

Abstract
EMA401 is an old molecule, synthesized by Parke-Davis in the last century and characterized at that time as an AT2R antagonist. Professor Maree Smith and her group from the University of Queensland (Australia) patented the drug and many related derivatives and other compounds with high affinity for the AT2R for the indication neuropathic pain in 2004, an example of drug repositioning. After some years of university work, the Australian biotech company Spinifex Pharmaceuticals took over further research and development and characterized the S-enantiomer, code name EMA401, and related compounds in a variety of animal models for neuropathic and cancer pain. EMA401 was selected as the lead compound, based on high selectivity for the AT2R and good oral bioavailability (33%). EMA401, however, was only administered once in a chronic neuropathic pain model, and no data have been published in other pain models, or during steady state, although such data were available for the racemate EMA400 and some related compounds (EMA200, EMA400). A pilot phase IIa study demonstrated the efficacy and safety of the drug taken twice daily as two capsules of 50 mg (400 mg/day). In 2015, Novartis took over the clinical development. Two phase IIb studies designed by Spinifex Pharmaceuticals were put on hold, probably because Novartis wanted to improve the clinical design or collect additional preclinical data. Further data are eagerly awaited, especially since EMA401 is first-in-class in neuropathic pain.
Keywords: angiotensin II type 2 receptor, antagonist, neuropathic pain, EMA401, novel, development, drug repositioning
Introduction
In the last decade, various findings supported the existence of a separate angiotensinergic system in the nervous system in various parts of the brain, the dorsal root ganglia and the trigeminal ganglia.1,2 These were identified as pointers for developing putative analgesic drugs acting via this system. The University of Queensland, Australia, filed a patent in 2004 based on the work of Drs Maree Smith and Bruce Wyse from the Centre for Integrated Preclinical Drug Development for a number of AT2R antagonists as analgesics for neuropathic pain (the so-called “use patent”, claiming a new use for an old drug). One specific racemate (EMA400; Figure 1) was especially promising in preclinical studies. Its S-enantiomer, EMA401, has a binding affinity in the nanomolar range for the murine and human AT2R.3 Among others based on this high receptor affinity, EMA401 was taken into development by Spinifex Pharmaceuticals, a private Australian biotechnology company. In 2012, this company stated that they had strong efficacy data in multiple animal and human tissue models of neuropathic pain.4 Accordingly, they initiated further development and formulated EMA401 sodium salt capsules of 50 mg for b.i.d. dosing for the first human proof of principle studies in postherpetic neuralgia (PHN). Spinifex further announced in 2012 that EMA401 had demonstrated good human safety and pharmacokinetics in five phase I clinical studies and in a subsequent randomized clinical trial (RCT; the results of the clinical trial were published in 2014, see ‘EMA401: drug candidate for neuropathic pain’ section). The compound was found to be significantly better compared to placebo in terms of relieving PHN. EMA401 is unique in its class; there are no other AT2R antagonists as advanced in clinical development. It seems, however, that some hurdles related to patent and drug development issues might slow its further development in neuropathic pain.
Figure 1.
