitochondrial fission in PRKAA-deficient cells, indicating that autophagy deficiency in PRKAA-deficient cells is a major mechanism for DNM1L accumulation and mitochondrial fission. This conclusion was also supported by our in vivo observation that the chronic administration of rapamycin-activated autophagy attenuated DNM1L accumulation and inhibited mitochondrial fission in prkaa2−/− mouse aortas. Similarly, gene silencing of MTOR has been shown to increase mitochondrial elongation in MEFs.32 Increased DNM1L protein levels and mitochondrial fragmentation occur in endothelial cells that are exposed to hyperglycemia.5 During hyperglycemia, PRKAA is inhibited, and autophagy is suppressed.49 Autophagy and lysosome inhibitors induce mitochondrial fragmentation that is accompanied by increased DNM1L protein levels. In addition, increased DNM1L protein levels are observed in atg7−/− MEFs and HEK-293T cells that are exposed to lysosome inhibition.11 As a result, PRKAA inactivation may inhibit the autophagy-dependent degradation of DNM1L, thereby triggering mitochondrial fission.
Our results also demonstrate that PRKAA promote the autophagy-dependent degradation of DNM1L by regulating the interaction between SQSTM1 and DNM1L. Autophagy is a conserved lysosomal self-digestion process that is essential for cellular homeostasis and survival. Autophagy is not only responsible for the nonspecific degradation of cytoplasmic components but also specifically degrades polyubiquitinated protein aggregates. The autophagic turnover of protein aggregates is facilitated by the signaling receptor scaffold protein SQSTM1, which binds to both polyubiquitinated proteins in aggregates and LC3.50,51 This substrate-specific autophagy involving ubiquitin modification and SQSTM1 binding can also regulate the turnover of small molecules, as well as entire organelles, within cells.52 Endogenous DNM1L is partially associated with the mitochondrial membrane, as it binds to FIS1 and self-assembles to drive the membrane fission of mitochondria.6 Furthermore, cytosolic DNM1L is degraded by the autophagy-lysosome pathway in endothelial cells, and mitochondria-associated DNM1L can be sequestered to autophagosomes during autophagy. Thus, the autophagy-mediated degradation of DNM1L occurs regardless of its mitochondrial association. In the current study, we found that PRKAA deficiency increased the association between SQSTM1 and DNM1L. Gene silencing of SQSTM1 markedly reduced the association between SQSTM1 and DNM1L, impaired the degradation of DNM1L, and decreased the colocalization between DNM1L and the autophagy marker LC3B. The inhibition of DNM1L degradation was due to the suppression of autophagic flux, which suggests that SQSTM1 is required for increased DNM1L-mediated mitochondrial fission in PRKAA-deficient cells.
Another major finding of the present study is that increased mitochondrial fission contributed to endothelial dysfunction in vivo. First, the inhibition of DNM1L-dependent mitochondrial fission attenuated endothelial dysfunction in prkaa2−/− mice. Second, gene silencing of MFN2, a mitochondrial fusion-related protein, induced mitochondrial fragmentation, which was associated with superoxide overproduction, reduced NO bioavailability, and impaired endothelial-dependent vasodilation. Fragmented mitochondria are prone to generating ROS, which reduce NO bioavailability and impair endothelial-dependent vasodilation. Thus, inhibiting mitochondrial fission might be a new strategy to protect against the development of vascular disease.
Overall, the current study established a direct link between PRKAA deficiency and aberrant mitochondrial fission. Inhibition of PRKAA suppressed the autophagy-dependent degradation of DNM1L, thereby triggering mitochondrial fragmentation and endothelial dysfunction. Furthermore, the interaction between SQSTM1 and DNM1L regulated the degradation of DNM1L via the autophagy-lysosome pathway. Inhibition of SQSTM1 impaired the degradation of DNM1L in PRKAA-deficient cells. Our results provide new insights into the molecular mechanisms controlling mitochondrial dynamics in the development of vascular dysfunction and suggest that the specific inhibition of mitochondrial fission by AMPK activation may represent a novel approach for the treatment of cardiovascular diseases.
