

Abstract
Objective:
Serum levels of nitric oxide (NO) are decreased in patients with atherosclerosis and also are a risk factor for the development of atherosclerosis. Endothelial dysfunction and diffuse atherosclerosis have been proposed for the etiology of coronary artery ectasia (CAE). The purpose of this clinical trial was to determine the relationship between CAE and serum NO levels.
Methods:
This prospective controlled study was conducted between January 2008 and March 2012. Serum levels of NO were compared in 40 patients with CAE (mean age 60.1±7.3 years) and 40 patients with normal coronary arteries (mean age 57.6±5 years) as a control group. CAE was diagnosed when a segment of coronary artery was more than 1.5 times the diameter of the adjacent healthy segment. Patients with stenotic atherosclerotic plaques, slow coronary flow, previous history of revascularization, acute coronary syndromes, left ventricular dysfunction, valvular heart disease, and systemic diseases were not included in the study. The effect of NO on the outcome was studied by constructing a receiver operating characteristic (ROC) curve with CAE as the primary variable. Effects of different variables on CAE were calculated using binary logistics regression analysis.
Results:
Serum NO concentrations were significantly lower in patients with CAE than in the control group (42.1±20.1 µmol/L vs. 77.3±15.7 µmol/L, p<0.001). According to the results of the multivariate regression analysis, LDL and NO levels were identified as independent factors associated with CAE (OR=1.02, 95% CI 1–1.04, p=0.02 and OR=0.88, 95% CI 0.83–0.93, p=0.001, respectively). ROC analysis revealed that using a cut-off point of 63.3, NO level predicts CAE with a sensitivity of 87.5% and specificity of 90%.
Conclusion:
Our study indicates that decreased levels of NO are present in patients with CAE compared to patients with normal coronary arteries, supporting the hypothesis that decreased levels of NO might be associated with CAE development. (Anatol J Cardiol 2016; 16: 947-52)
Keywords: coronary artery, ectasia, nitric oxide
Introduction
Coronary artery ectasia (CAE) is defined as diffuse or localized enlargement of the coronary arteries, with the luminal diameter becoming 1.5-fold larger or more than the normal adjacent segment (1). It is a relatively rare pathology in all of coronary artery anomalies, with the incidence varying from 0.3% to 4.9% in different cardiac catheterization studies (2–5). The etiopathogenesis of this clinical entity is poorly understood. However, CAE is frequently seen in association with atherosclerotic coronary artery disease (CAD), indicating an overlap in pathogenesis and risk factors. Despite the fact that atherosclerosis is accepted as the major clinical factor leading to the development of CAE in approximately 50% of the patients, connective tissue, inflammatory, and congenital pathologies have been proposed as potential causes (2, 5–9).
Chest pain is encountered as the main symptom of ectasia; however dysrhythmias, acute coronary syndromes, and sudden cardiac death are other possible clinical situations observed in ectasia (10–12). The most valuable method for diagnosing CEA is cardiac catheterization. The location, size, and extent of the pathology can be easily determined by angiography.
The endothelium plays a critical function in regulating vascular structure and physiology, primarily by producing nitric oxide (NO), which protects the vascular system as an endogenous defense against the development of atherosclerosis and its adverse clinical results. Failure of the physiological integrity of the endothelium, which is commonly seen as a result of an exposure to cardiovascular risk factors, results in decreased NO synthesis from the endothelium (13). Loss of endothelial integrity, characterized by diminished production of NO, is accepted as an important factor in the process of atherosclerosis and thrombosis (14). Although there are several studies in the literature investigating that decreased endothelial NO synthase leads to atherosclerosis via the proliferation of smooth muscle cells and adhesion of leukocytes, a direct relationship between CAE and NO levels is not examined by any previous study. Therefore, the objective of our study was to conduct a comparative analysis of serum NO concentrations in subjects with CAE and healthy controls.