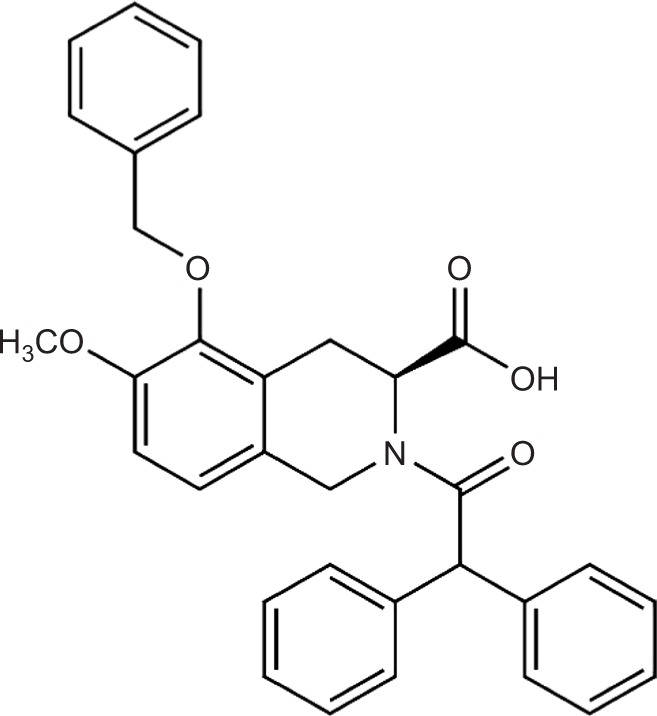
EMA401: structure of From the S-enantiomer of EMA400: (S)-2-(diphenylacetyl)-1,2,3,4-tetrahydro-6-methoxy-5-(phenylmethoxy)-3-isoquinoline carboxylic acid.
Note: Available from: http://www.chemspider.com/Chemical-Structure.8112918.html.
Discovery of AT2 antagonists as analgesics for neuropathic pain
Within the angiotensin system, two receptors have been recognized: angiotensin II type 1 receptor (AT1R) and AT2R. These receptors are phylogenetically old and have been identified in all vertebrate classes.5 Angiotensin II has similar binding properties at both receptors, but the receptors differ in their structure and function. The role of angiotensin II signaling via the AT1R was already established many years ago for the cardiovascular system.6 Drugs that antagonize the AT1R belong to the well-known category of antihypertensives.
In the 90s of the last century, both AT1R and AT2R were identified in the central nervous system,7,8 which led to suggestions to treat pain via this system.9 As early as 1993, AT2R antagonists were identified as having potential utility in the treatment and diagnosis of numerous neurological disorders, resulting in a patent by Warner–Lambert. The precise functions of the AT2R remained somewhat of an enigma and were difficult to explore at that time due to the fact that many ligands were mixed agonists of mixed antagonists.9,10 The Swedish group from the Department of Medicinal Chemistry, BMC, Uppsala University, claimed in 2004 to have synthesized the first pure AT2R agonist, codename M024/C21.11 Interestingly, AT2R agonists were found to have neuroprotective potential in a stroke model.12 It was indeed only after the development of pure agonists and antagonists that the field could further move forward.13 In 2013, AT2R expression was described, localized in small- and medium-sized cultured neurons of human and rat dorsal root ganglion, and EMA401 was found to be effective in a capsaicin-induced in vitro pain model.14
EMA400 (previously known as the Parke-Davis compound PD-126055) was originally described and synthesized as a derivative of a chemical structure named “compound 1” in a Warner–Lambert composition of matter (product) patent in 1993.15 EMA401, among a number of other derivatives, was identified in 2004 by the University of Queensland as relevant for the treatment of neuropathic pain. This group of antagonists were at that time claimed (in a method of use patent) as AT2R antagonists for “the treatment, prophylaxis, reversal and/or symptomatic relief of neuropathic pain, including mechanical hyperalgesia, thermal or mechanical allodynia, diabetic pain and entrapment pain.”16 The patent described how over a period of 15 years since the first identification of the AT2R, no clear insight into the function and the therapeutic utility of this receptor emerged. In the patent, AT2R antagonists were demonstrated and claimed to prevent or attenuate painful symptoms associated with neuropathic conditions and all such receptor antagonists were claimed in the patent, including small molecules, peptides, polypeptides, peptidomimetics, carbohydrates, and lipids. More recently, however, an international patent examiner identified a number of problems with several key claims in the patent, which were neither innovative nor inventive, due to prior findings in older patents from the pharmaceutical companies Warner–Lambert, Merck, and Bristol-Myers Squibb.17 In these older patents, a number of AT2R antagonists were disclosed, which were claimed to be of use in the treatment of peripheral neuropathies. Hopefully, this claim will not lead to premature abortion of the clinical project EMA401 in neuropathic pain. This would mean that the existing prior art could arrest further development of the compound in neuropathic pain, due to the absence of protection (of its intellectual property).