Materials and methods
Reagents
Antibodies against PRKAA, ATG5, ATG7, LC3B, ACACA/ACC, phospho-MTOR (Ser2248), phospho-ACACA/ACC (Ser79), phospho-RPS6KB (Thr389), NOS3/eNOS, phospho-NOS3/eNOS (Ser1177), PRKAA2 siRNA, and horseradish peroxidase-linked secondary antibodies were purchased from Cell Signaling Technology (2532, 12994, 8558, 4108, 4190, 5536, 3661, 9234, 9572, 9574, 6620, 7076, and 7074, respectively). Antibodies against SQSTM1/p62, MFN1, MFN2, OPA1, COX4I1/COX IV and PECAM1/CD31 were obtained from Abcam Company (Catalog numbers were 56416, 57602, 56889, 42364, 14744, and 28364 respectively). Antibodies against MFF, DNM1L, poly-ubiquitin, PRKAA1/AMPKα1, PRKAA2/AMPKα2, TOMM20/Tom20, and ACTB/β-actin were acquired from Santa Cruz Biotechnology (398617, 271583, 8017, 130394, 19129, and 47778, respectively). PRKAA1, PRKAA2, PRKAA, DNM1L, and MFN2 siRNA were obtained from Santa Cruz Biotechnology (29673, 38923, 45312, 43732, and 43928, respectively). PRKAA1 siRNA (AM51331, ID:767), Mfn2 siRNA (Silencer®, AM16708, ID:172635) for ex vivo transfection, and Dnm1l siRNA (Stealth RNAi™, 1320001, ID: MS232189, MSS292597, and MSS292598) for in vivo injection were purchased from ThermoFisher Scientific. The enhanced chemiluminescence (ECL) detection kit was obtained from ThermoFisher Scientific (RC227810). Mdivi-1 and bafilomycin A1 (BafA) were purchased from EMD Millipore (475856 and 196000). Chloroquine was purchased from Life Technologies (P36240). Cycloheximide, MG132, and A23187 were purchased from Sigma-Aldrich (C1988, M8699, and C7522).
Experimental animals
prkaa1−/− and prkaa2−/− mice were generated and characterized as previously described.53 C57BL/6J wild-type mice and mice expressing the Cre recombinase under control of the VE-cadherin promoter/enhanced (VE-CAD-Cre) were obtained from the Jackson Laboratory (Bar Harbor, ME). Prkaa2-floxed (Prkaa2Flox) mice were obtained as a gift from Dr. Benoit Viollet at the Paris Descartes University in France. Mice were housed in temperature-controlled cages under 12-h light-dark cycles. To generate endothelial cell-specific prkaa2 knockout mice, Prkaa2Flox mice were crossed with VE-CAD-Cre mice. The mice with genotype of Prkaa2Flox/Cre+ were referred to as endothelial cell-specific prkaa2 knockout mice (prkaa2Endoth−/−). The Prkaa2Flox mice were used as control (Prkaa2FloxCtrl). The animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Georgia State University.
To determine the role of mitochondrial fission in PRKAA deficiency-induced endothelial dysfunction, we treated male prkaa2−/− mice (3 mo of age) with mdivi-1 (1.2 mg/kg/d) by subcutaneously implanting Alzet mini-osmotic pumps (Model 2002, Cupertino, CA, USA) for 14 d. We also retro-orbitally injected prkaa2−/− mice with Dnm1l siRNA (5 mg/kg). To examine whether PRKAA inhibition induces mitochondrial fission by inhibiting autophagy, we fed prkaa2−/− mice a microencapsulated rapamycin-containing food (14 mg/kg in food) for 1 wk, as described previously.39 After treatment, thoracic aortas were collected for analyses of endothelial function, autophagy activity, and mitochondrial dynamics.