Methods
The study was designed as a prospective protocol. This study was accepted by the institutional Ethics Committee, and all participants gave informed consent to participate in the trial.
A total of 3658 consecutive selective coronary angiograms were retrospectively evaluated between January 2008 and March 2012. The patients’ coronary angiograms were performed who presented to our outpatient clinic in Cardiology Department of Ankara University School of Medicine because of stable angina pectoris. We decided whether the patient had undergone coronary angiography according to a positive noninvasive test or high pretest probability for CAD. Totally, the total number of patients with CAE was 298, and 258 patients with CAE were excluded. Forty consecutive patients with CAE and without stenotic plaques as a study group (31 male and 9 female; mean age 60.1±7.3 years) and 40 patients with normal coronary angiographic findings as a control group (29 men and 11 women; mean age 57.6±5 years) were included in the study. Patients with stenotic atherosclerotic plaques and slow coronary flow were not included in the study. Stenotic plaques were accepted as lesions causing at least 50% obstruction in one of the coronary arteries. The other exclusion criteria were as follows: previous history of revascularization, acute coronary syndromes, LV dysfunction (LVEF <50%) and hypertrophy, valvular heart disease, cardiac rhythm other than sinus, history of liver (ALT and AST levels more than twice the upper limits) or renal pathology (serum creatinine >1.4 mg/dL), and active malignancy or systemic diseases.
Baseline characteristics, vital signs, laboratory findings, and coronary angiogram and echocardiography results were obtained from the hospital records. All patients were asked questions regarding any cardiovascular drug use and smoking habit prospectively. The body mass index (BMI) was recorded for all patients.
We categorized our patients as hypertensive if the blood pressure was ≥140/90 mm Hg or if they used antihypertensive medications. Chronic obstructive pulmonary disease was characterized as necessity medical therapy for chronic pulmonary pathology accompanied by moderate to severe obstruction. Diabetes mellitus and family history for CAD were also examined in all patients.
Laboratory analyses
Approximately 10 mL of venous blood sample was collected from each patient after a 12-h fasting period by venipuncture to analyze total blood counts, biochemical parameters, and NO levels 1 day after coronary angiography. Complete blood count, glucose, urea, creatinine, uric acid, and lipid profile were determined by standard methods.
For the vast majority of convenient methods, it is impossible to detect NO because of its volatile and transient character. However, as a result of NO oxidizing to nitrite (NO2–) and nitrate (NO3–), we used these anions’ concentrations as a quantitative measurement of NO production. We performed the spectrophotometric measurement of NO2– by performing the Griess Reaction after the conversion of NO3– to NO2–.
NO can be determined via this assay, which is achieved by nitrate reductase activity converses nitrate to nitrite. After the reaction, nitrite is detected by colorimetric methods as an azo dye product of the Griess reaction. NO concentration is indirectly measured by identifying both nitrite and nitrate levels in the specimen.
Coronary angiography
Standard selective coronary angiography was performed in all patients in the study. The coronary angiographies of the patients were evaluated by two experienced interventional consultants who were completely blind to the study. CAE was diagnosed when a segment of the coronary artery was more than 1.5 times the diameter of the adjacent healthy reference segment by visual assessment. The quantitative assessment was used very rarely, particularly in controversial cases.
Statistical analyses
Analyses were performed using the Statistical Package for the Social Sciences (SPSS) 13.0 (SPSS Inc., Chicago, IL, USA). Categorical variables were summarized as percentages, continuous variables were defined as mean±standard deviation (SD), and skewed data were reported as median (interquartile range). Groups were compared using chi-square tests for categorical variables, independent-samples student t-tests for normally distributed continuous variables, and Mann–Whitney U tests when the distribution was skewed. Pearson’s or Spearman’s correlation tests were used to evaluate correlations. The effect of NO on the outcome was studied by constructing a receiver operating characteristic (ROC) curve with CAE as the primary variable. Effects of different variables on CAE were calculated using binary logistics regression analysis. Variables that had an unadjusted p value <0.1 in binary logistic regression analysis were identified as potential risk markers and included in the full model. The model was reduced using stepwise multivariate logistic regression analyses, and we eliminated potential risk markers using likelihood ratio tests. A p value <0.05 was considered statistically significant, and the confidence interval was 95%.
