

ABSTRACT
Spns1 (Spinster homolog 1 [Drosophila]) in vertebrates, as well as Spin (Spinster) in Drosophila, is a hypothetical lysosomal H+-carbohydrate transporter, which functions at a late stage of macroautophagy (hereafter autophagy). The Spin/Spns1 defect induces aberrant autolysosome formation that leads to developmental senescence in the embryonic stage and premature aging symptoms in adulthood. However, the molecular mechanism by which loss of Spin/Spns1 leads to the specific pathogenesis remains to be elucidated. Using chemical, genetic and CRISPR/Cas9-mediated genome-editing approaches in zebrafish, we investigated and determined a mechanism that suppresses embryonic senescence as well as autolysosomal impairment mediated by Spns1 deficiency. Unexpectedly, we found that a concurrent disruption of the vacuolar-type H+-ATPase (v-ATPase) subunit gene, atp6v0ca (ATPase, H+ transporting, lysosomal, V0 subunit ca) led to suppression of the senescence induced by the Spns1 defect, whereas the sole loss of Atp6v0ca led to senescent embryos similar to the single spns1 mutation. Moreover, we discovered that the combined stable defect seen in the presence of both the spns1 and atp6v0ca mutant genes still subsequently induced premature autophagosome-lysosome fusion marked by insufficient acidity, while extending developmental life span, compared with the solely mutated spns1 defect. Our data suggest that Spns1 and the v-ATPase orchestrate proper autolysosomal biogenesis with optimal acidification that is critically linked to developmental senescence and survival.
KEYWORDS: aging, autophagy, lysosome, senescence, spinster, vacuolar-type H+-ATPase, zebrafish
Introduction
Cellular senescence is a part of a normal physiological, as well as pathophysiological, process involved in development.1-6 Senescence is, therefore, not only a phenomenon observed in the later aging process but is also detectable during embryogenesis of vertebrates; as such, this process might have evolved to regulate embryonic development. However, the underlying genetic and molecular basis in embryonic development and senescence remains poorly understood. It is also unknown how homeostatic regulation of genes and proteins that are critical for developmental senescence might have an impact on subsequent aging later in life.
Previously our genetic screening based on the embryonic senescence phenotype in zebrafish has identified several genes including spns1 that play an essential role in autolysosomal function.,4,7 Spns1 in vertebrates, and Spin in Drosophila, are thought to function as a lysosomal H+-carbohydrate symporter, which functions at a late and terminal stage of autophagy.7-9 Mutations in the corresponding genes results in aberrant autolysosome formation, leading to embryonic senescence and premature aging symptoms in Drosophila and zebrafish.4,10 We recently have shown in zebrafish that Becn1/Beclin 1 suppression ameliorates developmental senescence and autolysosomal impairment associated with Spns1 deficiency, whereas loss of Tp53 exacerbates these characteristics in the Spns1-defective zebrafish embryos.7 We also demonstrate that basal Tp53 activity has a certain protective role(s) against the Spns1 defect by suppressing autolysosome biogenesis and/or autophagosome-lysosome fusion. Moreover, we find that chemical inhibition of the v-ATPase prevents the appearance of the Spns1-deficient hallmarks including aberrant autolysosomal formation and excessive autophagosome-lysosome fusion. However, the precise mechanism by which autolysosomal acidification and biogenesis are regulated by the balance between v-ATPase and Spns1 has not been fully elucidated. This has been particularly problematic because some defects of the v-ATPase itself can also cause developmental senescence in zebrafish as we have demonstrated previously.4 Moreover, cumulative lines of evidence suggest that autophagosome-lysosome fusion can still occur without v-ATPase-mediated acidification during endo-lysosomal biogenesis.11-13
We hypothesized that the distinct phenotypes resulting from each single mutation, namely loss of acidification with atp6v0ca and loss of symport activity due to spns1, when combined result in a counteracting (i.e., suppressing) effect at the cellular and organismal levels. Therefore, because we could not rule out the possibility that the previous pharmacological approach may have had off-target effects, we concurrently knocked down and/or mutated both the spns1 and atp6v0ca genes in zebrafish embryos to validate their epistasis as well as biological endpoints such as autophagy, senescence and survival of the subject animals during early development. We found that the dual defect of Spns1 and v-ATPase was indeed counteractive for senescence induction during development and extended the animal survival or life span. In addition, we found that the permanent defect of both the spns1 and atp6v0ca genes still resulted in the premature fusion of autophagosomes with lysosomes (and presumably endosomes) with insufficient acidity. Our findings therefore suggest that Spns1 and the v-ATPase function at a balance point that is critically involved in the regulation of developmental senescence, and is accompanied by proper lysosomal acidification during autophagy.
Results
Chemical and genetic modulations of v-ATPase in Spns1-deficient zebrafish
According to our recent finding of autolysosomal abnormality observed in spns1-mutant (spns1hi891/hi891) zebrafish, we performed chemical and genetic approaches with the mutant animals to rescue or ameliorate the phenotypes of yolk opacity and developmental senescence.7 It should noted that the hallmark observation of an opaque yolk phenotype was first originally described by Young et al.;14 we have confirmed this representative and unique phenotype at the gross morphological level and histologically, showing that it is a marker for the defect associated with the spns1 mutation.4 Furthermore, we have established the senescence-associated Glb1/β-galactosidase (SA-Glb1) assay, which is commonly used to monitor senescence in mammalian cells, in zebrafish embryos and larvae as well as adults.4,7,15-17 The appearance of yolk opaqueness accompanied by increased SA-Glb1 activity in spns1-mutant fish embryos or larvae supports the notion that the interruption of the intrinsic nutrient supply, supposedly from autophagy-dependent catabolism of the yolk in zebrafish embryos and larvae,18 may lead to profound energetic exhaustion under the aberrant autolysosomal condition resulting from Spns1 deficiency. As reported previously, we found that a specific inhibitor of the v-ATPase, bafilomycin A1 (BafA), and several other U.S. Food and Drug Administration-approved proton-pump inhibitors such as omeprazole, lansoprazole and pantoprazole could significantly suppress the phenotypes induced by Spns1 deficiency (Fig. S1 and S2).7
With any pharmacological approach, there is always a concern about off-target effects. Nonetheless, chemical inhibition is often the first method used because of its technical facility, especially in the zebrafish system where it used to be quite difficult to knock out specific genes. To extend our studies, we initially relied on a morpholino approach. Because BafA specifically binds to the Atp6v0ca/subunit c of the v-ATPase and thereby inhibits its enzymatic and proton-pump activity, we examined a specific knockdown of the atp6v0ca gene by using an antisense morpholino oligonucleotide (MO; atp6v0ca MO1),7,19,20 and found that the morphants recapitulated the phenotype of the BafA-treated animals resulting in reduced yolk opacity and SA-Glb1 activity compare with control spns1 mutants.7
The v-ATPase is a lysosomal proton transporter,21 and a proton-coupled symport permease function is proposed for Spns1.8 Furthermore, as noted above, mutations that block the function of either enzyme alone result in premature senescence.4 Thus, it was surprising that concurrent defects in both enzymes reverse or suppress this phenotype. We recently have proposed that this “counteractive” effect (i.e., suppression of the spns1 phenotype by inhibiting v-ATPase function) is due to “prior” alkalinization and neutralization in lysosomes and autolysosomes and reduction of their biogenesis by blocking v-ATPase activity.7 Nonetheless, the mechanistic nature of the interaction between Spns1 and Atp6v0ca had not been determined, including any potential role of autophagy.
To gain a detailed understanding of the relationship between these 2 lysosomal transporters, we examined the autophagic process in the atp6v0ca morphants as well as spns1 mutants by monitoring EGFP-Lc3b, as a marker of autophagosomes, and used LysoTracker Red DND-99, which accumulates in acidic compartments, to follow the biogenesis of lysosomes. The atp6v0ca MO1 itself significantly increased the formation of cellular EGFP-Lc3B puncta in wild-type (WT) zebrafish embryos (Fig. 1A and B). The spns1 mutant fish also displayed an elevated level of EGFP-Lc3B puncta; however, no additive induction of Lc3B puncta was observed at the cellular level in spns1-mutant fish injected with atp6v0ca MO1 versus control MO (Fig. 1A and B). In contrast to the basal level of lysosomal acidification seen in WT fish, the spns1 mutation resulted in elevated LysoTracker Red-positive compartments (Fig. 1A and B). As expected, this increase in acidified lysosomes was suppressed by the atp6v0ca knockdown. The schematic illustration in Fig. 1C illustrates the location and function of the v-ATPase and Spns1 in the lysosome membrane; a defect in Spns1 would theoretically lower the lysosomal pH, but a concomitant v-ATPase mutation would counteract this effect. At any rate, any pH change resulting from the spns1 mutation was apparently not substantial because the EGFP-Lc3B signal was not quenched, which should happen at the normal lysosomal pH.
Figure 1.
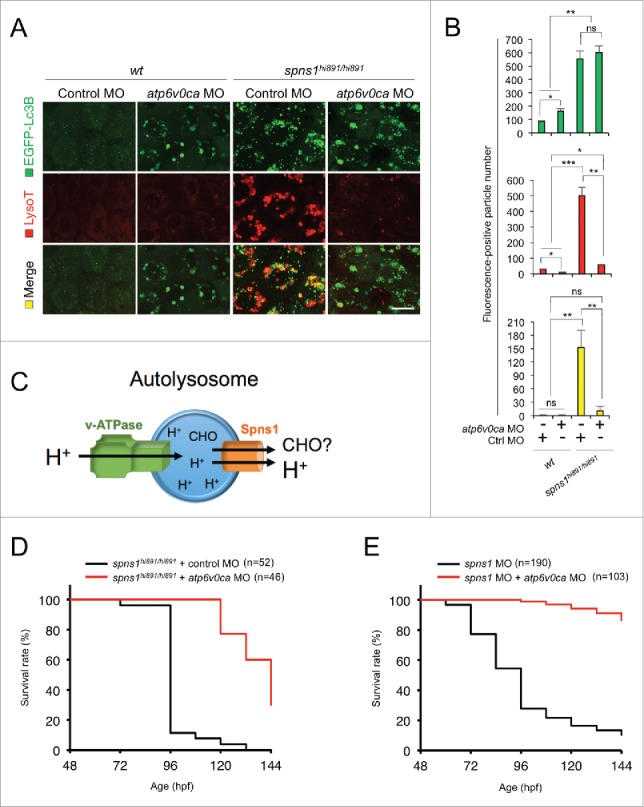
Atp6v0ca knockdown suppresses Spns1 deficiency and prolongs the survival of the defective animals. (A) Intracellular autolysosome formation and lysosomal biogenesis monitored by EGFP-Lc3B and LysoTracker at 76 hpf in wild-type (wt) and spns1-mutant animals injected with atp6v0c MO (4 ng/embryo). The samples were observed by using confocal microscopy at high magnification (x600). Scale bar: 10 µm. (B) Quantification of the EGFP-Lc3B (green), LysoTracker (red) and merged (yellow) fluorescence-positive particle numbers shown in (A). Quantification of images (and 4 additional sets of data) presented in the top, middle and bottom rows (red, LysoTracker Red; green, EGFP; yellow, merge) in panel (A) is shown (the number [n] of animals for each genotype with MO = 5). Error bars represent the mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.005; ns, not significant. (C) The schematic illustrates the location and function of the v-ATPase (which acidifies the lysosome and allows the activation of lysosomal hydrolases) and Spns1 (which is proposed to function in carbohydrate [CHO] efflux) in the lysosome membrane. (D) Survival curve for spns1-mutant animals injected with atp6v0ca MO or control MO (log rank test: χ2 = 82.71 on one degree of freedom; P < 0.0001). (E) Survival curve for spns1;atp6v0ca-double morphant larvae or spns1-single morphants (log rank test: χ2 = 151.6 on one degree of freedom; P < 0.0001).
