

Abstract
The cellular concentrations of certain metabolites are assiduously monitored to achieve appropriate levels of gene expression. While proteins have long been known to act as sensors in this capacity, metabolite-binding RNAs, or riboswitches, also play an important role. More than 20 distinct classes of riboswitches have been identified to date, and insights to the molecular recognition strategies of a significant subset of these have been provided by detailed structural studies. This diverse set of metabolite-sensing RNAs is found to exploit a variety of distinct mechanisms to regulate genes that are fundamental to metabolism.
Keywords: aptamer, expression platform, ribozyme, binding pocket
INTRODUCTION
The functions of the major biopolymers have historically been sorted neatly into general categories — nucleic acids as sets of instructions, proteins as agents of catalysis and molecular recognition, and carbohydrates as structural matrices and units of energy storage. With the discoveries of novel roles for some of these polymers, however, the boundaries separating their narrowly defined categories are becoming increasingly blurred. Our understanding of the functional capacity of RNA molecules in particular has recently undergone a tremendous expansion. The notion of RNA as merely a messenger having long been dispelled, this class of biopolymer is now appreciated as serving critical structural and catalytic roles in ribonucleoprotein complexes such as the ribosome, the spliceosome, the signal recognition particle, and telomerase (1–5). Extensive biochemical and structural studies of the small self-cleaving ribozymes, RNase P, and the group I and group II self-splicing introns have established firmly that RNA can achieve rate enhancements comparable to those of protein enzymes (6). Further, the catalytic repertoire of natural RNA enzymes is not limited only to transesterification and hydrolysis of the phosphate backbone, as evidenced by atomic resolution structures of the ribosome revealing the presence only of RNA at the peptidyl transferase center (7, 8).
The capabilities of RNA molecules are thus not partitioned as strictly as previously considered, and this realization has lent strong support to the notion that this polymer was the predominant macromolecule during a more primitive era in the evolution of life on earth, a period widely referred to as the RNA world. Having a chemical composition that predisposes it to the transmission of genetic information, but that also allows for the formation of complex three-dimensional structures that can mediate chemical transformations, RNA by itself satisfies two of the major requirements for a system capable of Darwinian evolution. But catalysis without substrate specificity is futile; any robust network of self-sustaining chemical reactions relies as much on accurate molecular recognition as it does on rate enhancement. Contemporary protein enzymes and regulatory factors have mastered this ability, fashioning active sites and binding pockets that recognize metabolites and other substrates with exquisite specificity. It seems unlikely, then, that an RNA world of even moderate complexity would lack this fundamental capability.
In fact, there were early indications from a limited number of natural examples that specific interactions could occur between relatively large RNAs and small molecules. Group I introns, for example, were known selectively to utilize free guanosine as a cofactor in self-splicing reactions (9, 10), and different classes of antibiotic compounds had been demonstrated to interact with distinct sets of phylogenetically conserved nucleotides in 16S ribosomal RNA (11). Corroboration of the molecular recognition capability of RNA subsequently emerged from demonstrations that RNA molecules synthesized in the laboratory could serve as highly selective receptors for small molecules. Using the powerful techniques of in vitro selection, individual molecules with desired binding properties could be isolated from large random-sequence pools. And RNA proved quite versatile in this capacity: many such receptor RNAs, commonly referred to as aptamers, were selected that targeted a variety of compounds, including dyes, coenzymes, amino acids and nucleotides (12).
Yet the relative ease with which aptamers could be identified in the laboratory was seemingly at odds with the apparent paucity of such RNAs in nature. It was as if metabolite-binding RNAs, presumably abundant during the RNA world era, had been lost during the course of evolution as a result of competition with protein factors, in much the same way that most primordial roles of RNA catalysts are thought to have been supplanted by protein enzymes.
Over the past several years, however, a large repository of naturally occurring RNA aptamers has been revealed in eubacterial mRNA. Typically residing in 5′ untranslated regions (UTRs), these RNA receptors serve as components of cis-acting genetic control modules called riboswitches (13). More than 20 distinct classes of these natural aptamers are currently known, recognizing a diverse array of fundamental metabolites including coenzymes, amino acids, nucleobases and an aminosugar. Many riboswitch classes are widespread among eubacteria, and are generally utilized in the feedback repression of genes involved in the synthesis or import of the cognate metabolite. To control gene expression, the aptamer domain interacts with a portion of the riboswitch termed the expression platform, which in turn provides the interface with various other components of the gene expression apparatus. Necessarily, these two domains partially overlap each other, in a way that allows the presence of the cognate metabolite to influence which of two mutually exclusive riboswitch structures is adopted.
One of the more common expression platforms employed by riboswitches is an intrinsic transcription terminator, a stable stem-loop structure typically followed by between five and nine consecutive uridines (14, 15). Upon encountering such structures, ternary elongation complexes of RNA polymerase rapidly dissociate, halting transcription. Because the occupation state of the aptamer domain dictates whether the intrinsic terminator or a competing RNA structure will be formed, the concentration of the cognate metabolite directs the decision whether to abort transcription prematurely or to read through into the open reading frame. Another frequently encountered type of expression platform among eubacteria involves RNA structures that modulate the efficiency of translation initiation. Riboswitches can harness metabolite binding to regulate the accessibility of the Shine-Dalgarno (SD) sequence, a short, purine-rich span that recruits 30S ribosomal subunits to mRNA sites near AUG start codons.
Thus, one of the attributes of RNA polymers generally considered a liability — their propensity to sample alternative folds (16) — is exploited by riboswitches as the very crux of their mechanism. The folding pathway decision is contingent upon structure stabilization provided by molecular contacts with the target, so that the intracellular concentration of free ligand tips the balance in favor of one regulatory outcome or the other.
STRUCTURES OF NATURAL APTAMERS
The evolutionary constraints shaping riboswitch aptamer domains and expression platforms differ markedly. Components of the latter, such as intrinsic terminators and SD sequestors, generally require certain secondary structures to be maintained but exhibit very little sequence conservation. As a result, expression platforms of various types may easily be combined with the myriad classes of natural aptamers in a modular fashion, since interdomain communication tends to rely primarily on simple Watson-Crick pairing interactions.
In stark contrast to the expression platforms with which they interface, natural metabolite-binding RNAs are exceedingly conserved elements. Each class of riboswitch aptamer exhibits a characteristic arrangement of secondary structure elements and joining sequences that are arrayed precisely in three dimensions, typically using highly conserved residues to mediate tertiary interactions as well as direct contacts to the ligand. The high degree of conservation of natural aptamers is partly attributable to the immutability of the compounds they recognize. In addition, these RNA motifs are likely to represent evolutionary optima, at least with regard to neighboring sequences on the fitness landscape, since they have been subjected to persistent and stringent evolutionary pressure. Therefore, significant penalties would be expected for aptamers in which mutations occur in nucleotides critical to the organization of the binding pocket.
In the past several years, a number of atomic-resolution models have been generated for natural aptamers complexed with their ligands, revealing an abundance of molecular recognition strategies available to RNA (17, 18). Not surprisingly, these RNA receptors bind their targets by relying on many of the chemical strategies employed by proteins in substrate recognition, including hydrogen-bonding, electrostatic, van der Waals, and stacking interactions. The roles of riboswitches as physiological sensors, where their responses must be tuned uniquely to a single cognate ligand amid a complex milieu, demand stringent selectivity. Crystallographic and other biochemical evidence has demonstrated that riboswitch aptamers achieve this level of selectivity by interacting with nearly all available functional groups of the metabolite. This high degree of ligand envelopment by natural aptamers implies that sufficient local disorder or flexibility exists in the unliganded state to permit access to the binding pocket. Indeed, a number of studies have shown that metabolite binding is concomitant with folding. Interestingly, the extent of the structural changes that accompany the transition to the bound state has been observed to vary depending on the aptamer class (19). While some RNA receptors are largely pre-organized, others undergo far more extensive structural rearrangements upon ligand docking. According to a classification system that distinguishes between these two general types, aptamers for which metabolite binding induces only modest, local structural adjustments are referred to as Type I, while those that experience more global conformational changes are categorized as Type II (19).
Overview of Riboswitch Aptamer Atomic-Resolution Structures
Natural aptamers recognizing the nucleobases guanine and adenine possess essentially identical architectures (20, 21). Each of these two types consists of a three-way junction in which phylogenetically conserved nucleotides reside both in the joining region and in the terminal loops (Figure 1a). Biochemical and genetic studies had implicated a pyrimidine in the J3/1 strand as a key determinant in nucleobase selectivity, functioning presumably to contact the ligand via a canonical Watson-Crick pairing interaction (21). A series of analyses using X-ray crystallographic (22, 23) and NMR spectroscopic methods (24) confirmed this component of molecular recognition and, furthermore, revealed a binding pocket in which nucleotides of the conserved aptamer core form hydrogen bonds with every available functional group of the purine ligand. Additional stability of the liganded RNA derives from extensive stacking interactions among multiple layers of base triples, with the central tier containing the nucleobase, itself part of a triplet interaction with the discriminator pyrimidine (Y74) and a conserved uridine (U51). The P2 and P3 elements serve as a scaffold, existing in a parallel arrangement where they are closely apposed and tethered by an intricate network of hydrogen bonds between the capping loops (Figure 1a). Overall, the structure of the bound state of the purine riboswitch aptamer shows almost the entire surface of the ligand to be solvent-inaccessible, highlighting a general characteristic of natural metabolite-binding RNAs.
Figure 1.

Atomic resolution structures for representatives of eight riboswitch aptamer classes. Aptamer structure models are shown as ribbon diagrams and ligands are represented as spheres. Pairing elements and joining regions are indicated. (a) The purine riboswitch aptamer (PDB ID 1Y26, 1Y27, 1U8D) (22, 23). (b) The TPP riboswitch aptamer (PDB ID 2GDI, 2HOJ, 2CKY) (32–34). (c) The SAM-I riboswitch aptamer (PDB ID 2GIS) (44); KT, kink-turn. (d) The SAM-II riboswitch aptamer (PDB ID 2QWY) (45). (e) The SAM-III/SMK riboswitch aptamer (PDB ID 3E5C) (46). (f) The lysine riboswitch aptamer (PDB ID 3D0U, 3DIL) (56, 57); a specifically bound K+ ion (57) is depicted as a purple sphere. (g) The GlcN6P-responsive glmS ribozyme (PDB ID 2HO7, 2N74) (65, 66). (h) The Mg2+-responsive M-box riboswitch aptamer (PDB ID 2QBZ) (75); six crystallographically observed Mg2+ ions are shown.
Among eubacteria, the riboswitch class responding to the coenzyme thiamine pyrophosphate (TPP) is one of the most widespread, its representatives having been identified in organisms from a diverse array of taxonomic groups (25–27). Moreover, members of this class have been uncovered in archaeal and eukaryotic organisms (25, 28–31), and thus represent the only known riboswitch examples not restricted to the eubacterial domain. Despite the distant evolutionary relationships among some of the organisms with TPP riboswitches, the aptamer domains are remarkably conserved in primary and secondary structure. Furthermore, detailed structural views from X-ray crystallographic studies have shown the TPP-binding domains from eubacterial and plant riboswitches to be nearly identical in architecture (32–34).
