

ABSTRACT
The endoplasmic reticulum (ER) is exploited by several diverse viruses during their infectious life cycles. Flaviviruses, including dengue virus (DENV) and Zika virus (ZIKV), utilize the ER as a source of membranes to establish their replication organelles and to facilitate their assembly and eventual maturation along the secretory pathway. To maintain normal homeostasis, host cells have evolved highly efficient processes to dynamically regulate the ER, such as through reticulophagy, a selective form of autophagy that leads to ER degradation. Here, we identify the ER-localized reticulophagy receptor FAM134B as a host cell restriction factor for both DENV and ZIKV. We show that RNAi-mediated depletion of FAM134B significantly enhances both DENV and ZIKV replication at an early stage of the viral life cycle. Consistent with its role as an antiviral host factor, we found that several flaviviruses including DENV, ZIKV, and West Nile virus (WNV), utilize their NS3 virally-encoded proteases to directly cleave FAM134B at a single site within its reticulon homology domain (RHD). Mechanistically, we show that NS3-mediated cleavage of FAM134B blocks the formation of ER and viral protein-enriched autophagosomes, suggesting that the cleavage of FAM134B serves to specifically suppress the reticulophagy pathway. These findings thus point to an important role for FAM134B and reticulophagy in the regulation of flavivirus infection and suggest that these viruses specifically target these pathways to promote viral replication.
KEYWORDS: autophagy, dengue virus, ER-phagy, FAM134B, flavivirus, reticulon, reticulophagy, Zika virus
Introduction
Flaviviruses, such as dengue (DENV) and Zika (ZIKV), are mosquito-borne pathogens that present a major risk to global public health. DENV infection can cause severe hemorrhagic fever leading to death, whereas ZIKV infection has historically been associated with relatively mild disease accompanied by low-grade fevers and skin rash.1-3 The recent introduction of ZIKV into naïve populations of French Polynesia and several countries in South America has led to a significant increase in congenital anomalies, including microcephaly.4-9 Over the past several decades, regions harboring the mosquitoes responsible for the spread of DENV and ZIKV have expanded and are predicted to continue,10 which will inevitably lead to the spread of these viruses to new and naïve populations. Thus, it is critical to better understand the viral and cellular components that facilitate flavivirus replication, which represent new targets for the development of antivirals and/or vaccines.
Autophagy is a degradative process that functions to sequester and break down cellular components, including organelles, within double-membrane vesicles at least partly derived from the ER to maintain cellular homeostasis.11-14 Several flaviviruses, including DENV and ZIKV, potently induce autophagy, which allows for efficient viral replication and virion assembly.15-22 Interestingly, DENV infection relies on selective autophagy of lipid droplets (lipophagy) to provide energy required for viral replication.15 A number of other forms of selective autophagy have been described including degradation of ribosomes (ribophagy), peroxisomes (pexophagy), mitochondria (mitophagy), and ER (reticulophagy).23 However, the molecular mechanisms that regulate many of these processes have not been well characterized for mammalian cells and, thus, have not been investigated during viral infection.
The ER is a dynamic organelle composed of a vast contiguous network of interconnected membranous sheets and tubules that facilitates protein/lipid biosynthesis, endosomal/secretory vesicle trafficking, and ion homeostasis. Recently, ER-localized members of the family with sequence similarity 134 (FAM134) proteins, which serve as receptors for components of the autophagic machinery, have been shown to regulate continuous turnover of the ER in mammalian cells.14 Thus, reticulophagy has been suggested to function as a constitutive pathway associated with the ER-quality control (ERQC) system that functions to clear overexpressed and misfolded proteins from the ER.14,24-26 Interestingly, flaviviruses replicate their positive-sense RNA genomes in virus-induced vesicular invaginations of the ER membrane and utilize the ER for both their assembly and maturation through the secretory pathway.27-29 Thus, the availability of ER membranes to support flavivirus replication would be expected to profoundly affect viral infection.
Given the close association between flavivius replication and the ER, we explored the interplay between reticulophagy and DENV and ZIKV replication. To identify whether reticulophagy plays a role in flavivirus replication, we determined the effects of RNAi-mediated silencing of FAM134B, an essential reticulophagy receptor, on DENV and ZIKV replication and found that depletion of FAM134B greatly enhanced viral replication. Moreover, we show that DENV and ZIKV, as well as West Nile virus (WNV), all utilize virally encoded proteases to attenuate FAM134B-mediated reticulophagy by cleaving FAM134B at a single site, which blocks the formation of reticulophagy-specific autophagosomes and sequestration of viral proteins within these structures. Thus, our data show that whereas canonical macroautophagy serves as a proviral pathway for flaviviruses, reticulophagy serves as a potent antiviral pathway and is inhibited by the viral protease-mediated cleavage of FAM134B.
Results
FAM134B expression restricts DENV and ZIKV infection
Given the intimate association between the ER and flavivirus replication/assembly, we hypothesized that FAM134B-mediated reticulophagy would have an impact on viral infection. To test this, we used 2 siRNAs against FAM134B, a previously characterized reticulophagy receptor,14 in human brain microvascular endothelial cells (HBMEC), an in vitro model of the blood-brain barrier, that are highly permissive for DENV and ZIKV infection30 and represent an important in vivo target for ZIKV given its association with neuronal dysfunction in the developing fetus. Consistent with a previous study using a different cell type, we found that depletion of FAM134B in HBMEC resulted in a modest degree of ER expansion (Fig. S1),14 particularly the smooth ER. Interestingly, we found that silencing of FAM134B enhanced the generation of DENV and ZIKV viral RNA (vRNA), with the extent of FAM134B knockdown correlating with the magnitude of this increase (Fig. 1A). FAM134Bsi-1 decreased transcript levels by ∼90%, which resulted in a 7-fold increase in both DENV and ZIKV vRNA (Fig. 1A and 1C) and a more than one-log increase in DENV titers and a statistically significant increase in ZIKV titers (Fig. 1B and 1D); thus, this siRNA was used for all subsequent experiments. To determine whether FAM134B restricted other viruses, we used vesicular stomatitis virus (VSV), a negative-strand RNA virus, and found that depletion of FAM134B had no effect on VSV infection (Fig 1E). These results indicate that FAM134B may act as a specific restriction factor for flaviviruses and not as a pan-viral host restriction factor.
