

Abstract
Mephedrone (MEPH) is a β-ketoamphetamine stimulant drug of abuse that is often a constituent of illicit bath salts formulations. Although MEPH bears remarkable similarities to methamphetamine (METH) in terms of chemical structure, as well as its neurochemical and behavioral effects, it has been shown to have a reduced neurotoxic profile compared with METH. The addition of a β-keto moiety and a 4-methyl ring substituent to METH yields MEPH, and a loss of direct neurotoxic potential. In the present study, two analogs of METH, methcathinone (MeCa) and 4-methylmethamphetamine (4MM), were assessed for their effects on mouse dopamine (DA) nerve endings to determine the relative contribution of each individual moiety to the loss of direct neurotoxicity in MEPH. Both MeCa and 4MM caused significant alterations in core body temperature as well as locomotor activity and stereotypy, but 4MM was found to elicit minimal dopaminergic toxicity only at the highest dose. By contrast, MeCa caused significant reductions in all markers of DA nerve-ending damage over a range of doses. These results lead to the conclusion that ring substitution at the 4-position profoundly reduces the neurotoxicity of METH, whereas the β-keto group has much less influence on this property. Although the mechanism(s) by which the 4-methyl substituent reduces METH-induced neurotoxicity remains unclear, it is speculated that this effect is mediated by a loss of DA-releasing action in MEPH and 4MM at the synaptic vesicle monoamine transporter, an effect that is thought to be critical for METH-induced neurotoxicity.
Introduction
β-Ketoamphetamines (bath salts) are an increasingly popular class of abused stimulants that represent an emerging public health concern. These compounds, including mephedrone (MEPH), methylone, and methylenedioxypyrovalerone (MDPV), are the β-keto analogs to the classic amphetamine drugs. Although now illegal, bath salts are still being frequently abused, primarily owing to their subjective effects, which include euphoria and increased sex drive (Johnson and Johnson, 2014). These effects are thought to be elicited by the release of dopamine (DA), serotonin (5-HT), and norepinephrine through their respective reuptake transporters, as the β-ketoamphetamines are known to have potent reuptake inhibition and releasing effects on these targets (Baumann et al., 2012; López-Arnau et al., 2012; Cameron et al., 2013; Eshleman et al., 2013; Simmler et al., 2013). The synthetic cathinones found in bath salts are less apt to produce lasting neurotoxicity compared with 3,4-methylenedioxymethamphetmine (MDMA) and methamphetamine (METH) (Angoa-Pérez et al., 2012; Baumann et al., 2012; den Hollander et al., 2013; Motbey et al., 2013; Anneken et al., 2015). This is despite a striking similarity in acute neurochemical and behavioral effects, including the aforementioned monoamine release, thermoregulation (Baumann et al., 2012; Fantegrossi et al., 2013; López-Arnau et al., 2014; Martínez-Clemente et al., 2014; Aarde et al., 2015; Kiyatkin et al., 2015; Shortall et al., 2016), hyperlocomotion (Baumann et al., 2012; López-Arnau et al., 2012; Marusich et al., 2012; Motbey et al., 2012; Wright et al., 2012; Aarde et al., 2013, 2015; Fantegrossi et al., 2013; Gatch et al., 2013), and measures of abuse potential (Hadlock et al., 2011; Lisek et al., 2012; Watterson et al., 2012; Aarde et al., 2013; 2015; Motbey et al., 2013; Karlsson et al., 2014).
These classic abused stimulants, MDMA and METH, share structures highly similar to the bath salts but have long been known to elicit lasting neurotoxicity. In mouse studies, this toxicity primarily impacts dopaminergic axons in the forebrain (Moratalla et al., 2015). METH elicits a pronounced depletion in both tissue DA content and in protein markers of dopaminergic terminals, including the dopamine transporter (DAT) and the synthetic enzyme tyrosine hydroxylase (TH) (McConnell et al., 2015). This toxicity is widely thought to be mediated by oxidative stress, as evidenced by increases in reactive oxygen and nitrogen species, resulting in damage to cellular components (Yamamoto and Raudensky, 2008). However, the key triggering mechanism for this cascade of damage has remained elusive. It has been proposed that these effects could be mediated by excessive DA release, mitochondrial dysfunction, or inflammation, among other inter-related processes (Halpin et al., 2014).
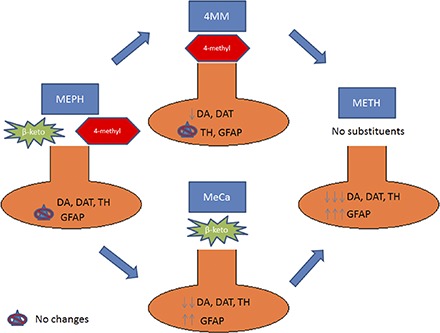
The divergence in persistent monoaminergic depletion between bath salts and amphetamines provides a unique opportunity to identify more precisely the key mechanism(s) responsible for long-term neurotoxicity. To this end, the bath salt MEPH can be particularly useful in studying METH toxicity. Although DA depletion when MEPH is given at elevated ambient temperatures has been reported, as well as a potentially toxic in vitro effect (den Hollander et al., 2014; Martínez-Clemente et al., 2014), numerous studies conducted in this laboratory and others under ambient conditions known to produce METH toxicity have reported no significant long-term in vivo dopaminergic neurotoxicity when MEPH is administered in a binge regimen (Angoa-Pérez et al., 2012, 2013; Baumann et al., 2012; den Hollander et al., 2013; Motbey et al., 2013; Anneken et al., 2015). As shown in Fig. 1, MEPH differs from METH by two structural modifications: 1) a β-keto substitution, and 2) the presence of a 4-methyl group on the phenyl ring. By utilizing two compounds, each carrying one of the two modifications, 4-methylmethamphetamine (4MM) and methcathinone (MeCa), we thought it may be possible to identify which of these substituents is responsible for the observed lack of direct toxicity in MEPH, and additionally to provide insight into the key mechanism that initiates the toxic effects of METH.