The TPP riboswitch aptamer is constructed essentially as a three-way helical junction, with the coaxially stacked P1 and P2/P3 elements arranged in parallel with P4/P5 (Figure 1b), and thus on a rudimentary level resembles the organization of the purine aptamer (32–34). In contrast to the purine-specific motif, however, sequences comprising the helix junction do not participate directly in TPP recognition, though they are nonetheless critical to the global fold. Instead, TPP is positioned through contacts with internal loops occurring within the irregular helices P2/P3 and P4/P5. The metabolite, which exists in an extended conformation, spans the distance between these helices, with the pyrimidine and pyrophosphate moieties docking into the J2/3 and J4/5 bulges, respectively (Figure 1b). Pyrimidine recognition is mediated in part by stacking interactions with highly conserved residues of J2/3, which are extruded from the major groove in a T-loop-like turn (35), while the pyrophosphate group contacts residues of the P4/P5 internal bulge both directly and via coordination of bridging Mg2+ ions. Although there appear to be no significant contacts to the thiazole ring, the overall length of the TPP ligand is likely to be critical for the optimally productive interaction with the sensor helices of the aptamer.
Key structural elements distal from the binding pocket also contribute to the stability of the aptamer-metabolite complex. A network of tertiary interactions among nucleotides within the hub of the three-way junction assists in stabilizing a kink in J2/4 that permits the sensor helices to pivot into parallel alignment. This orientation of the P2/P3 and P4/P5 elements is anchored additionally by an array of contacts between loop L5 and the base of helix P3 (Figure 1b).
Three discrete classes of natural aptamers have been identified that target the S-adenosylmethionine (SAM) cofactor (36–40). In addition, motifs related to two of these major classes have been uncovered that appear to use structurally distinct ancillary domains to organize highly similar ligand binding pockets (41–43). Atomic-resolution structures have been obtained for representatives of the SAM-I, SAM-II, and SAM-III, or SMK box riboswitch aptamer classes, providing detailed views of the different molecular strategies employed by RNA to recognize this compound (44–46).
The ligand-binding domain of the SAM-I riboswitch describes a four-way junction in which two pairs of coaxially stacked helices pack closely against one another in a splayed arrangement (Figure 1c) (44). The metabolite is ensconced between the minor grooves of the P1 and P3 elements, with additional molecular contacts contributed by junction residues. The existence of a kink-turn motif within the P2 stem enables the corresponding loop L2 to dock with J3/4 via a pseudoknot interaction, thereby lending stability to the global fold (Figure 1c). On a coarse level, this architecture is quite similar to that of the P4-P6 and P3-P7 domains of the group I intron, where similarly interwoven stacked helices, also stabilized by distal tertiary interactions, comprise the conserved catalytic core (44, 47–49). The use of a comparable arrangement of obliquely packed helical elements to assemble both the active site of the group I intron and the binding pocket of the SAM-I riboswitch aptamer reinforces the notion that RNA, in the same manner as proteins, can employ certain stable folds as scaffolds, which may be elaborated in various ways to generate distinct functions.
When complexed with the SAM-I aptamer, the metabolite itself assumes a cis configuration in which the methionine moiety overlays the nucleobase, stabilized in part by a cation-π interaction between the amino group and the adenine ring (Figure 1c) (44). This aspect of metabolite self-recognition may partially account for the weaker affinity exhibited toward S-adenosylcysteine (38), whose main chain is shorter than that of SAM by a single methylene group, since the difference in length could disrupt the alignment between the protruding amino group and the π electron cloud. Moreover, the negatively charged carboxylate of the methionyl group is contacted by a conserved guanosine in J1/2 through hydrogen bonds with its Watson-Crick edge, and this interaction would also be adversely affected by alterations in side chain length. The positively charged sulfur atom is recognized by electrostatic interactions with carbonyl oxygens of phylogenetically conserved uridine residues in P1, in accord with previous biochemical studies demonstrating the importance of this positive charge to binding affinity (50).
Although they share the same target metabolite, the SAM-I and SAM-II aptamer classes differ in their consensus primary and secondary structures, and a comparison of their three-dimensional structures has revealed distinct global folds (44, 45). The SAM-II ligand-binding domain adopts a classical, or H-type pseudoknot structure (Figure 1d), a recurring RNA motif with diverse roles in an assortment of biological processes (51). Loops L1 and L3 engage in triplex interactions with pairing elements P2a/b and P1, respectively, and these individual triple helical domains are stacked one upon the other to form a rod-like structure (Figure 1d). Direct contacts with a series of base pairs and triples along the major groove face of the P2b-L1 triplex mediate docking of the ligand in a trans, or extended configuration. This orientation of the metabolite differs from the more compact geometry adopted by SAM when complexed with the SAM-I aptamer.
The SAM-III riboswitch represents a third, distinct class of natural aptamers specific for SAM (40). The ligand is positioned by conserved residues within a three-way helical junction, resulting in an overall Y-shaped structure (Figure 1e) (46). Tertiary interactions involving the J3/2 trinucleotide bulge simultaneously stabilize this junction and provide a ceiling for the binding pocket. Further stabilization is derived from π-stacking interactions resulting from the intercalation of the adenosine moiety of SAM between the P1 and P2 helices. Interestingly, the SAM ligand itself is constrained by intramolecular interactions, recalling a conceptual similarity to the ligand configuration observed in the structural model of the SAM-I riboswitch aptamer (44). The SAM-III aptamer class, however, orients its cognate ligand in such a way that intramolecular electrostatic interactions with ribose and base atoms of SAM participate in stabilization of the sulfonium ion positive charge (46). The lack of observed electron density for methionine functional groups beyond the sulfonium ion suggests that this moiety is not specifically docked, clearly distinguishing the recognition strategy of the SAM-III riboswitch class from those of the SAM-I and SAM-II classes.
Despite the distinctiveness of their architectures, SAM-I, SAM-II, and SAM-III motifs exploit some of the same molecular strategies to recognize individual moieties and functional groups of their common target. One such shared approach is the use of base stacking and base triple interactions in the recognition of the adenine heterocycle of SAM. The modes of recognition of the sulfonium moiety are also strikingly similar, with each of the aptamer classes employing carbonyl oxygen groups from conserved uridine residues to interact with the positive charge.
Although SAM contains a methionine moiety, only two amino acids, lysine and glycine, are known to be detected in the free form by riboswitches (52–55). The three-dimensional structure of a representative of the lysine aptamer class has been determined crystallographically, revealing a five-way helix junction in which individual stem elements extending from the central hub pack together in a roughly parallel orientation (Figure 1f) (56, 57). Certain of the stems form coaxial stacks, so that the overall architecture can be described essentially as a three-helix bundle. This arrangement is girded by a number of tertiary interactions. The relative positions of the P2 and P3 stems, for example, are enforced by a kissing-loop-type interaction between their terminal loops (Figure 1f). This association requires two abrupt turns within the extended P2 element, one abutting a loop-E motif (58, 59) and the other resulting from a type of kink-turn structure (60). As might be expected from the intricacy of this tertiary sub-domain, each of the structural elements contributing to maintenance of the appropriate register between P2 and P3 appears to be important for lysine-dependent riboswitch function (61, 62). Additional stabilization of the helix bundle derives from contacts between L4 and a P2 internal loop, which bear some similarity to a GNRA-tetraloop-receptor interaction.
In the liganded complex, a single lysine molecule is buried within the helical junction, where the majority of the phylogenetically conserved nucleotides reside. The amino acid is held by the aptamer in an extended conformation, sensed mainly by hydrogen bonding interactions with the carboxylate and the α- and ε-amino groups. With the ligand spanning the binding pocket in this way, it ensures that only amino acids with methylene linkers of appropriate length will engage in the full complement of productive interactions. This is likely to account in part for the observed discrimination against related compounds with side chains of different lengths (53, 62). Stacking interactions between binding pocket nucleobases and the hydrophobic methylene groups of the lysine side chain also contribute to the specificity of metabolite recognition.
Interestingly, the detailed structure of the lysine receptor has revealed a role for a single K+ ion, which assists in organizing the junctional zone (Figure 1f). The presence specifically of this cation, which is coordinated both directly and through water molecules by multiple core nucleotides as well as a lysine carboxyl oxygen, has been demonstrated to be critical to the stability of the ligand-bound structure (57).
The mechanisms of most known riboswitch classes are based upon structural changes driven by association with their cognate metabolites, with strict target specificity resulting from exquisite complementarity of the corresponding binding pockets. In contrast, the riboswitch responsive to glucosamine 6-phosphate (GlcN6P) functions by undergoing ligand-induced, site-specific self-cleavage, leading to reduced gene expression through destabilization of the mRNA (54, 63, 64). This mechanistically unusual riboswitch is often referred to as the glmS ribozyme class, after the only gene known to be under its control, which is involved in the synthesis of GlcN6P.
X-ray crystallographic analyses have revealed the architecture of this autocatalytic regulatory RNA as two sets of coaxially stacked helices surrounding a third, shorter helical segment (Figure 1g) (65–67). The ribozyme core consists of the smaller, central P2.1 segment and the neighboring P2.2 helix, which exist as part of a doubly pseudoknotted structure containing both the scissile phosphate and the ligand-binding site. Sequences 3′ to this compact core comprise the P3/P4 peripheral domain, which has been shown to be necessary for optimal self-cleavage rates at physiological Mg2+ concentrations (68, 69). This accessory domain is anchored in position by a third pseudoknot at one end and a tertiary interaction between P1 and the L4 GNRA tetraloop at the other (Figure 1g). Extensive contacts between the A-rich internal loop of the P4 element and the P2.1 minor groove serve further to buttress the catalytic core of the glmS ribozyme.
A number of structure-probing experiments have indicated that the glmS ribozyme is largely preorganized, with very few conformational changes occurring upon association with the target metabolite (63, 70, 71). Consistent with these data, the atomic resolution structures of the free and ligand-bound forms of the glmS ribozyme are nearly superimposable. Collectively, the accumulated evidence for a rigid fold suggested that GlcN6P functions not as an allosteric effector of ribozyme action but as a coenzyme, participating directly in the catalytic mechanism. In this regard, particular attention was focused on the amine group of the target ligand, since prior biochemical studies had indicated a crucial role for this functional group in promoting strand scission (63, 72, 73).
Indeed, the three-dimensional structure of the glmS RNA element in the ligand-bound state revealed the proximity of the GlcN6P docking site to the labile phosphate linkage, corroborating the idea that the cognate ligand could be directly involved in catalysis (65, 66). Nucleotides comprising the GlcN6P binding pocket contact the hexosamine ring primarily through hydrogen bonding interactions with its exocyclic functional groups, while recognition of the phosphate moiety depends heavily on the coordination of fully hydrated Mg2+ ions. Importantly, the C2-amine of GlcN6P is located within 3 Å of the 5′ oxygen of G1, consistent with a model in which the ammonium form of this group functions as a general acid to activate the leaving group. Moreover, another GlcN6P functionality, the C1-hydroxyl group, is positioned appropriately to donate a hydrogen bond to the pro-SP non-bridging oxygen of the scissile phosphate, and is predicted to stabilize the developing negative charge on this moiety (66). Metabolite-dependent structure stabilization is therefore not a significant feature of the glmS regulatory RNA, which instead utilizes key functional groups of the cognate ligand for direct contributions to the catalytic mechanism.