Both the progression of yolk opaqueness and SA-Glb1 activity in spns1 mutants during the time period of 36 to 72 h postfertilization (hpf) are suppressed by atp6v0ca MO.7 The phenotype induced by the atp6v0ca knockdown during early development was particularly evident based on hypopigmentation observed at 48 hpf.,7,19,20 By further performing atp6v0ca MO1 injections into spns1-mutant embryos we demonstrated an increased number of viable larvae with temporally ameliorated yolk phenotype through 120 hpf (Fig. 1D). After this time, the mutant animals subsequently relapsed into deterioration, presumably due to the persistent impact of the Spns1 mutation and/or the transient activity of the atp6v0ca MO1. Conversely, the concurrent suppression of both spns1 and atp6v0ca by MO targeting (i.e., so that inhibition of Spns1 and v-ATPase is temporally correlated rather than using the spns1hi891/hi891 mutant fish) strikingly diminished the yolk opaqueness seen with the spns1 morphants and produced more than 80% viable larvae that survived beyond 6 d postfertilization, eventually reaching adulthood (Fig. 1E). These results support the notion that suppression of proper lysosomal acidification and biogenesis due to the atp6v0ca knockdown can offset the deleterious effect of Spns1 deficiency that occurs at the late stage of autophagy, thus prolonging animal survival.
The v-ATPase as a context-dependent senescence regulator in zebrafish embryos
Our previous genetic screening based on the embryonic senescence phenotype in zebrafish identified several genes including spns1. Among those genes, we also identified 2 atp6v genes, atp6v0ca and atp6v1h, as senescence suppressors; for both of these genes, an individual loss of function induces developmental senescence.4 Therefore, solely disrupted gene function of either spns1 or atp6v0ca induces embryonic senescence (Fig. 2A), whereas it appears that a concurrent suppression of both genes by MOs or a permanent disruption or mutation of spns1 with atp6v0c MO counteracts their respective effects (Fig. 1).7 Accordingly, we next investigated how the temporal suppression of the spns1 gene has an impact on the heritable defect of the atp6v0ca gene.
Figure 2.
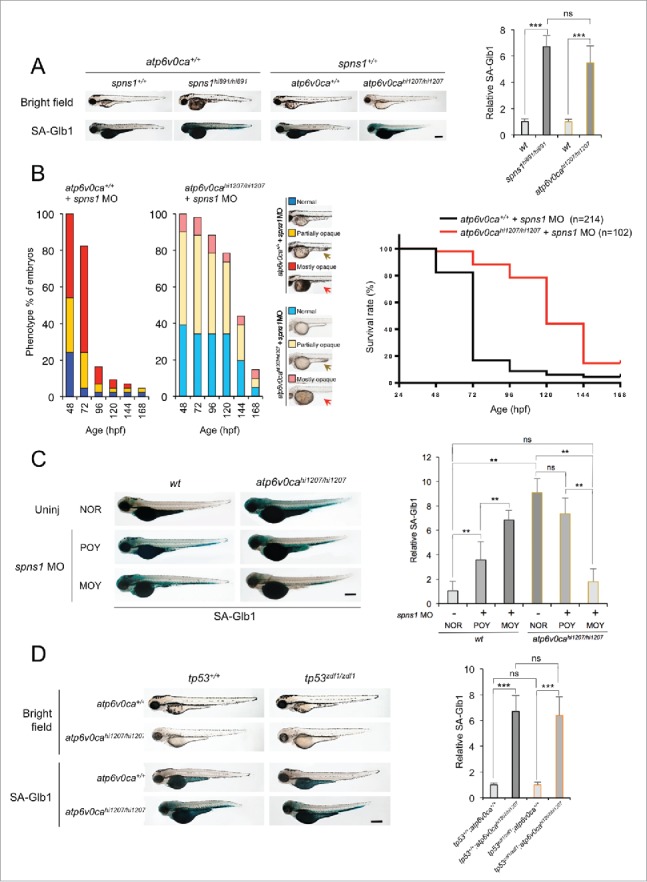
Counteractive balancing of developmental senescence induced by the defect of Spns1 and Atp6v0ca. (A) Induction of developmental senescence with the defect of either Spns1 or Atp6v0ca at 84 hpf. Scale bar: 500 µm. Quantification of the SA-Glb1 intensities is shown in the right graph (the number [n] of animals for each genotype = 6). Error bars represent the mean ± SD, ***P < 0.0005; ns, not significant. (B) Phenotype appearance of yolk opaqueness and survival of offspring cohorts from intercrosses of atp6v0cahi1207/+ fish. Comparisons of survival rates between wt (atp6v0ca+/+) and atp6v0cahi1207/hi1207 fish injected with spns1 MO are shown in the right survival curves (log rank test: χ2 = 99.36 on one degree of freedom; P < 0.0001). The arrows mark the yolk extension, which has largely regressed in embryos displaying the MOY phenotype. (C) Inverse correlation of strength of yolk opacity (NOR, normal; POY, partially opaque yolk; MOY, mostly opaque yolk) and SA-Glb1 activity in atp6v0ca-mutant animals injected with spns1 MO at 84 hpf. Scale bar: 500 µm. Quantification of the SA-Glb1 intensities is shown in the right graph (the number of animals for each genotype and phenotype = 6). Error bars represent the mean ± SD, **P < 0.001; ns, not significant. (D) Effect of a tp53 mutation on embryonic SA-Glb1 activity in the atp6v0ca mutant. The heritable impact of Tp53 and Atp6v0ca on SA-Glb1 induction was tested in each single-gene mutant (atp6v0cahi1207/hi1207 or tp53zdf1/zdf1) and double mutant atp6v0cahi1207/hi1207;tp53zdf1/zdf1 (atp6v0ca−/−;tp53 zdf1/zdf1) compared with WT animals at 84 hpf. Scale bar: 500 µm. Quantification of the SA-Glb1 intensities in WT, tp53zdf1/zdf1, atp6v0cahi1207/hi1207 and atp6v0cahi1207/hi1207;tp53zdf1/zdf1 animals, shown in the right graph (n = 10); the number (n) of animals is for each genotype. Error bars represent the mean ± SD, ***P < 0.0005; ns, not significant.
The loss of v-ATPase function results in a hypopigmentation phenotype (Fig. 2A; comparison of atp6v0cahi1207/hi1207 to atp6v0ca+/+ embryos in the bright field panel). Retrovirus-insertional atp6v0cahi1207/hi1207 mutant embryos were injected with spns1 MO, and hypopigmented homozygous mutant embryos were sorted according to their severity of yolk opaqueness, which is a hallmark of the spns1 defect, in 3 categories: normal (NOR), partially opaque yolk (POY), and mostly opaque yolk (MOY) phenotypes. The mild POY phenotype usually starts from the posterior (tip) yolk extension, and the subsequent severe MOY phenotype is generally accompanied by apparent regression or almost complete loss of the yolk extension that existed behind the yolk and under the trunk (Fig. 2B; compare the yellow and red arrows marking the yolk extension). In WT (atp6v0ca+/+) embryos spns1 MO induced yolk opaqueness (Fig. 2B). By comparison, in homozygous atp6v0cahi1207/hi1207 (atp6v0ca−/−) mutant embryos, the spns1 MO had a greatly reduced effect. In addition, there was an increased number of viable larvae that survived beyond 72 hpf in the atp6v0ca−/− background (Fig. 2B).
Hypopigmented MOY embryos, that is, those embryos that still had the dual-defective phenotypes, showed much lower SA-Glb1 intensity than hypopigmented POY ones (Fig. 2C; compare bar 5 to 6), almost comparable to that of uninjected WT animals (Fig. 2C; compare bar 1 to 6). In other words, when the MO-dependent knockdown of Spns1 was less effective in the v-ATPase mutant fish, there was still detectable SA-Glb1 activity. Hence, the more severe MOY zebrafish display a greater suppression of the senescence phenotype than seen with milder POY fish in combination with the atp6v0ca mutation.
A pivotal tumor suppressor, Tp53, plays critical roles in both autophagy and senescence,22,23 and we have previously found that either a decrease or loss of basal Tp53 (which results in an increase in autophagy activity24) enhances the phenotype resulting from Spns1 impairment.7 This additive effect may be the result of augmenting autophagy progression and/or lysosomal biogenesis in the absence of the Tp53 function, subsequently leading to autolysosomal malformation, inadequate termination of autophagy and embryonic senescence in the presence of the spns1 mutation. Therefore, we wondered if Tp53 has a similar or differential impact on autophagy or senescence in the presence of the Atp6v0ca defect, and hence may account for any part of the observed phenotype of the double spns1 and atp6v0ca mutant defects. Thus, we generated atp6v0cahi1207/hi1207 mutant fish harboring a tp53 mutation (tp53zdf1/zdf1), atp6v0cahi1207/hi1207;tp53zdf1/zdf1, and found that there was no apparent change of SA-Glb1 activity caused by the constitutive loss of normal Tp53 (Fig. 2D). Thus, there was no essential involvement for Tp53 function for the induction of embryonic senescence resulting from the v-ATPase deficiency. These results suggest that the reciprocally counteractive phenotype resulting from the combined defect is likely independent of Tp53.
Sustained autophagosome-lysosome fusion in the absence of Atp6v0ca or Spns1
We wondered if the suppressive effect of Atp6v0ca deficiency on the Spns1 defect is due to the loss of endosome and/or lysosome fusion with autophagosomes and/or an otherwise balanced acidification of the proton efflux (i.e., loss of H+ import by or through the v-ATPase into, and loss of export by or through Spns1 from, the lysosomal lumen; as shown by the schematic in Fig. 1C). A mutation in the v-ATPase should result in a reduction in the acidity of lysosomes, making their detection by LysoTracker Red staining problematic. Therefore, we confirmed the existence of mCherry-tagged Lamp1-positive lysosomal compartments and investigated if and how EGFP-Lc3B-positive vesicles (i.e., autophagosomes) were localized with the Lamp1-positive ones. For this purpose, we used Tg(CMV:EGFP-lc3b;eef1a1l1/EF1α:mCherry-lamp1);spns1hi891/+;atp6v0cahi1207/+ fish to intercross and concurrently obtain homozygous animals for either the spns1 or atp6v0ca defect (note that the CMV and eef1a1l1 promoters are both constitutive and ubiquitous). Intensities of intracellular compartments with red-only (Lamp1, lysosome), green-only (Lc3B, autophagosome) and red plus green (yellow, autolysosome; colocalization by fusion) vesicles were assessed in atp6v0cahi1207/hi1207 or spns1hi891/hi891 animals, compared with WT controls.