Figure 1.

FAM134B restricts DENV and ZIKV infection. (A and C) HBMEC transfected with the indicated siRNAs were infected with (A) DENV or (C) ZIKV (MOI of 1) for 24 h. Infection and host gene expression were determined by RT-qPCR. Data are presented as the fold change of the control (CONsi). (B and D) Extracellular (B) DENV and (D) ZIKV titer from HBMEC transfected with CONsi or FAM134Bsi, determined by fluorescent focus assay, data are presented as the focus-forming units per mL. (E) HBMEC transfected with CONsi and FAM134Bsi were infected with VSV expressing a GFP reporter (MOI of 1) for 7-8 h. Infection was determined by immunofluorescence (VSV). Data are presented as the fold change of CONsi. (F) HeLa cells stably propagating a DENV subgenomic replicon (DENVrep) were transfected with CONsi and FAM134Bsi. Replicon RNA and host gene expression were assessed at 48 h post transfection by RT-qPCR. Data are presented as the fold change of CONsi. All data represent the mean ± SD of at least 3 independent experiments. Significance was determined by Student t test, *p < 0.05 and ***p < 0.0005.
To determine the stage at which FAM134B restricted viral replication, we used cells that stably propagate a DENV subgenomic replicon (DENVrep), which encodes for only the viral nonstructural proteins and is thus capable of autonomous replication that can be used to monitor viral replication in the absence of productive infection. We found that depletion of FAM134B had no effect on the levels of viral RNA in these cells (Fig. 1F). Together, these results suggest that FAM134B restricts DENV and ZIKV replication at an early stage of infection, possibly at the time of the establishment of replication vesicles.
FAM134B is cleaved by the flavivirus proteases
Previous work has shown that flavivirus infection results in expansion of the ER to accommodate the formation of replication vesicles.31-33 Consistent with these findings, we observed the presence of DENV replication vesicles and immature virions within expanded ER of infected HBMEC (Fig. 2A and 2B). Additionally, we observed enhanced staining of the ER-sheet marker, CKAP4 (cytoskeleton associated protein 4), and ER-tubule marker, RTN4 (reticulon 4), at sites of DENV and ZIKV viral replication, suggesting a localized expansion of the ER (Fig. 2C–F). Coupled with our findings that FAM134B expression restricted DENV and ZIKV replication, these data suggest that flaviviruses might manipulate the activity of reticulophagy to enhance their replication.
Figure 2.
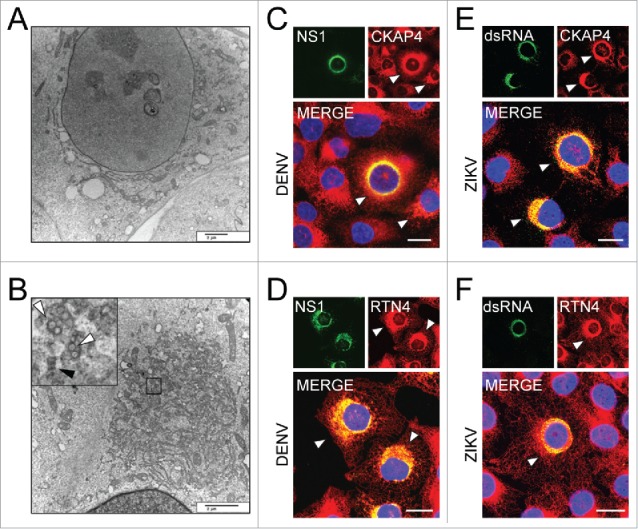
Flavivirus infection of HBMEC results in ER expansion. Transmission electron micrographs of (A) mock and (B) DENV (MOI of 3)-infected HBMEC 24 hpi. Black box shows area of zoomed-in region showing replication vesicles (white arrows) and immature virions (black arrows) within expanded ER. Scale bars: 2 µm. (C-F) Confocal microscopy of HBMEC infected with DENV (C and D) or ZIKV (E and F) at a MOI of 1; sites of ER expansion are marked with white arrowheads. Samples were stained 24 hpi for CKAP4, an ER sheet marker (red, C and E), and RTN4, an ER tubule marker (red, D and F). Sites of viral replication were determined by staining for DENV NS1 (green, C and D) or ZIKV dsRNA (green, E and F). Regions of colocalization are shown in yellow in merged images, with enhanced staining of ER markers shown at sites of viral replication. Scale bars: 20 µm.
To further characterize the role of reticulophagy during DENV and ZIKV infection, we created a FAM134B construct with a C-terminal HA epitope tag. Consistent with a previous study, FAM134B-HA migrated at ∼75 kDa14 by immunoblotting and was detected with a FAM134B-specific antibody (Fig. 3A); however, we were not able to detect endogenous FAM134B using this antibody. In addition to the major ∼75-kDa species, we also observed a lower molecular mass species that may represent a degradation product. Interestingly, we found that expression of FAM134B in DENVrep cells resulted in the appearance of a stable ∼45-kDa C-terminal fragment of FAM134B by immunoblotting, with a corresponding ∼80% decrease in the levels of full-length protein (Fig. 3B). Importantly, we found that this cleavage was specific to FAM134B, as other ER-membrane proteins such as members of the atlastin family (ATL2 and ATL3), ZFYVE27, and RTN4, were not cleaved in DENVrep cells (Fig. 3C). Together, these results indicate that DENV replication leads to the specific cleavage of FAM134B.
Figure 3.