Although the majority of characterized riboswitches target small organic compounds, evidence of Mg2+-sensing activity has been provided for two distinct classes of regulatory RNAs that modulate the expression of divalent ion transporters (74, 75). Insights to the detailed mechanism of the M-box motif, which appears to be the structurally more elaborate and phylogenetically more widespread of the two classes, have recently been provided using X-ray crystallographic methods (75). The global architecture is established by three helical elements, which are aligned in nearly parallel formation (Figure 1h). Of the six crystallographically observed Mg2+ ions, at least four appear to facilitate the close apposition of these helical elements at one end of the three-helix bundle, employing both inner-sphere and outer-sphere contacts with the RNA to mediate long-range tertiary interactions. Specifically, these buried Mg2+ ions assist in fastening together loops L4 and L5 with the P2 helix, resulting in an overall compaction of the receptor RNA (Figure 1h). Biochemical and biophysical evidence suggests that the divalent ions are bound in a cooperative manner, which would be consistent with the high degree of structural complexity exhibited by the M-box motif and would permit a robust regulatory response over a narrower concentration range (75).
MECHANISMS OF RIBOSWITCH-MEDIATED GENE CONTROL
Eubacterial Expression Platforms
Among eubacteria, where the vast majority of known riboswitches occur, there are two predominant types of expression platforms that translate the occupation states of aptamer domains into appropriate genetic responses. One of these mechanisms exerts control at the level of transcription termination by linking the metabolite binding event to the stability of an intrinsic terminator stem, while the other regulatory apparatus relies on ligand-induced structural changes to influence accessibility of the SD sequence, thereby modulating translation initiation. There is a wealth of experimental evidence to indicate that diverse aptamer classes rely on these prevalent control mechanisms (13, 76–78). Also, operative expression platforms can often be reliably inferred by searching riboswitch sequences for pertinent secondary structure features such as terminator stems or SD sequences with their corresponding sequestors (25).
The interaction between an aptamer domain and its associated expression platform is critical for establishing the ligand concentration to which a riboswitch responds. A key component in this relationship is the dissociation constant (KD) for the target metabolite. A KD value reflects the ratio of the concentrations of free ligand and aptamer to that of the aptamer-ligand complex, measured at equilibrium. Several riboswitch aptamers have been shown to require many seconds or even minutes to fold and equilibrate with their cognate ligands (79–84). In fact, the amount of time needed to reach thermodynamic equilibrium will, in certain instances, exceed the time available for this gene control decision.
Because KD values are determined under conditions of thermodynamic equilibrium, they should not necessarily be considered accurate gauges of the ligand concentrations that induce riboswitch responses in vivo. Communication between a metabolite-binding domain and an expression platform is dynamic, occurring concomitantly with elongation of the RNA chain. Thus, a comprehensive understanding of riboswitch function requires consideration of time-sensitive parameters, such as rates of RNA folding and polymerization (Figure 2).
Figure 2.
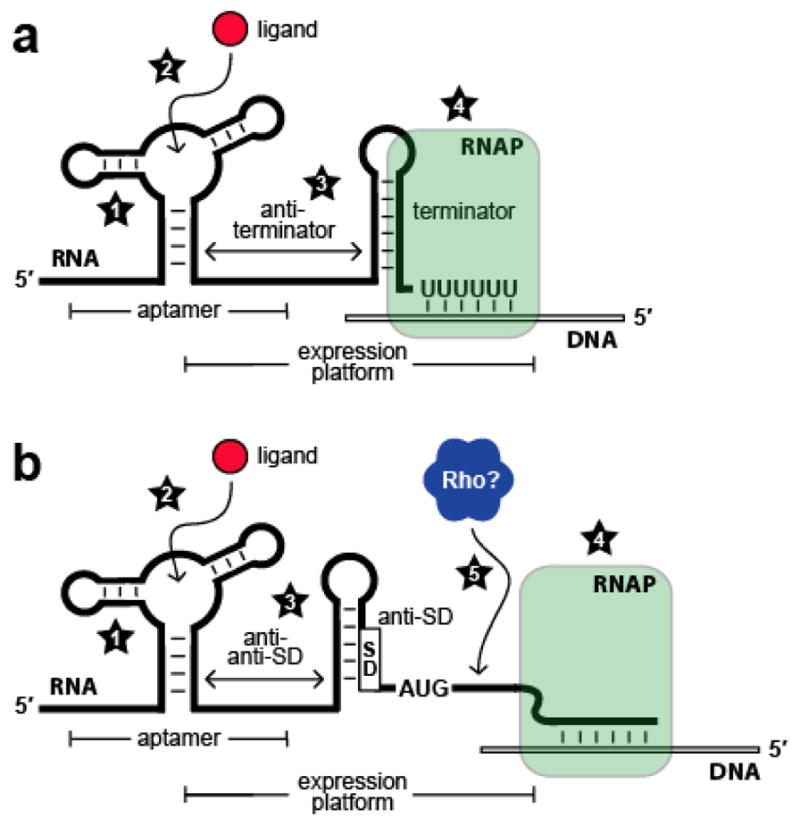
Kinetic and thermodynamic factors that influence riboswitch function. (a) Schematic representation of a riboswitch that represses gene expression upon ligand binding by controlling transcription termination. Factors that affect riboswitch function include (1) rate constants for aptamer folding and unfolding; (2) rate constants for ligand association and dissociation; (3) rate constants for expression platform folding and unfolding; (4) speed of transcription elongation by RNA polymerase (RNAP). (b) Schematic representation of a riboswitch that represses gene expression upon ligand binding by controlling translation initiation. Numbers 1 through 4 are as described in (a); (5) speed of Rho-dependent transcription termination.
There are several factors that can influence whether a particular riboswitch functions in a kinetically or thermodynamically driven manner. For example, in the case of a riboswitch controlling transcription termination, both the correct folding of the aptamer domain and subsequent interplay with the expression platform are required to occur before RNA polymerase progresses beyond the intrinsic terminator, since ligand binding events occurring after this time will not influence gene control (Figure 2a). Thus, kinetic parameters involved in RNA folding, metabolite binding, and transcription can be more influential than binding affinities measured at thermodynamic equilibrium (81, 82, 85).
Even some riboswitches that control translation initiation might be affected more by kinetic parameters than by thermodynamic ones (Figure 2b). Metabolite-binding RNAs that regulate gene expression by modulating SD accessibility occur more commonly in organisms that make extensive use of the protein factor Rho (25). Rho is a hexameric RNA-binding complex (86) that promotes termination of transcripts in regions that are not masked by ribosomes (87). In principle, it seems that a riboswitch should maintain ligand-dependent control of translation initiation even for a full-length mRNA, in which case the aptamer domain would reach thermodynamic equilibrium with the cognate ligand. In actuality, however, the prompt action of Rho complexes on transcripts not subject to active translation would likely preclude such a scenario. Rho-mediated transcription termination also explains how a riboswitch could efficiently control the expression of multiple genes within an operon despite occluding only the SD sequence corresponding to the first open reading frame (76, 88).
Although transcription termination and SD sequestration are the chief mechanisms employed by eubacterial riboswitches, additional types of expression platforms have been reported. For example, the glmS ribozyme effects genetic control by undergoing GlcN6P-induced self-cleavage (63). Interestingly, though, this metabolite-dependent strand scission leaves the open reading frame of the corresponding glmS gene intact, and the subsequent action of a protein ribonuclease is required to repress gene expression. The 3′ cleavage product resulting from GlcN6P-induced cleavage contains a 5′ hydroxyl terminus (63), thereby rendering this RNA fragment susceptible to degradation by the widespread exonuclease RNase J1 (64). There is also evidence that one class of Mg2+-responsive riboswitch (74), which controls expression of the Mg2+ transporter MgtA in certain γ-proteobacteria, functions in part by regulating susceptibility of the mRNA to RNase-E-mediated degradation (89). Together with observations that certain riboswitches are cleaved by RNase P (90, 91), these studies collectively suggest that the modulation of transcript vulnerability to the action of nucleases might represent a significant mechanism of gene control available to riboswitches.
Riboswitches nearly always occur in cis to the mRNAs whose expression they control. In Clostridium acetobutylicum, however, a SAM-I riboswitch encoded on the antisense strand is located downstream of an operon involved in converting methionine to cysteine (92). This unusually positioned riboswitch acts by controlling production of antisense transcripts in cis, which in turn interact with the mRNA sense strand to down-regulate gene expression (93).
Eukaryotic Expression Platforms
Given the diversity of riboswitch classes in eubacteria, it seems reasonable to speculate that metabolite-binding RNAs could be similarly exploited by eukaryotes. And since eukaryotes express far greater amounts of non-coding RNA than eubacteria (94), there would seem to be ample opportunity to accommodate such regulatory elements. Introns in particular provide large spans of non-coding RNA that could harbor riboswitches participating in the modulation of pre-mRNA splicing. Despite these predictions, only the TPP riboswitch class has been confirmed in eukaryotes, its mechanisms of ligand recognition and genetic control having been rigorously detailed (29–32, 95–99). The existence in fungi of an arginine-sensing riboswitch has been proposed (100), although verification of its physiological relevance awaits further analysis.
Eukaryotic TPP riboswitches contain aptamer domains that are strikingly similar to those occurring in bacteria, which facilitated their discovery via comparative sequence analysis (29, 31). However, as mechanisms of post-transcriptional genetic control differ extensively between eubacteria and eukaryotes, it would not unexpected for TPP riboswitches occurring in these two domains of life to rely on different expression platforms. Indeed, the few eukaryotic TPP riboswitches that have been examined in greater detail employ regulatory strategies distinct from those of their prokaryotic counterparts (31, 96–98). In many fungi, for example, TPP-binding RNAs appear to regulate alternative splicing of introns residing in the 5′ UTRs of thiamine metabolism genes NMT1 and THI4 (Figure 3) (30, 96). Control of these splicing steps subsequently modulates gene expression through either the elimination or retention of upstream open reading frames (uORFs) (101), which serve as decoys to prevent ribosomes from initiating at the start codon of the principal ORF.
Figure 3.

Gene control by a eukaryotic TPP riboswitch. (a) Secondary structure model of the TPP aptamer residing in the 5′ UTR of Neurospora crassa NMT1. Sequences of the aptamer P4/P5 domain are complementary (orange shading) to a region adjoining a key splice site, illustrating one way in which the availability of TPP influences splice site selection. Similar types of base-pairing interactions between aptamer domains and expression platforms are exploited by other eukaryotic TPP riboswitches. (b) Control of alternative splicing by TPP riboswitches regulates NMT1 gene expression in fungi. The main features are depicted of the riboswitch from N. crassa NMT1, including splice sites, pairing elements of the TPP aptamer (P1 through P5), and uORFs that compete with translation of the primary ORF to reduce gene expression. Green lines and red lines refer to splicing determinants that are activated or inhibited, respectively, depending on the occupancy state of the aptamer domain.