Results
A comparison of patients with CAE (n=40) and those with normal angiographic findings (n=40) regarding baseline characteristics and laboratory findings is demonstrated in Table 1 and Table 2. There were no significant differences between the two groups in terms of age, sex, presence of hypertension, chronic obstructive pulmonary disease, smoking, diabetes mellitus, family history of CAD, BMI, and cardiovascular drug use. In addition, systolic and diastolic blood pressure; fasting plasma glucose; total LDL and HDL cholesterol; triglyceride and hemoglobin levels; platelet count; mean platelet volume; urea; creatinine, and uric acid levels; and LVEF did not significantly differ between the groups. However, serum NO concentrations were significantly lower in patients with CAE than in the control group (42.1±20.1 µmol/L vs. 77.3±15.7 µmol/L, p<0.001; Table 2).
Table 1.
Baseline characteristics of the study and control group. Groups were compared using chi-square tests for categorical variables, independent-samples student t-tests for normally distributed continuous variables, and Mann–Whitney U tests when the distribution was skewed
| Variable | Patients with CAE (n=40) | Control group (n=40) | P |
|---|---|---|---|
| Age, years | 60.1±7.3 | 57.6±5 | 0.07 |
| Male gender, % | 31 (77.5%) | 29 (72.5%) | 0.61 |
| Hypertension, % | 24 (60%) | 27 (67.5%) | 0.48 |
| COPD, % | 3 (7.5%) | 4 (10%) | 0.69 |
| Diabetes mellitus, % | 6 (15%) | 8 (20%) | 0.56 |
| Current smoker, % | 13 (32.5%) | 16 (40%) | 0.48 |
| Family history of CAD, % | 9 (22.5%) | 5 (12.5%) | 0.24 |
| Body mass index, kg/m2 | 29.1±4.2 | 28.6±4.3 | 0.61 |
| Beta blocker use, % | 22 (55%) | 20 (50%) | 0.65 |
| ACE-I use, % | 20 (50%) | 19 (47.5%) | 0.82 |
| ARB use, % | 5 (12.5%) | 8 (20%) | 0.36 |
| Statin use, % | 8 (20%) | 9 (22.5%) | 0.78 |
| ASA use, % | 2 (5%) | 5 (12.5%) | 0.43 |
ACE-I - angiotensin-converting enzyme inhibitors; ARB - angiotensin II receptor blockers; ASA - acetylsalicylic acid; CAD - coronary artery disease; CAE - coronary artery ectasia; COPD - chronic obstructive pulmonary disease
Table 2.