The flavivirus protease cleaves FAM134B. (A) Immunoblot of FAM134B and GAPDH from U2OS cells transfected with empty plasmid or FAM134B-HA. (B) Immunoblot of HA, GFP, and GAPDH from HEK 293T cells or HEK 293T cells expressing a subgenomic DENV replicon (DENVrep) with a GFP-Zeocin reporter fusion protein (GFP-Zeo) transfected with a plasmid encoding FAM134B-HA. The black arrowhead indicates the FAM134B-HA cleavage fragment. (C) Immunoblot of HA, GFP, and endogenous RTN4 from HEK 293T cells or HEK293T DENVrep cells. The black arrowhead indicates the FAM134B-HA cleavage fragment. (D) Schematics of FAM134B showing predicted topology and scaled linear model (drawn to scale); SH, short-hairpin membrane domain; RHD, reticulon-homology domain; and LIR, LC3-interacting region. The black arrowhead indicates the exposed cytoplasmic loop. (E) Immunoblot of V5 and HA from HEK 293T cells transfected with FAM134B-HA and V5-tagged DENV NS3, DENV NS2B3, WNV NS2B3, or ZIKV NS2B3. (F) Immunoblot for V5 and HA from U2OS cells transfected with a plasmid encoding FAM134B-HA and V5-tagged WT DENV NS2B3 or active site mutant S135A. (G) Confocal image of DENV NS2B3-V5 (green) and FAM134B-HA (red) colocalized in HeLa cells with DAPI-stained nuclei. Pearson's correlation coefficient is indicated in the merged panel as the average ± SD of 10 individual cells. Scale bar: 30 µm (H) Schematics of FAM134B Q145X-HA (top) and FAM134BR142A-HA (bottom, residue 142 is underlined) mutations used to determine viral protease cleavage site. (I) Immunoblot for V5, HA, and GAPDH from U2OS cells transfected with empty vector or a plasmid encoding DENV NS2B3-V5 and WT FAM134B, truncation mutant Q145X-HA, or point mutant R142A-HA.
FAM134B contains a reticulon-homology domain (RHD), which is responsible for creating membrane curvature via 2 short hairpin membrane regions, and a C-terminal MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-interacting region (LIR) that targets sections of the ER to autophagosomes.14 Analysis of the size of the lower molecular weight fragment generated by DENV replication suggested that cleavage might be occurring in the cytoplasmic loop of the RHD (schematic, Fig. 3D). Because the viral protease complex (NS2B3) is active on the cytoplasmic membrane of the ER, we tested whether expression of NS2B3 was sufficient to cleave FAM134B. Indeed, we observed an ∼45-kDa fragment of FAM134B in cells expressing DENV NS2B3 in the absence of other viral proteins, but not in cells expressing only the NS3 protease in the absence of the NS2B cofactor, which is consistent with a requirement for protease activity (Fig. 3E). Furthermore, we also observed FAM134B cleavage upon expression of WNV and ZIKV NS2B3, indicating a conserved role for FAM134B cleavage among several flaviviruses (Fig. 3E). Additionally, NS2B3-mediated cleavage of FAM134B disrupted the formation of oligomers (Fig. 3E), which facilitate membrane curvature for reticulon proteins.34 Importantly, we also found that the catalytic activity of the NS3 protease was required for FAM134B cleavage, as a catalytically inactive mutant of the NS3 protease domain (S135A) abolished cleavage (Fig. 3F).
Using confocal microscopy, we observed colocalization between DENV NS2B3 and HA-tagged FAM134B, consistent with their localization in the ER (Fig. 3G). However, we did not observe cytoplasmic staining of FAM134B using an antibody directed against the C-terminal HA tag, suggesting that the C-terminal cleavage fragment remains associated with the ER membrane after proteolysis. Analysis of the primary FAM134B sequence did not reveal a conserved flavivirus protease cleavage site (R/K/Q-R/K-↓-G/S/A). However, nonconsensus sites may be targeted by the DENV protease.35 Therefore, we generated a C-terminal HA-tagged FAM134B truncation protein (Q145X) that retains the N-terminal 144 amino acids, including the first short hairpin membrane domain and 11 amino acids of the cytoplasmic loop, to determine where cleavage might be occurring (schematic, Fig. 3H). Expression of the viral protease decreased the amount of detectable Q145X using an antibody directed against the HA-tag on the C terminus, suggesting that protease cleavage occurs within the 11 amino acid cytoplasmic loop (Fig. 3I). Sequence analysis of this region revealed a degenerate protease recognition site at position 142 (R-T-R142-↓-G). Indeed, mutation of this putative recognition site (R142A) abolished viral protease-dependent cleavage of FAM134B (Fig. 3I). We also found that overexpression of FAM134B significantly decreased DENV infection (Fig. S2); however, overexpression of the R142A cleavage mutant did not further decrease infection. These results may suggest that the stoichiometry between the levels of viral protease and FAM134B are critical for the virus to overcome the decreased susceptibility of the non-consensus cleavage site to subvert the antiviral effects of this host protein. Together, these results indicate that flaviviruses target a nonconsensus protease recognition site to disrupt FAM134B oligomerization, which may lead to inhibition of reticulophagy.
Cleavage of FAM134B disrupts reticulophagy
Given that we found that DENV and other flaviviruses directly target FAM134B for cleavage, we next sought to determine the impact of cleavage of FAM134B on reticulophagy. Consistent with previous results,14 transient overexpression of FAM134B significantly induced reticulophagy, indicated by the formation of FAM134B-positive mRFP-LC3B-containing autophagosomes (Fig. 4A and B). Mutation of the LC3-interacting region (FAM134B-mutLIR) abolished the localization of FAM134B to mRFP-LC3B-positive autophagosomes (Fig. 4A and B), which is also consistent with a previous report.14 Similarly, a C-terminal fragment of FAM134B that is produced as a consequence of flaviviral protease cleavage also exhibited decreased localization to LC3B-positive autophagosomes (Fig. 4A and B), which suggests that viral protease-mediated cleavage of FAM134B subverts reticulophagy.
Figure 4.

The FAM134B C-terminal fragment is unable to mediate reticulophagy. (A) Representative confocal images of U2OS cells transfected with a plasmid encoding mRFP-LC3B and GFP or GFP tagged FAM134B construct, and DAPI-stained nuclei. Insets show zoomed in region outlined by white boxes. White arrowheads point to mRFP- and GFP-positive puncta. Scale bars: 20 µm. (B) Quantification of the number of mRFP-LC3B and GFP fusion protein co-expressing cells with mRFP- and GFP-positive puncta. Data are presented as a percentage and represent the average ± SD of 3 independent experiments (≥ 100 cells analyzed/experiment). Significance was determined using the Student t test, ***p <0.0001.