The TPP riboswitch from the NMT1 gene of Neurospora crassa (96) is similar in its features to those of other fungi. The aptamer domain occurs in an intron upstream of the start codon for the main ORF. This intron contains two potential 5′ splice sites upstream of the aptamer and one 3′ splice site downstream of the aptamer. In the absence of TPP, a conserved segment of the aptamer domain involved in contacting the pyrophosphate moiety of the ligand is instead base-paired with complementary sequence near the second 5′ splice site (Figure 3a). As a result, only the first 5′ splice site is accessible to the spliceosome, so that a processed mRNA is generated that lacks the decoy uORFs. It has previously been observed in a fungus that 5′ splice sites occurring closest to the 3′ splice site are preferred (102). Therefore, when the aptamer domain exists in the TPP-bound state, the now accessible second 5′ splice site is preferentially utilized, yielding a processed mRNA that retains the decoy uORFs (Figure 3b). A similar mechanism appears to operate in the regulation of fungal THI4 genes, whereas a third TPP riboswitch in N. crassa resides within an intron interrupting the main ORF, and therefore must rely on an alternative mechanism for gene control (96).
TPP-binding RNAs occur also in many plant species, where they are involved in regulation of the THIC gene. These aptamers domains reside in introns that are situated in the THIC 3′ UTRs (29, 97, 98), in contrast to the 5′ UTR locations of homologous riboswitches in fungal species. In Arabidopsis thaliana, the mode of the interplay between the TPP aptamer domain and a proximal 5′ splice site closely resembles the analogous interaction for the NMT1-associated riboswitch in N. crassa (Figure 3a) (97, 98). In the absence of TPP, this splicing event is disfavored, resulting in retention of a major RNA processing site located in the intron. RNAs that are cleaved at this processing site are subsequently polyadenylated, yielding transcripts with short 3′ UTRs that drive high levels of gene expression. In contrast, TPP binding unmasks the 5′ splice site, leading to excision of the intron and its interior processing site. This yields mRNAs with long 3′ UTRs, resulting in reduced levels of protein expression, which perhaps are due to decreased transcript stability stemming from less efficient processing and polyadenylation.
The THI4 and THIC genes of the green alga Chlamydomonas reinhardtii are associated with TPP riboswitches similar to those of fungi and plants (31). The THI4 riboswitch controls alternative splicing of an intron in the 5′ UTR, leading to retention of a uORF when TPP levels are high. In contrast, the TPP aptamer controlling THIC expression resides in an alternatively spliced cassette exon, which is retained as a result of ligand binding. Because the aptamer contains a stop codon in frame with the main ORF, retention of the cassette exon results in premature termination of translation. Although the root mechanism involves splicing regulation in every case described above, these examples showcase the diversity of genetic control mechanisms subsequent to the splicing event that are available to eukaryotic riboswitches.
EVOLUTIONARY PLASTICITY OF RIBOSWITCH APTAMERS
Recognition of One Metabolite by Multiple, Distinct Riboswitch Aptamer Classes
In its capacity as a metabolite receptor, RNA assumes a variety of structures in order to accommodate a diverse array of targets. There is no reason, however, to expect that the challenge of binding a particular metabolite should be met by one RNA motif alone. The vastness of sequence space from which unique riboswitch aptamers emerge is equally likely to contain multiple, discrete structural solutions for recognizing the same metabolite. Certainly this has been demonstrated to be the case among artificial RNA aptamers, with several distinct motifs selective for ATP having been selected in vitro (103–105).
Among natural riboswitch aptamers, there are at least three major classes of SAM receptors, which are largely non-overlapping in their phylogenetic distributions (25, 36–40). The SAM-I, SAM-II, and SAM-III motifs have distinct consensus structures (Figure 4), and so probably represent evolutionarily independent solutions to SAM recognition. Furthermore, atomic-resolution structures corresponding to these motifs have confirmed the distinctiveness both of their global architectures and ligand docking sites (44–46). Despite their different structures, representatives of each of these three classes exhibit high levels of discrimination against compounds closely related to SAM (38–40, 50), underscoring the ability of RNA to utilize wholly unique folds in the manufacture of binding pockets that are exquisitely complementary in shape to a single target.
Figure 4.

Conserved sequences and secondary structure models corresponding to different classes of SAM riboswitch aptamers. The SAM-I, SAM-II, and SAM-III classes have distinct architectures, while the SAM-IV aptamer is related in sequence and secondary structure to the SAM-I class. Nucleotides highlighted in yellow are observed to contact the SAM ligand directly (44–46). Note that, with only a single exception, the SAM-IV aptamer consensus structure contains these same conserved nucleotides in analogous positions, suggesting that the binding pockets are highly similar. Distinct but positionally analogous pseudoknots in the SAM-I and SAM-IV motifs are indicated by orange shading, while an additional predicted pseudoknot unique to the SAM-IV class is denoted by blue shading.
The second organic metabolite for which distinct classes of RNA receptors are known to exist is the modified nucleobase 7-aminomethyl-7-deazaguanine, or pre-queuosine-1 (preQ1). This molecule is a metabolic intermediate in the biosynthesis of queuosine, a hypermodified nucleoside occurring widely among eubacteria and eukaryotes that occupies the anticodon wobble position in certain tRNAs (106). Two structurally distinct natural aptamer motifs, termed the preQ1-I and preQ1-II classes, have been shown selectively to recognize this metabolite (107, 108). Of the two motifs, the preQ1-I riboswitch is the more widely distributed, having been identified in Firmicutes, Fusobacteria, and Proteobacteria (25, 107). In contrast, representatives of the preQ1-II class appear to be confined to the Streptococcaceae family and thus comprise a more limited phylogeny (41, 108). Interestingly, the distribution of preQ1-II representatives resembles quite closely that of SAM-III riboswitches, suggesting that there might be additional regulatory RNAs occurring exclusively in Streptococcaceae (40, 108).
The minimal aptamer domain of the preQ1-I class is unusually small compared to those of other riboswitches, consisting simply of a stem-loop and a short, adenosine-rich tail sequence (107), while the preQ1-II motif is predicted to adopt a pseudoknot structure (41, 108). Although each structural class recognizes the cognate ligand specifically and with high affinity, it appears that there are differences in the degrees to which certain functional groups of the ligand are contacted by the distinct binding pockets (107, 108). Moreover, biochemical analyses indicate that the selectivity of the preQ1-I aptamer class relies in part on a canonical Watson-Crick pairing interaction with the ligand, while the preQ1-II class appears to eschew this strategy. Collectively, these observations reinforce the notion that distinct aptamer classes targeting the same metabolite need not employ identical strategies for molecular recognition.
Structured RNAs are known to exploit the dense positive charge of divalent cations in order to adopt compact folds (109). As the most abundant intracellular divalent cation (110), Mg2+ can serve as a delocalized electrostatic shield to permit the close approach of negatively charged phosphate groups, or it can promote tertiary structure formation through specific contacts with RNA functional groups (111). As mentioned previously, two different classes of RNA structures have been identified that harness this natural metal ion affinity for the purposes of gene control. One class occurs in a subset of Gram-negative eubacteria, where it is associated with genes encoding the magnesium transporter MgtA (74). This RNA element employs a mechanism in which one of two competing stem-loop structures is preferentially stabilized in the presence of higher Mg2+ concentrations, leading to the interruption of transcription. The second class of regulatory RNA whose primary ligand is Mg2+ is the M-box, which is also commonly associated with metal ion transport genes (75). In comparison to the mgtA element, the M-box motif appears to be structurally more elaborate and phylogenetically more widespread. The atomic-resolution structure of the M-box RNA indicates that the global architecture relies on at least four Mg2+ ions to mediate long-range tertiary interactions among conserved domains (Figure 1h) (75).
Based on the intrinsic affinity of nucleic acids for cations, RNAs that function as metal ion sensors might be predicted to occur relatively frequently in sequence space. Furthermore, it is anticipated that this natural tendency toward association would obviate the requirement for the RNA sensor to form intricate, highly conserved metal-ion-binding pockets. These preconceptions concerning cation-responsive RNAs appear to coincide with characteristics of the mgtA RNA element, which has a relatively simple structure and a limited phylogenetic distribution. However, attributes of the M-box motif, including its widespread occurrence and high level of sequence and structure conservation, provide a stark contrast with the mgtA RNA element. It is likely that the more elaborate construction of the M-box motif is necessary for the observed cooperative binding of multiple Mg2+ ligands, which might afford greater sensitivity in genetic control (75).
Recognition of One Metabolite by Riboswitch Aptamer Variants within a Class
The architectural versatility of RNA applies to the construction not only of whole aptamers, but also of isolated structural elements within a given aptamer class. As evidence of this, certain riboswitch aptamer families contain representatives in which selected subdomains are deleted, substituted, or permuted without resulting in altered target selectivity. Despite the significant deviations from the consensus that can occur in these cases, enough conserved structural features remain for the variant aptamers to be recognizable as members of the same broad class.
The degree to which individual structural elements of a riboswitch class can be diversified while maintaining essential core features is illustrated by the SAM-IV RNA motif. Occurring predominantly in Actinomycetales, where they are associated with sulfur metabolism genes, SAM-IV representatives bear strong similarities in sequence and secondary structure to the ligand-binding core of the SAM-I aptamer class (Figure 4) (42). In fact, the two motifs conserve the identities and relative positions within the core domain of five of the six nucleotides proposed to contact the SAM ligand (Figure 4) (42, 44). The peripheral substructures, however, differ considerably between the two motifs. SAM-IV RNAs lack the interior P4 element of the SAM-I consensus, and instead position novel structural elements, including an additional predicted pseudoknot, 3′ to the P1 stem. Further, the kink-turn in P2 of the SAM-I motif is absent in SAM-IV, replaced with a distinct and perhaps functionally analogous internal loop (Figure 4). Despite these and other differences in their accessory domains, SAM-I and SAM-IV RNAs target SAM specifically and discriminate similarly against related compounds, supporting the conclusion that diverse peripheral domains can be utilized as scaffolds to assemble highly similar metabolite docking sites (42). Due to the apparently identical organization of their binding pockets, it seems plausible that SAM-I and SAM-IV RNAs evolved divergently from a single ancestral motif, and that they can be considered to represent distinct types of aptamers within the same general class. Interestingly, the generation of multiple aptamer types through structural diversification has also been observed in another SAM riboswitch class. A family of RNAs that appears quite similar in its core structure to that of the SAM-II class, but that diverges substantially in other respects, comprises a distinct aptamer type referred to as SAM-V (43).
Structural variations that distinguish aptamers by type need not be extensive or pronounced. For example, representatives of the preQ1-I riboswitch class are sorted into two types based solely upon alternative signature sequences occurring in one stem-loop (107).
Another example highlighting the degree to which aptamer substructures are interchangeable is provided by an unusual member of the adenosylcobalamin (AdoCbl) riboswitch class (112). Although it lacks a substantial block of consensus features, this variant retains its capacity for selective recognition of the AdoCbl ligand. Interestingly, however, a 3′ terminal sequence element occurring uniquely in the truncated version is required for metabolite binding, suggesting that this alternatively configured riboswitch aptamer could exemplify the wholesale exchange of modular, functionally equivalent structural domains.