Baseline laboratory findings of the study and control group. Groups were compared using chi-square tests for categorical variables, independent-samples student t-tests for normally distributed continuous variables, and Mann–Whitney U tests when the distribution was skewed
| Variable | Patients with CAE (n=40) | Control group (n=40) | P |
|---|---|---|---|
| SBP, mm Hg | 108.1±19.3 | 113.9±21.7 | 0.28 |
| DBP, mm Hg | 59.7±9.2 | 63.4±12.8 | 0.15 |
| FPG, mg/dL | 105 (84–128) | 105 (87–144.75) | 0.27 |
| LDL, mg/dL | 97.7±37.9 | 88.8±44.2 | 0.33 |
| HDL, mg/dL | 38 (28.25–51.75) | 36 (33–39) | 0.32 |
| Triglycerides, mg/dL | 127 (87–194) | 114 (110.5–141.25) | 0.92 |
| Hemoglobin, mg/dL | 14.5±1.7 | 15.2±1.9 | 0.11 |
| MPV, fL | 8.8 (8.5–9.3) | 8.65 (8.5–9) | 0.35 |
| Platelet count, x 109/L | 217.8±48.9 | 227.6±34.3 | 0.46 |
| Uric acid, mg/dL | 6.3±1.6 | 7±1.8 | 0.07 |
| Urea, mg/dL | 13 (12–17) | 14.5 (12–19.75) | 0.38 |
| Creatinine, mg/dL | 0.79 (0.74–1.14) | 0.84 (0.79–0.92) | 0.35 |
| NO level, μM | 42.1±20.1 | 77.3±15.7 | P<0.001 |
| LVEF, % | 56.3±10.4 | 57.1±9.2 | 0.73 |
DBP - diastolic blood pressure; FPG - fasting plasma glucose; HDL - high-density lipoprotein cholesterol; LDL - low-density lipoprotein cholesterol; LVEF - left ventricular ejection fraction; MPV - mean platelet volume; NO - nitric oxide; SBP - systolic blood pressure
There were no significant correlations between the mean serum NO level and the other data categories (p>0.05) (Table 3).
Table 3.
Correlation between serum NO level and baseline characteristics and laboratory parameters of patients. Pearson’s or Spearman’s correlation tests were used to evaluate correlations
| Variable | NO level | |
|---|---|---|
| r | P | |
| Age | -0.06 | 0.61 |
| Gender | 0.01 | 0.95 |
| Hypertension | 0.09 | 0.43 |
| Diabetes mellitus | 0.03 | 0.78 |
| COPD | 0.01 | 0.99 |
| Current smoking | 0.06 | 0.59 |
| Family history of CAD | -0.14 | 0.23 |
| Body mass index | 0.07 | 0.54 |
| Beta blocker use | -0.08 | 0.47 |
| ACE-I use | 0.01 | 0.97 |
| ARB use | -0.02 | 0.86 |
| Statin use | 0.15 | 0.19 |
| ASA use | 0.14 | 0.22 |
| SBP | 0.21 | 0.07 |
| DBP | 0.11 | 0.35 |
| FPG | 0.04 | 0.75 |
| LDL | 0.08 | 0.47 |
| HDL | -0.02 | 0.88 |
| Triglycerides | -0.07 | 0.56 |
| Hemoglobin | 0.17 | 0.14 |
| MPV | 0.01 | 0.91 |
| Platelet count | 0.1 | 0.37 |
| Uric acid | 0.11 | 0.36 |
| Urea | 0.02 | 0.84 |
| Creatinine | -0.07 | 0.5 |
| LVEF | 0.09 | 0.4 |
ACE-I - angiotensin-converting enzyme inhibitors; ARB - angiotensin II receptor blockers; ASA - acetylsalicylic acid; CAD - coronary artery disease; COPD - chronic obstructive pulmonary disease; DBP - diastolic blood pressure; FPG - fasting plasma glucose; HDL - high-density lipoprotein cholesterol; LDL - low-density lipoprotein cholesterol; LVEF – left ventricular ejection fraction; MPV - mean platelet volume; SBP - systolic blood pressure
In binary logistic regression analysis, age (OR=1.17, 95% CI 0.98–1.39, p=0.09), LDL level (OR=1.02, 95% CI 1–1.04, p=0.07), and serum NO level (OR=0.88, 95% CI 0.82–0.93, p=0.001) revealed a significant correlation with CAE. When these three variables were included in a multivariate regression model, LDL and NO levels remained as independent factors associated with CAE (OR=1.02, 95% CI 1–1.04, p=0.02 and OR=0.88 95% CI 0.83–0.93, p=0.001, respectively) (Table 4).
Table 4.