To determine if FAM134B mediates the sequestration of viral proteins in reticulophagy-specific autophagosomes, a process that would be expected to limit flavivirus replication, we transiently co-expressed the NS2B3 virally-encoded protease complex with FAM134B, FAM134B-mutLIR, or FAM134BR142A, a mutant that is resistant to protease cleavage (Fig. 3I), in U2OS cells. Consistent with previous results, expression of FAM134B, but not FAM134B-mutLIR, induced reticulophagy, as indicated by cells containing FAM134B-positive puncta (Fig. 5A and 5B). Interestingly, we found that expression of the cleavage-resistant FAM134B mutant (FAM134BR142A) resulted in significantly more cells containing FAM134B-positive and NS3-positive puncta compared with wild-type (WT) FAM134B (Fig. 5A and 5B), which is consistent with our previous results indicating that cleavage of FAM134B disrupts formation of reticulophagy-associated autophagosomes (Fig. 4). Image analysis indicated that there were significantly more cells containing FAM134B- and NS3-positive puncta in cells expressing FAM134BR142A compared with WT-expressing cells (Fig. 5A and 5B). Thus, NS2B3-mediated cleavage of FAM134B disrupts the sequestration of viral proteins in reticulophagy-specific autophagosomes. Together, these findings are consistent with a model whereby the cleavage of FAM134B by the flavivirus protease generates a LIR-containing C-terminal fragment that is able to interact with LC3B, but is unable to induce the necessary ER membrane curvature to facilitate reticulophagy of ER-localized viral and host proteins (schematic, Fig. 6).
Figure 5.
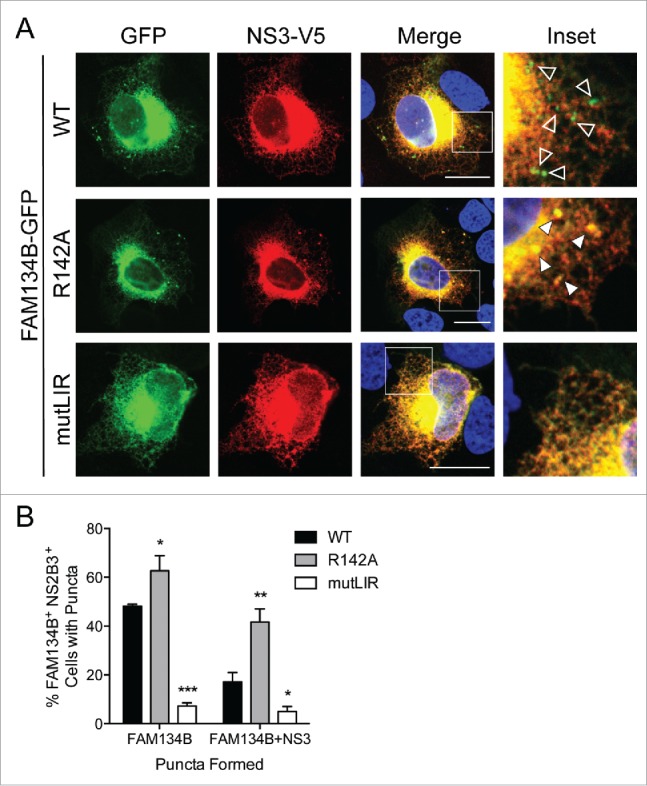
DENV NS2B3 disrupts viral protein sequestration in reticulophagy-derived autophagosomes. (A) Representative confocal images of U2OS cells co-transfected with a plasmid encoding DENV NS2B3-V5 (red) and the indicated GFP-tagged FAM134B constructs (green), and DAPI-stained nuclei. Insets show zoomed-in region outlined by white boxes. Empty white arrowheads point to GFP-positive puncta and filled white arrowheads point to V5- and GFP-positive puncta. Scale bars: 20 µM. (B) Quantification of the number of NS2B3-V5 and FAM134B-GFP construct co-expressing cells with GFP- or V5- and GFP-positive puncta. Data are presented as a percentage and represent the average ± SD of 3 independent experiments (≥ 100 cells analyzed/experiment). Significance was determined by the Student t test, *p < 0.05, **p < 0.005, and ***p < 0.0005.
Figure 6.

Working model for flavivirus inhibition of reticulophagy. In uninfected cells (top), ER-localized FAM134B (blue) forms oligomers that induce membrane curvature. FAM134B recruits phagophore membranes via LIR interaction with LC3B (red), which leads to the budding and scission of ER-derived vesicles that are eventually degraded after subsequent fusion between the autophagosome and lysosome. During flavivirus infection (bottom), the virally-encoded protease complex (NS2B3) cleaves FAM134B within its RHD, producing 2 ER-localized fragments (N-term and C-term). The LIR-containing C-terminal fragment is unable to form oligomers, which inhibits FAM134B-induced membrane curvature and subsequent budding of ER-derived vesicles required for reticulophagy.
Discussion
The ER is a highly structured organelle that facilitates many cellular processes. Consequently, this organelle is intimately involved in many aspects of RNA virus replication, including for flaviviruses. Here, we show that DENV and ZIKV infection is restricted by expression of FAM134B, an ER-localized protein that promotes membrane curvature and ER degradation via reticulophagy.14 Interestingly, we also show that DENV, ZIKV, and WNV utilize their NS2B3 virally-encoded proteases to specifically cleave FAM134B at a single site, inhibiting its oligimerization and ability to function as a reticulophagy receptor. Collectively, our data point to a prominent antiviral role for the reticulophagy receptor FAM134B in the life cycle of DENV and ZIKV and suggest that these viruses utilize their NS2B3 virally-encoded proteases to suppress this process (schematic, Fig. 6).