Cell culture
HUVECs were obtained from the American Type Culture Collection (PCS-100–013) and grown in EBM medium (Lonza, CC-3121), which was supplemented with 10% fetal bovine serum (Sigma, 12303C) and endothelial cell growth supplement (Lonza, CC-4133). U2OS cells were gifts from Dr. Khalid Salaita (Emory University). atg5−/− and atg7−/− MEFs were gifts from Dr. Noboru Mizushima (Japan Science and Technology Agency)54 and Dr. Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science),55 respectively. U2OS and MEFs were cultured in DMEM medium (Mediatech, 10–013-CV), which was supplemented with 10% fetal bovine serum. All culture media were supplemented with penicillin (100 Units/ml) and streptomycin (100 μg/ml) (Gibco, 15140–122). Cultured cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air at 37°C. HUVECs were used between passages 3 and 8.
Transmission electron microscopy
Freshly isolated aortas were cut into 1-mm aortic rings and fixed overnight in 0.1 M sodium phosphate buffer (pH 7.4, PBS; Sigma, D5652–10L) containing 4% formaldehyde (Fisher Chemical, F79) plus 1% glutaraldehyde (Fisher Chemical, O2957). The aortic rings were washed, dehydrated, and embedded, according to a standard protocol.56 Ultrathin sections (90 nm) were cut, mounted in nickel grids, and stained with uranyl acetate-lead citrate. The sections were observed, and digital images with scale bars were collected using a Hitachi H-7600 transmission electron microscope (Hitachi High Technology America, Inc., Schaumburg IL, USA). The length of individual mitochondria in a cell was measured using a Bioquant analysis system (Bioquant, Nashville, TN, USA). For each mouse, approximately 100 mitochondria were measured.
Measurement of endothelial function
Mouse aortas were isolated, and the adherent fat and connective tissues surrounding the aortas were cleaned. Each aorta was cut into 3-mm aortic rings, which were equilibrated in oxygenated Krebs-buffer (118.3 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, 0.026 mM EDTA, 11 mM glucose) for 1 h under a tension of 0.8 g in organ chambers (Radnoti LLC, Monrovia, CA, USA). Endothelial function of carotid artery was assessed using DMT wire myograph systems (DMT 620M, Denmark). Aortic rings were precontracted with 30 nM U46619 (Cayman, 16450). At the plateau of contraction, endothelium-dependent and endothelium-independent vasodilation responses were determined in the presence of acetylcholoine (Sigma, A6625; 10−9 to 10−5 M) and sodium nitrorousside (Fluka, 71778; 10−10 to 10−6 M), respectively.
Immunohistochemistry and immunofluorescence
Thoracic aortas were isolated, fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 5-µm sections. The sections were deparaffinized, rehydrated, and microwaved in citrate buffer to unmask the antigens. Aortic sections were incubated with primary antibodies against DNM1L (ThermoFisher, PA1–16986) or SQSTM1 overnight at 4°C. After rinsing in wash buffer, sections were incubated with a horseradish peroxidase-labeled polymer detection system (DAKO Envision+Dual link, K4061) and stained with DAB chromogen (DAKO, K3468).57 The semiquantitative analysis of tissue staining was performed using an arbitrary grading system from a score of 1 to 5 (score 1: 0 to 20%; score 2: 21 to 40%; score 3: 41 to 60%; score 4: 61 to 80%; score 5: 81 to 100% positive staining in the intima). Frozen sections from carotid arteries and thoracic aortas were fixed in cold acetone (Fisher Scientific, A949) and blocked with 2% goat serum (BioGenex, HK112). The sections were incubated with a primary antibody against MFN2, PECAM1, or PRKAA2/AMPKα2 overnight at 4°C, after which they were incubated with Alexa-Fluor® secondary antibody (Life Technologies, A11008, A11056, and A21422) for 30 min. The sections were mounted with Shur/Mount™ fluorescence mounting media containing DAPI (Life Technology, P36931) and analyzed using an Olympus BX-51 microscope (Olympus, Genter Valley, PA, USA).