Binary logistics regression analysis of possible predictors of coronary artery ectasia. Effects of different variables on CAE were calculated using binary logistics regression
| Binary logistics regression analysis | |||
|---|---|---|---|
| Variable | OR | 95% CI | P |
| Age | 1.17 | (0.98–1.39) | 0.09 |
| Gender | 1.42 | (0.13–15.4) | 0.77 |
| Hypertension | 0.37 | (0.04–3.46) | 0.38 |
| Diabetes mellitus | 0.51 | (0.05–5.23) | 0.57 |
| Family history of CAD | 0.68 | (0.05–8.88) | 0.77 |
| Current smoking | 0.21 | (0.02–2.51) | 0.22 |
| Body mass index | 1.09 | (0.88–1.34) | 0.43 |
| SBP | 1.01 | (0.96–1.05) | 0.73 |
| DBP | 0.94 | (0.86–1.03) | 0.19 |
| LDL level | 1.02 | (1–1.04) | 0.07 |
| HDL level | 1.02 | (0.98–1.06) | 0.23 |
| NO level | 0.88 | (0.82–0.93) | 0.001 |
CAD - coronary artery disease; DBP - diastolic blood pressure; HDL - high-density lipoprotein; LDL - low-density lipoprotein; NO - nitric oxide; SBP - systolic blood pressure
ROC analysis revealed that using a cut-off point of 63.3, NO level predicted CAE with a sensitivity of 87.5% and specificity of 90%. The area under the curve for this relationship was 0.93, and the 95% CI was 0.87–0.98 (Fig. 1).
Figure 1.
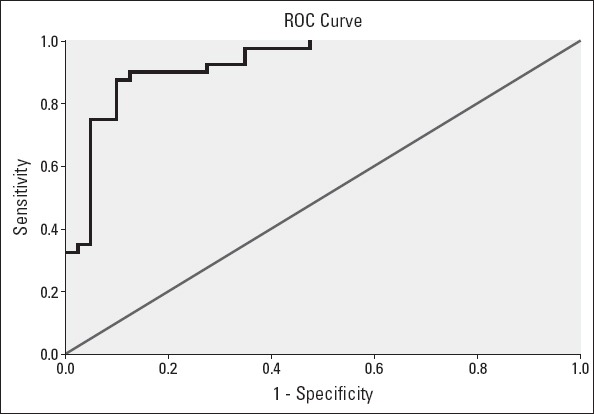
Receiver operating characteristic (ROC) curve for the determination of the cut-off for the NO level and coronary artery ectasia
Discussion
Our study demonstrates for the first time that serum concentrations of NO are decreased in patients who have CAE compared with control subjects. We also found that decreased NO levels remained as an independent risk factor associated with CAE.
The endothelium of the vascular tissue is involved in many different aspects of vascular physiology for the continuation of normal physiology. Endothelium-derived NO is a potent endogenous vasodilator, acting through the activation of the soluble guanylate cyclase by producing cyclic GMP (15–17). NO, playing a crucial role in flow-mediated vasodilation, is released from the endothelium mainly by the effect of endothelial shear stress.
A genetic deficiency or pharmacological inhibition of endothelial NO synthase (eNOS) can give rise to decreased endothelium-dependent vasodilation, leading to an increase in vascular resistance (18–20). Vascular actions of NO not only include regulating the vascular tone but also suppressing vascular smooth muscle proliferation, inhibiting platelet adhesion to the vascular endothelium, and interfering with leukocyte-endothelial cell interaction (21–26). In the process of vascular pathology, decreased NO activity is the precursor event leading to abnormal vasoconstriction and ischemia (27).
Apart from the direct measurement of NO, the serum concentrations of asymmetric dimethylarginine (ADMA), an endogenous competitive inhibitor of NO synthase, are studied as a novel risk factor for endothelial dysfunction in recent clinical trials. Increased ADMA levels are associated with reduced activities of NO synthase because NO synthesis from L-arginine is inhibited by endogenous methylated L-arginine analogues such as ADMA (28).