In the patent from the University of Queensland, various neuropathic pain models were described and used to screen a number of compounds, such as the old Parke-Davis compounds PD-123319 (EMA 200) and PD-126055 (EMA400). The AT2R-selective antagonist PD-126055 had been used earlier as a pharmacological tool to study apoptosis.18 Interestingly, the compound was described explicitly in the literature as an AT2R selective antagonist as early as 1994.19
PD-126055 or EMA400 is a racemate, and this compound was tested in different paradigms such as in diabetic rats and in the unilateral sciatic nerve chronic constriction injury (CCI) model. In the latter model, EMA400 was dosed as a bolus intraperitoneal (i.p.; in the range of 3–30 μg/kg) versus the vehicle. This led to a dose-dependent onset of anti-allodynia in the ipsilateral hind paw with peak responses observed at 0.75–1 hour post dosing, a duration of action of 1.5 hours at the lowest dose tested (3 μg/kg) and >3 hours for doses >3 μg/kg. Oral administration of 30 μg/kg to CCI rats resulted in comparable effects with a duration of action of ~3 hours.
EMA401: drug candidate for neuropathic pain
The first PubMed-indexed preclinical publication on the effects of EMA401 in an in vitro model was published in 2013 by two consultants and members of the Spinifex Scientific Advisory Board, together with scientists from the Imperial College (London, UK) and two other British institutes.3 EMA401 was presented as a member of the tetrahydroisoquinoline class of AT2R antagonists. The AT2R was found in that study to be expressed in human peripheral somatic and visceral nerves and was partly co-localized together with TRPV1 receptors. Sensitization of the TRPV1 ion channel increased after application of angiotensin II and could be reduced by the administration of EMA401. This inhibition of the capsaicin responses by 100 nmol/L EMA401 was comparable to the inhibitory effects of 1 mmol/L gabapentin and 10 mmol/L morphine. Angiotensin II is known to stimulate neurite outgrowth, and consistent with this observation, EMA401 leads to the opposite effects in a neurite outgrowth assay: both the neurite density and the length of the neurites decreased after administering the compound in concentrations of 10 and 100 nmol/L. However, how to interpret the effects on neurite branching and length, for instance in the light of patients suffering from small fiber neuropathic pain, is unclear. The absence of neurite vesiculation and disintegration under EMA401 treatment was seen as supportive for the absence of a neurotoxic effect. One might argue, however, that inhibition of neurite regeneration can be regarded as a neurotoxic effect. This clearly seems an issue that needs to be further explored.
In 2013, the investigators from the University of Queensland who designed the patent from 2004 published the first in vivo data on EMA401 and a series of EMA compounds characterized by high affinity for the AT2R (AT2R antagonists with >1000-fold selectivity over the AT1R). These compounds included EMA200, EMA300, EMA400, and the sodium salt S-enantiomer EMA401.20 Both the pharmacokinetics of these compounds and the results of the sciatic nerve CCI model were presented. Unfortunately, the authors only assessed the analgesic efficacy of single i.p. bolus doses of EMA200, EMA300, and EMA400 in the rat CCI model of the sciatic nerve and did not assess the effects of the drugs in steady state. Furthermore, they did not explore the lead compound EMA401 as a stand-alone compound in these studies. The S-enantiomer EMA401 had a 20- to 30-fold higher binding affinity than the R-enantiomer EMA402 for the AT2R. EMA401 was claimed to have better oral bioavailability (33%), but it was not clear from the text to what EMA compound the bioavailability was compared. The analgesic effect of EMA400 was ~60-fold higher compared to EMA300 and 250-fold higher than that of EMA200. Based on these data, EMA401 was selected as the lead compound for further development.