Visualization of autophagic vacuoles
Using electroporation, HUVECs were cotransfected with PRKAA2 siRNA and adenovirus encoding GFP-LC3B. Later, cells were plated on coverslips. After 24 h of transfection, the cells were incubated with 5 µM CQ for 16 h. Fluorescence images were obtained using the LSM-800 confocal microscope (Carl Zeiss, Inc., Jena, Germany). Autophagy was measured by quantifying the average number of autophagosomes per cell for each sample. A minimum of 50 cells per sample was counted.
Quantification of mitochondrial morphology and mitochondrial fission events in cultured cells
HUVECs were incubated with 100 nM MitoTracker® Deep Red (Invitrogen, M22426) for 30 min to visualize mitochondrial morphology. Then, live HUVECs were transferred into 37°C chamber of a Zeiss LSM-800 time-lapse confocal microscope (63X oil immersion objective lens) and cultured in phenol red-free EBM medium for 2 h. The live cells were observed, and images were collected using a Zeiss LSM-800 confocal microscope. Analysis was limited to regions of interest in the periphery of cells, where individual mitochondria were readily resolved. Mitochondrial morphology, length, and number in cells were analyzed by ImageJ software (NIH) with “Mitochondrial Morphology” plugin, supported by Ruben K. Dagda, Ph.D. (University of Nevada School of Medicine).58 At least 100 cells were analyzed in each sample to determine the percentage of cells undergoing mitochondrial fragmentation. Time-lapse images were collected at 1 min intervals for 10 min by excitation at 644 nm. Mitochondrial fusion and fission events were quantified by manual review of captured fluorescence images using Zen software. 15 cells each group were analyzed the mitochondrial fission events and the ratio of fission/(fission + fusion).
Adenovirus infection and siRNA transfection
HUVECs were inflected with an adenovirus encoding ATG7 (Ad-ATG7) for 48 h. An adenovirus encoding GFP (Ad-GFP) served as a control. These conditions typically produced infection efficiency of more than 80%, as determined by the percentage of GFP-positive cells.
HUVECs were transfected with PRKAA1, PRKAA2, or ATG7 siRNA in OPTI-MEM reduced-serum media (GIBCO, 31985) using Lipofectamine™ RNAiMax transfection reagent (Life Technologies, 13778150). HUVECs were transfected with plasmid coding for cherry-DNM1L (Addgene, 49152; Gia Voeltz Lab) and GFP-LC3B (Life Technologies, P36240) using electroporation, and then transfected with GFP-SQSTM1 (Life Technologies, P36240) or GFP-LC3B (ThermoFisher Scientific, P36235).
Carotid arteries that were isolated from WT mice were transfected with siRNA using electroporation, according to a previous protocol.35 Briefly, the isolated carotid artery was mounted onto 2 small glass cannulas in the bath chamber of the culture myograph system (DMT 204CM, Denmark) and perfused with Mfn2 siRNA (20 µM), which was diluted with P5 primary cell 4D-Nucleofector™ solution (Lonza, PBP5–00675). The artery was transferred into an electroporation cuvette, and electroporation was performed using a program for HUVECs on the Nucleofector™ 4D device (Lonza, Allendale, NJ). After the treatment, the artery was cultured in oxygen-saturated medium, which was supplemented with 10% fetal bovine serum and 50 µg/mL penicillin, at 37°C for 2 d.
To determine whether gene silencing of DNM1L can inhibit PRKAA deficiency-induced mitochondrial fission, we purchased Stealth RNAi™ Dnm1l siRNA from Invitrogen and retro-orbitally injected prkaa2−/− mice with Dnm1l siRNA (5 mg/kg) or control siRNA-containing liposomes (Life Technologies, Invivofectamine® 2.0, 1377505). After 4 d of siRNA injection, aortas were harvested for the analyses of vascular function and mitochondrial dynamics.