It has been shown that ADMA was elevated in young hypercholesterolemic individuals, and elevation of ADMA was associated with impaired endothelium-dependent vasodilatation in the forearm and reduced urinary nitrate excretion (29). Our study results also demonstrate that LDL levels remained as independent factors associated with the development of CAE as well as decreased NO levels. However, in our study, there was no significant difference between the groups concerning hyperlipidemia and cholesterol levels.
ADMA levels-CAE relationship has also been reported. Increased levels of ADMA are present in patients with CAE compared with patients with normal coronary arteries, supporting the hypothesis that ADMA might be a determinant factor of endothelial and structural dysfunction in the process of CAE development (28).
A polymorphism in the eNOS gene might be a contributing factor for the development of coronary heart diseases by reducing eNOS activity. Although the association of eNOS gene polymorphisms with the development and progression of CAD has been evaluated in several studies, data regarding its role in CAE are very scarce.
The c.894G>T polymorphism has been investigated in patients with CAE among the Turkish population (30). However, in this clinical trial, the effect of polymorphism on serum NO concentrations, which are believed to reflect endogenous production of NO, is not mentioned. In our study, direct measurements of serum NO levels are performed rather than investigating the possible responsible gene polymorphisms.
On the contrary, chronic overstimulation of the endothelium by NO is accepted as a causative mechanism in the development of CAE. Enhanced NO production is supplied by some chemical compounds that inhibits acetylcholinesterase, resulting in increased levels of acetylcholine and NO production (31). In these clinical entities, NO production is provided by the iNOS pathway; however, our trial results demonstrate that decreased NO levels in CAE patients occur because of the decreased eNOS activity associated with endothelial dysfunction.
The evidence for a causal association between oxidative stress and CAE development has been found in another study (32). The total anti-oxidant status decreased in patients with CAE, and it was also observed that the total oxidant status and the oxidative status index values were significantly higher in the study group than in the control group. Increased NO synthesis by directly enhancing the expression and activity of eNOS has been shown to significantly reduce oxidative stress and improve endothelial dysfunction (33).
The specific causative mechanisms of CAE are essentially unknown. However, CAE can be accepted as a type of atherosclerotic CAD because endothelial dysfunction plays a major role in the development of CAE. In contrast, NO is a protective agent against inflammation, oxidative stress, and endothelial dysfunction, which are possible causative entities in the pathogenesis of CAE. Therefore, increased bioavailability of NO can play a critical role in the prevention of CAE development.
Study limitations
This result is not supported by any clinical test demonstrating the relationship between endothelial dysfunction and decreased serum NO levels. Additionally, the small patient population is another limitation of this study. The diagnosis and exclusion of patients were performed mainly by visual assessment rather than using any quantitative methods such as intravascular ultrasound. We did not study any additional inflammatory markers such as CRP to analyse whether there is a correlation between the formation of CAE and inflammatory status.
Conclusion
In conclusion, our study indicates that decreased levels of NO are present in patients with CAE compared with those in patients with normal coronary arteries, supporting the hypothesis that decreased levels of NO might be associated with CAE development. These results provide the rationale for further studies with larger sample sizes that investigate the prognostic evaluation of decreased NO levels in patients with CAE during long-term follow-up.
Footnotes
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – A.G., K.E.; Design – Ç.E., Ö.U.Ö.; Supervision – Ö.F.Ç.; Fundings – P.A.A., G.Ö.K.; Materials – P.A.A., G.Ö.K.; Data collection &/or processing – Ö.U.Ö., P.A.A.; Analysis &/or interpretation – Ö.F.Ç., G.Ö.K.; Literature search – Ö.U.Ö., Ö.F.Ç.; Writing – A.G., Ç.E., K.E.; Critical review – A.G., Ç.E., K.E.