In homozygous atp6v0ca-mutant animals both mCherry-Lamp1 and EGFP-Lc3B intensities were enhanced; there was a sustainable increase in merged-yellow compartments compare with Tg(CMV:EGFP-lc3b;eef1a1l1:mCherry-lamp1) fish embryos with the wt background, indicating a small but significant increase in the number of fused autolysosomal structures (Fig. 3A). Moreover, in spns1-mutant embryos, we observed an apparently even much higher level of autophagosomes (green) and autolysosomal (merged mCherry-Lamp1 and EGFP-Lc3B, i.e., yellow) signals (Fig. 3A). Next, we examined endogenous Lc3A/B by western blotting. We detected a cytosolic form of Lc3A/B (Lc3A/B-I) and the phosphatidylethanolamine-conjugated form (Lc3A/B-II) that is recruited to phagophore (the autophagosome precursor) membranes, and confirmed the appearance of slightly increased Lc3A/B-II in atp6v0cahi1207/hi1207 and highly accumulated Lc3A/B-II in spns1hi891/hi891 fish (Fig. 3A, lower right). The accumulation of Lc3A/B-II could correspond to an increase in autophagosomes and autophagy activation. However, it was not clear if this Lc3A/B-II accumulation occurred because autophagosomes could not fuse with lysosomes, or if undegraded autolysosomal Lc3A/B-II might be accumulated after fusion.
Figure 3.

Premature autolysosomal fusion resulting from Atp6v0ca deficiency, as well as Spns1 deficiency. (A) Appearance of EGFP-Lc3B- and mCherry-Lamp1-positive autolysosomal fusion with Atp6v0ca deficiency, as well as Spns1 deficiency, was detectable. Embryos of EGFP-Lc3B- and mCherry-Lamp1-double transgenic spns1-mutant (Tg[CMV:EGFP-Lc3B;mCherry-Lamp1];spns1hi891/hi891) fish and EGFP-Lc3B- and mCherry-Lamp1-double transgenic atp6v0ca-mutant (Tg[CMV:EGFP-lc3b;eef1a1l1:mCherry-lamp1];atp6v0cahi1207/hi1207) fish were examined to confirm autophagosome-lysosome fusion at 76 hpf. Scale bar: 10 µm. Quantification of the EGF (green), mCherry (red) and merged (yellow) fluorescence-positive particle numbers is shown in the right graph (n = 6); the number (n) of animals is for each genotype and phenotype. Error bars represent the mean ± SD, *P < 0.005, **P < 0.001. Three independent areas (periderm or basal epidermal cells above the eye) were selected from individual animals. Western blot analysis using anti-Lc3A/B antibody shows endogenous Lc3A/B protein levels, which can confirm an increase of the total amount of Lc3A/B in wt, atp6v0cahi1207/hi1207- and spns1hi891/hi891-mutant fish. Increased Lc3A/B-II conversion or accumulation was detected slightly in atp6v0cahi1207/hi1207 and more significantly in spns1 mutants. (B) Most of the EGFP-Lc3B-positive autolysosomes were not LysoTracker Red-positive compartments. LysoTracker Red DND-99 staining of EGFP-Lc3B-transgenic spns1-mutant (Tg[CMV:EGFP-lc3b];spns1hi891/hi891) or atp6v0ca-mutant (Tg[CMV:EGFP-lc3b];atp6v0cahi1207/hi1207) embryos was performed at 76 hpf. Scale bar: 10 µm. Quantification of the EGFP (green) and LysoTracker (red) fluorescence-positive particle numbers is shown in the right graph (the number of animals for each genotype and phenotype = 6). Error bars represent the mean ± SD, **P < 0.001, ***P < 0.0005; ns, not significant. Three independent areas (periderm or basal epidermal cells above the eye) were selected from individual animals.
We next determined that the LysoTracker Red staining (corresponding to acidified lysosomes, rather than total lysosomal compartments detected by mCherry-Lamp1) was only slightly visible but was not significantly merged with EGFP-Lc3B-positive compartments in cells from atp6v0ca-mutant embryos (similar to that in WT), whereas EGFP-Lc3B-positive vesicles were still increased reproducibly (Fig. 3B). Taken together, these results indicate that genetic disruption of atp6v0ca does not lead to a substantial block in the fusion of autophagosomes with lysosomes (and/or potentially even less acidic endosomes; mCherry-Lamp1-positive but LysoTracker Red-negative vesicles). These results therefore also suggest that the suppression of autolysosomal fusion is not the primary reason for the inhibition of the Spns1 defect seen with the concomitant loss of v-ATPase activity.
Concurrent heritable defects of both Spns1 and Atp6v0ca in zebrafish
Both the spns1 and atp6v0ca genes are in the same linkage group of zebrafish chromosome 3, having a distance of 584 kgbase pairs between the genes (Fig. 4A). Zebrafish, as diploid organisms, have 2 alleles at each genetic locus, with one allele inherited from each parent. After multiple test crosses, however, we were unable to generate double homozygous mutant progeny with meiotic crossover (i.e., any breakage and rejoining process through meiosis). That is, the proximity of the 2 genes on the same chromosome would make it extremely difficult to generate homozygous double mutant offspring through standard crossing of mutant parental fish; the offspring could become ‘trans-heterozygote’ with single mutations, one on each copy of chromosome 3 (Fig. 4B, left), but could not be ‘cis-heterozygote’ with 2 mutations on the same allele side (Fig. 4B, right); the latter would be needed to generate double homozygote offspring from a subsequent cross.
Figure 4.
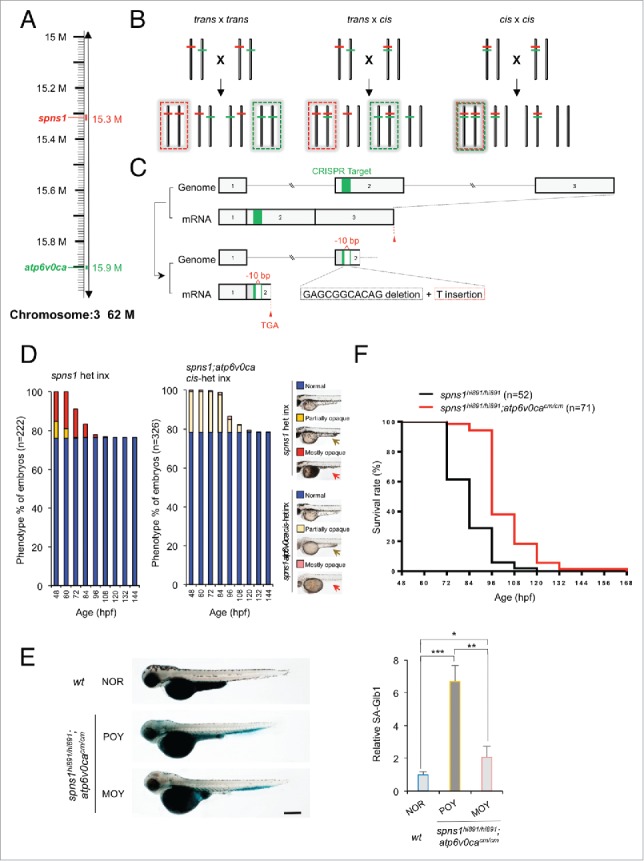
CRISPR/Cas9-mediated knockout of the atp6v0ca gene in spns1-mutant zebrafish. (A) Distance of the spns1 and atp6v0ca loci at the same linkage group of chromosome 3. M, mega-base pairs. (B) Schemes for trans- and cis-heterozygosity of the spns1 and atp6v0ca loci generated by Crisper/Cas9-mediated genome editing at chromosome 3. Chromosome pairs in the dashed “red” squares contain homozygous spns1 mutations with opaque-yolk phenotype and lethality. Chromosome pairs in the dashed “green” squares contain homozygous atp6v0ca mutations with hypopigmented phenotype and lethality. Double homozygous mutations generated by cis x cis concurrently show both opaque-yolk and hypopigmented phenotypes. (C) A schematic presentation of CRISPR/Cas9-mediated genome editing for the atp6v0ca gene. (D) Phenotype appearance of yolk opaqueness and survival of offspring cohorts from intercrosses (inx) of spns1hi891/+fish and spns1hi891/+;atp6v0cacm/+ fish. (E) SA-Glb1 images of spns1;atp6v0ca-double mutants with partially opaque yolk (POY) and mostly opaque yolk (MOY) phenotypes compared with a normal wild-type (WT) phenotype animal. Quantification of the SA-Glb1 intensities is shown in the right graph (n = 8); the number (n) of animals is for each genotype and phenotype. Error bars represent the mean ± SD, *P < 0.005, **P < 0.001, ***P < 0.0005. (F) Comparisons of survival rates between Spns1-deficient and double Spns1- and Atp6v0ca-deficient animals (log rank test: χ2 = 54.16 on one degree of freedom; P < 0.0001).
To generate double-homozygous mutant animals for both of the genes, we extended the CRISPR/Cas9-mediated genome editing approach to the atp6v0ca gene in spns1 mutant fish. The Cas9 mRNA and the atp6v0ca-specific single guide (sg) RNA were both injected into embryos of the Tg(CMV:EGFP-lc3b);spns1hi891/+ fish line. We then identified a targeted atp6v0ca mutant allele that occurred in the same chromosome with the spns1 mutant allele (cis allele) by crossing the existing trans-heterozygous animals for the 2 genes (Fig. 4B, middle). DNA sequencing of the targeted genome region demonstrated that there was a specific nucleotide deletion, which generated a premature stop codon in the atp6v0ca gene (Fig. 4C and Fig. S3). We also confirmed that the occurrence of the hypopigmented atp6v0ca-mutant phenotype was apparently consistent with the expected Mendelian inheritance ratio at 48 hpf when the resulting offspring were intercrossed (Fig. 4D and Table S1). Subsequently, yolk opaqueness gradually and slowly appeared only in the hypopigmented embryos, approaching 100% of by 84 hpf (Fig. 4D and Table S1), suggesting that our CRISPR/Cas9-mediated targeting successfully disrupted atp6v0ca within the same cis-allele of spns1hi891 (cis-spns1hi891/hi891;atp6v0cacm/cm); the two phenotypes (yolk opaqueness and hypopigmentation) were only displayed together and not separately in individual fish.
Hypopigmented homozygous cis-spns1hi891/hi891;atp6v0cacm/cm-mutant embryos were sorted according to their severity of yolk opaqueness in the 3 different categories (NOR, POY and MOY phenotypes), and there was apparent suppression of severe yolk opaqueness (MOY), compared with that in single spns1hi891/hi891 mutants (Fig. 4D). Similar to the results with morpholino or chemical inhibition of the v-ATPase, SA-Glb1 activity induced by disruption of each gene individually was counteracted in the double homozygous mutants with the MOY rather than the POY phenotype (Fig. 4E). Note that these homozygous mutant fish have been generated by intercrossing heterozygotes and accordingly still have maternally derived mRNA for each gene (see below); the loss of such mRNA is stochastic, which may affect the presence of the differing MOY versus POY phenotypes particularly at the early developmental stage. However, longitudinally the concurrent double defect of both spns1 and atp6v0ca strikingly diminished the progression of yolk opaqueness seen with the spns1 mutation and did not show a severe MOY phenotype (Fig. 4D). In fact, the double-defect animals produced an increased number of viable larvae that survived beyond 72 hpf (Fig. 4F). These results indicate that the original mutant phenotypes of the atp6v0ca and spns1 defects (i.e., hypopigmentation and yolk opaqueness) counteracted, although not completely, under the heritable genetic condition.
