

Abstract
Heart has long been considered a terminally differentiated organ. Recent studies, however, have suggested that there is a modest degree of cardiomyocyte (CM) turnover in adult mammalian heart, albeit not sufficient for replacement of lost CMs following cardiac injuries. Cardiac regeneration studies in various model organisms including zebrafish, newt, and more recently in neonatal mouse, have demonstrated that CM dedifferentiation and concomitant proliferation play important roles in replacement of lost CMs and restoration of cardiac contractility. Further studies with neonatal cardiac regeneration mouse model suggested that major source of new CMs is existing CMs, with the possibility of involvement of cardiac stem cells. Numerous studies have now been conducted on induction of cardiac regeneration and have identified various cardiogenic factors, cardiogenic micro ribonucleic acid and cardiogenic small molecules. This report is a review of studies regarding generation of CM and prospects for application.
Keywords: cardiac regeneration, cardiogenic factors, cardiogenic small molecules, cardiomyocyte proliferation, cardiomyocyte renewal, cell cycle activators
Introduction
Cardiovascular disorders are leading cause of death worldwide, with death rates up to 40% in some developing countries (1). Heart failure (HF) is a progressive and debilitating form of cardiovascular disorder with penetrance that rises exponentially with age. There are various causes of HF, but depressed ejection fraction following myocardial infarction (MI) is among the most common and can progressively lead to end-stage cardiomyopathy. Myocardium may be stunned and hibernate during ischemic MI or heart attack. If these cardiac muscle cells cannot recover their function, lack of viable myocardium leads to death in half of these patients within 5 years of diagnosis (2). Heart has been thought to be a terminally differentiated organ due lack of regeneration after HF, but there is some controversy, as recovery has also been observed following myocardial necrosis (3).
Heart transplantation is postulated as definitive treatment for HF. However, heart transplantation is limited by scarcity of human leukocyte antigen-compatible donors, as well as immunological complications that occur following transplantation (4). Thus, researchers seek alternative solutions for HF including induction of cardiac regeneration. In the last decade, promising studies related to cardiac regeneration have gained enormous momentum and have demonstrated cardiomyocyte (CM) renewal, either spontaneously or following cell-based therapies (5–7).
Different species have distinct regenerative capacities following various cardiac injury (8). Newts and zebrafish, for instance, regenerate their hearts following myocardial injury in as little as 60 days (9). Even though adult mammalian heart does not possess full regenerative capacity, it does has regenerative potential at a certain age and after specific stimulation (8) (Fig. 1). HF commonly originates from systolic HF due to lack of sufficient CM renewal following cardiac injury (10, 11). Recent landmark studies indicate that adult heart can produce new CMs (5, 6). The source of newly produced CMs is not yet evident, but pre-existing CMs or resident cardiac stem cells are thought to have a role in CM regeneration (5). During embryonic development and early postnatal period, CM proliferation (hyperplesia) is considered the main mechanism of cardiac growth. In the adult heart, cardiac hypertrophy becomes major growth mechanism subsequent to CM cell cycle arrest. Interestingly, cardiac development in Xenopus genus requires Hif-1a signaling, which acts upstream of Nkx2.5 (12). Concomitantly, it has also recently been demonstrated that adult mouse heart possesses hypoxic microenvironment where Hif-1a+ CMs are localized (labeled with TnnT) and glycolytic cardiac progenitors are housed in subepicardium (13). These hypoxic progenitors and CMs need to be further analyzed with lineage tracing studies to determine if they contribute to newly formed CMs. In addition, underlying mechanisms of CM turnover, role of CM proliferation, and contribution of extracardiac or resident stem cell population still remain to be determined (Fig. 1). Cardiogenic factors, micro ribonucleic acid molecules, antimiRs, and small molecules have been shown to induce cardiomyocyte proliferation and improvement of cardiac function following myocardial infarctions. In addition, studies showed that cellular therapies with stem cells could enhance renewal of cardiomyocytes.
Figure 1.