Fig. 1.

Comparative structures of METH, 4MM, MeCa, and MEPH. This figure demonstrates the structural differences between neurotoxic METH, non-neurotoxic MEPH, and their intermediates.
To date, no study has investigated the toxic potential of 4MM. A limited number of studies examining MeCa have documented dopaminergic toxicity, although there is variation in the effect sizes reported (Gygi et al., 1996, 1997; Sparago et al., 1996). Therefore, it was the objective of this study to compare the neurotoxicity of these two intermediates. Both compounds significantly altered body temperature and locomotion, but only MeCa evoked significant dopaminergic toxicity. These results suggest that ring substituents in bath salt components have more impact on lessening neurotoxic potential than does the β-keto moiety.
Materials and Methods
Drugs and Reagents.
(R,S)-N-Methcathinone HCl was obtained from the NIDA Research Resources Drug Supply Program. Racemic 4MM was synthesized as described by Davis et al. (2012) from methylamine HCL and 4-methylphenylacetone purchased from Alfa Aesar (ThermoFisher Scientific, Tewksbury, MA), and its structure was confirmed by NMR and mass spectrometry. DA, polyclonal antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and all buffers and high-performance liquid chromatography reagents were purchased from Sigma-Aldrich (St. Louis, MO). Bicinchoninic acid protein assay kits were obtained from Pierce (Rockford, IL). Polyclonal antibodies against rat TH were produced as previously described (Kuhn and Billingsley, 1987). Monoclonal antibodies against rat DAT were generously provided by Dr. Roxanne A. Vaughan (University of North Dakota, Grand Forks, ND). Polyclonal antibodies against glial fibrillary acidic protein (GFAP) were obtained from ThermoFisher Scientific (Rockford, IL). IRDye secondary antibodies for Odyssey Imaging Systems were purchased from LiCor Biosciences (Lincoln, NE).
Animals.
Female C57BL/6 mice (Harlan, Indianapolis, IN) weighing 18–25 g at the time of experimentation were housed 5–7 per cage in large shoe-box cages in a light- (12-hour light/dark) and temperature-controlled room. Female mice were used, as they are very sensitive to neurotoxicity induced by amphetamines and to maintain consistency with our previous studies of METH and β-ketoamphetamine interactions (Angoa-Pérez et al., 2012, 2013, 2014; Anneken et al., 2015). Mice had free access to food and water. The mice used were randomly divided into treatment groups. The Institutional Care and Use Committee of Wayne State University approved the animal care and experimental procedures. All procedures were also in compliance with the NIH Guide for the Care and Use of Laboratory Animals and were conducted in compliance with Animal Research: Reporting of In Vivo Experiments guidelines and under Institutional Animal Care and Use Committee–approved protocols.
Drug Treatments.
Mice were treated intraperitoneally with a total of four injections of drug in a binge-like regimen, with 2-hour intervals between each administration. Each injection contained either 4MM (2.5, 5, 10, 20, or 40 mg/kg), MeCa (10, 20, 40, or 80 mg/kg), or saline vehicle, given in 200 μl volume. The cumulative doses for 4MM over four injections were 10, 20, 40, 80, or 160 mg/kg, and for MeCa, they were 40, 80, 160, or 320 mg/kg. This binge treatment regimen has been established by multiple prior studies in this laboratory and others, and elicits significant neurotoxicity for amphetamine compounds. Doses of MeCa used fall within the range used in similar published studies (Gygi et al., 1996, 1997; Sparago et al., 1996), whereas the range of 4MM doses used were comparable to studies involving METH and other β-ketoamphetamines (Angoa-Pérez et al., 2012, 2013, 2014; Anneken et al., 2015). Mice were sacrificed 48 hours after the last drug treatment when amphetamine-associated neurotoxicity was known to reach maximal levels.
Determination of Striatal DA Content.
Striatal tissue was dissected from the brain after treatment and stored at –80°C. Frozen tissues were weighed and sonicated in 10 volumes of 0.16 N perchloric acid at 4°C. Insoluble protein was removed by centrifugation and DA was determined by high-performance liquid chromatography with electrochemical detection, as previously described (Angoa-Pérez et al., 2012, 2013).
Determination of DAT, TH, and GFAP Protein Levels by Immunoblotting.
The effects of drug treatments on striatal DAT and TH levels, markers highly specific for striatal DA nerve endings, were determined by immunoblotting as an index of toxicity. GFAP levels were determined as a marker of neuroinflammation, as described previously (Angoa-Pérez et al., 2012). Mice were sacrificed by decapitation 48 hours after treatment, and striata were dissected bilaterally. Tissue was stored at –80°C. Frozen tissue was disrupted by sonication in 1% SDS at 95°C, and insoluble material was removed by centrifugation. The concentration of soluble protein was determined by the bicinchoninic acid method, and equal amounts of protein (70 μg/lane) were resolved by SDS-PAGE and then electroblotted to nitrocellulose. Blots were blocked in Odyssey blocking buffer (phosphate-buffered saline) for 1 hour at room temperature. Primary antibodies against DAT (1:1000), TH (1:1000), GFAP (1:2000), or GAPDH (1:10,000) were added to blots and allowed to incubate overnight at 4°C. Blots were washed three times in Tris-buffered saline to remove unreacted antibodies and then incubated with IRDye secondary antibodies (1:4000) for 1 hour at room temperature. Immunoreactive bands were visualized by enhanced fluorescence, and the relative densities of TH-, DAT-, GFAP-, and GAPDH-reactive bands were determined by imaging with an Odyssey CLx Infrared Image System (LiCor Biosciences) and quantified using ImageJ software (NIH). TH, DAT, and GFAP relative densities were normalized to the GAPDH level for each lane to control for loading error.