Tipping the balance between the v-ATPase and Spns1 for lysosomal biogenesis and autophagy
During their development the cis-double-mutant fish embryos showed an apparent reduction in SA-Glb1 activity along with the advance of yolk opaqueness (Fig. 4E). That is, the stronger the defect in Spns1 function (and increased opacity), the greater the suppression of the senescence phenotype due to the disruption (mutation) of atp6v0ca. However, as we still found premature autolysosomal fusion even in the absence of Atp6v0ca (the increase in the merged yellow signal in Fig. 3A), prevention of autolysosomal fusion does not appear to be the reason for the suppression of the Spns1 deficiency phenotype. As noted above, autophagy is involved in early development, utilizing the yolk to supply nutrients, and is dependent on proper lysosomal function. To investigate a potential role of autophagy in the double-mutant phenotype, we further observed the autophagic process of the cis-double mutant embryos through monitoring EGFP-Lc3B and LysoTracker Red. By utilizing Tg(CMV:EGFP-lc3b) animals carrying spns1hi891/hi891, atp6v0cacm/cm or double cis-spns1hi891/hi891;atp6v0cacm/cm mutations, concomitant LysoTracker Red staining revealed significantly reduced numbers of intracellular yellow (both GFP/green- and LysoTracker/red-positive) puncta that indicated fewer acidic autolysosomal compartments in cis-spns1hi891/hi891;atp6v0cacm/cm fish embryos compared with spns1hi891/hi891 (Fig. 5A). There was a larger number of yellow puncta in cis-spns1hi891/hi891;atp6v0cacm/cm fish embryos with MOY than those with POY phenotypes. By contrast, as expected, increased EGFP-Lc3B puncta but basically normal (less visible) LysoTracker Red signals were detected in atp6v0ca-mutant animals (Fig. 5A) Thus, the defect in lysosomal acidification resulting from the defect in v-ATPase activity can suppress the loss of Spns1 function, and the extent of the suppression correlated with the degree of yolk opacity due to the Spns1 deficiency.
Figure 5.
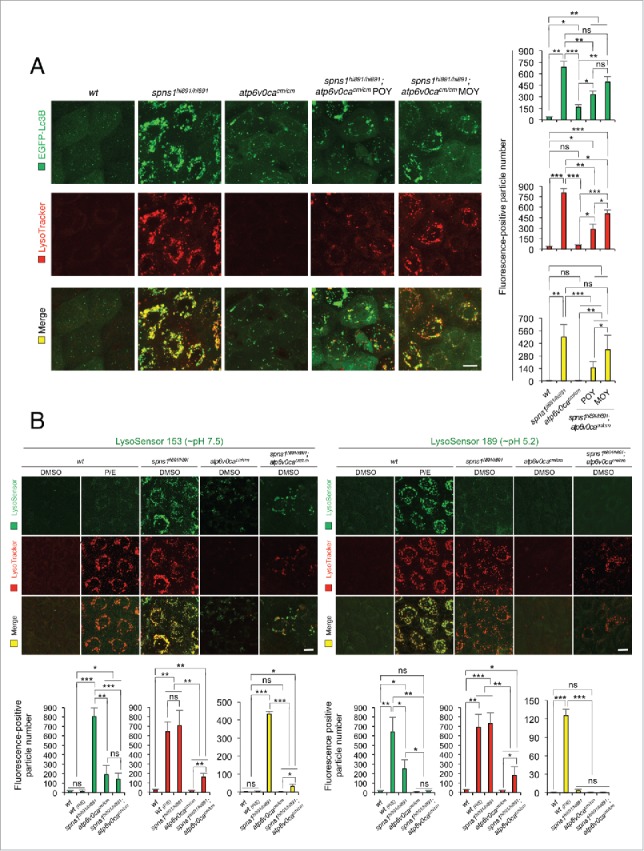
Autolysosomal biogenesis with the concurrent defect of both Atp6v0ca and Spns1 in zebrafish. (A) Subsequent formation of aberrant premature autolysosomes with concurrent deficiency of both Atp6v0ca (atp6v0cacm/cm) and Spns1 (spns1hi891/hi891) in double-homozygous mutant zebrafish (atp6v0cacm/cm;spns1hi891/hi891). Scale bar: 10 µm. Quantification of the EGFP (green), LysoTracker (red) and merged (yellow) fluorescence-positive particle numbers is shown in the right graph (the number of animals for each genotype and phenotype = 9). Three independent areas (periderm or basal epidermal cells above the eye) were selected from individual animals. Error bars represent the mean ± SD, *P < 0.005, **P < 0.001, ***P < 0.0005; ns, not significant. Three independent areas (periderm or basal epidermal cells above the eye) were selected from individual animals. (B) Acidity-dependent autolysosomal biogenesis is rate limiting in spns1- and atp6v0ca-mutant animals; intracellular constituents still have insufficient acidity in the double spns1;atp6v0ca mutants. Using 2 different acidic-sensitive probes, LysoSensor Green 189 and neutral-sensitive LysoSensor Green 153 (green), in combination with LysoTracker Red (red), WT and spns1-mutant animals showed detectable signals when stained at 72 hpf. In spns1-mutant animals, autolysosomal and/or lysosomal compartments were more prominently detectable by LysoSensor Green 153 than by LysoSensor Green 189, at the cellular level with enhanced signal intensity of these enlarged compartments. In stark contrast, the cellular compartments in WT fish treated with pepstatin A and E-64-d (P/E) (12-h treatment from 60 hpf through 72 hpf) were more prominently detectable by LysoSensor Green 189 than by LysoSensor Green 153 under the identical LysoTracker-staining conditions. Scale bar: 10 µm. Quantification of the LysoSensor (green), LysoTracker (red) and merged (yellow) fluorescence-positive particle is shown in the right graph (the number (n) of animals for each genotype and phenotype = 9). Three independent areas (periderm or basal epidermal cells above the eye) were selected from individual animals. Error bars represent the mean ± SD, *P < 0.005, **P < 0.001, ***P < 0.0005; ns, not significant. Three independent areas (periderm or basal epidermal cells above the eye) were selected from individual animals.
We further confirmed the counteractive effect of the 2 mutations on autolysosome formation by generating 2 additional double-transgenic zebrafish; Tg(CMV:EGFP-lc3b;eef1a1l1:mCherry-lamp1) and Tg(CMV:EGFP-lc3b; eef1a1l1:mCherry-sqstm1/p62) with the spns1hi891/hi891 or cis-spns1hi891/hi891;atp6v0cacm/cm backgrounds. Lamp1 as a lysosomal marker and Sqstm1/p62 (sequestosome 1) as an autophagy receptor and substrate were found extensively colocalized with Lc3B in puncta of spns1hi891/hi891 fish (Fig. S4A and B), whereas such puncta were significantly diminished in fish with the cis-spns1hi891/hi891;atp6v0cacm/cm genotype. These results indicate that there is apparently a counteractive relationship between the combined genetic defects to suppress the aberrant autolysosome biogenesis in the spns1hi891/hi891;atp6v0cacm/cm-double mutants, compared with the spns1hi891/hi891-single mutants. This observation also leads us to propose that an explanation for the decrease in SA-Glb1 activity in the double mutants may reflect partially restored autolysosomal catabolism or alternatively an inability to retain the enzyme due to increasing cellular catastrophe.
To extend our analysis we utilized the LysoSensor Green dye to monitor acidification levels in lysosomes and autolysosomes; this would allow us to determine whether the suppression of the spns1-mutant phenotypes by the atp6v0ca deficiency could be due to prealkalinization or neutralization of the lysosomes. In contrast to the LysoTracker probes, which exhibit fluorescence that is largely independent of the precise pH level (i.e., LysoTracker Red accumulates in any acidified compartment), the LysoSensor reagents can show a pH-dependent increase in fluorescence intensity upon acidification.7 The neutral pH-sensitive LysoSensor Green 153 fluoresces optimally at pH 7.5, whereas the acidic pH-sensitive LysoSensor Green 189 fluoresces optimally at pH 5.2. When these probes (green) were used in combination with LysoTracker Red, we found a much stronger signal with LysoSensor Green 153 (pH 7.5) than with LysoSensor Green 189 (pH 5.2) at the cellular level in spns1-mutant animals (Fig. 5B; yellow bar). By contrast, treatment of WT animals with lysosomal protease inhibitors, pepstatin A and E-64-d (P/E), which allows the retention of intact autolysosomal or lysosomal acidity while preventing autolysosomal maturation and turnover, showed highly acidic vesicles stained by LysoSensor Green 189 (pH 5.2), rather than by LysoSensor Green 153 (pH 7.5) (Fig. 5B). While the neutral LysoSensor Green 153 (pH 7.5) signal became lower in double cis-spns1hi891/hi891;atp6v0cacm/cm fish compared with spns1hi891/hi891 fish, the acidic LysoSensor Green 189 (pH 5.2) signal was not restored in the double mutants (Fig. 5B). Thus, spns1 mutant fish showed a dramatic increase in lysosomes (e.g., mCherry-Lamp1 positive as in Fig. 3A) compare with WT fish, but these compartments were not properly acidified; however, the acidification defect was partially corrected (i.e., there was a reduction in neutral pH detected by LysoSensor Green 153) by a subsequent disruption of the v-ATPase V0ca (also refer to Fig. S6A). We do not know the reason for the partial suppression of the pH defect that accompanies loss of v-ATPase activity; however, there are several other permeases in the lysosome that may be able to exchange protons with other ions.21 Collectively, these results demonstrate that the appearance of deleterious changes with aberrant autophagic progression in spns1 animals due to impaired or suboptimal acidification in malformed autolysosomes was temporarily attenuated by the v-ATPase defect; however, the defect in a subsequently essential process resulting in both lysosomal and autolysosomal pathogenesis in Spin1 deficiency was not corrected.
Next, by performing quantitative RT-PCR (qRT-PCR), we checked and compared the gene (mRNA) expression of senescence-associated biomarkers including the glb1 gene (that could correspond to SA-Glb1 activity).25-28 First, in spns1hi891/hi891 fish embryos, our qRT-PCR experiments showed that the expression of glb1 was more than 2-fold enhanced, and serpine1/pai-1 expression, which can also be elevated in cellular and organismal senescence,29-31 became more than 3-fold through 8-fold higher, with the advancing opaque yolk phenotype (Fig. 6A and B). Second, in hypopigmented atp6v0cacm/cm embryos, the glb1 and serpine1 expression were more than 1.5-fold and 3-fold higher, respectively, than that seen in normal-phenotype animals (Fig. 6A and B). We then examined the dual cis-spns1hi891/hi891;atp6v0cacm/cm-mutant animals with yolk opaqueness and found that expression of both glb1 and serpine1 was suppressed relative to those in spns1hi891/hi891 mutants, and were fairly similar to those in atp6v0cacm/cm mutants (Fig. 6A and B). Surprisingly, the glb1 (and serpine1) mRNA level was higher in the MOY double-mutant zebrafish compare with the POY counterparts even though SA-Glb1 activity was reduced (Fig. 4E), indicating that the transcript levels were not directly comparable to the enzyme activity.