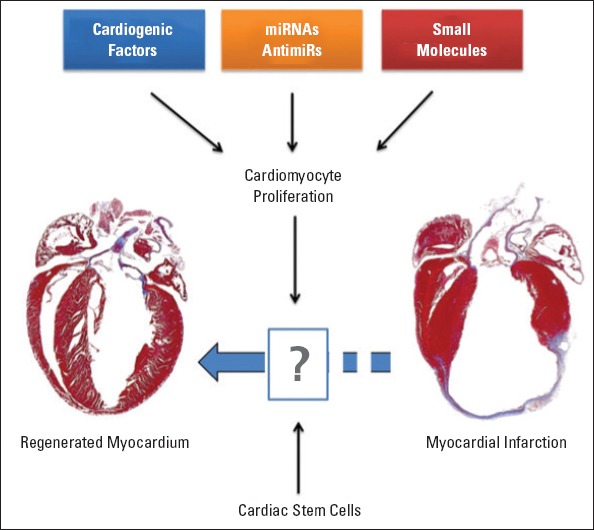
Therapeutic stimulation of resident cardiomyocyte cell cycle.
Cardiomyocyte renewal
Regeneration capacity varies from organism to organism. Zebrafish and newt demonstrate remarkable heart regeneration after 20% amputation of the ventricular apex. Regeneration capacity of both organisms has been reported as capability for complete restoration over 2-month period (14). Restoration of cardiac function has been associated with proliferation of dedifferentiated CMs, characterized by dissolution of sarcomeric structures (15). In newts, however, regeneration of heart involves blastema formation with accumulation of dedifferentiated cells near edge of lesion (16). In mammals, on the other hand, regeneration capacity of adult mammalian heart is highly restricted; instead, it responds to cardiac injury with scarring (fibrosis). However, fate mapping techniques have provided evidence that low rates of CM turnover occur following cardiac injury (6). Several studies have suggested that myocardial recovery and CM renewal after heart injury might originate in stem cells. One study of patients with heart transplants indicated that vascular cells and small percentage (0.016–0.04%) of newly formed CMs were host-derived (7).
Rate of CM renewal has been estimated using a number of approaches such as autoradiographic measurements of deoxyribonucleic acid (DNA) synthesis (17). According to Bergmann et al. (5) CM turnover rates were estimated at 1% and 0.4% per year at the age of 20 and 75 years of age, respectively. On the other hand, studies by Anversa group demonstrated that CM turnover rates were specified respectively at 7%, 12%, and 32% per year at 20, 60, and 100 years of age in men, while higher rates were estimated in females (18). Although these studies provided further evidence of existence of CM turnover in human heart, whether the source of CMs is existing or newly produced CMs is not completely understood. To this end, 5-Bromo-2’-deoxyuridine (BrdU), which incorporates into newly synthesized DNA, has been used to assess DNA synthesis in CMs. BrdU pulse-chase analysis and lineage tracing studies in neonatal mouse heart regeneration model indicated that origin of newly formed CMs is likely to be pre-existing CMs (19).
Neonatal heart regeneration
CM cell cycle and activity change over course of cardiac growth. Growth phases of CMs can be categorized developmentally as fetal life, postnatal, and adult period. During fetal life, CMs proliferate rapidly. In early postnatal period of heart, murine CMs become binucleated around postnatal day 7 to 10. However, hypertrophy becomes major form of growth in adult heart. When regeneration capacity was analyzed, neonatal and adult hearts demonstrated unequal capacity following injury (20). Recent studies have demonstrated that neonatal heart could regenerate without noticeable fibrosis or cardiac dysfunction after removal of up to 15% of the ventricle apex or MI at postnatal day 1 (P1) (21). However, at P7, regeneration capacity of the heart appears to be lost. Rather, there is adult-like response to heart injury with scar formation and cardiac dysfunction. Renewal of CMs that occurs following myocardial injury is not sufficient in adult mammalian heart. This deficiency has been considered the primary limiting factor for adult heart regeneration. Therefore, a number of studies have sought means to reactivate intrinsic proliferative capacity of adult CMs.