Body Temperature.
Core body temperatures were monitored in treated animals by telemetry using IPTT-300 implantable temperature transponders from Bio Medic Data Systems, Inc. (Seaford, DE) inserted subcutaneously. Temperatures were recorded noninvasively every 20 minutes starting 60 minutes before the first drug injection and continuing for 9 hours thereafter using the DAS-5001 console system from Bio Medic. The averaged mean difference across all time points was calculated for each treatment compared with controls.
Locomotor Activity and Stereotypy.
Mice were treated with either vehicle, MeCa (20, 40, or 80 mg/kg), or 4MM (2.5, 5, 10, 20, or 40 mg/kg) using the same binge regimen described above. Immediately following each injection, mice were placed in locomotor activity monitoring cages (Omnitech Electronics, Inc., Columbus, OH), divided into 20 cm × 20 cm quadrants with accompanying inserts. Parameters including locomotor activity as measured in horizontal beam breaks, movement time, and stereotypy, as measured by vertical beam breaks, were tracked for 60 minutes using Fusion photo beams and software (Omnitech Electronics, Inc.). Animals were then placed into the home cage for 60 minutes of food and water access. This process was repeated for each animal throughout drug administration. The data acquired over 4 hours of monitoring were pooled for each animal, and averaged for each treatment group.
Data Analysis.
Group sizes, given in the figure legends, were chosen on the basis of prior experiments in this laboratory. Certain control groups are larger owing to pooling of multiple experiments. In Fig. 2, the group sizes given are for animals treated, but there is some variation in the group size analyzed for some markers owing to sample loss during processing. None of the data presented reflect replicates, only discretely treated individuals. The effects of drug treatments on core body temperature over time were analyzed using two-way analysis of variance (ANOVA), and post-hoc comparisons were carried out using Bonferroni’s test. One-way ANOVA was performed to analyze the dose effects of 4MM and MeCa on striatal levels of DA, DAT, TH, and GFAP, with post-hoc comparisons carried out using Bonferroni’s test. Locomotor effects of each drug were also analyzed using one-way ANOVA, and post-hoc tests were conducted with Bonferroni’s test. Differences were considered significant if p < 0.05. All statistical analyses were carried out using GraphPad Prism version 6.01 for Windows (GraphPad Software, San Diego, CA).
Fig. 2.

Effects of 4MM and MeCa on markers of dopaminergic toxicity. Mice were treated with 4MM (4 × 2.5, 5, 10, 20, or 40 mg/kg), MeCa (4 × 10, 20, 40, or 80 mg/kg), or saline vehicle every 2 hours for a total of four injections at the given dose. Levels of DA (A), DAT (B), TH (C), and GFAP (D) were determined 48 hours after drug exposure. Data are means ± S.E.M. (for 4MM: control, n = 18; 2.5 mg/kg, n = 8; 5 mg/kg, n = 8; 10 mg/kg, n = 9; 20 mg/kg, n = 12; 40 mg/kg, n = 12; for MeCa: control, n = 15; 10 mg/kg, n = 6; 20 mg/kg, n = 6; 40 mg/kg, n = 11; 80 mg/kg, n = 10). Each experiment was performed three times, with differing doses, and results were then pooled for analysis. METH and MEPH data were reprinted for comparison from Anneken et al. (2015) with permission from John Wiley and Sons, Inc. These data are not statistically analyzed in the present figure, but METH elicited highly significant toxic effects on each measure in the original experiments, whereas MEPH had no significant effects. One-way ANOVA, *P < 0.05 **P < 0.01 ***P < 0.001 ****P < 0.0001 compared with controls, with Bonferroni’s post-hoc test.
Results
Toxic Effects of 4MM and MeCa on Markers of Striatal Dopaminergic Nerve Endings.
The effects of 4MM and MeCa on DA, DAT, TH, and GFAP are shown in Fig. 2. Typical toxicity over a range of doses for METH (2.5–10 mg/kg) as well as MEPH (20–40 mg/kg) is also included from a prior publication and supplemental experiments from our laboratory for comparison (Anneken et al., 2015). There was very little toxicity elicited by 4MM, with only the highest dose evoking a significant reduction (20%) in striatal DA (Fig. 2A; F(5,53) = 3.32, P < 0.05), whereas all doses of MeCa with the exception of 10 mg/kg resulted in significant DA reductions (Fig. 2A; F(4,42) = 32.21, P < 0.0001). However, although 4MM at the highest dose showed only a nonsignificant trend of 10% reduction in DAT levels, MeCa reduced this marker significantly at the three highest doses assessed (Fig. 2B; F(4,39) = 25.72, P < 0.0001). 4MM had no significant effect on TH at any dose, but MeCa evoked a significant reduction at 20–80 mg/kg (Fig. 2C; F(4,39) = 21.90, P < 0.0001). Finally, no dose of 4MM had a significant effect on the inflammatory marker GFAP, but all doses of MeCa caused significant elevation in GFAP levels (Fig. 2D; F(4,39) = 18.44, P < 0.0001).