Measurement of ROS
Freshly isolated aortas were immersed in tissue freezing medium (O.C.T. compound; Torrance, 4583) and snap frozen in liquid nitrogen. Frozen sections (8 µm) were prepared and incubated in PBS containing 5 µM dihydroethidium (DHE; Life Technologies, D11347) in a light-proof humidified chamber at 37°C for 30 min. Sections were then viewed under an Olympus BX-51 fluorescence microscope using the same imaging settings in each experimental condition.59 The grade of fluorescence intensity for DHE in the whole artery was shown with color lookup table using ImageJ software.
Immunoprecipitation and western blotting
Proteins were extracted from HUVECs and mouse aortas using RIPA lysis buffer containing 1 mM Na3VO4, 1 mg/ml leupeptin (Sigma, L2884), and 1 mM phenylmethanesulfonylfluoride (Santa Cruz Biotechnology, sc-24948A). Protein concentrations were determined using the bicinchoninic acid protein assay reagent (Thermo Scientific, 23223, 23224). For immunoprecipitation, proteins (500 μg) were incubated with anti-DNM1L antibody and protein A sepharose beads (GE Healthcare, 17–0780–01) overnight at 4°C. The sepharose beads were washed 4 times with lysis buffer and subjected to western blot analysis.
For western blot analysis, 30 μg of protein were resolved by SDS-PAGE, transferred to PVDF membranes (Millipore Corp., IPVH00010), and probed with specific antibodies. The signals were visualized using the ECL detection system. The intensity (area x density) of individual bands was measured by densitometry (model GS-700, Imaging Densitometer; Bio-Rad, Hercules, CA), and the background was subtracted from the calculated area.
Statistical analysis
Data were expressed as mean ± standard error of the mean. Statistical differences were analyzed by one-way or 2-way analysis of variance, followed by the Bonferroni post hoc analysis. Comparisons between 2 groups were assessed using the Student t test. P < 0.05 was considered statistically significant.
Supplementary Material
Abbreviations
- ACACC/ACC
acetyl-coenzyme A carboxylase α
- Ach
acetylcholine
- ACTB
actin β
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- AMPK
5′ AMP-activated protein kinase
- ATG7
autophagy-related 7
- BafA
bafilomycin A1
- CHX
cycloheximide
- CQ
chloroquine
- DHE
dihydroethidium
- DNM1L
dynamin 1-like
- FIS1fission
fissionmitochondrial 1
- prkaa2Endoth−/−
endothelial-specific Prkaa2 knockout mice
- HUVEC
human umbilical vein endothelial cell
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- MEF
mouse embryonic fibroblast
- MFF
mitochondrial fission factor
- MFN2
mitofusin 2
- MIEF1/2
mitochondrial elongation factor 1/2
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NOS3/eNOSnitric oxide synthase 3
nitric oxide synthase 3 endothelial cell
- OPA1
optic atrophy 1
- PPARGC1A/PGC-1α
peroxisome proliferative activated receptor, gamma, coactivator 1 α
- PRKAA1/AMPKα1
protein kinase, AMP-activated, α 1 catalytic subunit
- PRKAA2/AMPKα2
protein kinase, AMP-activated α 2 catalytic subunit
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- SNP
sodium nitroprusside
- ULK1
unc51-like kinase 1
- WT
wild type.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by grants (HL079584, HL080499, HL089920, HL110488, CA213022, and AG047776 to M.H. Zou) and (HL128014, HL132500, and HL137371 to Z. Xie and M. H. Zou) from the National Institutes of Health.