Reactivating cardiomyocyte cell cycle with cardiogenic factors
MI causes loss of CMs through apoptosis and necrosis. Ideal cardiovascular therapies aim to both reduce CM death and induce proliferation by manipulation of CM cell cycle. Cell cycle is a highly regulated, complex process; prominent regulators include cyclins, cyclin-dependent kinases (CDKs), CDK inhibitors (CDKIs), CDK-activating kinases (CAKs), and retinoblastoma (Rb) family. Cyclin/CDK complex is activated through periodic phosphorylation by CAKs. In addition, cell cycle is regulated by CDKIs including Cip/Kip family (p21Cip1, p27Kip1, p57Kip2) and Ink4 family members (p15Ink4b, p16Ink4a, p18Ink4c, p19Ink4d). Cell cycle comprises 4 phases: G1, S, G2, and M phases. There is also a Go phase when cells exit the cell cycle. Majority of adult CMs are arrested in Go/G1 cell cycle and marked by low proliferation index. Even though CDKIs are highly expressed and cell cycle activators are decreased in adult CMs, several reports have suggested that adult CMs can divide in injured heart (22). Studies have indicated that S phase progression in CMs can be induced with immunodepletion of p21Cip1 or deletion of CDKI p27Kip1 (23). In addition, overexpression or activation of E2F-1, cyclin D2, and cyclin D1 leads to DNA synthesis and CM mitosis, to some extent (Table 1) (19, 23–27, 28–36). Moreover, several studies have reported that growth factors and cytokines such as periostin, neuregulin, fibroblast growth factor 1, and oncostatin-M, as well as induction of Hippo pathway affect CM cell cycle progression (25–27, 37).
Table 1.
Factors involved in cardiomyocyte proliferation
| Factors | Examples | References |
|---|---|---|
| Growth factors | Periostin, neuregulin FGF1, oncostatin-M | (25–27, 28) |
| Cell cycle activators | Cdk2, c-myc, cyclin D2, Cyclin A4, cyclin D1, E2F-1 | (24, 29–32) |
| Gene deletion or inhibition | Meis1, antimiR-15, p27Kip1 and p21Cip1 | (19, 23, 33–36) |
Cdk2 - cyclin-dependent kinase 2; FGF1 - fibroblast growth factor 1; MEIS1 - meis homeobox 1
Cardiogenic miRNAs and anti-miRs
Micro ribonucleic acid (miRNA) molecules are small, single-strand, 22-nucleotide-long, non-coding RNAs. The most important role of miRNAs is target-specific inhibition of translation. This inhibition occurs through base pairing with specific binding sites located in the 3’ untranslated region (UTR) of specific mRNA targets. MicroRNAs have significant role in many cellular and biological processes such as cell proliferation, differentiation, and apoptosis, as well as cardiac development. Moreover, miRNAs act as dynamic regulators in cardiomyopathy and HF (38). In study of mice, CM-specific deletion of miRNA processing-associated genes such as Dicer and Dgcr8 with a-myosin heavy chain promoter-driven Cre recombinase and muscle creatine kinase promoter-driven Cre-recombinase (MCK–Cre) led to lethality through P0 and P4, respectively (38). When miRNA expressions are analyzed in the mammalian heart, miR-1 was found to be highly expressed (Table 2) (19, 37, 38–41). miR133 and miR-1 deletion in CMs negatively affects CM proliferation and differentiation. This effect occurs through modulation of various myogenic transcription factors including serum-response factor, myocyte-enhancer factor 2, myogenic differentiation factor D, and Nkx2.5 (42). An important cluster of miRNAs in heart development is miR-17Ø92 cluster. Overexpression of miR-17 leads to growth retardation of various organs including the heart. MiR-17Ø92 cluster also affects myocardial differentiation of cardiac progenitors (43). In addition, miRNA-dependent therapeutic strategies, especially using miR-199a and miR-590, induce CM proliferation though stimulation of cell cycle re-entry without inducing CM apoptosis (44). On the other hand, miR-15 family downregulates cell cycle genes and induces cell cycle arrest postnatally (45). miR-15 family not only affects heart regeneration, but also regulates mitochondrial functions. For instance, overexpression of miR195 inhibits numerous mitochondrial and cell cycle genes. All of these effects suggest that repression of miR-15 family could lead to delay in CM mitotic arrest with administration of antimiRs (44).