Figure 6.

Gene expression analyses of senescence markers and/or mediators (glb1, serpine1 and rgn) in WT, spns1hi891/hi891 and spns1hi891/hi891;atp6v0cacm/cm (POY and MOY), and atp6v0cacm/cm (Mu; mutant) embryos from intercrosses (inx) of spns1hi891/+fish and spns1hi891/+;atp6v0cacm/+ fish at 72 hpf. Data are mean ±SD (n = 4 samples [3 embryos/sample] per genotype). Error bars represent the mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.005; ns, not significant.
In contrast to glb1 and serpine1, rgn/smp30 (regucalcin) is normally downregulated during senescence.,7,32-34 However, the reduction of rgn/smp30 seen in the spns1hi891/hi891 mutant was not suppressed in cis-spns1hi891/hi891;atp6v0cacm/cm double-mutant animals; there was only moderate or merginal recovery of the rgn expression in cis-spns1hi891/hi891;atp6v0cacm/cm mutants, compared with spns1hi891/hi891 mutants, but not when compared with atp6v0cacm/cm fish (Fig. 6C). These results indicate that some senescence aspects, but not others, represented by these biomarkers are consistently suppressed in the double-mutant animals, at least for the gene expression levels, when they are compared with spns1hi891/hi891 mutants.
Developmental life-span models for regulation by the balance between v-ATPase and Spns1
While permanent disruption by the mutation of atp6v0ca could extend the original survival or life span of Spns1-mutant zebrafish embryos and larvae (Fig. 4F), it was less effective for life-span extension compared with the MO-based transient knockdown of Atp6v0ca in Spns1-defective animals (Fig. 1D). Therefore, we assumed that timing for suppression of v-ATPase activity could be critical and essential for life-span control with regard to Spns1 deficiency.
The heritable cis-double zygotic defect condition of Spns1 and Atp6v0ca could still be affected by maternal RNAs derived from the egg stochastically, while displaying both mutant phenotypes (yolk opaqueness and hypopigmentation) zygotically. Initial attenuation of either mutant phenotype may potentially depend on penetrance, affecting the amounts and balance of such maternal RNAs. For the purpose of shutoff of Atp6v0ca throughout the developmental process, we knocked down the maternal atp6v0ca RNA in cis-spns1hi891/hi891;atp6v0cacm/cm mutants, by using the 2 different translation-block MOs (MO1 and MO2) (Fig. 7A and B; MO1, and Fig. S5A and B; MO2).7,19,20 We found that further knockdown of atp6v0ca by the MOs was more effective in suppressing the opaque yolk phenotype in cis-spns1hi891/hi891;atp6v0cacm/cm animals compare with the single spns1hi891/hi891 mutant (Fig. 7A and B, and Fig. S5A and B), whereas it was less effective in extending the life span of the cis-double mutants (Fig. 7C and D, and Fig. S5C and D). In fact, the survival ratio was better for the single spns1hi891/hi891 mutant injected with MOs compare with the cis-spns1hi891/hi891;atp6v0cacm/cm double mutant similarly injected with the MOs (Fig. 7C to E, and Fig. S5C and D), suggesting an effect of the maternal WT atp6v0ca RNA that apparently interferes with the suppressive effect of the atp6v0cacm/cm mutation introduced by mating heterozygous individuals.
Figure 7.

Survival control by balancing the deficiency between Spns1 and Atp6v0ca. (A) Appearance of an opaque-yolk phenotype (NOR; normal, POY; partially opaque yolk, MOY; mostly opaque yolk) in embryonic and larval spns1hi891/hi891 fish with the impact of atp6v0ca MO1. (B) Appearance of an opaque-yolk phenotype in embryonic and larval spns1hi891/hi891;atp6v0cacm/cm fish with the impact of atp6v0ca MO1. (C) Survival of embryonic and larval spns1hi891/hi891 fish with the impact of atp6v0ca MO1 (log rank test: χ2 = 139.9 on one degree of freedom; P < 0.0001). Inset shows Gompertz curves from survival data of embryo and larval zebrafish. (D) Survival of embryonic and larval spns1hi891/hi891;atp6v0cacm/cm fish with the impact of atp6v0ca MO1 (log rank test: χ2 = 56.43 on one degree of freedom; P < 0.0001). Inset shows Gompertz curves from survival data of embryo and larval zebrafish. (E) Comparisons of survival rates between Spns1-deficient and double Spns1- and Atp6v0ca-deficient animals with or without atp6v0ca MO (2 groups with atp6v0ca MO1; log rank test: χ2 = 7.735 on one degree of freedom; P < 0.0054, 4 groups with and without atp6v0ca MO1; log rank test: χ2 = 233.3 on 3 degrees of freedom; P < 0.0001).
We tested whether the increase in survival or life span that occurs in the spns1 mutant zebrafish embryos or larvae with MO-based transient inactivation of atp6v0ca was the result of a change in the rate of developmental survival or initial mortality rate (IMR), analogous to a Gompertz analysis of typical mortality data. Through this analysis, we found that the more robust inactivation of atp6v0ca by MO1 in spns1hi891/hi891-intercrossed embryos resulted in a dramatic 2.2-fold reduction in the slope, which is often referred to as the rate of aging (P < 0.01), with a negligible change in initial mortality rate (P = 0.536, the intercept) (Table 1 and 2, and Figure 7C, inset and Figure S5C). Similarly, the weaker MO2 inactivation of atp6v0ca had a modest yet significant (P < 0.01) reduction in the rate of aging without changing the IMR (P = 0.841). Additionally, both atp6v0ca MOs in spns1hi891/hi891;atp6v0cacm/cm animals also significantly (P < 0.01) reduced the rate of aging without altering the IMR, despite the fact that a less dramatic increase in life span was observed (Table 1 and 2, and Figure 7D, inset and Figure S5D). This finding is consistent with the notion that the increased survival with atp6v0ca MO in conjunction with the loss of spns1, or the combination of the spns1 and atp6v0ca mutations might be the result of delayed developmental senescence rather than any impact of initial mortality (which is age-independent, in this case developmental age). Taken together, these results suggest that timing of the gene regulation between spns1 and atp6v0ca can differentially influence biological phenotypes and animal survival during the developmental process.
Table 1.
Gompertz analysis and fit parameters based on the survival curve and mortality rate of the genetic background and condition.
| Fit parameters |
||||||
|---|---|---|---|---|---|---|
| Genetic background and condition | Abbreviation | IMR/G | G | IMR | Median | logIMR |
| spns1hi891/hi891 (n = 94) | spns1 | 0.00434 | 0.06081 | 0.00026 | 83.5 | −8.24003 |
| spns1hi891/hi891 + atp6v0ca MO 1 (n = 103) | spns1.atpMO1 | 0.01781 | 0.02768 | 0.00049 | 133.2 | −7.61489 |
| spns1hi891/hi891 + atp6v0ca MO 2 (n = 78) | spns1.atpMO2 | 0.00452 | 0.04505 | 0.00020 | 111.9 | −8.49901 |
| spns1hi891/hi891;atp6v0cacm/cm (n = 84) | spns1.atpcm | 0.00012 | 0.08902 | 0.00001 | 97.5 | −11.46625 |
| spns1hi891/hi891;atp6v0cacm/cm + atp6v0ca MO 1 (n = 51) | spns1.atpcm.atpMO1 | 0.00178 | 0.05018 | 0.00009 | 118.9 | −9.32280 |
| spns1hi891/hi891;atp6v0cacm/cm + atp6v0ca MO 2 (n = 54) | spns1.atpcm.atpMO2 | 0.00232 | 0.05381 | 0.00012 | 106.0 | −8.99050 |
IMR/G; Convenient parameter for curve fitting, G; Gompertz slope, IMR; Gompertz intercept “Initial Mortality Rate,” Median; Time (hours) of 50% survival, logIMR; the y-intercept on Force of Mortality plots. The values were derived from the survival curves.
Table 2.
P values for differences in parameters between the 2 different compared conditions.
| Two conditions compared |
P values for differences in parameters |
|||||
|---|---|---|---|---|---|---|
| Condition 1 | Condition 2 | IMR/G | G | IMR | Median | logIMR |
| spns1 | spns1.atpMO1 | 0.584 | 0 | 0.536 | 0 | 0.007 |
| spns1 | spns1.atpMO2 | 0.988 | 0.003 | 0.841 | 0 | 0.631 |
| spns1.atpcm | spns1.atpcm.atpMO1 | 0.699 | 0.004 | 0.633 | 0 | 0.038 |
| spns1.atpcm | spns1.atpcm.atpMO2 | 0.186 | 0.001 | 0.176 | 0.002 | 0.009 |
Discussion
Our current study demonstrates for the first time that the heritable genetic impact of loss of both Spns1 and v-ATPase can be counteractive, resulting in suppression of the pathogenesis related to autophagy and senescence during development, and able to extend organismal survival (larval life span) at the end point. Thus, Spns1 and v-ATPase play critical roles in developmental senescence as well as autolysosomal homeostasis, as a proper balancing control of acidification via Spns1 and v-ATPase is necessary for normal biogenesis and function in the hybrid organelles generated by fusion of lysosomes (or endosomes) and autophagosomes (Fig. S6B).
The v-ATPase has been suggested to promote lysosomal fusion with endocytic and autophagic vesicles.35,36 However, we also demonstrated here that Atp6v0ca-deficient lysosomes and/or possible endosomes in zebrafish remain at least partially competent to fuse with autophagosomes, resulting in a time-dependent formation of enlarged autolysosomes, which are still smaller than the ones generated by Spns1 deficiency. Several lines of recent evidence actually support the notion that the v-ATPase is not essential for autophagosome-lysosome fusion.11-13 Enhanced Lc3 and Lamp1 colocalization following either knockdown or knockout of atp6v0ca, as well as of spns1, suggests that autophagic flux was inhibited primarily due to improper autolysosomal degradation rather than by a simple block in vesicular fusion. Nevertheless, Spns1 deficiency that also inhibits autophagic flux can be rescued by the loss or decrease of Atp6v0ca partially but significantly.