Our results indicate that flavivirus infection is associated with expansion of the ER rather than its degradation, which is consistent with previous reports.31-33 This expansion is possibly facilitated by the DENV NS3-induced relocalization of FASN (fatty acid synthase) and de novo lipid synthesis at sites of viral replication, which likely aids in the formation of replication vesicles.32 Our work describes a mechanism by which several flaviviruses actively disrupt reticulophagy, a process that would function to restrict virus-induced ER-expansion and eliminate ER-associated viral proteins resulting in restriction of viral replication. Additionally, our results show that NS3 serves a crucial role in replication vesicle formation both by promoting ER expansion via recruitment of FASN32 and inhibiting reticulophagy through cleavage of FAM134B.
The mechanisms regulating reticulophagy in mammalian cells are not well characterized, but have been extensively studied in yeast. Reticulophagy in yeast requires core autophagy-related proteins including Atg9, the Rab family GTPase Ypt1, and Atg40, which is a FAM134B ortholog that also functions as a reticulophagy receptor.24,25 Reticulophagy is suggested to be a constitutive process that functions as a part of the ERQC system to remove excess ER membrane proteins.24 Interestingly, there are 21 ER-membrane interacting regions in the flavivirus polyprotein precursor that facilitate the accumulation of the mature viral proteins in the ER, which are required for replication and virion assembly. Furthermore, a recent study has shown that Ebola virus infection is also restricted by FAM134B expression.36 However, the mechanism regarding this restriction remains unknown. Interestingly, expression of the Ebola virus glycoprotein, which is heavily modified with glycans in the ER, results in accumulation in the ER and induces cytotoxicity.37,38 Thus, it is not surprising that regions of the ER containing excess viral proteins would be targeted for degradation via reticulophagy, which would significantly restrict viral replication. However, the mechanisms by which reticulophagy receptors might target specific regions of the ER have not been characterized.
Several studies have demonstrated the importance of autophagy during flavivirus infection.15,17-21 Autophagy is a degradative process that is activated in response to a variety of cellular stresses, including viral infection, to maintain cellular homeostasis. This results in a high turnover of cytoplasmic proteins and organelles, which would be hypothesized to be detrimental to the expression of viral proteins required for replication and assembly. The precise signaling pathways that lead to induction of selective autophagy are currently unknown. Thus, it is difficult to determine which of these processes are specifically activated during DENV and/or ZIKV infection. However, a previous report suggests DENV-induced autophagy leads to the degradation of lipid droplets, a selective process termed lipophagy, which results in an increase in β oxidation that provides energy required for efficient viral replication.15 Our work shows the reticulophagy receptor, FAM134B, restricts DENV and ZIKV replication at early stages of replication. Current literature suggests reticulophagy constitutively functions at a low basal level as a part of the ERQC to remove excess ER membrane proteins24 and would thus present an obstacle for the establishment of viral replication vesicles in the ER at early stages of infection when viral proteins are being produced at high levels. Our results thus suggest that the accumulation of the NS2B3 protease complex results in cleavage of FAM134B at sites of viral polyprotein processing, which inhibits reticulophagy to allow for the efficient formation of replication vesicles and subsequent genome replication. Taken together, our findings presented here and the work of others15 support both proviral (lipophagy) and anti-viral (reticulophagy) roles for different selective autophagy pathways in regulating flavivirus replication. Furthermore, our work suggests that the development of therapeutics that inhibit the ability of the virally-encoded protease to target anti-flaviviral host cell pathways and proteins, such as reticulophagy and FAM134B, could be an efficient approach to suppress viral replication.
Materials and methods
Cells and viruses
HBMEC were maintained in RPMI 1640 (HyClone Laboratories, SH30027.01) supplemented with 10% fetal bovine serum (FBS; Gibco, 26140-079), 10% NuSerum (Corning, 355500), 1x minimum essential medium vitamins (Gibco, 11120-052), 1x non-essential amino acids (HyClone Laboratories, SH30238.01), 1% sodium pyruvate (HyClone Laboratories, SH30239.01), and 1% antibiotics (Lonza, 17-602E). Human osteosarcoma U2OS cells (ATCC, HTB-96), Vero cells (ATCC, CCL-81), HEK 293T cells (ATCC, CRL-11268), and Aedes albopictus midgut C6/36 cells (ATCC, CRL-1660) were cultured in DMEM (Corning, 10-017-CV) supplemented with 10% FBS and antibiotics. C6/36 cells were maintained at 28°C in a 5% CO2 atmosphere. HeLa cells (ATCC, CCL-2) were maintained in MEM (Lonza, 12-611F) supplemented with 10% FBS, nonessential amino acids, sodium pyruvate, and antibiotics. Development of HeLa and HEK 293T cells stably propagating a DENV subgenomic replicon has been described previously,30,39 using plasmids provided by Theodore Pierson (Viral Pathogenesis Section Laboratory of Viral Diseases, NIH/NIAID).
DENV2 16881 and ZIKV FSS13025 were propagated in C6/36 cells, as described previously.40 Viral titers were determined by fluorescent focus assay, as described previously,41 using flavivirus anti-E monoclonal antibody 4G2 (provided by Margaret Kielian, Albert Einstein College of Medicine) for DENV and recombinant anti-dsRNA monoclonal antibody (provided by Abraham Brass, University of Massachusetts) for ZIKV. VSV-GFP was propagated in Vero cells and titrated by plaque assay. Experiments measuring infection were performed with a multiplicity of infection (MOI) of 1 and infection was quantified by RT-qPCR (24 h post-infection [hpi], DENV and ZIKV) or immunofluorescence (8 hpi, VSV-GFP).