Table 2.
miRNAs and antimiRs in cardiac regeneration
| miRNA/AntimiR | Effect on cardiomyocytes | Targets | References |
|---|---|---|---|
| miR-199a KO | Proliferation | Hopx, Homer1c | (40) |
| miR-590 KO | Proliferation | Hopx, Homer1c | (40) |
| mir-17-92 cluster KO | Proliferation | PTEN | (41) |
| miR-15 antimiR treatment | Proliferation | miR-15 | (19) |
| miR-133 KO | Inhibition of proliferation | Ccnd2, SRF, Hand2 | (38) |
| miR-1 KO | Inhibition of proliferation | Hand2, PTEN | (38) |
| miR-15 KO | Inhibition of proliferation | Chek1, Arl2 | (38) |
Arl2 - adipose-ribosylation factor-like 2; Ccnd2 - cyclin D2; Chek1 - checkpoint kinase 1; Hand2 - heart and neural crest derivatives expressed 2; HOP homeobox; KO-knockout; miRNA/miR - micro ribonucleic acid; PTEN - phosphatase and tensin homolog; SRF – serum response factor
Cardiogenic small molecules
Small molecules are chemically defined as low molecular weight organic compounds with upper limit of 900 Da. Given that many drugs are small molecules, they possess many advantages in terms of diffusion through the cell membrane, flexibility, ease of production and storage. In addition, one of the appealing characteristics of small molecules is lack of immune response against them. Thus, they are more conceivable than recombinant proteins or nucleic acid reagents. Small molecules rapidly influence a variety of cellular compartments in a reversible manner. They have been shown to modulate processes such as self-renewal, differentiation, and reprogramming mechanisms (46).
Small molecules demonstrate multiple effects by manipulating target proteins and modulating enzymatic activities and signaling pathways. The identification of small molecules has been accelerated with improved understanding of their cellular mechanisms. High throughput screening has been utilized to characterize small molecules for a specific phenotype or activity. For instance, Sadek et al. (47) characterized activators of cardiac phenotype in stem cells (Table 3) (13, 46–50). 5-Azacytidine (5-AzaC) has been widely used for CM differentiation of various cell types including glycolytic cardiac progenitors and Sca-1+ cardiac progenitors. In addition, 5-AzaC and oxytocin-treated Sca-1+ cardiac progenitors demonstrated an increase in expression of cardiac factors and spontaneous beating in vitro. 6-bromoindirubin-3’-oxime (BIO), defined as an inhibitor of glycogen synthase kinase-3 (GSK-3) pathway, is one of the most important small molecules that play a role in CM cell cycle. BIO induces maintenance of self-renewal in embryonic stem cells and proliferation of differentiated CMs (48). BIO treatment increases cardiac proliferation of rat neonatal CMs through stimulation of S phase entrance in dose-dependent manner, which increases expression levels of Ki67, cyclin D1, cyclin A, and also enhances activity of ß-Catenin. In search for cardiogenic small molecules, screening tests were applied to various heart models and 4 small molecules were identified: NBI-31772, an insulin-like growth factor; smoothened agonist (SAG), an activator of the smoothened protein that plays a role in Hedgehog signaling pathway; SB203580, an inhibitor of p83 mitogen-activated protein kinase; and CHIR99021, a GSK-3ß inhibitor. CHIR99021 was also used to generate primitive streak cells from human embryonic stem cells that would then undergo differentiation into CMs (49). Another small molecule, dorsomorphin, increases expression of cardiogenic markers such as Nkx2.5, troponin-T and Mhy6 (50). Various members of sulfonyl-hydrazone (Shz) family were applied on human mobilized peripheral blood mononuclear cells, and Shz-1 and Shz-3 treatments were found to stimulate expression of CM markers such as Nkx2.5 in a dose-dependent manner (47). Lastly, skeletal muscle stem cells (skeletal myoblasts) were reprogrammed into skeletal myoblast-derived induced pluripotent stem (SiPS) cells after treatment with RG108, a DNA methyltransferase inhibitor. Transplantation of simulated SiPS from skeletal myoblasts without any genetic modification induced repair in damaged myocardium (46).
Table 3.