It should be noted that the most versatile and widely used senescence marker SA-Glb1 is a lysosomal glycoside hydrolase (an acid β-D-galactosidase), which catalyzes the hydrolysis of β-galactosides into monosaccharides,37 and its substrates also include various glycoproteins, gangliosides (glycosphingolipids), lactose, and lactosylceramidases.38,39 The aberrant increase of the in situ SA-Glb1 activity may indicate that such a hydrolase product itself can still be preserved in autolysosomes and/or lysosomes, but might not function properly in vivo when there is an unbalanced condition for the v-ATPase and an essential permease(s) such as Spns1, which are needed to allow the digestion of substrates (via proper activation of lysosomal hydrolases including glycosidases) and the subsequent transport of the degradation products into the cytoplasm as energy sources. Therefore, given the defect of Spin/Spns1, the breakdown products from a certain lysosomal carbohydrate(s) digested by such glycosidases, including Glb1, cannot be exported from the lumen into the cytoplasm, which could subsequently lead to the accumulation of enlarged autolysosomes. Presumably, the Glb1 induction, as a senescence marker, reflects a certain adaptive response to execute hydrolysis of various accumulated glycosylated substrates.
spns1hi891/hi891 animals injected with atp6v0ca MO as well as co-injected morphants with both spns1 MO and atp6v0ca MO showed a correlation in counteractive effect between their representative morphological phenotypes (yolk opaqueness by the spns1 defect and hypopigmentation by the atp6v0ca defect) and SA-Glb1 activity.7 Thus, we propose that such counteraction of the 2 different abnormal phenotypes could reduce SA-Glb1 activity. By contrast, in cis-spns1hi891/hi891;atp6v0cacm/cm animals (Fig. 4E), as well as atp6v0cahi1207/hi1207 zebrafish injected with spns1 MO (Fig. 2C), the counteractive effect of the 2 different phenotypes was not merely correlative with the SA-Glb1 levels but depended on the strength of the yolk opacity phenotype. Thus, hypopigmented MOY fish, which concurrently strongly showed both the mutant hallmarks, displayed lower SA-Glb1 activity than hypopigmented POY fish (Fig. 4E), even though the glb1 gene expression was higher in the hypopigmented MOY fish than hypopigmented POY fish (Fig. 6A), and the expression levels were similar to those in sole atp6v0cacm/cm mutants. We suspect that posttranscriptional regulation of the glb1 gene could explain this discrepancy. We also propose that the initial timing for the suppression of the atp6v0ca gene expression by the MO was essential in this regard because maternal RNA of atp6v0ca could influence the vulnerability of the Spns1-deficient animals later on during development.
Perpetual restoration of survival in Spns1 deficiency by the atp6v0ca depletion or suppression was essentially impossible, whereas a significant extension of temporal life span in the dual-deficit animals was robustly confirmed in our current model system. We propose that this implies the possibility to develop a simplified life-span model through developmental senescence by epistasis. We can hypothetically apply the idea for genetic schemes in haploinsufficient (or heterozygote-advantageous) adult animals with advancing age because many genetic or environmental manipulations that affect life span in organisms also affect survival following chemical and genetic stresses during development.4,40 For instance, whereas heterozygous spns1 adult fish showed accelerated aging and shortened life span, double heterozygosity of both spns1 and atp6v0ca might attenuate the symptoms observed in either single-heterozygote animals in adulthood. The potential coupling of developmental senescence and biological aging mechanisms may prove a useful tool in identifying new genes that affect the aging process without the need for performing lengthy adult life-span analyses. Therefore, it is likely that this approach may also be applied to the high-throughput identification of pharmacological agents that control aging and life span through enhanced resistance to various stressors including oxygen radicals. Moreover, cumulative lines of evidence are consistent with the point of view that cellular senescence might evolve to optimize embryogenesis, and that its beneficial postnatal functions, to balance tumor suppression and tissue repair or regeneration, could arise and be sustained later in evolution.1-6 We expect that our approach of analyzing evolutionary developmental mechanisms of senescence in zebrafish will accelerate the discovery of new pharmacological interventions in aging, stress-mediated disorders and cancers.
Having consistent outcomes for rescuing the Spns1-mutant phenotypes by v-ATPase suppression, our chemical- and genetic-based studies could provide insights into the relevance of particular molecular targets and functions of chemical compounds. Our findings suggest that the effect of the genetic atp6v0ca depletion on lysosomal pH plays a limited role in fusion for preventing the Spns1 defect, but apparently postpones the actual progression of the deteriorative event. This counteractive impact is theoretically being considered for a substrate reduction strategy to treat lysosomal storage diseases such as Pompe disease,4142 and may also be applicable to other age-associated degenerative diseases linked to autophagic inability at the late stages. Associated disorders could result from an imbalance in the capacity of the lysosomal compartment to degrade substrates. Substrate reduction therapy addresses the pathological imbalance between the rate of synthesis of committed precursors of cellular macromolecules and their rate of breakdown in situ. Continuously prolonged suppression of both the Spns1 and v-ATPase molecules would not be relevant, but imbalanced downregulation of one could be treated by a temporal suppression of the other. The use of appropriate inhibitors, selective for key steps in the biosynthesis of cellular macromolecules in general, may restore normal dynamics in the autolysosomal compartment and correct the pathological storage that is the ultimate cause of the disease phenotype. Since Spns1 is a symporter, it presumably has 2 different substrates and solutes; one is a proton and the other is hypothetically a carbohydrate. This fact highlights the need to fully consider the effect(s) of both substrates (not only the proton) when developing novel interventions by targeting Spns1 and the v-ATPase.
Materials and methods
Zebrafish maintenance and ethics
Zebrafish (AB and casper strains) were maintained under a 14:10 h light/dark cycle and fed living brine shrimp twice daily. Brine shrimp were presented using 1-mL pipettes (approximately 0.75 mL brine shrimp per 20 fish). Flake food was also given semiquantitatively every other day in accordance with the number of fish in the tanks. A continuously cycling aquarium system was used to maintain water quality (Aquatic Habitats Inc.). Zebrafish embryos were collected from pairwise matings of adults and raised at 28.5°C. The embryos to be used in the experiments were then staged by hpf at 28.5°C and also by morphological criteria for experiments.43 All animal experiments were approved by and conducted in accordance with the guidelines established by the Institutional Animal Care and Use Committee (IACUC) at The Scripps Research Institute, IACUC approval numbers 15–008 and 14–035.
Confocal microscopy and imaging
Microscopy and imaging were performed essentially as described previously.7 Zebrafish embryos (in the case of the AB fish line) were transferred into 0.003% (w/v) 1-phenyl-2-thiourea (Sigma, P7629) prior to 24 hpf to prevent pigmentation. Embryos or larvae were then mounted live in water containing 0.16 mg/ml tricaine (Sigma, A5040) for imaging. Images were taken using the FluoView™ FV1000 confocal laser scanning microscope system (Olympus, NY, USA) with a 60x objective lens). The settings used to capture the images were standardized by adjusting sensitivity (of HV with gain value as 1) and reducing noise (by offset), in addition to standardizing the threshold values. We also made sure that the images were not oversaturated by controlling the original capturing system and program provided by Olympus. We then examined the captured images to quantify intracellular fluorescent compartments in several different parameters (such as “positive particle number”) by using the Fiji-ImageJ programs <https://fiji.sc/> for image processing. Threshold values for standardization were set to adjust the cytosolic background and identify the more intense signals. The same threshold value was applied for the examined samples at the same time in each indicated experiment. The extent of colocalization between different fluorescent colors was quantified in 3 independent visual fields from 3 independent embryos. All values are represented as mean ± standard deviation (SD). Mounted animals were photographed using each specific fluorescent signal by confocal laser microscopy. For analyses of cells within the zebrafish embryos, these regions were selected in each actual embryo only and not in the yolk. Following pixel selection, a fuzziness setting of 64 was used, and the chosen pixel number was determined using the image histogram calculation.
Morpholino oligonucleotides
Morpholino oligonucleotides were designed and synthesized by Gene Tools, LLC (Philomath, OR). The sequences of the atp6v0ca MO1 and MO2 are 5′- GTTCGGGAGACTAAAACTAAAGGCA-3′ and 5′-CTTCTTTTGGCTGACAGGTCTTGCA-3′, respectively, which bound to the 5′-untranslated regions of zebrafish gene atp6v0ca mRNA, blocking translation.7,19,20 The spns1 MO sequence 5′-ATCTGCTTGTGACATCACTGCTGGA-3′ overlaps the 5′ untranslated region-exon boundary at the 5′-splice junction of exon 1 in the zebrafish spns1 gene. The sequence of the standard control MO is 5′-CCTCTTACCTCAGTTACAATTTATA-3′. MOs were resuspended in sterile water at a concentration of 1 mM as stock solutions. For microinjection into embryos, the stock solutions were diluted to 125, 250, 500, and 750 µM. A 10 nl volume of each MO solution was injected into the yolk during the one-cell stage. All other MO sequences have been reported previously.4,15,19,44
Western blotting
Embryos were dechorionated, deyolked and homogenized in RIPA buffer. Protein concentrations of embryo lysates were determined using the bicinchoninic acid (BCA) protein assay. The lysates were mixed with equal volumes of 2× SDS sample buffer, heated at 95°C for 2 min, and resolved on 12.5% or 15% gels. After transfer, the polyvinylidene difluoride membranes were incubated with primary antibodies (anti-Lc3A/B [Cell Signaling Technology, Inc., 4108], anti-ACTB/β-actin [Cell Signaling Technology, 4967], or anti-GFP [Life Technologies, A11122]), diluted in TBST (50 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH 7.5 [Fisher, 28360]) overnight at 4°C. After washing, the blot was then incubated with a secondary anti-rabbit horseradish peroxidase-conjugated antibody (Cell Signaling Technology, 7074) at room temperature for 1 h and visualized using an Western Lightning® Plus-ECL (Perkin Elmer, NEL103E001EA) in accordance with the manufacturer's instructions.
Generation of the spns1hi891/+;atp6v0cacm/+ cis-heterozygote fish line using the CRISPR/Cas9 system
For generating the atp6v0ca specific gRNA, the 20-base pair (bp) coding sequence 5′-GGGACTGCTAAGAGCGGCAC-3′ (Exon 2) was cloned into the pT7-gRNA vector (Addgene, plasmid 46759, deposited by Dr. Wenbiao Chen) through the PCR cloning method.45,46 Primers and their sequences are shown in Table S2. The resulting plasmid was digested with BamHI, and atp6v0ca gRNA was transcribed by T7 RNA polymerase in vitro, using the Ambion mMESSAGE mMACHINE T7 Kit (Ambion, AM1344). For making Cas9 RNA, pT3TS-nCas9n (Addgene plasmid 46757, deposited by Dr. Wenbiao Chen) vector was linearized by XbaI digestion and Cas9 RNA was synthesized using the T3 Kit (Ambion, AM1348). After transcription, the gRNA (∼100 bp) and Cas9 RNA (∼4500 bp) were cleaned up using an RNeasy® Mini Kit (QIAGEN, 74104), and the quality of the RNA was confirmed by electrophoresis through a 1.5% agarose gel. Subsequently, the atp6v0ca gRNA (100 ng/μl) and Cas9 RNA (100 ng/μl) were co-injected into one-cell-stage embryos. The injected embryos were raised to adulthood and outcrossed to WT fish. The F1 embryo genomic DNA was extracted and confirmed by T7 endonuclease I assay as well as a high-resolution agarose gel (3%) to confirm the germ line was positively transmitted. Primers and their sequences are shown in Table S2. The DNA fragment amplified for T7E1 assays was purified using EXOSAP-IT™ (Affymetrix Usb, AF78200200) and sent for sequencing (GENEWIZ, South Plainfield, NJ, USA).
Identification of cis-heterozygote spns1hi891/+;atp6v0cacm/+ zebrafish
To search for “cis”-heterozygote animals that have both atp6v0ca and spns1 mutations on the same side of chromosome 3, putatively “positive” founders (F0), of which their germ cells are still mosaic in the targeted atp6v0ca mutation(s), were identified by crossing with atp6v0ca- and spns1-double “trans”-heterozygote animals (Fig. 4B, left and middle). For offspring that had a hypopigmented phenotype, presumably due to the atp6v0ca defect, the hypopigmented embryos (∼50 embryos) were genotyped for the spns1 mutation. If they had the spns1 mutation, as well as atp6v0ca mutation, at least the founder would have/contain the cis-condition germ cell(s) for both the atp6v0ca and spns1 genes (Fig. 4B, middle), and they were raised as F1 generations. Next, we performed individual outcrosses of F1 candidates with atp6v0ca- and spns1-double “trans”-heterozygote animals. We performed the procedure for the identification of the hypopigmented embryos (∼50 embryos) genotyped for the spns1 mutation. Then, in addition, if one of the candidates of the F1 cross existed in the cis condition, subsequently surviving healthy offspring (without yolk opaqueness and with pigmentation) should be solely either atp6v0ca or spns1 heterozygotes (Fig. 4B, middle). By contrast, if the candidates were still in the trans condition, the normal offspring should be all in the trans-heterozygote condition by genotyping (Fig. 4B, left).