Effects of 4MM and MeCa on Core Body Temperature.
The body temperature effects of 4MM and MeCa were monitored for the duration of the experiments for the same cohorts of mice evaluated for dopaminergic toxicity. As shown in Fig. 3A, 4MM elicited a dose-dependent increase in body temperature over the course of binge treatment. A two-way ANOVA revealed highly significant main effects of treatment (F(5,19) = 144.0, P < 0.0001) and time (F(30,570) = 4.99, P < 0.0001), as well as a significant interaction of the two (F(150, 570) = 1.94, P < 0.0001). The results of a post-hoc comparison over the duration of the entire experiment using Bonferroni’s test is shown in Fig. 3B, with significance indicated over the mean difference of each treatment dose compared with controls across all time points. As demonstrated, all doses elicited significant hyperthermia compared with control animals with magnitudes increasing in a dose-dependent manner.
Fig. 3.

Effects of 4MM and MeCa on core body temperature. Mice were treated with (A) 4MM (4 × 2.5, 5, 10, 20, or 40 mg/kg), (C) MeCa (4 × 20, 40, or 80 mg/kg), or saline vehicle every 2 hours for a total of four injections. Body temperature was monitored beginning 40 minutes prior to the first injection via telemetry, and was measured at 20-minute intervals for the duration of the experiment. Data are mean temperature values with S.E.M. (<5% of the mean for each group) omitted for clarity (4MM: control, n = 4; 2.5–20 mg/kg, n = 4; 40 mg/kg, n = 5; for MeCa: control, n = 9; 20 mg/kg, n = 6; 40 mg/kg, n = 10; 80 mg/kg, n = 8). This experiment was performed once for 4MM, and repeated twice with differing doses for MeCa. The mean difference from control animals over the course of the entire experiment for each dose is shown in (B) and (D) ± S.E.M. Two-way repeated-measures ANOVA, ****P < 0.0001 compared with controls, with Bonferroni’s post-hoc test.
As shown in Fig. 3C, MeCa also significantly impacted core body temperature in a dose-dependent, albeit unusual, manner. Highly significant main effects of treatment (F(3,29) = 53.86, P < 0.0001) and time (F(30, 870) = 5.63, P < 0.0001) were revealed by two-way ANOVA, as was a significant interaction between the two factors (F(90,870) = 7.17, P < 0.0001). The results of a post-hoc comparison over the duration of the entire experiment using Bonferroni’s test is shown in Fig. 3D, with significance indicated over the mean difference of each treatment dose compared with controls across all time points. There was a significant hyperthermia elicited by 20 and 40 mg/kg, and at the 80 mg/kg dose there was a profound, persistent hypothermia following each injection of MeCa.
Effects of 4MM and MeCa on Locomotor Activity and Stereotypy.
As most stimulants are known to increase the amount of locomotor activity via increased DA release, separate cohorts of mice were administered the binge regimen of 4MM and MeCa, and were monitored for parameters of horizontal beam breaks, movement time, and stereotypy time over 4 hours total, measured for 1 hour at a time following each of the four injections.
As shown in Fig. 4, 4MM given at 5–20 mg/kg induced a significant increase in horizontal activity, as quantified by horizontal beam breaks (Fig. 4A; F(5,28) = 6.94, P < 0.001), whereas there were no significant changes in this parameter elicited by any dose of MeCa. Furthermore, all doses with the exception of the lowest of 4MM significantly increased the time spent in motion (Fig. 4B; F(5,28) = 12.48, P < 0.0001), but only the lowest dose assessed for MeCa increased this parameter (Fig. 4B; F(3,12) = 5.05, P < 0.05). Finally, there was a trend for increased stereotypy (i.e., rearing, grooming, etc.) as measured by time spent in vertical motion for most doses of 4MM, but this only achieved significance at 5 and 40 mg/kg (Fig. 4C; F(5,28) = 6.94, P < 0.01). All administered doses of MeCa (Fig. 4C; F(3,12) = 7.69, P < 0.01) caused a significant increase in stereotyped behavior, and these increases were of greater magnitude than those seen in 4MM.
Fig. 4.

Effects of 4MM and MeCa on locomotor behavior. Mice were treated with 4MM (4 × 2.5, 5, 10, 20, or 40 mg/kg), MeCa (4 × 20, 40, or 80 mg/kg), or saline vehicle every 2 hours for a total of four injections. Horizontal activity (A), movement time (B), and stereotypy time (C) were monitored for 60 minutes following each injection. Data are mean values ± S.E.M. (4MM: control, n = 10; 2.5 mg/kg, n = 4; 5–40 mg/kg, n = 5; for MeCa: all groups: n = 4). This experiment was performed twice for 4MM and once for MeCa with differing doses, though owing to equipment size constraints, each experiment was performed over consecutive days. One-way ANOVA, *P < 0.05 **P < 0.01 ****P < 0.0001 compared with controls, with Bonferroni’s post-hoc test.
Discussion
The studies presented in this report sought to dissect the individual contributions of two substituents, a 4-methyl group and a β-keto group, to observed differences in neurotoxicity between METH and its structural analog, the bath salt MEPH. The availability of structural analogs carrying the individual substituents, 4MM and MeCa, provided a means to probe the relative contributions of each group.