References
- [1].Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta 2008; 1777:1092-7; PMID:18519024; http://dx.doi.org/ 10.1016/j.bbabio.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 2009; 89:799-845; PMID:19584314; http://dx.doi.org/ 10.1152/physrev.00030.2008 [DOI] [PubMed] [Google Scholar]
- [3].Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A 2006; 103:2653-8; PMID:16477035; http://dx.doi.org/ 10.1073/pnas.0511154103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 2015; 11:11-24; PMID:25486875; http://dx.doi.org/ 10.1038/nrneurol.2014.228 [DOI] [PubMed] [Google Scholar]
- [5].Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, et al.. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011; 124:444-53; PMID:21747057; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.110.014506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 2013; 24:659-67; PMID:23283981; http://dx.doi.org/ 10.1091/mbc.E12-10-0721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wells RC, Picton LK, Williams SC, Tan FJ, Hill RB. Direct binding of the dynamin-like GTPase, Dnm1, to mitochondrial dynamics protein Fis1 is negatively regulated by the Fis1 N-terminal arm. J Biol Chem 2007; 282:33769-75; PMID:17884824; http://dx.doi.org/ 10.1074/jbc.M700807200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 2003; 23:5409-20; PMID:12861026; http://dx.doi.org/ 10.1128/MCB.23.15.5409-5420.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, et al.. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 2009; 11:958-66; PMID:19578372; http://dx.doi.org/ 10.1038/ncb1907 [DOI] [PubMed] [Google Scholar]
- [10].Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta 2013; 1833:150-61; PMID:22580041; http://dx.doi.org/ 10.1016/j.bbamcr.2012.05.002 [DOI] [PubMed] [Google Scholar]
- [11].Purnell PR, Fox HS. Autophagy-mediated turnover of dynamin-related protein 1. BMC Neurosci 2013; 14:86; PMID:23937156; http://dx.doi.org/ 10.1186/1471-2202-14-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hardie DG, Carling D. The AMP-activated protein kinase–fuel gauge of the mammalian cell? Eur J Biochem 1997; 246:259-73; PMID:9208914; http://dx.doi.org/ 10.1111/j.1432-1033.1997.00259.x [DOI] [PubMed] [Google Scholar]
- [13].Hardie DG, Carling D, Halford N. Roles of the Snf1/Rkin1/AMP-activated protein kinase family in the response to environmental and nutritional stress. Semin Cell Biol 1994; 5:409-16; PMID:7711289; http://dx.doi.org/ 10.1006/scel.1994.1048 [DOI] [PubMed] [Google Scholar]
- [14].Schulz E, Anter E, Zou MH, Keaney JF Jr.. Estradiol-mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinase. Circulation 2005; 111:3473-80; PMID:15967841; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.105.546812 [DOI] [PubMed] [Google Scholar]
- [15].Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes 2006; 55:496-505; PMID:16443786; http://dx.doi.org/ 10.2337/diabetes.55.02.06.db05-1064 [DOI] [PubMed] [Google Scholar]
- [16].Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y, Zhu Y, DeFea K, Pan S, Tsai MD, et al.. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res 2009; 104:496-505; PMID:19131647; http://dx.doi.org/ 10.1161/CIRCRESAHA.108.187567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab 2001; 281:E1340-6; PMID:11701451 [DOI] [PubMed] [Google Scholar]
- [18].Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A 2002; 99:15983-7; PMID:12444247; http://dx.doi.org/ 10.1073/pnas.252625599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al.. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999; 98:115-24; PMID:10412986; http://dx.doi.org/ 10.1016/S0092-8674(00)80611-X [DOI] [PubMed] [Google Scholar]
- [20].Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, WGt Wiles, Schlattner U, Neumann D, Brownlee M, Freeman MB, Goldman MH. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem 2004; 279:43940-51. [DOI] [PubMed] [Google Scholar]
- [21].Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. Activation of 5′-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role of peroxynitrite. J Biol Chem 2003; 278:34003-10; http://dx.doi.org/ 10.1074/jbc.M300215200 [DOI] [PubMed] [Google Scholar]