RNA extraction, cDNA synthesis, and RT-qPCR
RNA extraction was performed with Tri-Reagent LS (Molecular Research Center, TS 120) according to the manufacturer's protocol. Isolated RNA was treated with RNase-free DNase (QIAGEN, 79254) before reverse transcription. Total cellular RNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, 1708891) according to the manufacturer's protocol. RT-qPCR was performed using iQ SYBR Green Supermix (Bio-Rad Laboratories, 1708882) in a StepOne Plus real-time system (Applied Biosystems, Foster City, CA, USA) or CFX96 Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA). Viral infection and host gene expression was calculated using the 2-ΔΔ CT method normalized to ACTB. The following primer sequences were used for qPCR: ACTB (5′-ACTGGGACGACATGGAGAAAA-3′ and 5′-GCCACACGCAGCTC-3′), DENV2 (5′-AGTTGTTAGTCTACGTGGACCGA-3′ and 5′-CGCGTTTCAGCATATTGAAAG-3′), ZIKV (5′-AGATGACTGCGTTGTGAAGC-3′ and 5′-GAGCAGAACGGGACTTCTTC-3′), and FAM134B (5′-AGACTTTTCAGCTCTTTGTC-3′ and 5′-GATCATCAGAAGTGTCTGTC-3′).
siRNAs, Plasmids, and Transfections
Cells were reverse transfected with 25 nM Mission siRNA Univeral Control (Sigma-Aldrich, SIC001) or individual siRNAs, based on previously characterized sequences targeting FAM134B14 (FAM134Bsi-1: GAGGUAUCCUGGACUGAUA[dT][dT] and FAM134Bsi-2: AGAUGACAGUGAAUUAGAC[dT][dT]) using Dharmafect 1 (GE Healthcare Dharmacon, T-2001). Cells were infected ∼36 h post transfection and RNA was isolated at 24 hpi.
A vector containing the FAM134B cDNA was purchased from transOMIC (TCH1003). The full- length FAM134B ORF, the first 432 nucleotides (Q145X truncation mutant), or the last 1065 nucleotides (C-terminal fragment) preceded by an ATG, methionine codon, were PCR-amplified and inserted into the BamHI-EcoRI sites of pcDNA3.1(+) (Invitrogen, V79020) with a flexible linker and HA epitope tag (GGSGYPYDVPDYA) or GFP fluorescent protein tag inserted at the EcoRI-XhoI sites. The FAM134B R142A mutation was introduced using QuikChange site-directed mutagenesis (Agilent, 200518) according to the manufacturer's protocol followed by Sanger sequencing. DENV and WNV nonstructural protein expression plasmids were generated by PCR amplification of protein coding regions from subgenomic replicon plasmids (provided by Theodore Pierson, NIH/NIAID). The ZIKV NS2B3 protein-coding region was amplified from cDNA produced from HBMEC infected with ZIKV MR766. PCR products were cloned into the pcDNA3.1 V5-His TOPO TA vector (Invitrogen, K480001) according to the manufacturer's protocol. Expression plasmids containing human ATL2 and ATL3 with C-terminal HA epitope tags were provided by Craig Blackstone (NIH/NINDS). The ZFYVE27-HA construct was produced by PCR amplification of the ORF from a vector purchased from the Havard PlasmID Database (HsCD00333117) and inserted into pcDNA3.1. The mRFP-LC3B plasmid has been described previously.42 U2OS cells and HEK 293T cells were reverse transfected with plasmids using X-tremeGENE 9 (Sigma-Aldrich, XTG9-RO) or X-tremeGENE HP (Sigma-Aldrich, XTGHP-RO) according to the manufacturer's protocol. Transfected cells were infected, immunostained, or lysed for immunoblots 48 h post transfection.
Antibodies
Rabbit polyclonal antibodies directed against GAPDH (FL-335, sc-25778), HA epitope tag (Y-11, sc-473818), and GFP (FL, sc-805) were purchased from Santa Cruz Biotechnology. Mouse monoclonal anti-V5 epitope tag was purchased from Invitrogen (R960-25). Rabbit polyclonal antibodies against CKAP4 (16686-1-AP), RTN4 (10950-1-AP), and FAM134B (21537-1-AP) were purchased from ProteinTech. Mouse monoclonal anti-DENV NS1 (DN3) was purchased from Abcam (ab41616). Recombinant mouse monoclonal anti-dsRNA was provided by Abraham Brass (University of Massachusetts) and mouse monoclonal anti-DENV E 4G2 was provided by Margaret Kielian (Albert Einstein College of Medicine).
Microscopy
Cells cultured in 8-well chamber slides (Thermo Scientific, 154534) were fixed in 4% paraformaldehyde followed by permeabilization in 0.1% Triton X-100 (Fisher Scientific, BP151-500) or ice-cold methanol for experiments using anti-E, anti-NS1 and anti-dsRNA antibodies. Following fixation samples were incubated with primary antibody for 1 h, followed by Alex Fluor-conjugated secondary antibodies (Invitrogen, A11034, A11037, A11029, or A11032) for 30 min, and mounted with VectaShield (Vector Laboratories, H-1200) containing 4′-6-diamino-2-phenylindole (DAPI). Images were captured using a FV1000 confocal laser scanning microscope (Olympus, Tokyo, Japan) and contrasted/merged using Photoshop (Adobe Systems, San Jose, CA USA). Images from 4 replicate wells (4 fields/well) of VSV-GFP-infected samples were captured on an IX83 inverted microscope (Olympus, Tokyo, Japan) and infection was quantified using cellSens software (Olympus, Tokyo, Japan). Images used for quantification of U2OS cells containing the FAM134B-GFP construct and mRFP-LCB puncta, at least 100 cells for each condition per experiment, were captured with an IX83 inverted microscope. Samples were prepared for transmission electron microscopy as described previously42 and micrographs were captured using a JEOL 1011 transmission electron microscope (JEOL USA, Peabody, MA USA). Pearson's correlation coefficients were calculated for 10 individual cells with the Coloc2 plugin for ImageJ/Fiji (NIH, Bethesda, MD, USA).
Immunoblots
Cells lysates were prepared on ice in cold RIPA buffer (Millipore, 20-188: 50 mM Tris-HCl, pH7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented with 1x Pierce protease inhibitor cocktail (AEBSF, aprotinin, bestatin, E64, leupeptin, pepstatin A; Thermo Scientific, 88266). Lysates were clarified by centrifugation at 13,000 xg for 15 min at 4°C and protein concentration was measured by bicinchoninic assay (Thermo Scientific, 23235). Lysates (10–30 µg) were separated on 4-20% Tris-HCl gels (Bio-Rad, 4561093 or 3450033) and transferred to nitrocellulose membranes. Membranes were blocked with 10% nonfat milk before probing with the indicated primary antibodies followed by incubation with IR-dye-conjugated secondary antibodies (LI-COR Biosciences, 926-32211, 926-32210, 926-68070, or 926-68021). Immunoblots were visualized using an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Statistics
Data were analyzed with Prism 6 software (GraphPad Software, La Jolla, CA, USA) using the two-tailed unpaired Student t test, unless otherwise indicated.