In Fig. 2, 4MM exhibited only a slight reduction in DA at the highest dose given (40 mg/kg), with no significant changes observed in DAT, TH, or GFAP at any dose. To our knowledge, this is the first investigation of the neurotoxic potential of this compound. Conversely, MeCa evoked a significant reduction in DA, DAT, and TH at 20–80 mg/kg, as well as a significant increase in GFAP at 10–80 mg/kg. The neurotoxicity of MeCa demonstrated here is in agreement with earlier binge studies that found depletions of dopaminergic markers at similar doses in the literature (30–120 mg/kg) (Gygi et al., 1996, 1997; Sparago et al., 1996). The depletions observed exceeded the MEPH effects, which were nonsignificant at 20–40 mg/kg but were not as pronounced as those evoked by METH in prior work in this laboratory, and required doses much higher than the 2.5- to 10-mg/kg binge dosing for METH needed for toxicity (Anneken et al., 2015).
Simmler et al. (2013) reported blood-brain barrier permeability for both MEPH and MeCa at nearly identical in vitro rates as METH, but METH is reported to have five times the brain-to-plasma ratio in vivo compared with MEPH, so it is possible that in vivo, decreased drug availability owing to the polarity of the β-keto substitution could account for the required higher doses and lessened toxicity. Alternatively, MEPH has lessened potency at the DAT compared with METH, owing to the presence of the β-keto group negatively impacting the binding affinity of these compounds for the neurotransmitter transporters, and thus requiring higher doses to elicit transmitter release (Simmler et al., 2013). As this excessive DA release is thought to contribute to the toxicity of METH by increasing the amount of oxidative stress and free radicals through DA metabolism, promoting persistent inflammatory activation and eventual synaptic damage (Thomas et al., 2008), it was important to investigate the other DA-dependent behavioral and physiologic effects of these drugs, such as body temperature and locomotor activity.
As shown in Fig. 3, a dose-dependent increase in core body temperature was observed with 4MM, and was significant for all doses tested. This effect peaked at 1.5–2°C, which is similar to but lower in magnitude than METH (Anneken et al., 2015). In animals given MeCa, there were also dose-dependent changes to core body temperature, but the patterns were different for either 4MM or METH. In the lower-dose groups, there was a significant increase in core temperature peaking at 1.5–2.0°C. However, at 80 mg/kg, marked hypothermia followed each drug injection, dropping to 3–4°C below control levels, and slowly recovering to baseline. MEPH, when administered to mice at 20 or 40 mg/kg as a binge regimen, causes a similar immediate 2–3°C reduction in body temperature (Angoa-Pérez et al., 2012). However, unlike MeCa, this recovers quickly and the overall effect of MEPH is to elicit a significant hyperthermia. It has been demonstrated by Shortall et al. (2016) that the initial hypothermia elicited by MEPH is attributable to serotonergic signaling through 5-HT1 receptors, as both preemptive 5-HT depletion and blockade of 5-HT1 receptors attenuated the hypothermic response. Thus it is possible that MeCa at 80 mg/kg interacts with the serotonergic system in a manner not possible at lower drug concentrations. In support of this hypothesis, Simmler et al. (2013) reported that MeCa has a relatively lower affinity for the 5-HT transporter, which could account for such a difference at higher dosage.
Hyperlocomotion and increased stereotypy are both hallmarks of stimulant drugs, including METH. Horizontal hyperlocomotion is primarily mediated by the increase in DA signaling elicited by METH treatment, as mice lacking in DAT expression or treated with pharmacological agents interfering with the ability of METH to release DA have been shown to attenuate the acute motor stimulant effect of the drug (Fukushima et al., 2007; Matsumoto et al., 2008; Yoo et al., 2008; Kaushal et al., 2011). Interestingly, it has been demonstrated by Glickstein and Schmauss (2004) that an intermediate dose of METH (5 mg/kg) in mice initially elicited an increase in horizontal locomotor activity, but after multiple injections in a binge regimen, mice exhibited predominantly stereotyped behavior, a change not observed in mice that lacked D2 DA receptors. Supporting the differential roles of D1 and D2 receptors, Kelly et al. (2008) reported a diminished but still significant increase in horizontal locomotor activity in D2 receptor-deficient mice, indicating a primary role for these receptors in mediating stereotypy but only a contributory role in hyperlocomotion. This is in agreement with other studies that implicate D2-receptor activation in stereotypy by abolishing the behaviors through pharmacological reduction of DA release (Mori et al., 2004; Tatsuta et al., 2006; Kitanaka et al., 2015).
As MEPH has also been shown by multiple groups to promote both hyperlocomotion and stereotypy (Kehr et al., 2011; Angoa-Pérez et al., 2012; Baumann et al., 2012), the intermediate compounds used in the present study were expected to elicit the same type of behaviors. As shown in Fig. 4, MeCa increased horizontal movement time significantly only at the lowest dose tested, whereas it greatly increased stereotypy at all doses. On the other hand, 4MM significantly increased horizontal activity at several doses, and although there was a trend toward increased stereotyped behavior, this only achieved significance at 5 mg/kg and 40 mg/kg, and was lesser in magnitude than the increases seen in MeCa. If, in fact, predominance of stereotypy over hyperlocomotion reflects greater DA release and preferential D2-receptor activation, then it would be reasonable to conclude that MeCa releases DA in greater amounts compared with 4MM since it elicits less horizontal and more stereotyped activity. In support of this conclusion, although there are no studies on 4MM release, an in vitro study found the magnitude of DA release mediated by MeCa to be similar to METH, and greater than that of MEPH (Simmler et al., 2013). Consequently, a greater release in DA would promote the hypothesis that MeCa would induce greater neurotoxicity than MEPH and/or 4MM, since it has been shown in multiple studies that increasing the amount of DA available for release enhances METH toxicity (Kita et al., 1995, Thomas et al., 2008, 2009). Indeed, the depletion levels reported above support this hypothesis.