Cardiogenic small molecules
| Small molecules | Effect on cardiomyocytes | Reference |
|---|---|---|
| BIO | GSK-3 inhibitor. Induces proliferation of mammalian cardiomyocytes | (48) |
| 5-azacytidine | Induces cardiomyocyte differentiation of glycolytic cardiac progenitors | (13) |
| SAG, NBI-31772, SB-203580, and CHIR99021 | Drive cardiomyocyte proliferation | Reviewed in (49) |
| Dorsomorphin | Inhibits the BMP signaling and induces cardiomyocyte differentiation in mouse ESCs | (50) |
| sulfonyl-hydrazone | Induces cardiac differentiation in M-PBMCs | (47) |
| RG108 | Conversion of skeletal muscle stem cells into pluripotent state and use in cardiac regeneration | (46) |
BIO - 6-bromoindirubin-3’-oxime; BMP - bone morphogenetic proteins; ESCs - embryonic stem cells, GSK-3 - glycogen synthase kinase 3; M-PBMCs - mouse peripheral blood mononuclear cells; SAG - smoothened agonist
Approaches to cardiogenic small molecule discovery
Development of therapeutics targeting CM renewal in the injured myocardium may be achieved with discovery of CM-specific cell cycle modulators (Table 4). Thus, recent studies have sought to determine cardiogenic small molecules that provide reactivation of cell cycle in adult heart. Integration of a variety of scientific approaches is required to identify cardiogenic small molecules. Candidate small molecules may first be identified using in silico methods. These methods rely on identification of active residues of target protein and utilization of molecular docking programs. However, crystal structure of target protein must be known for in silico screening approaches. Thus, target proteins that are druggable with small molecules may be computationally screened and selected hits may be further validated with in vitro protein or cell- based assays. Microarray and proteomics techniques may be utilized during validation and target identification. In addition, flow cytometric or fluorescent microscopy techniques may be used in ex-vivo neonatal CM cultures in order to determine effects of small molecules on neonatal CM proliferation. However, target validations require preclinical studies in animals or humans. Ultimately, identified small molecules could be further validated in vivo using neonatal mouse cardiac regeneration model as well as adult myocardial injury models to assess effect on CM renewal and improvement of diastolic function.
Table 4.
Approaches to cardiogenic small molecule discovery
| Approaches | Method | Hurdles | Advantages | |
|---|---|---|---|---|
| In silico | Molecular docking of small molecules to active site of target protein | Requires crystal structure of targeted protein | A large library of druggable small molecules may be computationally screened for in vitro verification | |
| In vitro | Screening of small molecules against expressed target protein | Requires development of in vitro assay | A high throughput screening may be designed | |
| Ex vivo | Screening of small molecules inducing neonatal rat CM proliferation | Neonatal proliferating CMs are used instead of adult CMs | Flow cytometric or fluorescent microscopy techniques may be used to determine proliferating CMs using markers such as Nkx2.5 and Phospho-H3 | |
| In vivo | Injection of small molecule into mouse | Costly. Requires use of a large number of animals | Provides in vivo stimulation effect of injected small molecules toward CM renewal |
CM - cardiomyocyte
Conclusion
CM renewal has been documented in adult mammalian heart, albeit inadequate for restoration of cardiac function following cardiac injury. Cardiac regeneration in zebrafish, newt, and neonatal mouse is associated with reactivation of CM cell cycle. Discovery of CM cell cycle modulators provided a new platform for development of cardiovascular therapeutics targeting CM cell cycle. Studies have demonstrated that CM cell cycle could be induced with small molecules.
Use of small molecules or miRNA to stimulate cardiac cell proliferation brings up questions regarding their involvement in the induction of tumor formation. It is noteworthy that small molecule treatments are designed to be short term; thus, their effect will be transient. During this period, if intended CM proliferation achieved, then small molecule treatment could be halted to avoid any side effects such as unwanted induction of cellular proliferation in other cell types or uncontrolled cell growth in other tissues. Small molecules must be mutagenic to cause cancer formation and tumuorogenicity in any tissue; however, it is possible that long-term exposure to such stimulating small molecules along with exposure to mutagens could lead to accumulated mutations in various cell types and eventually raise issues of tumor formation. More studies are needed to determine timing, dose, and route of administration of small molecules.
De novo CM proliferation and differentiation are thought to be a prospect for cardiac regeneration. Manipulations used for CM cell cycle modulation have yielded DNA synthesis, karyokinesis and cytokinesis in the heart to some extent. Inducible knockout systems used in adult mouse models further demonstrated that CM cell cycle re-entry may be achieved in adult mammalian heart. Discovery of small molecules that trigger and promote differentiation of stem cells into CMs and induce CM cell cycle re-entry brought further excitement for development of therapies targeting MI and HF. Overall, studies have proven feasibility of resident CMs and stem cell recruitment following therapeutic stimulation in heart regeneration.