Riboswitch Aptamer Variants with Altered Ligand Specificities
Structural disparities within an aptamer class often represent modified architectures that result nonetheless in recognition of the identical metabolite. Occasionally, though, departures from a riboswitch aptamer consensus reflect an alteration in ligand specificity. Binding pocket adjustments that elicit specificity changes can be highly focused within the aptamer core, as evidenced by representatives of the purine riboswitch class (21). Members of this class specifically bind either guanine or adenine, despite possessing nearly identical global architectures (22, 23). A key determinant of ligand selectivity involves a Watson-Crick pairing interaction between the purine ligand and a critical pyrimidine residue (Y74) within the conserved core (22–24); in fact, the guanine- and adenine-specific versions of this RNA motif are distinguished solely by the identity of the single discriminator residue. Thus, the use of Watson-Crick complementarity as a component of target recognition allows the purine riboswitch class, with only minimal changes to the core, to interconvert evolutionarily between two ligand selectivities. More generally, however, it is expected that the emergence of altered target specificities within a given aptamer framework will require more extensive deviations from the consensus.
Interestingly, examples of more substantial revisions to an aptamer binding pocket are provided also by derivatives of the purine riboswitch class. One of these variant subclasses, which conforms only loosely to the purine aptamer consensus structure, has been shown specifically to bind the nucleoside 2′-deoxyguanosine (113). This distinctive subclass has a secondary structure nearly identical to that of the nucleobase-specific riboswitch, though a significant number of violations to the consensus occur in joining sequences comprising the aptamer core as well as in the terminal loops of the P2 and P3 elements. Not surprisingly, the discriminator cytidine at position 74 remains unaltered, presumably to maintain a Watson-Crick pairing interaction with the guanine moiety of the nucleoside ligand. Importantly, however, there are deviations from the consensus at several positions that correspond, in the atomic resolution structure of the purine aptamer (22, 23), to residues in the immediate vicinity of the ligand sugar edge. Ostensibly, these mutations from the consensus sequence are necessary for the variant RNA to accommodate the deoxyribose moiety protruding from the N9 position of the nucleobase.
Because the association between the capping loops of the P2 and P3 stems is distal to the binding pocket, the nucleotide changes in these loops probably reflect a different mode of tertiary interaction. However, these altered loop sequences are observed in purine-specific aptamers that in all other respects adhere to the consensus (113), indicating that this structural component of the aptamer can function in scaffolding both nucleobase and nucleoside binding pockets.
To date, these variant, nucleoside-responsive versions of purine riboswitches have been identified only in Mesoplasma florum, a species of eubacteria belonging to the Mollicute class. As an obligate parasite, this organism has pared down its genome through reductive evolution, relying in large part on host metabolic pathways (114). M. florum harbors a total of eight purine riboswitch variants, many of which appear to regulate transporters with undefined substrate specificities (113). Of these eight RNA elements, only two are specific for 2′-deoxyguanosine, while the remainder selectively bind other purine nucleoside and nucleobase derivatives. For an organism that depends on its host for a wide array of metabolites, perhaps the evolution of a battery of RNA receptors targeting specific purine derivatives is advantageous, permitting finely tuned genetic control in response to intracellular concentrations of individual compounds. Whatever the provenance of this variant subclass, the 2′-deoxyguanosine riboswitch illustrates the remarkable degree to which a binding site can be revised to accommodate related ligands and, furthermore, reveals that certain metabolite-binding RNAs can be severely restricted in their phylogenetic distributions, due either to recent evolutionary origin or niche-specific utility.
There is some evidence to suggest that structurally distinct forms of another riboswitch class also have different ligand specificities. Highly structured RNA elements that modulate gene expression in response to intracellular concentrations of molybdenum cofactor (Moco) are broadly distributed among eubacteria (41, 115). Moco, a tricyclic pyranopterin with a single molybdenum atom coordinated by dithiolene sulfurs, is a nearly ubiquitous coenzyme utilized in a wide assortment of redox reactions (116). An analogous cofactor differing primarily in the identity of the coordinated metal exists predominantly in archaea but also among eubacteria (117). But for the insertion of tungsten, a transition metal with properties similar to those of molybdenum, tungsten cofactor (Wco or Tuco) is nearly identical structurally and functionally to Moco. Notably, however, Wco is consistently observed in the bispterin form, while Moco and its derivatives exist in versions with one or two pterin equivalents (Figure 5) (118).
Figure 5.

Conserved sequences and secondary structure models corresponding to Moco and Wco RNAs. These RNAs exhibit characteristics of riboswitches, including a complex aptamer-like structure and gene control function. Many representatives reside immediately upstream of an AUG start codon and are predicted to function by sequestering the SD sequence (blue shading). The phylogenetic distributions and specific gene associations of these motifs suggest that RNAs containing the P3 element recognize Moco, while those lacking this stem bind the related coenzyme Wco.
This widely occurring RNA motif is likely to represent the aptamer component of a Moco riboswitch class, and is associated with genes that encode proteins involved in cofactor biosynthesis, transporters of the metal constituent, and apoenzymes that utilize Moco (41, 115). Based on sequence alignment data, the RNA is predicted to comprise a multi-stem junction in which most of the phylogenetically conserved nucleotides reside within the junction and in two subdomains proposed to form a GNRA-tetraloop-receptor interaction (Figure 5). Representatives of the Moco RNA class can be divided into two categories according to whether the P3 pairing element is present (115). Interestingly, examples of the motif that contain the P3 stem are associated exclusively with genes encoding enzymes that either employ Moco as a cofactor or that are involved in its biosynthesis. In contrast, RNAs lacking the P3 structure appear most often to regulate the expression of enzymes that utilize Wco or that participate in synthesis of the pterin precursor common to Moco and Wco. Thus, there appears to be a strong correlation between the presence or absence of the P3 stem and association of the corresponding RNA element with genes relating to Moco or Wco metabolism, respectively (Figure 5). This raises the possibility that the structural disparity between these subclasses reflects different ligand selectivities. Such an interpretation is consistent with the observation that a reporter gene under the control of a Moco element containing P3 is derepressed under conditions expected to result in an elevated concentration of Wco but not of Moco (115).
In contrast to the other predicted stems of the putative Moco riboswitch class, which possess significant degrees of nucleotide conservation proximal to the core, the P3 helix exhibits no such constraints in sequence (Figure 5) (115). It therefore seems unlikely that this substructure is involved in direct contacts with the ligand. Rather, it is possible that this stem acts as a steric hindrance that permits docking only of single equivalents of metal-bound pterins, which represent a commonly occurring form of molybdopterin cofactors. Bulkier bispterin derivatives, such as the type consistently adopted by Wco, would then be tolerated only in the absence of P3. Conceivably, then, the presence of the P3 stem could serve as a structural basis for the abilities of these putative aptamer types to discriminate between the closely related ligands Moco and Wco.
APTAMERS AND RIBOSWITCHES ARRANGED IN TANDEM
The vast majority of riboswitches occur singly, controlling gene expression through the binding of one metabolite by one aptamer domain. In certain cases, however, riboswitches or their component aptamers are arranged in series, resulting in more complex modes of ligand-dependent gene regulation (Figure 6). Sequentially arranged RNA elements with identical ligand specificities, for example, can regulate gene expression with enhanced sensitivity to fluctuating metabolite concentrations. Furthermore, the placement in series of aptamers or riboswitches with distinct target specificities can be exploited to integrate multiple chemical inputs in the modulation of a single genetic response.
Figure 6.

Tandem arrangements of aptamers and riboswitches. The schematic diagrams illustrate some of the distinct ways in which riboswitches or their component aptamers can be combined to effect more sophisticated genetic control.
Cooperative Binding by the Glycine Riboswitch
The metabolite-binding RNAs that are observed to occur in tandem with the highest frequency are representatives of the glycine riboswitch class (Figure 6) (54, 55). This motif typically contains two consecutively arranged aptamer domains, which are highly similar to one another in sequence and secondary structure, and are separated by an intervening linker. Each aptamer serves as a receptor for a single glycine molecule and, importantly, the regulatory effects of these paired motifs are exerted via a single downstream expression platform. This is achieved through cooperative ligand recognition, with docking of glycine at one site positively influencing the binding event at the other (55). As a result, the maximum regulatory effect of this riboswitch can be elicited over a narrower range in glycine concentration. Because the genes typically associated with this motif encode enzymes of the glycine cleavage system, which catabolize excess glycine as an energy source, the increased sensitivity to small fluctuations in ligand concentration provides a clear benefit.
The structural changes that are induced by cooperative binding of glycine have been examined in some detail using small-angle X-ray scattering and hydroxyl radical footprinting, and suggest a minimal three-state thermodynamic model for folding of the glycine riboswitch aptamers (119). Under conditions of low salt, an RNA containing both aptamers exists in an extended conformation, which is likely to represent an ensemble of unfolded structures. In the presence of millimolar concentrations of Mg2+, however, a transition occurs to a partially folded form, as evidenced both by footprinting assays that indicate regional protection from chain cleavage, and by overall structural compaction. Upon addition of glycine, still further packing is observed, probably reflective of ligand-induced tertiary structure formation. These analyses also predict that the binding constant for glycine is dependent on Mg2+ concentration, a characteristic that has been reported for other aptamer-ligand interactions (22, 120).
Investigations into the molecular basis for cooperativity of the glycine riboswitch have implicated key regions that are likely to be involved in tertiary interactions between the two aptamers (121). Nucleotide analog interference mapping was used to identify specific functional groups required for proper folding, as assessed by the rate of migration on a native polyacrylamide gel. The positions in which incorporation of nucleotide analogs disrupted folding were then classified as occurring either symmetrically or asymmetrically with respect to the two aptamer domains. Because the putative interface between the two glycine-binding modules is expected to be at least partially asymmetric, interference sites that were observed in one aptamer but not at the analogous position in the other were considered among the most likely to be involved in cooperative interactions. Using this approach, sites that appear to enable interdomain tertiary contacts were identified within the minor groove of the P1 helix from aptamer I and in the major grooves of the P3a helices from both aptamers (121).
Tandem Riboswitches
Known natural examples of tandemly arranged aptamers that interact in a cooperative fashion are limited to the glycine riboswitch class, but less exotic cases of sequentially occurring metabolite-binding RNAs are common. Many cases exist in which two complete riboswitches, each specific for the same ligand, are juxtaposed in 5′ UTRs. Riboswitch classes occurring in such contexts include those specific for TPP (122, 123), AdoCbl (123), Moco/Wco (41, 115), Mg2+ (M-box) (123), and cyclic di-GMP (41, 124). Consistently among these examples, there is an expression platform corresponding to each aptamer domain, suggesting that individual riboswitches arranged in tandem operate independently of one another, a conclusion that has been verified for TPP riboswitches organized in this way (Figure 6) (125).
The tandem placement of riboswitches with identical target specificities appears to allow achievement of the maximum regulatory effect in response to smaller changes in ligand concentration than would otherwise be required (125). Although such regulatory behavior approaches that of the glycine riboswitch, basic tandem architectures fail to afford the full benefit of a cooperatively acting system. Nevertheless, because duplication of a motif is presumably less complicated than the evolution of cooperatively interacting aptamer domains, tandemly arrayed riboswitches represent a simpler means to achieve a compressed dynamic range of target concentration.