The most unusual aspect of the observed MeCa toxicity is lesser toxicity at 80 mg/kg compared with 40 mg/kg. The most probable reason for this effect is the marked hypothermia observed at this dosage. Reductions in core body temperature have long been known to be protective against METH neurotoxicity (Ali et al., 1994, 1996). The drastic drop in body temperature elicited by MeCa may provide a slight level of neuroprotection in a similar manner at this higher dose.
The temperature and drug availability factors that may impact MeCa toxicity are not as relevant for 4MM, which almost completely lacks the toxicity of METH or even MeCa. It still elicits hyperthermia, and should readily cross the blood-brain barrier like METH, as it lacks the polar β-keto group. As its toxic profile is most similar to MEPH, it seems that the substitution on the phenyl ring is more crucial to MEPH’s lack of direct toxic potential than the β-keto substitution seen in MeCa.
It has been demonstrated by Eshleman et al. (2013) that MEPH lacks binding affinity for vesicular monoamine transporter-2 (VMAT2) compared with METH. METH binds this transporter in much the same way it does DAT, inhibiting and reversing its function, releasing vesicular DA into the cytoplasm where it can subsequently be released via DAT. Whereas METH can release both cytosolic and vesicular stores, MEPH is only able to access the newly synthesized DA in the cytosol, limiting the amount of neurotransmitter it can release. Increasing the available pool of DA by either VMAT2 inhibition or exogenous supplementation is known to potentiate the toxicity of METH (Thomas et al., 2008, 2009). Thus, it is feasible that the inability of MEPH to release DA via VMAT2 would blunt its neurotoxic potential. If this were the case, it would indicate that the crucial determining mechanism leading to lasting neurotoxicity is the release of a threshold amount of DA. It is tempting to speculate that the methyl substituent in MEPH and 4MM reduces its binding affinity for VMAT2, and thus its ability to release sufficient DA to initiate the neurotoxic cascade. The contribution of reduced DA availability and release to the observed lack of direct toxicity in these compounds is the focus of ongoing investigation in this laboratory. Should this prove fruitful, then therapeutic intervention for METH toxicity could target a specific, initiating event in the process, and potentially reduce the health care burden associated with METH abuse.
Acknowledgments
The authors thank Dr. Roxanne Vaughan for her generous gift of DAT antibodies, and the NIDA Drug Supply Program for providing methamphetamine and methcathinone for these and prior studies. All experiments were performed in compliance with ARRIVE guidelines.
Abbreviations
- 4MM
4-methylmethamphetamine
- 5-HT
serotonin
- ANOVA
analysis of variance
- DA
dopamine
- DAT
dopamine transporter
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GFAP
glial fibrillary acid protein
- MDVP
methylenedioxypyrovalerone
- MeCa
methcathinone
- MEPH
mephedrone
- METH
methamphetamine
- TH
tyrosine hydroxylase
- VMAT2
vesicular monoamine transporter-2
Authorship Contributions
Participated in research design: Anneken, Angoa-Pérez, Kuhn.
Conducted experiments: Anneken.
Contributed new reagents or analytic tools: Sati, Crich.
Performed data analysis: Anneken.
Wrote or contributed to the writing of the manuscript: Anneken, Angoa-Pérez, Sati, Crich, Kuhn.
Footnotes
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant DA 039667]. The authors declare no conflicts of interest.
References
- Aarde SM, Creehan KM, Vandewater SA, Dickerson TJ, Taffe MA. (2015) In vivo potency and efficacy of the novel cathinone α-pyrrolidinopentiophenone and 3,4-methylenedioxypyrovalerone: self-administration and locomotor stimulation in male rats. Psychopharmacology (Berl) 232:3045–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarde SM, Huang PK, Creehan KM, Dickerson TJ, Taffe MA. (2013) The novel recreational drug 3,4-methylenedioxypyrovalerone (MDPV) is a potent psychomotor stimulant: self-administration and locomotor activity in rats. Neuropharmacology 71:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SF, Newport GD, Holson RR, Slikker W, Jr, Bowyer JF. (1994) Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res 658:33–38. [DOI] [PubMed] [Google Scholar]
- Ali SF, Newport GD, Slikker W., Jr (1996) Methamphetamine-induced dopaminergic toxicity in mice. Role of environmental temperature and pharmacological agents. Ann N Y Acad Sci 801:187–198. [DOI] [PubMed] [Google Scholar]
- Angoa-Pérez M, Kane MJ, Briggs DI, Francescutti DM, Sykes CE, Shah MM, Thomas DM, Kuhn DM. (2013) Mephedrone does not damage dopamine nerve endings of the striatum, but enhances the neurotoxicity of methamphetamine, amphetamine, and MDMA. J Neurochem 125:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angoa-Pérez M, Kane MJ, Francescutti DM, Sykes KE, Shah MM, Mohammed AM, Thomas DM, Kuhn DM. (2012) Mephedrone, an abused psychoactive component of ‘bath salts’ and methamphetamine congener, does not cause neurotoxicity to dopamine nerve endings of the striatum. J Neurochem 120:1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angoa-Pérez M, Kane MJ, Herrera-Mundo N, Francescutti DM, Kuhn DM. (2014) Effects of combined treatment with mephedrone and methamphetamine or 3,4-methylenedioxymethamphetamine on serotonin nerve endings of the hippocampus. Life Sci 97:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anneken