References
- 1.Seabra-Gomes R, Somerville J, Ross DN, Emanuel R, Parker DJ, Wong M. Congenital coronary artery aneurysms. Br Heart J. 1974;36:329–35. doi: 10.1136/hrt.36.4.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartnell GG, Parnell BM, Pridie RB. Coronary artery ectasia. Its prevalence and clinical significance in 4993 patients. Br Heart J. 1985;54:392–5. doi: 10.1136/hrt.54.4.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Markis JE, Joffe CD, Cohn PF, Feen DJ, Herman MV, Gorlin R. Clinical significance of coronary artery ectasia. Am J Cardiol. 1976;37:217–22. doi: 10.1016/0002-9149(76)90315-5. [DOI] [PubMed] [Google Scholar]
- 4.Oliveros RA, Falsetti HL, Carroll RJ, Heinle RA, Ryan GF. Arteriosclerotic coronary artery aneurysm. Report of five cases and a review of the literature. Arch Intern Med. 1974;134:1072–6. doi: 10.1001/archinte.134.6.1072. [DOI] [PubMed] [Google Scholar]
- 5.Swaye PS, Fisher LD, Litwin P, Vignola PA, Judkins MP, Kemp HG, et al. Aneurysmal coronary artery disease. Circulation. 1983;67:134–8. doi: 10.1161/01.cir.67.1.134. [DOI] [PubMed] [Google Scholar]
- 6.Kruger D, Stierle U, Herrmann G, Simon R, Sheikhzadeh A. Exercise-induced myocardial ischemia in isolated coronary artery ectasias and aneurysms (“dilated coronopathy”) J Am Coll Cardiol. 1999;34:1461–70. doi: 10.1016/s0735-1097(99)00375-7. [DOI] [PubMed] [Google Scholar]
- 7.Swanton RH, Thomas ML, Coltart DJ, Jenkins BS, Webb-Peploe MM, Williams BT. Coronary artery ectasia- a variant of occlusive coronary arteriosclerosis. Br Heart J. 1978;40:393–400. doi: 10.1136/hrt.40.4.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Befeler B, Aranda J, Embi A, Mullin FL, El-Sherif N, Lazzara R. Coronary artery aneurysms: study of the etiology, clinical course and effect on left ventricular function and prognosis. Am J Med. 1977;62:597–607. doi: 10.1016/0002-9343(77)90423-5. [DOI] [PubMed] [Google Scholar]
- 9.Schoenhagen P, Ziada KM, Vince DG, Nissen SE, Tuzcu EM. Arterial remodeling and coronary artery disease: The concept of “dilated” versus “obstructive” coronary atherosclerosis. J Am Coll Cardiol. 2001;38:297–306. doi: 10.1016/s0735-1097(01)01374-2. [DOI] [PubMed] [Google Scholar]
- 10.Sanyal S, Caccavo N. Is nitroglycerin detrimental in patients with coronary artery ectasia? A case report. Tex Heart Inst J. 1998;25:140–4. [PMC free article] [PubMed] [Google Scholar]
- 11.Mrdovic I, Jozic T, Asanin M, Perunicic J, Ostojic M. Myocardial reinfarction in a patient with coronary ectasia. Cardiology. 2004;102:32–4. doi: 10.1159/000077000. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg VD, Nepomnyashchikh LM. Pathomorphological peculiarities of coronary artery ectasias and their role in the pathogenesis of sudden cardiac death. Bull Exp Biol Med. 2004;138:515–21. doi: 10.1007/s10517-005-0085-9. [DOI] [PubMed] [Google Scholar]
- 13.Vincent MA, Montagnani M, Quon MJ. Molecular and physiologic actions of insulin related to production of nitric oxide in vascular endothelium. Curr Diab Rep. 2003;3:279–88. doi: 10.1007/s11892-003-0018-9. [DOI] [PubMed] [Google Scholar]
- 14.Sogo N, Magid KS, Shaw CA, Webb DJ, Megson IL. Inhibition of human platelet aggregation by nitric oxide donor drugs: relative contribution of cGMP-independent mechanisms. Biochem Biophys Res Commun. 2000;279:412–9. doi: 10.1006/bbrc.2000.3976. [DOI] [PubMed] [Google Scholar]