Composite riboswitches are not limited to examples in which the component aptamer domains belong to the same class. In certain cases, motifs with different ligand specificities occur in combination, resulting in the modulation of gene expression in response to two metabolites (123). In several isolates of Bacillus clausii, for example, a SAM-I riboswitch resides immediately upstream of a member of the AdoCbl class (Figure 6). There is a separate intrinsic terminator stem corresponding to each aptamer domain, which is consistent with the reported independent function of each riboswitch module (123). These tandemly arrayed riboswitches control the expression of MetE (126), which is one of two enzymes in B. clausii that catalyze the formation of methionine from homocysteine. The other catalyst, MetH, is the more efficient of the two enzymes and utilizes the coenzyme methylcobalamin, an AdoCbl derivative (127). MetE activity is therefore important only when AdoCbl concentrations are reduced, providing a rationale for why this less efficient route to methionine production is repressed when either SAM or AdoCbl is abundant.
CONCLUDING REMARKS
Riboswitches are responsible for assessing a significant proportion of the inventory of fundamental metabolites, especially among eubacteria. Despite possessing only four chemically similar building blocks, and in the face of presumably intense evolutionary competition from protein factors, RNA polymers accomplish this feat by fashioning exquisite binding pockets that grip their targets with remarkable specificities and affinities. In many cases, it appears that aptamers are constructed with a degree of modularity, as individual subdomains can be substituted or deleted. These changes sometimes generate altered receptors for the same ligand, but can also result in structural adaptations that modify the target specificity, highlighting the evolutionary plasticity of this polymer.
Contemporary organisms have harnessed this extensive collection of RNA-based metabolite receptors to govern an equally extensive array of genetic control processes in all domains of life. Moreover, additional layers of regulatory complexity can be derived by deploying combinations of riboswitches in tandem. In terms of the sheer scope of the processes they influence and the proficiency with which they perform, contemporary metabolite-binding RNAs suggest the possibility of an ancient, RNA-centered metabolism of staggering complexity.
SUMMARY POINTS.
More than 20 distinct riboswitch classes have been identified that respond specifically to a variety of fundamental metabolites, including amino acids, nucleobases, coenzymes, and metal ions. Each individual class of RNA receptor contains hallmark features of conserved sequence and structure.
Atomic resolution structures for representatives of a number of aptamer classes reveal intricately constructed binding pockets that make extensive contacts to the ligand.
The affinity of an aptamer for its cognate ligand, as determined in vitro under conditions of thermodynamic equilibrium, does not necessarily reflect metabolite concentrations to which the corresponding riboswitch responds in vivo. Because riboswitch action is dynamic, occurring concomitantly with transcript elongation, kinetic parameters such as the rate of ligand association and the speed of RNA polymerase can play critical roles in tuning riboswitch responses.
Riboswitches in eubacteria control gene expression using a variety of mechanisms, with the most common of these involving transcription termination and translation initiation. TPP-specific riboswitches in eukaryotes exert their effects on gene control by regulating splice site accessibility. Diverse secondary mechanisms subsequently account for differential protein expression from the alternatively spliced variants.
Multiple, distinct aptamer classes have been identified that recognize the same metabolite. Similarly, riboswitches within a given class can exhibit variability in their architectures while maintaining the same target specificity. In certain cases, though, changes to key nucleotides in the binding pocket result in altered ligand selectivities.
More complex modes of metabolite-dependent genetic control can be achieved through the use of aptamers or riboswitches in combination. The glycine riboswitch represents a special case in which cooperative metabolite binding by mutually interacting aptamer domains yields greater sensitivity to small changes in ligand concentration. More generally, however, independently operating riboswitches belonging to the same class can be arranged in series to achieve similar effects. Furthermore, combining riboswitches with different target specificities on the same mRNA allows gene expression to be controlled by multiple metabolites.
FUTURE ISSUES.
With more than 20 riboswitch families having been identified to date, it is apparent that metabolite-binding regulatory RNAs represent a major mechanism of genetic control in eubacteria. As increasingly sophisticated bioinformatics strategies are used to mine burgeoning databases of sequenced genomes, the full extent of such RNA-mediated regulation will begin to come into view. Novel riboswitch classes are perhaps most likely to be uncovered from eubacterial clades that are underrepresented in current databases.
It remains unclear what fraction of riboswitch classes is characterized by exceedingly narrow phylogenetic distribution. Although several motifs of this type have been found, it is expected to be difficult to discover rarely occurring RNA structures because their scarcity is likely to be an impediment to computational approaches based on pattern detection.
A growing number of candidate riboswitches identified using bioinformatics are associated with suites of disparate genes. Identification of the cognate ligands in these cases could therefore shed light on the biology pertaining to these potentially novel regulons.
Natural aptamers appear overwhelmingly to be used for the purposes of gene control. Intriguingly, however, several highly conserved RNA structures, including representatives of some riboswitch classes, have been identified in atypical genomic contexts where no putative target genes are obvious. Although more mundane explanations exist, this raises the possibility that metabolite-binding RNAs might be involved in processes other than gene control.
Acknowledgments
Studies of riboswitches in the Breaker laboratory are supported by grants from the NIH and by the Howard Hughes Medical Institute.
Acronyms and Abbreviations
- UTR
untranslated region
- SD
Shine-Dalgarno sequence
- TPP
thiamine pyrophosphate
- SAM
S-adenosylmethionine
- GlcN6P
glucosamine 6-phosphate
- KD
dissociation constant
- ORF
open reading frame
- uORF
upstream open reading frame
- ATP
adenosine triphosphate
- preQ1
pre-queuosine-1 (7-aminomethyl-7-deazaguanine)
- AdoCbl
adenosylcobalamin (coenzyme B12)
- Moco
molybdenum cofactor
- Wco
tungsten cofactor
- cyclic di-GMP
cyclic diguanosine monophosphate
Glossary
- Riboswitch
an RNA element that modulates gene expression in response to the direct binding of a metabolite
- Aptamer
a structured nucleic acid (typically RNA) capable of specifically binding a target molecule
- Intrinsic transcription terminator
stable stem-loop structure followed by several consecutive uridines, which causes RNA polymerase to halt transcription
- Shine-Dalgarno (SD) sequence
a short, purine-rich sequence that recruits 30S ribosomal subunits
- Expression platform
portion of a riboswitch that interfaces directly with cellular factors involved in gene expression
- H-type pseudoknot
RNA structural motif in which nucleotides within a hairpin-loop form Watson-Crick base-pairs with complementary sequence near the base of the stem
- Ribozyme
an RNA participating directly in the rate acceleration of a chemical transformation
- Non-coding RNA
RNA sequence that does not encode protein
Footnotes
DISCLOSURE STATEMENT
R.R.B. is a cofounder and member of the scientific advisory board of BioRelix, a company interested in using riboswitches as drug targets.
LITERATURE CITED
- 1.Steitz TA, Moore PB. RNA, the first macromolecular catalyst: the ribosome is a ribozyme. Trends Biochem Sci. 2003;28:411–8. doi: 10.1016/S0968-0004(03)00169-5. [DOI] [PubMed] [Google Scholar]
- 2.Yean SL, Wuenschell G, Termini J, Lin RJ. Metal-ion coordination by U6 small nuclear RNA contributes to catalysis in the spliceosome. Nature. 2000;408:881–4. doi: 10.1038/35048617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valadkhan S, Manley JL. Splicing-related catalysis by protein-free snRNAs. Nature. 2001;413:701–7. doi: 10.1038/35099500. [DOI] [PubMed] [Google Scholar]
- 4.Doudna JA, Batey RT. Structural insights into the signal recognition particle. Annu Rev Biochem. 2004;73:539–57. doi: 10.1146/annurev.biochem.73.011303.074048. [DOI] [PubMed] [Google Scholar]
- 5.Qiao F, Cech TR. Triple-helix structure in telomerase RNA contributes to catalysis. Nat Struct Mol Biol. 2008;15:634–40. doi: 10.1038/nsmb.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doudna JA, Cech TR. The chemical repertoire of natural ribozymes. Nature. 2002;418:222–8. doi: 10.1038/418222a. [DOI] [PubMed] [Google Scholar]
- 7.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–30. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 8.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–20. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 9.Bass BL, Cech TR. Specific interaction between the self-splicing RNA of Tetrahymena and its guanosine substrate: implications for biological catalysis by RNA. Nature. 1984;308:820–6. doi: 10.1038/308820a0. [DOI] [PubMed] [Google Scholar]
- 10.Bass BL, Cech TR. Ribozyme inhibitors: deoxyguanosine and dideoxyguanosine are competitive inhibitors of self-splicing of the Tetrahymena ribosomal ribonucleic acid precursor. Biochemistry. 1986;25:4473–7. doi: 10.1021/bi00364a001. [DOI] [PubMed] [Google Scholar]
- 11.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–94. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 12.Hermann T. Chemical and functional diversity of small molecule ligands for RNA. Biopolymers. 2003;70:4–18. doi: 10.1002/bip.10410. [DOI] [PubMed] [Google Scholar]
- 13.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 14.Gusarov I, Nudler E. The mechanism of intrinsic transcription termination. Mol Cell. 1999;3:495–504. doi: 10.1016/s1097-2765(00)80477-3. [DOI] [PubMed] [Google Scholar]
- 15.Yarnell WS, Roberts JW. Mechanism of intrinsic transcription termination and antitermination. Science. 1999;284:611–5. doi: 10.1126/science.284.5414.611. [DOI] [PubMed] [Google Scholar]
- 16.Russell R. RNA misfolding and the action of chaperones. Front Biosci. 2008;13:1–20. doi: 10.2741/2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edwards TE, Klein DJ, Ferre-D’Amare AR. Riboswitches: small-molecule recognition by gene regulatory RNAs. Curr Opin Struct Biol. 2007;17:273–9. doi: 10.1016/j.sbi.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Schwalbe H, Buck J, Furtig B, Noeske J, Wohnert J. Structures of RNA switches: insight into molecular recognition and tertiary structure. Angew Chem Int Ed Engl. 2007;46:1212–9. doi: 10.1002/anie.200604163. [DOI] [PubMed] [Google Scholar]
- 19.Montange RK, Batey RT. Riboswitches: emerging themes in RNA structure and function. Annu Rev Biophys. 2008;37:117–33. doi: 10.1146/annurev.biophys.37.032807.130000. [DOI] [PubMed] [Google Scholar]