Footnotes
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – R.D.T., G.S.A., D.Y., R.D., F.K.; Design – F.K., R.D.; Supervision – F.K., R.D.; Data collection &/or processing – R.D.T., G.S.A., D.Y., R.D., F.K.; Literature search – R.D.T., G.S.A., D.Y., R.D., F.K.; Writing – R.D.T., G.S.A., D.Y., R.D., F.K.; Critical review – R.D.T., G.S.A., D.Y., R.D., F.K.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–402. doi: 10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- 3.Nadal-Ginard B. Generation of new cardiomyocytes in the adult heart: Prospects of myocardial regeneration as an alternative to cardiac transplantation. Rev Esp Cardiol. 2001;54:543–50. doi: 10.1016/s0300-8932(01)76354-3. [DOI] [PubMed] [Google Scholar]
- 4.Jones DW, Peterson ED, Bonow RO, Masoudi FA, Fonarow GC, Smith SC, Jr, et al. Translating research into practice for healthcare providers: the American Heart Association’s strategy for building healthier lives, free of cardiovascular diseases and stroke. Circulation. 2008;118:687–96. doi: 10.1161/CIRCULATIONAHA.108.189934. [DOI] [PubMed] [Google Scholar]
- 5.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, et al. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–4. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laflamme MA, Myerson D, Saffitz JE, Murry CE. Evidence for cardiomyocyte repopulation by extracardiac progenitors in transplanted human hearts. Circ Res. 2002;90:634–40. doi: 10.1161/01.res.0000014822.62629.eb. [DOI] [PubMed] [Google Scholar]
- 8.Tsonis PA. Regenerative biology: the emerging field of tissue repair and restoration. Differentiation. 2002;70:397–409. doi: 10.1046/j.1432-0436.2002.700802.x. [DOI] [PubMed] [Google Scholar]
- 9.Singh BN, Koyano-Nakagawa N, Garry JP, Weaver CV. Heart of newt: a recipe for regeneration. J Cardiovasc Transl Res. 2010;3:397–409. doi: 10.1007/s12265-010-9191-9. [DOI] [PubMed] [Google Scholar]
- 10.Kortekaas KA, Lindeman JH, Versteegh MI, Stijnen T, Dion RA, Klautz RJ. Preexisting heart failure is an underestimated risk factor in cardiac surgery. Neth Heart J. 2012;20:202–7. doi: 10.1007/s12471-012-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fritzenwanger M, Lorenz F, Jung C, Fabris M, Thude H, Barz D, et al. Differential number of CD34+, CD133+and CD34+/CD133+cells in peripheral blood of patients with congestive heart failure. Eur J Med Res. 2009;14:113–7. doi: 10.1186/2047-783X-14-3-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagao K, Taniyama Y, Kietzmann T, Doi T, Komuro I, Morishita R. HIF-1alpha signaling upstream of NKX2.5 is required for cardiac development in Xenopus. J Biol Chem. 2008;283:11841–9. doi: 10.1074/jbc.M702563200. [DOI] [PubMed] [Google Scholar]
- 13.Kocabaş F, Mahmoud AI, Sosic D, Porrello ER, Chen R, Garcia JA, et al. The hypoxic epicardial and subepicardial microenvironment. J Cardiovasc Transl Res. 2012;5:654–65. doi: 10.1007/s12265-012-9366-7. [DOI] [PubMed] [Google Scholar]
- 14.Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool. 1974;187:249–53. doi: 10.1002/jez.1401870208. [DOI] [PubMed] [Google Scholar]
- 15.Jopling C, Sleep E, Raya M, Marti M, Raya A, Izpisua Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–9. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laube F, Heister M, Scholz C, Borchardt T, Braun T. Re-programming of newt cardiomyocytes is induced by tissue regeneration. J Cell Sci. 2006;119:4719–29. doi: 10.1242/jcs.03252. [DOI] [PubMed] [Google Scholar]
- 17.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–6. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- 18.Steinhauser ML, Lee RT. Regeneration of the heart. EMBO Mol Med. 2011;3:701–12. doi: 10.1002/emmm.201100175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110:187–92. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–80. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–53. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ Res. 2002;90:1044–54. doi: 10.1161/01.res.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- 23.Poolman RA, Gilchrist R, Brooks G. Cell cycle profiles and expressions of p21CIP1 AND P27KIP1 during myocyte development. Int J Cardiol. 1998;67:133–42. doi: 10.1016/s0167-5273(98)00320-9. [DOI] [PubMed] [Google Scholar]