Abbreviations
- ATL
atlastin GTPase
- CKAP4
cytoskeleton associated protein 4
- DENV
dengue virus
- DENVrep
dengue virus subgenomic replicon
- ER
endoplasmic reticulum
- ERQC
endoplasmic reticulum quality control
- FAM134B
family with sequence similarity 134 member B
- GFP
green fluorescent protein
- HBMEC
human brain microvascular endothelial cells
- hpi
hours post infection
- LC3
microtubule associated protein 1 light chain 3
- LIR
LC3-interacting region
- MOI mRFP
multiplicity of infection monomeric red fluorescent protein
- mutLIR
LC3-interacting region mutant
- NS
nonstructural protein
- RHD
reticulon homology domain
- RTN4
reticulon 4
- vRNA
viral RNA
- WNV
West Nile virus
- ZIKV
Zika virus
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Kwang Sik Kim (Johns Hopkins University) for HBMEC, Theodore Pierson (NIH/NIAID) for subgenomic replicon plasmids, Margaret Kielian (Albert Einstein College of Medicine) for providing the anti-E 4G2 antibody, and Abraham Brass (University of Massachusetts) for recombinant anti-dsRNA antibody.
Funding
This project was supported by NIH T32-AI049820 (N.J.L), NIH F32-AI122456 (N.J.L), NIH R01-AI081759 (C.B.C) and a Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award (C.B.C).
References
- [1].Fauci AS, Morens DM. Zika Virus in the Americas-Yet Another Arbovirus Threat. N Engl J Med 2016; 374:601-4; PMID:26761185; http://dx.doi.org/ 10.1056/NEJMp1600297 [DOI] [PubMed] [Google Scholar]
- [2].Simpson DI. Zika Virus Infection in Man. Trans R Soc Trop Med Hyg 1964; 58:335-8; PMID:14175744; http://dx.doi.org/ 10.1016/0035-9203(64)90201-9 [DOI] [PubMed] [Google Scholar]
- [3].Bearcroft WG. Zika virus infection experimentally induced in a human volunteer. Trans R Soc Trop Med Hyg 1956; 50:442-8; PMID:13380987; http://dx.doi.org/ 10.1016/0035-9203(56)90090-6 [DOI] [PubMed] [Google Scholar]
- [4].Cauchemez S, Besnard M, Bompard P, Dub T, Guillemette-Artur P, Eyrolle-Guignot D, Eyrolle-Guignot D, Salje H, Van Kerkhove MD, Abadie V, et al.. Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet 2016; 387(10033):2125-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Oliveira Melo AS, Malinger G, Ximenes R, Szejnfeld PO, Alves Sampaio S, Bispo de Filippis AM. Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet Gynecol 2016; 47:6-7; PMID:26731034; http://dx.doi.org/ 10.1002/uog.15831 [DOI] [PubMed] [Google Scholar]
- [6].Schuler-Faccini L, Ribeiro EM, Feitosa IM, Horovitz DD, Cavalcanti DP, Pessoa A, Doriqui MJ, Neri JI, Neto JM, Wanderley HY, et al.. Possible Association Between Zika Virus Infection and Microcephaly - Brazil, 2015. MMWR Morbidity Mortality Weekly Report 2016; 65:59-62; PMID:26820244; http://dx.doi.org/ 10.15585/mmwr.mm6503e2 [DOI] [PubMed] [Google Scholar]
- [7].Ventura CV, Maia M, Bravo-Filho V, Gois AL, Belfort R Jr. Zika virus in Brazil and macular atrophy in a child with microcephaly. Lancet 2016; 387:228; PMID:26775125; http://dx.doi.org/ 10.1016/S0140-6736(16)00006-4 [DOI] [PubMed] [Google Scholar]
- [8].Rasmussen SA, Jamieson DJ, Honein MA, Peterson LR. Zika Virus and Birth Defects-Reviewing the evidence for causality. N Engl J Med 2016; 374(20):1981-7. [DOI] [PubMed] [Google Scholar]
- [9].Sarno M, Sacramento GA, Khouri R, do Rosario MS, Costa F, Archanjo G, Santos LA, Nery N Jr, Vasilakis N, Ko AI, et al.. Zika virus infection and stillbirths: A case of hydrops fetalis, Hydranencephaly and fetal demise. PLoS Negl Trop Dis 2016; 10:e0004517; PMID:26914330; http://dx.doi.org/ 10.1371/journal.pntd.0004517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kraemer MU, Sinka ME, Duda KA, Mylne AQ, Shearer FM, Barker CM, Moore CG, Carvalho RG, Coelho GE, Van Bortel W, et al.. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife 2015; 4:e08347; PMID:26126267; http://dx.doi.org/ 10.7554/eLife.08347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685-701; PMID:18725538; http://dx.doi.org/ 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 2009; 11:1433-7; PMID:19898463; http://dx.doi.org/ 10.1038/ncb1991 [DOI] [PubMed] [Google Scholar]
- [13].Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al.. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495:389-93; PMID:23455425; http://dx.doi.org/ 10.1038/nature11910 [DOI] [PubMed] [Google Scholar]
- [14].Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, et al.. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015; 522:354-8; PMID:26040720; http://dx.doi.org/ 10.1038/nature14498 [DOI] [PubMed] [Google Scholar]
- [15].Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010; 8:422-32; PMID:21075353; http://dx.doi.org/ 10.1016/j.chom.2010.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mateo R, Nagamine CM, Spagnolo J, Mendez E, Rahe M, Gale M Jr, Yuan J, Kirkegaard K. Inhibition of cellular autophagy deranges dengue virion maturation. J Virol 2013; 87:1312-21; PMID:23175363; http://dx.doi.org/ 10.1128/JVI.02177-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, Lin YS, Yeh TM, Liu CC, Liu HS. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008; 374:240-8; PMID:18353420; http://dx.doi.org/ 10.1016/j.virol.2008.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J Biol Chem 2011; 286:22147-59; PMID:21511946; http://dx.doi.org/ 10.1074/jbc.M110.192500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li JK, Liang JJ, Liao CL, Lin YL. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect / Institut Pasteur 2012; 14:159-68; http://dx.doi.org/ 10.1016/j.micinf.2011.09.001 [DOI] [PubMed] [Google Scholar]