In 2014, supportive results of the first clinical phase II trial of EMA401 were published, supporting the efficacy and safety of EMA401 in patients with PHN.21 This study was a randomized, placebo-controlled study that included 183 participants from 29 centers across six countries. In the publication, the authors referred to data on file held by Spinifex Pharmaceuticals, related to the pharmacokinetic and pharmacodynamic results in animals and from human phase I studies. The authors maintained that these data, together with the in vitro functional assay data, supported a potentially efficacious dose of 10–50 mg/day in human beings. EMA401 was reported to have demonstrated sufficient safety in the phase I program up to a dose of 400 mg. Consequently, the authors chose a dose of 200 mg/day (administered as 200 mg b.i.d.) for the phase II trial. The change in mean pain intensity between baseline and the final week of treatment (days 22–28) was a 2.3-point reduction in the EMA401 group on the classical 11-point numerical rating scale (NRS-11), compared with a 1.6-point reduction in the placebo group, representing a statistically significant difference (p=0.0066). This difference was 0.7 points smaller than the 1 unit used in calculating the sample size for this study. However, statistical significant does not necessarily translate to clinical relevance. Therefore, the investigator conducted an explorative analysis, in which the “number needed to treat” to achieve 50% reduction on the NRS-11 was 6.7. There were no relevant side effects, including any cardiovascular safety concerns. There was one methodological issue identified by the authors: one patient at the day 1 pre dose had a blood plasma level of EMA401 of 1070 μg/L. The investigators were concerned about the mislabeling at the clinic site and called into question the labeling of the other samples. The study is expected to be followed by a phase IIb/III program, assessing efficacy and safety of doses of EMA401 >100 mg and EMA401 administered during a longer period of time. However, such a study (evaluating 200 and 600 mg/day EMA401 in PHN) was registered in ClinicalTrials.gov, but was withdrawn in 2015, as was a second clinical study of diabetic neuropathy. The information on the site stated for both studies was as follows: “study has been withdrawn prior to enrollment”.
A review article written by the chief executive officer of Spinifex Pharmaceuticals in 2014 discussed the mechanism of action and the phase II results, but did not present any preclinical in vivo data.22 Other data evidently are not available. In 2016, a review was published by Smith et al.23 The 10,000-fold specificity of EMA401 at the AT2R over the AT1R and the results of single-dose administrations of EMA200, EMA300, and EMA400 in the unilateral CCI model were discussed. A second rat model was presented: peripheral neuropathy induced by the antiretroviral drug dideoxycytidine (ddC) resulted in mechanical allodynia. The administration of an i.p. bolus dose of EMA200 (0.3–10 mg/kg) resulted in dose-dependent relief of mechanical hypersensitivity, comparable to gabapentin at a dose of 30 mg/kg. EMA200 was also tested in two cancer pain models. Further explorative studies in animal models for neuropathic pain evaluating EMA401 were not reported in that article.
Post-phase IIa development
With the positive proof of principle results of the RCT in PHN, a first clinical milestone was reached. However, the study was a simple one-dose phase IIa study, and no dose-finding study compared at least two different doses (high dose and low dose) to placebo. In the absence of a clear dose–response curve and without the identification of a no-effect dose, isolated positive findings related to one dose do not yet bear much weight. A phase IIb (dose-finding) and confirmative phase III trials are required, as well as additional data from dose-finding studies, preferably including kinetics, in a number of neuropathic pain models.
Novartis took over Spinifex in 2015 in a $700M buyout deal. In the Novartis 2016 brochure, the company wrote “We have also added therapeutic depth in neuroscience: EMA401; Neuropathic Pain”.24 In the investor presentation of January 27, 2016, it was communicated that the US Food and Drug Administration filing date for EMA401 was scheduled after the year 2020, which is certainly vague.25 There is silence from Novartis since the withdrawal of the two phase IIb studies. Let us hope this is based on the insight of Novartis experts regarding the need for a better dose-finding phase, additional explorative animal studies, or a once-daily formulation, rather than based on the issues of non-novelty and absence of innovation in one of the major patents protecting the development of EMA401.