Lysotracker red and lysosensor green staining
The vital lysosomal dyes were diluted to final concentrations as follows: LysoTracker Red DND-99 (Invitrogen/Molecular Probes, L7528), 1 μM; LysoSensor Green DND-189 (Invitrogen/Molecular Probes, L7535), 10 μM; and LysoSensor Green DND-153 (Invitrogen/Molecular Probes, L7534), 1 μM, in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4), and prewarmed to 28.5°C. Each dye was then added to an equal volume of fresh water on embryos and incubated at 28.5°C in the dark for 30 min to 1 h. Embryos were then rinsed 4 times in fresh E3 medium before imaging.
RNA isolation and RT-PCR analysis for zebrafish glb1, serpine1/pai-1 and rgn/smp30
qRT-PCR analysis of zebrafish embryos was performed to check the mRNA level for the zebrafish glb1, serpine1 and rgn genes using the StepOnePlus Real-Time PCR system (Applied Biosystems, CA, USA). Total RNA was extracted from 72 hpf embryos of different groups, using the TRIzol reagent according to the manufacturer's protocol (Invitrogen, 15596026). Then, double-stranded cDNA was synthesized using M-MLV reverse transcriptase (Promega, M1705), followed by qPCR with 2x SYBR-Green PCR Master Mix (Applied Biosystems, Foster City, CA). In each qRT-PCR experiment, pools of three embryos were run in quadruplicate. PCR primers used to amplify the fragments of the zebrafish genes were designed using a web-based primer design tool, PrimerQuest (Integrated DNA Technology, Inc.): -Dr glb1 EX1 forward primer; 5′-ACTAGTCTCGGTTCTGCTCCTTATC-3′, Dr glb1 EX2 reverse primer; 5′-ATCTTGAGTAGCCGGTCTTTCC-3′; Dr serpine1 EX2-3 forward primer; 5′-AAGAAAGAGGAATGCCCAAA-3′, Dr serpine1 EX3 reverse primer; 5′-GATCTTCCGGTCAACCATGA-3′; Dr rgn EX4 forward primer; 5′-ACTATGACATCCAAACTGGAGGA-3′, Dr rgn EX5 reverse primer; 5′-CTTCTGTGTCTATGCACATACCG-3′. As controls for these PCR analyses, actb/-actin was examined and total RNA from each sample was normalized. The forward and reverse primers used to amplify actb were 5′-ACTGTATTGTCTGGTGGTAC -3′ and 5′- TACTCCTGCTTGCTAATCC -3′, respectively.
SA-Glb1/β-gal assay and quantification
Zebrafish embryos and larvae at 48 to 72 hpf were washed 3 times in phosphate-buffered saline (PBS; Fisher Scientific, M6506) and fixed overnight in 4% paraformaldehyde with PBS at 4°C. After fixation, the samples were washed 3 times in PBS, pH 7.5, twice again in PBS, pH 6.0 at 4°C, and then incubated at 37°C (in the absence of CO2) for 12 to 16 h with SA-Glb1 staining solution (5 mM potassium ferricyanide [Sigma, 702587], 5 mM potassium ferrocyanide [Sigma, P3289], 2 mM MgCl2 in PBS, pH 6.0).4,7,15 All animals were photographed under the same conditions using reflected light with a macro microscope, AZ100 (Nikon, Tokyo, Japan). SA-Glb1 activity in each animal was quantified using a selection tool in Adobe Photoshop software for a color range that was chosen using 25 additive color selections of regions that showed visual SA-Glb1 staining. For analyses of embryos, these regions were selected in each embryo proper only and not in the yolk in order to exclude variability in the initial yolk volume and yolk consumption levels over time. Since the yolk stains much more intensely for SA-Glb1 at all stages of development than any other embryonic tissues in general, it was desirable to eliminate the yolk staining as a source of variability. Following pixel selection, a fuzziness setting of 14 was used, and the chosen pixel number was determined using the image histogram calculation.
Chemical treatments
Bafilomycin A1 (LC Laboratories, B-1080), omeprazole, lansoprazole, and pantoprazole (Sigma, O104, L8533, and P0021, respectively) treatment was performed for 12 h (from 36 through 48 hpf or 48 through 60 hpf) in E3 medium at 28.5˚C in 12- or 6-well plates. The chemicals dissolved in DMSO were added to the embryo water (E3 medium) at the final concentrations of 200 nM for BafA and 100 μM for omeprazole, lansoprazole and pantoprazole. Pepstatin A (Fisher BioReagent, BP26715) and E-64-d (Enzo Life Sciences, BML-PI107) treatment was administered from 60 through 72 hpf for 12 h in E3 medium at 28.5˚C in 12- or 6-well plates. These reagents were both dissolved in DMSO and added to the embryo water (E3 medium) at the final concentration of 5 μg/ml.
Generating transgenic zebrafish
To generate transgenic zebrafish expressing N-terminal mCherry-tagged zebrafish Sqstm1 and C-terminal mCherry-tagged zebrafish Lamp1, the corresponding expression construct pT2-mCherry-Sqstm1 and pT2-Lamp1-mCherry were generated by PCR with specific primers (Table S3). Briefly, the mCherry gene was cloned at BamHI and ClaI sites of the pT2AL200R150G vector,47 replacing the EGFP gene (pT2-mCherry). Total RNA from whole zebrafish embryos was prepared using the Trizol Reagent. cDNA was synthesized using M-MLV reverse transcriptase and oligo(dT) as primers. Finally, DNA encoding full-length zebrafish Sqstm1 or Lamp1 were amplified with PfuUltra II Fusion HotStart DNA Polymerase (Agilent Technologies, 600670) and subcloned into the constructed pT2-mCherry vector by the methods previously described.48 Individual clones were sequenced to completion. For making Tol2 transposase-encoding mRNA, the pCS-TP construct was linearized by digestion with NotI, and transcribed with SP6 RNA polymerase using the mMESSAGE mMACHINE kit (Ambion, AM1340) to produce Tol2 transposase mRNA. Approximately 5 nl of the mixture of plasmid DNA (100 ng/ml; pT2-mCherry-Sqstm1 or pT2-Lamp1-mCherry) and 50 pg of Tol2 transposase mRNA (100 ng/ml) were coinjected into newly fertilized embryos at the one-cell stage to produce transgenic fish. Injected embryos were raised to adulthood and out-crossed to WT fish to identify germline-transmitted transgenic founders (F0) as described previously.7,49 Positive founders were determined by screening F1 embryos for visible mCherry expression. The mCherry-positive offspring were then allowed to grow to maturity for further experiments.
Survival and life-span experiments in embryonic and larval zebrafish
Kaplan-Meier survival analyses were performed in spns1h891/h891 and double-homozygous spns1 h891/h891;atp6v0cacm/cm embryos/larvae with or without atp6v0ca MOs (MO1 and MO2). Examined sole-homozygous spns1h891/h891 and double-homozygous spns1h891/h891;atp6v0cacm/cm embryos or larvae were offspring from intercrossing of spns1h891/+ and cis-spns1h891/+;atp6v0cacm/+ parents, respectively. Therefore, these offspring had maternal mRNA for spns1 and atp6v0ca from the heterozygous females. To knock down the maternal atp6v0ca mRNA, the translation-block MOs were injected into the yolk during the one-cell stage, and subjected animals were followed for their survival every 12 h until all of them with hypopigmented and/or opaque-yolk phenotype died.
Gompertz model fits from survival data of embryonic and larval zebrafish
For each of the 4 animal conditions, we fit a 2-parameter Gompertz curve. This model fitting treated each death event as occurring at an unknown time in the interval between when the individual was last measured alive, and when the individual was found dead. From these death intervals, we fit the parameters of the Gompertz distribution to maximize the likelihood of the observed data. For testing differences between assayed populations, we used a permutation approach. For any comparison, individuals were randomly swapped, curve fitting repeated, and the difference in resampled estimated parameters compare with the observed difference.50 For each test, 10,000 permutations were sampled and fit. For each tested parameter, P values listed reflect the proportion of resampled difference that exceeds the observed difference.
Quantitative analysis and statistics
Data processing and statistical analyses were performed using Statistical Package for the Social Sciences (SPSS) version 14.0. This software was used to generate each of the graphs shown in the text to perform statistical tests where appropriate. Data are presented as mean ± SEM. Comparisons between different groups of samples and animals were made with Student t test or Log Rank test.
Supplementary Material
Abbreviations
- atp6v0ca
ATPase, H+ transporting, lysosomal, V0 subunit ca
- BafA
bafilomycin A1
- bp
base pair
- gRNA
guide RNA
- hpf
hours postfertilization
- IMR
initial mortality rate
- MO
morpholino oligonucleotide
- MOY
mostly opaque yolk
- POY
partially opaque yolk
- SA-Glb1
senescence-associated β-galactosidase
- Spns1
spinster homolog 1 (Drosophila)
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge our laboratory members for constructive discussions and providing valuable comments. We are particularly grateful to Jie Qi, Delgado Valdez, Qing Xia, Tadi Tsujita, Masa Sugiyama, Delacia Igram, Kae Sasaki, and Junko Kishi for technical assistance. Finally, we greatly thank Matt Gill for critical reading of the manuscript and valuable comments.
Funding
This work was supported by grant of GM101508 from NIGMS/NIH. DJK was supported by grant GM053396 from NIGMS/NIH.