- [22].Cai Y, Martens GA, Hinke SA, Heimberg H, Pipeleers D, Van de Casteele M. Increased oxygen radical formation and mitochondrial dysfunction mediate beta cell apoptosis under conditions of AMP-activated protein kinase stimulation. Free Radic Biol Med 2007; 42:64-78; PMID:17157194; http://dx.doi.org/ 10.1016/j.freeradbiomed.2006.09.018 [DOI] [PubMed] [Google Scholar]
- [23].Xie Z, Zhang J, Wu J, Viollet B, Zou MH. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes 2008; 57:3222-30; PMID:18835932; http://dx.doi.org/ 10.2337/db08-0610 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [24].Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T, Araki E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006; 55:120-7; PMID:16380484; http://dx.doi.org/ 10.2337/diabetes.55.01.06.db05-0943 [DOI] [PubMed] [Google Scholar]
- [25].Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al.. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/ 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhu H, Foretz M, Xie Z, Zhang M, Zhu Z, Xing J, Leclerc J, Gaudry M, Viollet B, Zou MH. PRKAA1/AMPKalpha1 is required for autophagy-dependent mitochondrial clearance during erythrocyte maturation. Autophagy 2014; 10:1522-34; PMID:24988326; http://dx.doi.org/ 10.4161/auto.29197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bhatt MP, Lim YC, Kim YM, Ha KS. C-peptide activates AMPKalpha and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes 2013; 62:3851-62; PMID:23884890; http://dx.doi.org/ 10.2337/db13-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li J, Wang Y, Wang Y, Wen X, Ma XN, Chen W, Huang F, Kou J, Qi LW, Liu B, et al.. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J Mol Cell Cardiol 2015; 86:62-74; PMID:26196303; http://dx.doi.org/ 10.1016/j.yjmcc.2015.07.010 [DOI] [PubMed] [Google Scholar]
- [29].Wikstrom JD, Israeli T, Bachar-Wikstrom E, Swisa A, Ariav Y, Waiss M, Kaganovich D, Dor Y, Cerasi E, Leibowitz G. AMPK regulates ER morphology and function in stressed pancreatic beta-cells via phosphorylation of DRP1. Mol Endocrinol 2013; 27:1706-23; PMID:23979843; http://dx.doi.org/ 10.1210/me.2013-1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, Benigni A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest 2015; 125:715-26; PMID:25607838; http://dx.doi.org/ 10.1172/JCI77632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Toyama EQ, Herzig S, Courchet J, TL Lewis Jr., Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, et al.. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016; 351:275-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011; 13:589-98; PMID:21478857; http://dx.doi.org/ 10.1038/ncb2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stein DA, Perry ST, Buck MD, Oehmen CS, Fischer MA, Poore E, Smith JL, Lancaster AM, Hirsch AJ, Slifka MK, et al.. Inhibition of dengue virus infections in cell cultures and in AG129 mice by a small interfering RNA targeting a highly conserved sequence. J Virol 2011; 85:10154-66; PMID:21795337; http://dx.doi.org/ 10.1128/JVI.05298-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al.. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008; 14:193-204; PMID:18267088; http://dx.doi.org/ 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Straub AC, Lohman AW, Billaud M, Johnstone SR, Dwyer ST, Lee MY, Bortz PS, Best AK, Columbus L, Gaston B, et al.. Endothelial cell expression of haemoglobin alpha regulates nitric oxide signalling. Nature 2012; 491:473-7; PMID:23123858; http://dx.doi.org/ 10.1038/nature11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, et al.. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011; 60:1770-8; PMID:21562078; http://dx.doi.org/ 10.2337/db10-0351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013; 62:1270-81; PMID:23223177; http://dx.doi.org/ 10.2337/db12-0533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy 2008; 4:322-9; PMID:18216495; http://dx.doi.org/ 10.4161/auto.5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, Afonso LC, Aguilar-Pimentel JA, Becker L, Garrett L, et al.. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest 2013; 123:3272-91; PMID:23863708; http://dx.doi.org/ 10.1172/JCI67674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang Q, Liang B, Shirwany NA, Zou MH. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One 2011; 6:e17234; PMID:21386904; http://dx.doi.org/ 10.1371/journal.pone.0017234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 2008; 18:165-73; PMID:18296052; http://dx.doi.org/ 10.1016/j.tcb.2008.01.006 [DOI] [PubMed] [Google Scholar]