- [20].Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, et al.. Biology of Zika virus infection in human skin cells. J Virol 2015; 89:8880-96; PMID:26085147; http://dx.doi.org/ 10.1128/JVI.00354-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Metz P, Chiramel A, Chatel-Chaix L, Alvisi G, Bankhead P, Mora-Rodriguez R, Long G, Hamacher-Brady A, Brady NR, Bartenschlager R. Dengue virus inhibition of autophagic flux and dependency of viral replication on proteasomal degradation of the autophagy receptor p62. J Virol 2015; 89:8026-41; PMID:26018155; http://dx.doi.org/ 10.1128/JVI.00787-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, Ge J, Wang S, Goldman SA, Zlokovic BV, et al.. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 2016; 19(5):663-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Farre JC, Subramani S. Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol 2016; 17(9):537-52; PMID:27381245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lipatova Z, Segev N. A Role for Macro-ER-Phagy in ER Quality Control. PLoS Genet 2015; 11:e1005390; PMID:26181331; http://dx.doi.org/ 10.1371/journal.pgen.1005390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015; 522:359-62; PMID:26040717; http://dx.doi.org/ 10.1038/nature14506 [DOI] [PubMed] [Google Scholar]
- [26].Houck SA, Ren HY, Madden VJ, Bonner JN, Conlin MP, Janovick JA, Conn PM, Cyr DM. Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol Cell 2014; 54:166-79; PMID:24685158; http://dx.doi.org/ 10.1016/j.molcel.2014.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J Virol 1997; 71:6650-61; PMID:9261387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Junjhon J, Pennington JG, Edwards TJ, Perera R, Lanman J, Kuhn RJ. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J Virology 2014; 88:4687-97; PMID:24522909; http://dx.doi.org/ 10.1128/JVI.00118-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller , Antony C, Krijnse-Locker J, Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009; 5:365-75; PMID:19380115; http://dx.doi.org/ 10.1016/j.chom.2009.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr, Cherry S, Sadovsky Y, Coyne CB. Type III interferons produced by human placental trophoblasts confer protection against zika virus infection. Cell Host Microbe 2016; 19:705-12; PMID:27066743; http://dx.doi.org/ 10.1016/j.chom.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pena J, Harris E. Early dengue virus protein synthesis induces extensive rearrangement of the endoplasmic reticulum independent of the UPR and SREBP-2 pathway. PloS One 2012; 7:e38202; PMID:22675522; http://dx.doi.org/ 10.1371/journal.pone.0038202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, Randall G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci U S A 2010; 107:17345-50; PMID:20855599; http://dx.doi.org/ 10.1073/pnas.1010811107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yu CY, Hsu YW, Liao CL, Lin YL. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol 2006; 80:11868-80; PMID:16987981; http://dx.doi.org/ 10.1128/JVI.00879-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shibata Y, Voss C, Rist JM, Hu J, Rapoport TA, Prinz WA, Voeltz GK. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J Biol Chem 2008; 283:18892-904; PMID:18442980; http://dx.doi.org/ 10.1074/jbc.M800986200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yu CY, Liang JJ, Li JK, Lee YL, Chang BL, Su CI, Huang WJ, Lai MM, Lin YL. Dengue virus impairs mitochondrial fusion by cleaving mitofusins. PLoS Pathogens 2015; 11:e1005350; PMID:26717518; http://dx.doi.org/ 10.1371/journal.ppat.1005350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chiramel AI, Dougherty JD, Nair V, Robertson SJ, Best SM. FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of Ebola Virus Strains Makona and Mayinga. J Infect Dis 2016; 214(suppl 3):S319-S325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bhattacharyya S, Hope TJ. Full-length Ebola glycoprotein accumulates in the endoplasmic reticulum. Virol J 2011; 8:11; PMID:21223600; http://dx.doi.org/ 10.1186/1743-422X-8-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yang ZY, Duckers HJ, Sullivan NJ, Sanchez A, Nabel EG, Nabel GJ. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nature Med 2000; 6:886-9; PMID:10932225; http://dx.doi.org/ 10.1038/78645 [DOI] [PubMed] [Google Scholar]
- [39].Ansarah-Sobrinho C, Nelson S, Jost CA, Whitehead SS, Pierson TC. Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology 2008; 381:67-74; PMID:18801552; http://dx.doi.org/ 10.1016/j.virol.2008.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Medina F, Medina JF, Colón C, Vergne E, Santiago GA, Muñoz-Jordán JL. Dengue virus: isolation, propagation, quantification, and storage. Curr Protoc Microbiol 2012; Chapter 15:Unit 15D.2; http://dx.doi.org/ 10.1002/9780471729259.mc15d02s27 [DOI] [PubMed] [Google Scholar]
- [41].Payne AF, Binduga-Gajewska I, Kauffman EB, Kramer LD. Quantitation of flaviviruses by fluorescent focus assay. J Virological Methods 2006; 134:183-9; PMID:16510196; http://dx.doi.org/ 10.1016/j.jviromet.2006.01.003 [DOI] [PubMed] [Google Scholar]
- [42].Delorme-Axford E, Donker RB, Mouillet JF, Chu T, Bayer A, Ouyang Y, Wang T, Stolz DB, Sarkar SN, Morelli AE, et al.. Human placental trophoblasts confer viral resistance to recipient cells. Proc Natl Acad Sci U S A 2013; 110:12048-53; PMID:23818581; http://dx.doi.org/ 10.1073/pnas.1304718110 [DOI] [PMC free article] [PubMed] [Google Scholar]