- 20.Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003;113:577–86. doi: 10.1016/s0092-8674(03)00391-x. [DOI] [PubMed] [Google Scholar]
- 21.Mandal M, Breaker RR. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat Struct Mol Biol. 2004;11:29–35. doi: 10.1038/nsmb710. [DOI] [PubMed] [Google Scholar]
- 22.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–5. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 23.Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem Biol. 2004;11:1729–41. doi: 10.1016/j.chembiol.2004.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noeske J, Richter C, Grundl MA, Nasiri HR, Schwalbe H, Wohnert J. An intermolecular base triple as the basis of ligand specificity and affinity in the guanine- and adenine-sensing riboswitch RNAs. Proc Natl Acad Sci U S A. 2005;102:1372–7. doi: 10.1073/pnas.0406347102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrick JE, Breaker RR. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007;8:R239. doi: 10.1186/gb-2007-8-11-r239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazanov MD, Vitreschak AG, Gelfand MS. Abundance and functional diversity of riboswitches in microbial communities. BMC Genomics. 2007;8:347. doi: 10.1186/1471-2164-8-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J Biol Chem. 2002;277:48949–59. doi: 10.1074/jbc.M208965200. [DOI] [PubMed] [Google Scholar]
- 28.Miranda-Rios J, Navarro M, Soberon M. A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc Natl Acad Sci U S A. 2001;98:9736–41. doi: 10.1073/pnas.161168098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sudarsan N, Barrick JE, Breaker RR. Metabolite-binding RNA domains are present in the genes of eukaryotes. RNA. 2003;9:644–7. doi: 10.1261/rna.5090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kubodera T, Watanabe M, Yoshiuchi K, Yamashita N, Nishimura A, Nakai S, Gomi K, Hanamoto H. Thiamine-regulated gene expression of Aspergillus oryzae thiA requires splicing of the intron containing a riboswitch-like domain in the 5′-UTR. FEBS Lett. 2003;555:516–20. doi: 10.1016/s0014-5793(03)01335-8. [DOI] [PubMed] [Google Scholar]
- 31.Croft MT, Moulin M, Webb ME, Smith AG. Thiamine biosynthesis in algae is regulated by riboswitches. Proc Natl Acad Sci U S A. 2007;104:20770–5. doi: 10.1073/pnas.0705786105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thore S, Leibundgut M, Ban N. Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science. 2006;312:1208–11. doi: 10.1126/science.1128451. [DOI] [PubMed] [Google Scholar]
- 33.Serganov A, Polonskaia A, Phan AT, Breaker RR, Patel DJ. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature. 2006;441:1167–71. doi: 10.1038/nature04740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edwards TE, Ferre-D’Amare AR. Crystal structures of the thi-box riboswitch bound to thiamine pyrophosphate analogs reveal adaptive RNA-small molecule recognition. Structure. 2006;14:1459–68. doi: 10.1016/j.str.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Nagaswamy U, Fox GE. Frequent occurrence of the T-loop RNA folding motif in ribosomal RNAs. RNA. 2002;8:1112–9. doi: 10.1017/s135583820202006x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Epshtein V, Mironov AS, Nudler E. The riboswitch-mediated control of sulfur metabolism in bacteria. Proc Natl Acad Sci U S A. 2003;100:5052–6. doi: 10.1073/pnas.0531307100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDaniel BA, Grundy FJ, Artsimovitch I, Henkin TM. Transcription termination control of the S box system: direct measurement of S-adenosylmethionine by the leader RNA. Proc Natl Acad Sci U S A. 2003;100:3083–8. doi: 10.1073/pnas.0630422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat Struct Biol. 2003;10:701–7. doi: 10.1038/nsb967. [DOI] [PubMed] [Google Scholar]
- 39.Corbino KA, Barrick JE, Lim J, Welz R, Tucker BJ, Puskarz I, Mandal M, Rudnick ND, Breaker RR. Evidence for a second class of S-adenosylmethionine riboswitches and other regulatory RNA motifs in alpha-proteobacteria. Genome Biol. 2005;6:R70. doi: 10.1186/gb-2005-6-8-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuchs RT, Grundy FJ, Henkin TM. The SMK box is a new SAM-binding RNA for translational regulation of SAM synthetase. Nat Struct Mol Biol. 2006;13:226–33. doi: 10.1038/nsmb1059. [DOI] [PubMed] [Google Scholar]
- 41.Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, Wang JX, Lee ER, Block KF, Sudarsan N, Neph S, Tompa M, Ruzzo WL, Breaker RR. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 2007;35:4809–19. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weinberg Z, Regulski EE, Hammond MC, Barrick JE, Yao Z, Ruzzo WL, Breaker RR. The aptamer core of SAM-IV riboswitches mimics the ligand-binding site of SAM-I riboswitches. RNA. 2008;14:822–8. doi: 10.1261/rna.988608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang JX, Breaker RR. Riboswitches that sense S-adenosylmethionine and S-adenosylhomocysteine. Biochem Cell Biol. 2008;86:157–68. doi: 10.1139/O08-008. [DOI] [PubMed] [Google Scholar]
- 44.Montange RK, Batey RT. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441:1172–5. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- 45.Gilbert SD, Rambo RP, Van Tyne D, Batey RT. Structure of the SAM-II riboswitch bound to S-adenosylmethionine. Nat Struct Mol Biol. 2008;15:177–82. doi: 10.1038/nsmb.1371. [DOI] [PubMed] [Google Scholar]
- 46.Lu C, Smith AM, Fuchs RT, Ding F, Rajashankar K, Henkin TM, Ke A. Crystal structures of the SAM-III/SMK riboswitch reveal the SAM-dependent translation inhibition mechanism. Nat Struct Mol Biol. 2008;15:1076–83. doi: 10.1038/nsmb.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA. Crystal structure of a self-splicing group I intron with both exons. Nature. 2004;430:45–50. doi: 10.1038/nature02642. [DOI] [PubMed] [Google Scholar]
- 48.Guo F, Gooding AR, Cech TR. Structure of the Tetrahymena ribozyme: base triple sandwich and metal ion at the active site. Mol Cell. 2004;16:351–62. doi: 10.1016/j.molcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Golden BL, Kim H, Chase E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat Struct Mol Biol. 2005;12:82–9. doi: 10.1038/nsmb868. [DOI] [PubMed] [Google Scholar]
- 50.Lim J, Winkler WC, Nakamura S, Scott V, Breaker RR. Molecular-recognition characteristics of SAM-binding riboswitches. Angew Chem Int Ed Engl. 2006;45:964–8. doi: 10.1002/anie.200503198. [DOI] [PubMed] [Google Scholar]
- 51.Hilbers CW, Michiels PJ, Heus HA. New developments in structure determination of pseudoknots. Biopolymers. 1998;48:137–53. doi: 10.1002/(SICI)1097-0282(1998)48:2<137::AID-BIP4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 52.Grundy FJ, Lehman SC, Henkin TM. The L box regulon: lysine sensing by leader RNAs of bacterial lysine biosynthesis genes. Proc Natl Acad Sci U S A. 2003;100:12057–62. doi: 10.1073/pnas.2133705100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, Breaker RR. An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev. 2003;17:2688–97. doi: 10.1101/gad.1140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, Lee M, Roth A, Sudarsan N, Jona I, Wickiser JK, Breaker RR. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc Natl Acad Sci USA. 2004;101:6421–6. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–9. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 56.Garst AD, Heroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem. 2008;283:22347–51. doi: 10.1074/jbc.C800120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serganov A, Huang L, Patel DJ. Structural insights into amino acid binding and gene control by a lysine riboswitch. Nature. 2008 doi: 10.1038/nature07326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Correll CC, Freeborn B, Moore PB, Steitz TA. Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell. 1997;91:705–12. doi: 10.1016/s0092-8674(00)80457-2. [DOI] [PubMed] [Google Scholar]
- 59.Leontis NB, Westhof E. The 5S rRNA loop E: chemical probing and phylogenetic data versus crystal structure. RNA. 1998;4:1134–53. doi: 10.1017/s1355838298980566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klein DJ, Schmeing TM, Moore PB, Steitz TA. The kink-turn: a new RNA secondary structure motif. EMBO J. 2001;20:4214–21. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blouin S, Lafontaine DA. A loop-loop interaction and a K-turn motif located in the lysine aptamer domain are important for the riboswitch gene regulation control. RNA. 2007;13:1256–67. doi: 10.1261/rna.560307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blount KF, Wang JX, Lim J, Sudarsan N, Breaker RR. Antibacterial lysine analogs that target lysine riboswitches. Nat Chem Biol. 2007;3:44–9. doi: 10.1038/nchembio842. [DOI] [PubMed] [Google Scholar]
- 63.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281–6. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 64.Collins JA, Irnov I, Baker S, Winkler WC. Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev. 2007;21:3356–68. doi: 10.1101/gad.1605307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein DJ, Ferre-D’Amare AR. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science. 2006;313:1752–6. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- 66.Cochrane JC, Lipchock SV, Strobel SA. Structural investigation of the GlmS ribozyme bound to its catalytic cofactor. Chem Biol. 2007;14:97–105. doi: 10.1016/j.chembiol.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klein DJ, Wilkinson SR, Been MD, Ferre-D’Amare AR. Requirement of helix P2.2 and nucleotide G1 for positioning the cleavage site and cofactor of the glmS ribozyme. J Mol Biol. 2007;373:178–89. doi: 10.1016/j.jmb.2007.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roth A, Nahvi A, Lee M, Jona I, Breaker RR. Characteristics of the glmS ribozyme suggest only structural roles for divalent metal ions. RNA. 2006;12:607–19. doi: 10.1261/rna.2266506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilkinson SR, Been MD. A pseudoknot in the 3′ non-core region of the glmS ribozyme enhances self-cleavage activity. RNA. 2005;11:1788–94. doi: 10.1261/rna.2203605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hampel KJ, Tinsley MM. Evidence for preorganization of the glmS ribozyme ligand binding pocket. Biochemistry. 2006;45:7861–71. doi: 10.1021/bi060337z. [DOI] [PubMed] [Google Scholar]
- 71.Tinsley RA, Furchak JR, Walter NG. Trans-acting glmS catalytic riboswitch: locked and loaded. RNA. 2007;13:468–77. doi: 10.1261/rna.341807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McCarthy TJ, Plog MA, Floy SA, Jansen JA, Soukup JK, Soukup GA. Ligand requirements for glmS ribozyme self-cleavage. Chem Biol. 2005;12:1221–6. doi: 10.1016/j.chembiol.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 73.Lim J, Grove BC, Roth A, Breaker RR. Characteristics of ligand recognition by a glmS self-cleaving ribozyme. Angew Chem Int Ed Engl. 2006;45:6689–93. doi: 10.1002/anie.200602534. [DOI] [PubMed] [Google Scholar]
- 74.Cromie MJ, Shi Y, Latifi T, Groisman EA. An RNA sensor for intracellular Mg2+ Cell. 2006;125:71–84. doi: 10.1016/j.cell.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 75.Dann CE, 3rd, Wakeman CA, Sieling CL, Baker SC, Irnov I, Winkler WC. Structure and mechanism of a metal-sensing regulatory RNA. Cell. 2007;130:878–92. doi: 10.1016/j.cell.2007.06.051. [DOI] [PubMed] [Google Scholar]