Discussion
EMA401 is first in class as a high-affinity ligand for the AT2R. It is an old compound from the Parke-Davis laboratories and has been characterized in the previous century as a high-affinity AT2R antagonist. Preclinical experiments at the University of Queensland by Professor Maree Smith and colleagues supported the use of AT2R antagonists in neuropathic pain, and in a patent filed by the university in 2004, such antagonists were patented for the use in neuropathic pain (the so-called use patent). Subsequently, the patent examiner described “prior art” based on patents of pharmaceutical industries, and the novelty and innovation of a number of claims in the patent are now debated.
In preclinical experiments, supportive data for the entire EMA group of molecules in neuropathic pain and related models were generated, but only limited data have been presented for the lead compound EMA401. Data on chronic dosing and the dose–response curves in chronic neuropathic pain models have not yet been published. One issue for further exploration might be the fact that AT2R antagonists inhibit neuritic outgrowth, while agonists have been found to be neuroprotective. In a phase IIa study, EMA401 was significantly better compared to placebo after 3–4 weeks of oral administration of 100 mg b.i.d., although the numerical difference was <1 point on the NRS. It is anticipated that because Novartis took over development, extra preclinical and formulation work will be started, leading to a new design for the phase III program and possibly a once-daily dose formulation.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.McKinley MJ, Albiston AL, Allen AM, et al. The brain renin-angiotensin system: location and physiological roles. Int J Biochem Cell Biol. 2003;35(6):901–918. doi: 10.1016/s1357-2725(02)00306-0. [DOI] [PubMed] [Google Scholar]
- 2.Bohlender J, Imboden H. Angiotensinergic neurotransmission in the peripheral autonomic nervous system. Front Biosci. 2012;17:2419–2432. doi: 10.2741/4062. [DOI] [PubMed] [Google Scholar]
- 3.Smith MT, Wyse BD, Edwards SR. Small molecule angiotensin II type 2 receptor (AT2R) antagonists as novel analgesics for neuropathic pain: comparative pharmacokinetics, radioligand binding and efficacy in rats. Pain Med. 2013;14(5):692–705. doi: 10.1111/pme.12063. [DOI] [PubMed] [Google Scholar]
- 4.McCarthy T. Spinifex Pharmaceuticals EMA401: A Novel Treatment of Neuropathic Pain. Australasian Life Science Investment Summit. Melbourne, VIC, Australia: Nov 2, 2012. [Google Scholar]
- 5.Nishimura H. Renin-angiotensin system in vertebrates: phylogenetic view of structure and function. Anat Sci Int. 2016 Oct 7; doi: 10.1007/s12565-016-0372-8. Epub. [DOI] [PubMed] [Google Scholar]
- 6.Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol. 2006;20(5):953–970. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- 7.Tsutsumi K, Saavedra JM. Characterization and development of angiotensin II receptor subtypes (AT1 and AT2) in rat brain. Am J Physiol. 1991;261(1 pt 2):R209–R216. doi: 10.1152/ajpregu.1991.261.1.R209. [DOI] [PubMed] [Google Scholar]
- 8.Allen AM, Moeller I, Jenkins TA, et al. Angiotensin receptors in the nervous system. Brain Res Bull. 1998;47(1):17–28. doi: 10.1016/s0361-9230(98)00039-2. [DOI] [PubMed] [Google Scholar]
- 9.Pelegrini-da-Silva A, Martins AR, Prado WA. A new role for the renin-angiotensin system in the rat periaqueductal gray matter: angiotensin receptor-mediated modulation of nociception. Neuroscience. 2005;132(2):453–463. doi: 10.1016/j.neuroscience.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 10.Porrello ER, Delbridge LMD, Thomas WG. The angiotensin II type 2 (AT2) receptor: an enigmatic seven transmembrane receptor. Front Biosci. 2009;14:958–972. doi: 10.2741/3289. [DOI] [PubMed] [Google Scholar]