References
- [1].Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev 2003; 17(2):201; PMID:12533509; http://dx.doi.org/ 10.1101/gad.1050003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Keyes WM, Wu Y, Vogel H, Guo X, Lowe SW, Mills AA. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev 2005; 19(17):1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al.. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 2009; 35(4):534; PMID:19716796; http://dx.doi.org/ 10.1016/j.molcel.2009.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kishi S, Bayliss PE, Uchiyama J, Koshimizu E, Qi J, Nanjappa P, Imamura S, Islam A, Neuberg D, Amsterdam A, et al.. The identification of zebrafish mutants showing alterations in senescence-associated biomarkers. PLoS Genet 2008; 4(8):e1000152; PMID:18704191; http://dx.doi.org/ 10.1371/journal.pgen.1000152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al.. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013; 155(5):1119; PMID:24238961; http://dx.doi.org/ 10.1016/j.cell.2013.10.041 [DOI] [PubMed] [Google Scholar]
- [6].Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al.. Programmed cell senescence during mammalian embryonic development. Cell 2013; 155(5):1104; PMID:24238962; http://dx.doi.org/ 10.1016/j.cell.2013.10.019 [DOI] [PubMed] [Google Scholar]
- [7].Sasaki T, Lian S, Qi J, Bayliss PE, Carr CE, Johnson JL, Guha S, Kobler P, Catz SD, Gill M, et al.. Aberrant autolysosomal regulation is linked to the induction of embryonic senescence: differential roles of Beclin 1 and p53 in vertebrate Spns1 deficiency. PLoS Genet 2014; 10(6):e1004409; PMID:24967584; http://dx.doi.org/ 10.1371/journal.pgen.1004409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dermaut B, Norga KK, Kania A, Verstreken P, Pan H, Zhou Y, Callaerts P, Bellen HJ. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J Cell Biol 2005; 170(1):127; PMID:15998804; http://dx.doi.org/ 10.1083/jcb.200412001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rong Y, McPhee CK, Deng S, Huang L, Chen L, Liu M, Tracy K, Baehrecke EH, Yu L, Lenardo MJ. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci U S A 2011; 108(19):7826; PMID:21518918; http://dx.doi.org/ 10.1073/pnas.1013800108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nakano Y, Fujitani K, Kurihara J, Ragan J, Usui-Aoki K, Shimoda L, Lukacsovich T, Suzuki K, Sezaki M, Sano Y, et al.. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol Cell Biol 2001; 21(11):3775; PMID:11340170; http://dx.doi.org/ 10.1128/MCB.21.11.3775-3788.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mangieri LR, Mader BJ, Thomas CE, Taylor CA, Luker AM, Tse TE, Huisingh C, Shacka JJ. ATP6V0C knockdown in neuroblastoma cells alters autophagy-lysosome pathway function and metabolism of proteins that accumulate in neurodegenerative disease. PLoS One 2014; 9(4):e93257; http://dx.doi.org/ 10.1371/journal.pone.0093257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kissing S, Hermsen C, Repnik U, Nesset CK, von Bargen K, Griffiths G, Ichihara A, Lee BS, Schwake M, De Brabander J, et al.. Vacuolar ATPase in phagosome-lysosome fusion. J Biol Chem 2015; 290(22):14166; PMID:25903133; http://dx.doi.org/ 10.1074/jbc.M114.628891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mauvezin C, Nagy P, Juhasz G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun 2015; 6:7007; PMID:25959678; http://dx.doi.org/ 10.1038/ncomms8007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Young RM, Marty S, Nakano Y, Wang H, Yamamoto D, Lin S, Allende ML. Zebrafish yolk-specific not really started (nrs) gene is a vertebrate homolog of the Drosophila spinster gene and is essential for embryogenesis. Dev Dyn 2002; 223(2):298; PMID:11836794; http://dx.doi.org/ 10.1002/dvdy.10060 [DOI] [PubMed] [Google Scholar]
- [15].Koshimizu E, Imamura S, Qi J, Toure J, Valdez DM, Jr., Carr CE, Hanai J, Kishi S. Embryonic senescence and laminopathies in a progeroid zebrafish model. PLoS One 2011; 6(3):e17688; PMID:21479207; http://dx.doi.org/ 10.1371/journal.pone.0017688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kishi S, Uchiyama J, Baughman AM, Goto T, Lin MC, Tsai SB. The zebrafish as a vertebrate model of functional aging and very gradual senescence. Exp Gerontol 2003; 38(7):777; PMID:12855287; http://dx.doi.org/ 10.1016/S0531-5565(03)00108-6 [DOI] [PubMed] [Google Scholar]
- [17].Tsai SB, Tucci V, Uchiyama J, Fabian NJ, Lin MC, Bayliss PE, Neuberg DS, Zhdanova IV, Kishi S. Differential effects of genotoxic stress on both concurrent body growth and gradual senescence in the adult zebrafish. Aging Cell 2007; 6(2):209; PMID:17376146; http://dx.doi.org/ 10.1111/j.1474-9726.2007.00278.x [DOI] [PubMed] [Google Scholar]
- [18].Fan X, Klein M, Flanagan-Steet HR, Steet R. Selective yolk deposition and mannose phosphorylation of lysosomal glycosidases in zebrafish. J Biol Chem 2010; 285(43):32946; PMID:20729204; http://dx.doi.org/ 10.1074/jbc.M110.158295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pickart MA, Sivasubbu S, Nielsen AL, Shriram S, King RA, Ekker SC. Functional genomics tools for the analysis of zebrafish pigment. Pigment Cell Res 2004; 17(5):461; PMID:15357832; http://dx.doi.org/ 10.1111/j.1600-0749.2004.00189.x [DOI] [PubMed] [Google Scholar]
- [20].Pickart MA, Klee EW, Nielsen AL, Sivasubbu S, Mendenhall EM, Bill BR, Chen E, Eckfeldt CE, Knowlton M, Robu ME, et al.. Genome-wide reverse genetics framework to identify novel functions of the vertebrate secretome. PLoS One 2006; 1:e104; http://dx.doi.org/ 10.1371/journal.pone.0000104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol 2012; 74:69; PMID:22335796; http://dx.doi.org/ 10.1146/annurev-physiol-012110-142317 [DOI] [PubMed] [Google Scholar]
- [22].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40(2):280; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab 2013; 18(5):617; PMID:23954639; http://dx.doi.org/ 10.1016/j.cmet.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al.. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10(6):676; PMID:18454141; http://dx.doi.org/ 10.1038/ncb1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].van Deursen JM. The role of senescent cells in ageing. Nature 2014; 509(7501):439; PMID:24848057; http://dx.doi.org/ 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153(6):1194; PMID:23746838; http://dx.doi.org/ 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192(4):547; PMID:21321098; http://dx.doi.org/ 10.1083/jcb.201009094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev 2010; 24(22):2463; PMID:21078816; http://dx.doi.org/ 10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Eren M, Boe AE, Murphy SB, Place AT, Nagpal V, Morales-Nebreda L, Urich D, Quaggin SE, Budinger GR, Mutlu GM, et al.. PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci U S A 2014; 111(19):7090; PMID:24778222; http://dx.doi.org/ 10.1073/pnas.1321942111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Elzi DJ, Lai Y, Song M, Hakala K, Weintraub ST, Shiio Y. Plasminogen activator inhibitor 1–insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc Natl Acad Sci U S A 2012; 109(30):12052; PMID:22778398; http://dx.doi.org/ 10.1073/pnas.1120437109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yamamoto K, Takeshita K, Shimokawa T, Yi H, Isobe K, Loskutoff DJ, Saito H. Plasminogen activator inhibitor-1 is a major stress-regulated gene: implications for stress-induced thrombosis in aged individuals. Proc Natl Acad Sci U S A 2002; 99(2):890; PMID:11792849; http://dx.doi.org/ 10.1073/pnas.022608799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fujisawa K, Terai S, Hirose Y, Takami T, Yamamoto N, Sakaida I. Senescence marker protein 30 (SMP30)/regucalcin (RGN) expression decreases with aging, acute liver injuries and tumors in zebrafish. Biochem Biophys Res Commun 2011; 414(2):331; PMID:21951853; http://dx.doi.org/ 10.1016/j.bbrc.2011.09.067 [DOI] [PubMed] [Google Scholar]
- [33].Fujita T, Uchida K, Maruyama N. Purification of senescence marker protein-30 (SMP30) and its androgen-independent decrease with age in the rat liver. Biochim Biophys Acta 1992; 1116(2):122; PMID:1581340; http://dx.doi.org/ 10.1016/0304-4165(92)90108-7 [DOI] [PubMed] [Google Scholar]
- [34].Yamaguchi M, Isogai M. Tissue concentration of calcium-binding protein regucalcin in rats by enzyme-linked immunoadsorbent assay. Mol Cell Biochem 1993; 122(1):65; PMID:8350865; http://dx.doi.org/ 10.1007/BF00925738 [DOI] [PubMed] [Google Scholar]
- [35].Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23(1):33; PMID:9639028; http://dx.doi.org/ 10.1247/csf.23.33 [DOI] [PubMed] [Google Scholar]
- [36].Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 1991; 266(26):17707; PMID:1832676 [PubMed] [Google Scholar]
- [37].Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006; 5(2):187; PMID:16626397; http://dx.doi.org/ 10.1111/j.1474-9726.2006.00199.x [DOI] [PubMed] [Google Scholar]
- [38].Suzuki K, Tanaka H, Yamanaka T, Van Damme O. The specificity of beta-galactosidase in the degradation of gangliosides. Adv Exp Med Biol 1980; 125:307; PMID:6767344; http://dx.doi.org/ 10.1007/978-1-4684-7844-0_28 [DOI] [PubMed] [Google Scholar]
- [39].Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 2000; 113(Pt 20):3613. [DOI] [PubMed] [Google Scholar]
- [40].Gill MS, Olsen A, Sampayo JN, Lithgow GJ. An automated high-throughput assay for survival of the nematode Caenorhabditis elegans. Free Radic Biol Med 2003; 35(6):558; PMID:12957648; http://dx.doi.org/ 10.1016/S0891-5849(03)00328-9 [DOI] [PubMed] [Google Scholar]
- [41].Douillard-Guilloux G, Raben N, Takikita S, Batista L, Caillaud C, Richard E. Modulation of glycogen synthesis by RNA interference: towards a new therapeutic approach for glycogenosis type II. Hum Mol Genet 17(24):3876; PMID:18782850; http://dx.doi.org/ 10.1093/hmg/ddn290 [DOI] [PubMed] [Google Scholar]
- [42].Clayton NP, Nelson CA, Weeden T, Taylor KM, Moreland RJ, Scheule RK, Phillips L, Leger AJ, Cheng SH, Wentworth BM. Antisense oligonucleotide-mediated suppression of muscle glycogen synthase 1 synthesis as an approach for substrate reduction therapy of pompe disease. Mol Ther Nucleic Acids 3:e206; http://dx.doi.org/ 10.1038/mtna.2014.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn 1995; 203(3):253; PMID:8589427; http://dx.doi.org/ 10.1002/aja.1002030302 [DOI] [PubMed] [Google Scholar]
- [44].Imamura S, Uchiyama J, Koshimizu E, Hanai J, Raftopoulou C, Murphey RD, Bayliss PE, Imai Y, Burns CE, Masutomi K, et al.. A non-canonical function of zebrafish telomerase reverse transcriptase is required for developmental hematopoiesis. PLoS One 2008; 3(10):e3364; PMID:18846223; http://dx.doi.org/ 10.1371/journal.pone.0003364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jao LE, Wente SR, Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci U S A 2013; 110(34):13904; http://dx.doi.org/ 10.1073/pnas.1308335110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A, Montague TG, Zimmerman S, Richter C, Schier AF. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One 2014; 9(5):e98186; http://dx.doi.org/ 10.1371/journal.pone.0098186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Urasaki A, Morvan G, Kawakami K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics 2006; 174(2):639; PMID:16959904; http://dx.doi.org/ 10.1534/genetics.106.060244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li C, Wen A, Shen B, Lu J, Huang Y, Chang Y. FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol 2011; 11:92; http://dx.doi.org/ 10.1186/1472-6750-11-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].He C, Bartholomew CR, Zhou W, Klionsky DJ. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy 2009; 5(4):520; PMID:19221467; http://dx.doi.org/ 10.4161/auto.5.4.7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dwass M. Modified randomization tests for nonparametric hypotheses. Ann Math Stat 1957; 28:181; http://dx.doi.org/ 10.1214/aoms/1177707045 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.