JH, Angoa-Pérez M, Kuhn DM. (2015) 3,4-Methylenedioxypyrovalerone prevents while methylone enhances methamphetamine-induced damage to dopamine nerve endings: β-ketoamphetamine modulation of neurotoxicity by the dopamine transporter. J Neurochem 133:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Ayestas MA, Jr, Partilla JS, Sink JR, Shulgin AT, Daley PF, Brandt SD, Rothman RB, Ruoho AE, Cozzi NV. (2012) The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37:1192–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron K, Kolanos R, Vekariya R, De Felice L, Glennon RA. (2013) Mephedrone and methylenedioxypyrovalerone (MDPV), major constituents of “bath salts,” produce opposite effects at the human dopamine transporter. Psychopharmacology (Berl) 227:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Blakey K, Rands-Trevor K. (2012) GC-MS and GC-IRD analysis of 2-, 3- and 4-methylmethamphetamine and 2-, 3- and 4-methylamphetamine. Forensic Sci Int 220:67–73. [DOI] [PubMed] [Google Scholar]
- den Hollander B, Rozov S, Linden AM, Uusi-Oukari M, Ojanperä I, Korpi ER. (2013) Long-term cognitive and neurochemical effects of “bath salt” designer drugs methylone and mephedrone. Pharmacol Biochem Behav 103:501–509. [DOI] [PubMed] [Google Scholar]
- den Hollander B, Sundström M, Pelander A, Ojanperä I, Mervaala E, Korpi ER, Kankuri E. (2014) Keto amphetamine toxicity-focus on the redox reactivity of the cathinone designer drug mephedrone. Toxicol Sci 141:120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman AJ, Wolfrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A. (2013) Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol 85:1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantegrossi WE, Gannon BM, Zimmerman SM, Rice KC. (2013) In vivo effects of abused ‘bath salt’ constituent 3,4-methylenedioxypyrovalerone (MDPV) in mice: drug discrimination, thermoregulation, and locomotor activity. Neuropsychopharmacology 38:563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima S, Shen H, Hata H, Ohara A, Ohmi K, Ikeda K, Numachi Y, Kobayashi H, Hall FS, Uhl GR, et al. (2007) Methamphetamine-induced locomotor activity and sensitization in dopamine transporter and vesicular monoamine transporter 2 double mutant mice. Psychopharmacology (Berl) 193:55–62. [DOI] [PubMed] [Google Scholar]
- Gatch MB, Taylor CM, Forster MJ. (2013) Locomotor stimulant and discriminative stimulus effects of ‘bath salt’ cathinones. Behav Pharmacol 24:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickstein SB, Schmauss C. (2004) Effect of methamphetamine on cognition and repetitive motor behavior of mice deficient for dopamine D2 and D3 receptors. Ann N Y Acad Sci 1025:110–118. [DOI] [PubMed] [Google Scholar]
- Gygi MP, Fleckenstein AE, Gibb JW, Hanson GR. (1997) Role of endogenous dopamine in the neurochemical deficits induced by methcathinone. J Pharmacol Exp Ther 283:1350–1355. [PubMed] [Google Scholar]
- Gygi MP, Gibb JW, Hanson GR. (1996) Methcathinone: an initial study of its effects on monoaminergic systems. J Pharmacol Exp Ther 276:1066–1072. [PubMed] [Google Scholar]
- Hadlock GC, Webb KM, McFadden LM, Chu PW, Ellis JD, Allen SC, Andrenyak DM, Vieira-Brock PL, German CL, Conrad KM, et al. (2011) 4-Methylmethcathinone (mephedrone): neuropharmacological effects of a designer stimulant of abuse. J Pharmacol Exp Ther 339:530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, Collins SA, Yamamoto BK. (2014) Neurotoxicity of methamphetamine and 3,4-methylenedioxymethamphetamine. Life Sci 97:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PS, Johnson MW. (2014) Investigation of “bath salts” use patterns within an online sample of users in the United States. J Psychoactive Drugs 46:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson L, Andersson M, Kronstrand R, Kugelberg FC. (2014) Mephedrone, methylone and 3,4-methylenedioxypyrovalerone (MDPV) induce conditioned place preference in mice. Basic Clin Pharmacol Toxicol 115:411–416. [DOI] [PubMed] [Google Scholar]
- Kaushal N, Seminerio MJ, Shaikh J, Medina MA, Mesangeau C, Wilson LL, McCurdy CR, Matsumoto RR. (2011) CM156, a high affinity sigma ligand, attenuates the stimulant and neurotoxic effects of methamphetamine in mice. Neuropharmacology 61:992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehr J, Ichinose F, Yoshitake S, Goiny M, Sievertsson T, Nyberg F, Yoshitake T. (2011) Mephedrone, compared with MDMA (ecstasy) and amphetamine, rapidly increases both dopamine and 5-HT levels in nucleus accumbens of awake rats. Br J Pharmacol 164:1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Low MJ, Rubinstein M, Phillips TJ. (2008) Role of dopamine D1-like receptors in methamphetamine locomotor responses of D2 receptor knockout mice. Genes Brain Behav 7:568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita T, Wagner GC, Philbert MA, King LA, Lowndes HE. (1995) Effects of pargyline and pyrogallol on the methamphetamine-induced dopamine depletion. Mol Chem Neuropathol 24:31–41. [DOI] [PubMed] [Google Scholar]
- Kitanaka N, Kitanaka J, Hall FS, Kayama M, Sugimori H, Uhl GR, Takemura M. (2015) Pretreatment or posttreatment with aripiprazole attenuates methamphetamine-induced stereotyped behavior in mice. J Exp Neurosci 9 (Suppl 1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin EA, Kim AH, Wakabayashi KT, Baumann MH, Shaham Y. (2015) Effects of social interaction and warm ambient temperature on brain hyperthermia induced by the designer drugs methylone and MDPV. Neuropsychopharmacology 40:436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn DM, Billingsley ML. (1987) Tyrosine hydroxylase: purification from PC-12 cells, characterization and production of antibodies. Neurochem Int 11:463–475. [DOI] [PubMed] [Google Scholar]