- 11.Wan Y, Wallinder C, Plouffe B, et al. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J Med Chem. 2004;47(24):5995–6008. doi: 10.1021/jm049715t. [DOI] [PubMed] [Google Scholar]
- 12.McCarthy CA, Vinh A, Callaway JK, Widdop RE. Angiotensin AT2 receptor stimulation causes neuroprotection in a conscious rat model of stroke. Stroke. 2009;40(4):1482–1489. doi: 10.1161/STROKEAHA.108.531509. [DOI] [PubMed] [Google Scholar]
- 13.Alterman M. Development of selective non-peptide angiotensin II type 2 receptor agonists. J Renin Angiotensin Aldosterone Syst. 2010;11(1):57–66. doi: 10.1177/1470320309347790. [DOI] [PubMed] [Google Scholar]
- 14.Anand U, Facer P, Yiangou Y, et al. Angiotensin II type 2 receptor (AT2 R) localization and antagonist-mediated inhibition of capsaicin responses and neurite outgrowth in human and rat sensory neurons. Eur J Pain. 2013;17(7):1012–1026. doi: 10.1002/j.1532-2149.2012.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blankley CJ, Hodges JC, Klutchko S, inventors. Warner-Lambert Company 5246943 A. Patent: substituted 1,2,3,4-tetahydroisoquinolines with angiotensin II receptor antagonist properties US. 1992 May 19;
- 16.Smith MT, Wyse BD, inventors The University Of Queensland. WO 2006066361 A1. Patent: method of treatment or prophylaxis. 2004 Dec 24;
- 17.England KG. PCT Written Opinion of the International Searching Authority. Apr 26, 2006. [Google Scholar]
- 18.Papp M, Li X, Zhuang J, Wang R, Uhal BD. Angiotensin receptor subtype AT(1) mediates alveolar epithelial cell apoptosis in response to ANG II. Am J Physiol Lung Cell Mol Physiol. 2002;282(4):L713–L718. doi: 10.1152/ajplung.00103.2001. [DOI] [PubMed] [Google Scholar]
- 19.Mantlo NB, Kim D, Ondeyka D, et al. Potent imidazo[4,5-b]- pyridine angiotensin II antagonists which exhibit balanced affinity to the AT1 and AT2 receptor subtypes. Bioorg Med Chem Lett. 1994;4:17–22. [Google Scholar]
- 20.Smith MT, Wyse BD, Edwards SR. Small molecule angiotensin II type 2 receptor (AT2R) antagonists as novel analgesics for neuropathic pain: comparative pharmacokinetics, radioligand binding, and efficacy in rats. Pain Med. 2013;14(5):692–705. doi: 10.1111/pme.12063. [DOI] [PubMed] [Google Scholar]
- 21.Rice AS, Dworkin RH, McCarthy TD, et al. EMA401-003 study group. EMA401, an orally administered highly selective angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: a randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet. 2014;383(9929):1637–1647. doi: 10.1016/S0140-6736(13)62337-5. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy T. Development of EMA401 as an orally-administered, highly-selective angiotensin II type 2 receptor antagonist for the treatment of neuropathic pain. J Peripher Nerv Syst. 2014;19(Suppl 2):S13–S14. doi: 10.1111/jns.12080_4. [DOI] [PubMed] [Google Scholar]
- 23.Smith MT, Anand P, Rice AS. Selective small molecule angiotensin II type 2 receptor antagonists for neuropathic pain: preclinical and clinical studies. Pain. 2016;157(Suppl 1):S33–S41. doi: 10.1097/j.pain.0000000000000369. [DOI] [PubMed] [Google Scholar]
- 24.Novartis, editor. Reimagining Medicine. [Accessed January 27, 2016]. Available from: https://www.novartis.com/sites/www.novartis.com/files/2016-01-novartis-jpm-presentation.pdf.
- 25.Novartis Q4 and Full Year 2015 Results. [Accessed January 27, 2016]. Available from: https://www.novartis.com/sites/www.novartis.com/files/q4-2015-ir-presentation.pdf.