- 15.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–6. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 16.Murad F. The 1996 Albert Lasker Medical Research Awards. Signal transduction using nitric oxide and cyclic guanosine monophosphate. JAMA. 1996;276:1189–92. [PubMed] [Google Scholar]
- 17.Palmer RM, Ferrige AG, Moncada S. Nitric oxide accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–6. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 18.Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- 19.Cooke JP, Dzau VJ. Nitric oxide synthase: role in the genesis of vascular disease. Annu Rev Med. 1997;48:489–509. doi: 10.1146/annurev.med.48.1.489. [DOI] [PubMed] [Google Scholar]
- 20.Huang PL, Hyang Z, Mashimo H, Block KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–42. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 21.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromocyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:1774–7. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von der Leyen HE, Gibbons GH, Morishita R, Lewis NP, Zhang L, Nakajima M, et al. Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci U S A. 1995;92:1137–41. doi: 10.1073/pnas.92.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stamler J, Mendelsohn ME, Amarante P, Smick D, Andon N, Davies PF, et al. N-Acetylcysteine potentiates platelet inhibition by endothelium-derived relaxing factor. Circ Res. 1989;65:789–95. doi: 10.1161/01.res.65.3.789. [DOI] [PubMed] [Google Scholar]
- 24.Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;2:1057–8. doi: 10.1016/s0140-6736(87)91481-4. [DOI] [PubMed] [Google Scholar]
- 25.Bath PM, Hassall DG, Gladwin AM, Palmer RM, Martin JF. Nitric oxide and prostacyclin: Divergence of inhibitory effects on monocyte chemotaxis and adhesion to endothelium in vitro. Arterioscler Thromb. 1991;11:254–60. doi: 10.1161/01.atv.11.2.254. [DOI] [PubMed] [Google Scholar]
- 26.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–5. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nabel EG, Selwyn AP, Ganz P. Large coronary arteries in humans are responsive to changing blood flow: an endothelium-dependent mechanism that fails in patients with atherosclerosis. J Am Coll Cardiol. 1990;16:349–56. doi: 10.1016/0735-1097(90)90584-c. [DOI] [PubMed] [Google Scholar]
- 28.Çay S, Çağırcı G, Akçay M, Karakurt O, Kahraman E, Yazıhan N, et al. Asymmetric dimethylarginine levels in patients with coronary artery ectasia. Kardiol Pol. 2009;67:1362–8. [PubMed] [Google Scholar]
- 29.Böger RH, Bode-Böger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998;98:1842–7. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- 30.Arif Yalçın A, Faruk Aktürk I, Çelik O, Ertürk M, Sabri Hancer V, Yalçın B, et al. Coronary artery ectasia is associated with the c.894G>T (Glu298Asp) polymorphism of the endothelial nitric oxide synthase gene. Tohoku J Exp Med. 2014;232:137–44. doi: 10.1620/tjem.232.137. [DOI] [PubMed] [Google Scholar]
- 31.England JF. Herbicides and coronary artery ectasia (letter) M J Aust. 1981;68:260. doi: 10.5694/j.1326-5377.1981.tb100952.x. [DOI] [PubMed] [Google Scholar]
- 32.Sezen Y, Baş M, Polat M, Yıldız A, Büyükhatipoğlu H, Küçükdurmaz Z, et al. The relationship between oxidative stress and coronary artery ectasia. Cardiol J. 2010;17:488–94. [PubMed] [Google Scholar]
- 33.Cheang WS, Wong WT, Tian XY, Yang Q, Lee HK, He GW, et al. Endothelial nitric oxide synthase enhancer reduces oxidative stress and restores endothelial function in db/db mice. Cardiovasc Res. 2011;92:267–75. doi: 10.1093/cvr/cvr233. [DOI] [PubMed] [Google Scholar]