- 76.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chem Biol. 2002;9:1043. doi: 10.1016/s1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 77.Fuchs RT, Grundy FJ, Henkin TM. S-adenosylmethionine directly inhibits binding of 30S ribosomal subunits to the SMK box translational riboswitch RNA. Proc Natl Acad Sci U S A. 2007;104:4876–80. doi: 10.1073/pnas.0609956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ontiveros-Palacios N, Smith AM, Grundy FJ, Soberon M, Henkin TM, Miranda-Rios J. Molecular basis of gene regulation by the THI-box riboswitch. Mol Microbiol. 2008;67:793–803. doi: 10.1111/j.1365-2958.2007.06088.x. [DOI] [PubMed] [Google Scholar]
- 79.Wickiser JK, Winkler WC, Breaker RR, Crothers DM. The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol Cell. 2005;18:49–60. doi: 10.1016/j.molcel.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 80.Lemay JF, Penedo JC, Tremblay R, Lilley DM, Lafontaine DA. Folding of the adenine riboswitch. Chem Biol. 2006;13:857–68. doi: 10.1016/j.chembiol.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 81.Wickiser JK, Cheah MT, Breaker RR, Crothers DM. The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry. 2005;44:13404–14. doi: 10.1021/bi051008u. [DOI] [PubMed] [Google Scholar]
- 82.Gilbert SD, Stoddard CD, Wise SJ, Batey RT. Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J Mol Biol. 2006;359:754–68. doi: 10.1016/j.jmb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 83.Rieder R, Lang K, Graber D, Micura R. Ligand-induced folding of the adenosine deaminase A-riboswitch and implications on riboswitch translational control. Chembiochem. 2007;8:896–902. doi: 10.1002/cbic.200700057. [DOI] [PubMed] [Google Scholar]
- 84.Lang K, Rieder R, Micura R. Ligand-induced folding of the thiM TPP riboswitch investigated by a structure-based fluorescence spectroscopic approach. Nucleic Acids Res. 2007;35:5370–8. doi: 10.1093/nar/gkm580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Winkler W, Nahvi A, Breaker RR. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002;419:952–6. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- 86.Skordalakes E, Berger JM. Structure of the Rho transcription terminator: mechanism of mRNA recognition and helicase loading. Cell. 2003;114:135–46. doi: 10.1016/s0092-8674(03)00512-9. [DOI] [PubMed] [Google Scholar]
- 87.Ciampi MS. Rho-dependent terminators and transcription termination. Microbiology. 2006;152:2515–28. doi: 10.1099/mic.0.28982-0. [DOI] [PubMed] [Google Scholar]
- 88.Nou X, Kadner RJ. Adenosylcobalamin inhibits ribosome binding to btuB RNA. Proc Natl Acad Sci U S A. 2000;97:7190–5. doi: 10.1073/pnas.130013897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spinelli SV, Pontel LB, Garcia Vescovi E, Soncini FC. Regulation of magnesium homeostasis in Salmonella: Mg2+ targets the mgtA transcript for degradation by RNase E. FEMS Microbiol Lett. 2008;280:226–34. doi: 10.1111/j.1574-6968.2008.01065.x. [DOI] [PubMed] [Google Scholar]
- 90.Altman S, Wesolowski D, Guerrier-Takada C, Li Y. RNase P cleaves transient structures in some riboswitches. Proc Natl Acad Sci U S A. 2005;102:11284–9. doi: 10.1073/pnas.0505271102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seif E, Altman S. RNase P cleaves the adenine riboswitch and stabilizes pbuE mRNA in Bacillus subtilis. RNA. 2008;14:1237–43. doi: 10.1261/rna.833408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Comparative genomics of the methionine metabolism in Gram-positive bacteria: a variety of regulatory systems. Nucleic Acids Res. 2004;32:3340–53. doi: 10.1093/nar/gkh659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Andre G, Even S, Putzer H, Burguiere P, Croux C, Danchin A, Martin-Verstraete I, Soutourina O. S-box and T-box riboswitches and antisense RNA control a sulfur metabolic operon of Clostridium acetobutylicum. Nucleic Acids Res. 2008 doi: 10.1093/nar/gkn601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pheasant M, Mattick JS. Raising the estimate of functional human sequences. Genome Res. 2007;17:1245–53. doi: 10.1101/gr.6406307. [DOI] [PubMed] [Google Scholar]
- 95.Sudarsan N, Cohen-Chalamish S, Nakamura S, Emilsson GM, Breaker RR. Thiamine pyrophosphate riboswitches are targets for the antimicrobial compound pyrithiamine. Chem Biol. 2005;12:1325–35. doi: 10.1016/j.chembiol.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 96.Cheah MT, Wachter A, Sudarsan N, Breaker RR. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447:497–500. doi: 10.1038/nature05769. [DOI] [PubMed] [Google Scholar]
- 97.Wachter A, Tunc-Ozdemir M, Grove BC, Green PJ, Shintani DK, Breaker RR. Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mRNAs. Plant Cell. 2007;19:3437–50. doi: 10.1105/tpc.107.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bocobza S, Adato A, Mandel T, Shapira M, Nudler E, Aharoni A. Riboswitch-dependent gene regulation and its evolution in the plant kingdom. Genes Dev. 2007;21:2874–9. doi: 10.1101/gad.443907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thore S, Frick C, Ban N. Structural basis of thiamine pyrophosphate analogues binding to the eukaryotic riboswitch. J Am Chem Soc. 2008;130:8116–7. doi: 10.1021/ja801708e. [DOI] [PubMed] [Google Scholar]
- 100.Borsuk P, Przykorska A, Blachnio K, Koper M, Pawlowicz JM, Pekala M, Weglenski P. L-arginine influences the structure and function of arginase mRNA in Aspergillus nidulans. Biol Chem. 2007;388:135–44. doi: 10.1515/BC.2007.015. [DOI] [PubMed] [Google Scholar]
- 101.Vilela C, McCarthy JE. Regulation of fungal gene expression via short open reading frames in the mRNA 5′ untranslated region. Mol Microbiol. 2003;49:859–67. doi: 10.1046/j.1365-2958.2003.03622.x. [DOI] [PubMed] [Google Scholar]
- 102.Romfo CM, Alvarez CJ, van Heeckeren WJ, Webb CJ, Wise JA. Evidence for splice site pairing via intron definition in Schizosaccharomyces pombe. Mol Cell Biol. 2000;20:7955–70. doi: 10.1128/mcb.20.21.7955-7970.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sassanfar M, Szostak JW. An RNA motif that binds ATP. Nature. 1993;364:550–3. doi: 10.1038/364550a0. [DOI] [PubMed] [Google Scholar]
- 104.Huang Z, Szostak JW. Evolution of aptamers with a new specificity and new secondary structures from an ATP aptamer. RNA. 2003;9:1456–63. doi: 10.1261/rna.5990203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sazani PL, Larralde R, Szostak JW. A small aptamer with strong and specific recognition of the triphosphate of ATP. J Am Chem Soc. 2004;126:8370–1. doi: 10.1021/ja049171k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Harada F, Nishimura S. Possible anticodon sequences of tRNAHis, tRNAAsn, and tRNAAsp from Escherichia coli B. Universal presence of nucleoside Q in the first position of the anticodons of these transfer ribonucleic acids. Biochemistry. 1972;11:301–8. doi: 10.1021/bi00752a024. [DOI] [PubMed] [Google Scholar]
- 107.Roth A, Winkler WC, Regulski EE, Lee BW, Lim J, Jona I, Barrick JE, Ritwik A, Kim JN, Welz R, Iwata-Reuyl D, Breaker RR. A riboswitch selective for the queuosine precursor preQ1 contains an unusually small aptamer domain. Nat Struct Mol Biol. 2007;14:308–17. doi: 10.1038/nsmb1224. [DOI] [PubMed] [Google Scholar]
- 108.Meyer MM, Roth A, Chervin SM, Garcia GA, Breaker RR. Confirmation of a second natural preQ1 aptamer class in Streptococcaceae bacteria. RNA. 2008;14:685–95. doi: 10.1261/rna.937308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Draper DE, Grilley D, Soto AM. Ions and RNA folding. Annu Rev Biophys Biomol Struct. 2005;34:221–43. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 110.Romani AM, Scarpa A. Regulation of cellular magnesium. Front Biosci. 2000;5:D720–34. doi: 10.2741/romani. [DOI] [PubMed] [Google Scholar]
- 111.Pyle AM. Metal ions in the structure and function of RNA. J Biol Inorg Chem. 2002;7:679–90. doi: 10.1007/s00775-002-0387-6. [DOI] [PubMed] [Google Scholar]
- 112.Nahvi A, Barrick JE, Breaker RR. Coenzyme B12 riboswitches are widespread genetic control elements in prokaryotes. Nucleic Acids Res. 2004;32:143–50. doi: 10.1093/nar/gkh167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim JN, Roth A, Breaker RR. Guanine riboswitch variants from Mesoplasma florum selectively recognize 2′-deoxyguanosine. Proc Natl Acad Sci U S A. 2007;104:16092–7. doi: 10.1073/pnas.0705884104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hutchison CAI, Montague MG. In: Molecular Biology and Pathogenicity of Mycoplasmas. Razin S, Herrmann R, editors. New York: Kluwer Academic/Plenum; 2002. pp. 221–53. [Google Scholar]
- 115.Regulski EE, Moy RH, Weinberg Z, Barrick JE, Yao Z, Ruzzo WL, Breaker RR. A widespread riboswitch candidate that controls bacterial genes involved in molybdenum cofactor and tungsten cofactor metabolism. Mol Microbiol. 2008;68:918–32. doi: 10.1111/j.1365-2958.2008.06208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baugh PE, Collison D, Garner CD, Joule JA. Molybdenum metalloenzymes. In: Sinnott M, editor. Comprehensive Biological Catalysis. San Diego: Academic Press; 1998. pp. 377–400. [Google Scholar]
- 117.Kletzin A, Adams MW. Tungsten in biological systems. FEMS Microbiol Rev. 1996;18:5–63. doi: 10.1016/0168-6445(95)00025-9. [DOI] [PubMed] [Google Scholar]
- 118.Hille R. Molybdenum and tungsten in biology. Trends Biochem Sci. 2002;27:360–7. doi: 10.1016/s0968-0004(02)02107-2. [DOI] [PubMed] [Google Scholar]
- 119.Lipfert J, Das R, Chu VB, Kudaravalli M, Boyd N, Herschlag D, Doniach S. Structural transitions and thermodynamics of a glycine-dependent riboswitch from Vibrio cholerae. J Mol Biol. 2007;365:1393–406. doi: 10.1016/j.jmb.2006.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yamauchi T, Miyoshi D, Kubodera T, Nishimura A, Nakai S, Sugimoto N. Roles of Mg2+ in TPP-dependent riboswitch. FEBS Lett. 2005;579:2583–8. doi: 10.1016/j.febslet.2005.03.074. [DOI] [PubMed] [Google Scholar]
- 121.Kwon M, Strobel SA. Chemical basis of glycine riboswitch cooperativity. RNA. 2008;14:25–34. doi: 10.1261/rna.771608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rodionov DA, Dubchak I, Arkin A, Alm E, Gelfand MS. Reconstruction of regulatory and metabolic pathways in metal-reducing delta-proteobacteria. Genome Biol. 2004;5:R90. doi: 10.1186/gb-2004-5-11-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sudarsan N, Hammond MC, Block KF, Welz R, Barrick JE, Roth A, Breaker RR. Tandem riboswitch architectures exhibit complex gene control functions. Science. 2006;314:300–4. doi: 10.1126/science.1130716. [DOI] [PubMed] [Google Scholar]
- 124.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science. 2008;321:411–3. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Welz R, Breaker RR. Ligand binding and gene control characteristics of tandem riboswitches in Bacillus anthracis. RNA. 2007;13:573–82. doi: 10.1261/rna.407707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gonzalez JC, Peariso K, Penner-Hahn JE, Matthews RG. Cobalamin-independent methionine synthase from Escherichia coli: a zinc metalloenzyme. Biochemistry. 1996;35:12228–34. doi: 10.1021/bi9615452. [DOI] [PubMed] [Google Scholar]
- 127.Pejchal R, Ludwig ML. Cobalamin-independent methionine synthase (MetE): a face-to-face double barrel that evolved by gene duplication. PLoS Biol. 2005;3:e31. doi: 10.1371/journal.pbio.0030031. [DOI] [PMC free article] [PubMed] [Google Scholar]