- 24.Jackson T, Allard MF, Sreenan CM, Doss LK, Bishop SP, Swain JL. The c-myc proto-oncogene regulates cardiac development in transgenic mice. Mol Cell Biol. 1990;10:3709–16. doi: 10.1128/mcb.10.7.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci USA. 2006;103:15546–51. doi: 10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–70. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 27.Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, et al. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med. 2007;13:962–9. doi: 10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- 28.Braun T, Dimmeler S. Breaking the silence: stimulating proliferation of adult cardiomyocytes. Dev Cell. 2009;17:151–3. doi: 10.1016/j.devcel.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 29.Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96:110–8. doi: 10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1) EMBO J. 1999;18:5310–20. doi: 10.1093/emboj/18.19.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, et al. Adenoviral delivery of E2F-1 directs cell cycle reentry and p53-independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest. 1997;100:2722–8. doi: 10.1172/JCI119817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–9. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 33.Poolman RA, Brooks G. Expressions and activities of cell cycle regulatory molecules during the transition from myocyte hyperplasia to hypertrophy. J Mol Cell Cardiol. 1998;30:2121–35. doi: 10.1006/jmcc.1998.0808. [DOI] [PubMed] [Google Scholar]
- 34.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–61. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poolman RA, Li JM, Durand B, Brooks G. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res. 1999;85:117–27. doi: 10.1161/01.res.85.2.117. [DOI] [PubMed] [Google Scholar]
- 36.Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol. 2013;14:529–41. doi: 10.1038/nrm3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan X, Hughes BG, Ali MA, Chan BY, Launier K, Schulz R. Matrix metalloproteinase-2 in oncostatin M-induced sarcomere degeneration in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2016;311:H183–9. doi: 10.1152/ajpheart.00229.2016. [DOI] [PubMed] [Google Scholar]
- 38.Porrello ER. microRNAs in cardiac development and regeneration. Clin Sci. 2013;125:151–66. doi: 10.1042/CS20130011. [DOI] [PubMed] [Google Scholar]
- 39.Rao PK, Toyama Y, Chiang HR, Gupta S, Bauer M, Medvid R, et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ Res. 2009;11:585–94. doi: 10.1161/CIRCRESAHA.109.200451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pouliquin P, Dulhunty AF. Homer and the ryanodine receptor. Eur Biophys J. 2009;39:91–102. doi: 10.1007/s00249-009-0494-1. [DOI] [PubMed] [Google Scholar]
- 41.Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, et al. miR-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res. 2013;112:1557–66. doi: 10.1161/CIRCRESAHA.112.300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, et al. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc Natl Acad Sci Uni S A. 2007;104:20844–9. doi: 10.1073/pnas.0710558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danielson LS, Park DS, Rotllan N, Chamorro-Jorganes A, Guijarro MV, Fernandez-Hernando C, et al. Cardiovascular dysregulation of miR-17-92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. 2013;27:1460–7. doi: 10.1096/fj.12-221994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–81. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 45.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim WH, Jung DW, Williams DR. Making cardiomyocytes with your chemistry set: Small molecule-induced cardiogenesis in somatic cells. World J Cardiol. 2015;7:125–33. doi: 10.4330/wjc.v7.i3.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sadek H, Hannack B, Choe E, Wang J, Latif S, Garry MG, et al. Cardiogenic small molecules that enhance myocardial repair by stem cells. Proc Natl Acad Sci U S A. 2008;105:6063–8. doi: 10.1073/pnas.0711507105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tseng AS, Engel FB, Keating MT. The GSK-3 inhibitor BIO promotes proliferation in mammalian cardiomyocytes. Chem Biol. 2006;13:957–63. doi: 10.1016/j.chembiol.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 49.Langle D, Halver J, Rathmer B, Willems E, Schade D. Small molecules targeting in vivo tissue regeneration. ACS Chem Biol. 2014;9:57–71. doi: 10.1021/cb4008277. [DOI] [PubMed] [Google Scholar]
- 50.Firestone AJ, Chen JK. Controlling destiny through chemistry: small-molecule regulators of cell fate. ACS Chem Biol. 2010;5:15–34. doi: 10.1021/cb900249y. [DOI] [PMC free article] [PubMed] [Google Scholar]