- [42].Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J 2002; 21:1616-27; PMID:11927546; http://dx.doi.org/ 10.1093/emboj/21.7.1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yue W, Zheng X, Lin Y, Yang CS, Xu Q, Carpizo D, Huang H, DiPaola RS, Tan XL. Metformin combined with aspirin significantly inhibit pancreatic cancer cell growth in vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and Bcl-2. Oncotarget 2015; 6:21208-24; PMID:26056043; http://dx.doi.org/ 10.18632/oncotarget.4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Loubiere C, Goiran T, Laurent K, Djabari Z, Tanti JF, Bost F. Metformin-induced energy deficiency leads to the inhibition of lipogenesis in prostate cancer cells. Oncotarget 2015; 6:15652-61; PMID:26002551; http://dx.doi.org/ 10.18632/oncotarget.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rice S, Pellat L, Ahmetaga A, Bano G, Mason HD, Whitehead SA. Dual effect of metformin on growth inhibition and oestradiol production in breast cancer cells. Int J Mol Med 2015; 35:1088-94; PMID:25716282 [DOI] [PubMed] [Google Scholar]
- [46].Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 2010; 191:1141-58; PMID:21149567; http://dx.doi.org/ 10.1083/jcb.201007152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 2008; 19:2402-12; PMID:18353969; http://dx.doi.org/ 10.1091/mbc.E07-12-1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chiang YY, Chen SL, Hsiao YT, Huang CH, Lin TY, Chiang IP, Hsu WH, Chow KC. Nuclear expression of dynamin-related protein 1 in lung adenocarcinomas. Mod Pathol 2009; 22:1139-50; PMID:19525928; http://dx.doi.org/ 10.1038/modpathol.2009.83 [DOI] [PubMed] [Google Scholar]
- [49].Weikel KA, Cacicedo JM, Ruderman NB, Ido Y. Glucose and palmitate uncouple AMPK from autophagy in human aortic endothelial cells. Am J Physiol Cell Physiol 2015; 308:C249-63; PMID:25354528; http://dx.doi.org/ 10.1152/ajpcell.00265.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al.. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/ 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- [51].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- [52].Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A 2008; 105:20567-74; PMID:19074260; http://dx.doi.org/ 10.1073/pnas.0810611105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu C, Viollet B, Yan D, Zou MH. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res 2010; 106:1117-28; PMID:20167927; http://dx.doi.org/ 10.1161/CIRCRESAHA.109.212530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/ 10.1038/nature03029 [DOI] [PubMed] [Google Scholar]
- [55].Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al.. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/ 10.1083/jcb.200412022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, et al.. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 2012; 110:1484-97; PMID:22511751; http://dx.doi.org/ 10.1161/CIRCRESAHA.111.263848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liang B, Wang S, Wang Q, Zhang W, Viollet B, Zhu Y, Zou MH. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP-activated protein kinase-alpha2 in vivo. Arterioscler Thromb Vasc Biol 2013; 33:595-604; PMID:23288166; http://dx.doi.org/ 10.1161/ATVBAHA.112.300606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009; 284:13843-55; PMID:19279012; http://dx.doi.org/ 10.1074/jbc.M808515200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang M, Song P, Xu J, Zou MH. Activation of NAD(P)H oxidases by thromboxane A2 receptor uncouples endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol 2011; 31:125-32; PMID:20947827; http://dx.doi.org/ 10.1161/ATVBAHA.110.207712 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.