- Lisek R, Xu W, Yuvasheva E, Chiu YT, Reitz AB, Liu-Chen LY, Rawls SM. (2012) Mephedrone (‘bath salt’) elicits conditioned place preference and dopamine-sensitive motor activation. Drug Alcohol Depend 126:257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Arnau R, Martínez-Clemente J, Abad S, Pubill D, Camarasa J, Escubedo E. (2014) Repeated doses of methylone, a new drug of abuse, induce changes in serotonin and dopamine systems in the mouse. Psychopharmacology (Berl) 231:3119–3129. [DOI] [PubMed] [Google Scholar]
- López-Arnau R, Martínez-Clemente J, Pubill D, Escubedo E, Camarasa J. (2012) Comparative neuropharmacology of three psychostimulant cathinone derivatives: butylone, mephedrone and methylone. Br J Pharmacol 167:407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Clemente J, López-Arnau R, Abad S, Pubill D, Escubedo E, Camarasa J. (2014) Dose and time-dependent selective neurotoxicity induced by mephedrone in mice. PLoS One 9:e99002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusich JA, Grant KR, Blough BE, Wiley JL. (2012) Effects of synthetic cathinones contained in “bath salts” on motor behavior and a functional observational battery in mice. Neurotoxicology 33:1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto RR, Shaikh J, Wilson LL, Vedam S, Coop A. (2008) Attenuation of methamphetamine-induced effects through the antagonism of sigma (sigma) receptors: Evidence from in vivo and in vitro studies. Eur Neuropsychopharmacol 18:871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell SE, O’Banion MK, Cory-Slechta DA, Olschowka JA, Opanashuk LA. (2015) Characterization of binge-dosed methamphetamine-induced neurotoxicity and neuroinflammation. Neurotoxicology 50:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moratalla R, Khairnar A, Simola N, Granado N, García-Montes JR, Porceddu PF, Tizabi Y, Costa G, Morelli M. (2015) Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog Neurobiol DOI: 10.1016/j.pneurobio.2015.09.011. (published ahead of print). [DOI] [PubMed] [Google Scholar]
- Mori T, Ito S, Kita T, Sawaguchi T. (2004) Effects of dopamine- and serotonin-related compounds on methamphetamine-induced self-injurious behavior in mice. J Pharmacol Sci 96:459–464. [DOI] [PubMed] [Google Scholar]
- Motbey CP, Clemens KJ, Apetz N, Winstock AR, Ramsey J, Li KM, Wyatt N, Callaghan PD, Bowen MT, Cornish JL, et al. (2013) High levels of intravenous mephedrone (4-methylmethcathinone) self-administration in rats: neural consequences and comparison with methamphetamine. J Psychopharmacol 27:823–836. [DOI] [PubMed] [Google Scholar]
- Motbey CP, Karanges E, Li KM, Wilkinson S, Winstock AR, Ramsay J, Hicks C, Kendig MD, Wyatt N, Callaghan PD, et al. (2012) Mephedrone in adolescent rats: residual memory impairment and acute but not lasting 5-HT depletion. PLoS One 7:e45473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortall SE, Spicer CH, Ebling FJ, Green AR, Fone KC, King MV. (2016) Contribution of serotonin and dopamine to changes in core body temperature and locomotor activity in rats following repeated administration of mephedrone. Addict Biol 21:1127–1139. [DOI] [PubMed] [Google Scholar]
- Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME. (2013) Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol 168:458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparago M, Wlos J, Yuan J, Hatzidimitriou G, Tolliver J, Dal Cason TA, Katz J, Ricaurte G. (1996) Neurotoxic and pharmacologic studies on enantiomers of the N-methylated analog of cathinone (methcathinone): a new drug of abuse. J Pharmacol Exp Ther 279:1043–1052. [PubMed] [Google Scholar]
- Tatsuta T, Kitanaka N, Kitanaka J, Morita Y, Takemura M. (2006) Lobeline attenuates methamphetamine-induced stereotypy in adolescent mice. Neurochem Res 31:1359–1369. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. (2008) The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J Neurochem 105:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. (2009) Increases in cytoplasmic dopamine compromise the normal resistance of the nucleus accumbens to methamphetamine neurotoxicity. J Neurochem 109:1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson LR, Hood L, Sewalia K, Tomek SE, Yahn S, Johnson CT, Wegner S, Blough BE, Marusich JA, Olive MF. (2012) The reinforcing and rewarding effects of methylone, a synthetic cathinone commonly found in “bath salts”. J Addict Res Ther (suppl 9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MJ, Jr, Angrish D, Aarde SM, Barlow DJ, Buczynski MW, Creehan KM, Vandewater SA, Parsons LH, Houseknecht KL, Dickerson TJ, et al. (2012) Effect of ambient temperature on the thermoregulatory and locomotor stimulant effects of 4-methylmethcathinone in Wistar and Sprague-Dawley rats. PLoS One 7:e44652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Raudensky J. (2008) The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J Neuroimmune Pharmacol 3:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JH, Nam YS, Lee SY, Jang CG. (2008) Dopamine neurotransmission is involved in the attenuating effects of 5-HT3 receptor antagonist MDL 72222 on acute methamphetamine-induced locomotor hyperactivity in mice. Synapse 62:8–13. [DOI] [PubMed] [Google Scholar]