

Abstract
Welander distal myopathy is a rare autosomal dominant disorder characterized by muscle weakness in the hands and feet. Exome sequencing of affected families discovered a segregating p.Glu384Lys pathogenic variant in TIA-1 as the main genetic cause of Welander distal myopathy. TIA-1 encodes an RNA-binding protein which serves as a key component of stress granules. This protein also regulates splicing and translation of mRNA. Our patient developed progressive weakness in his hands and feet during his late 40s that was misdiagnosed as a neuropathy that caused muscle atrophy. Follow-up genetic testing revealed a p.Glu384Lys pathogenic variant in TIA-1, and he was then diagnosed with Welander distal myopathy. Our case report underlines the importance of electrodiagnostic and genetic testing of patients.
Key Words: distal myopathy, Welander distal myopathy, whole exome sequencing, next-generation sequencing, TIA-1
INTRODUCTION
Welander distal myopathy (WDM, OMIM: 604454) is characterized by weakness of the extensor muscles in the hands and feet, and in the dorsiflexors of the feet.1 This autosomal dominant disease was recently linked to a pathogenic missense variant, c.1150G>A (NM_022173.2, p.Glu384Lys), in TIA-1 (TIA1 cytotoxic granule-associated RNA binding protein).2,3 This variant is mainly found in patients from Sweden, Finland, and Great Britain.2 TIA-1 is involved in stress granule formation and pre-mRNA splicing.4 Molecular investigations have reported that both of these functions are altered in the p.Glu384Lys variant associated with WDM, which falls within the highly conserved RNA-binding domain in TIA-1.3
In this report, we describe a patient who underwent 2 years of extensive testing to finally be diagnosed with WDM. Our report demonstrates the importance of electrodiagnostic and genetic testing in patients to prevent additional stresses, unnecessary tests, delays in correct diagnosis, increased financial burden, and potentially harmful and unwarranted therapeutic interventions before establishing the correct diagnosis.
CASE REPORT
A 60-year-old right-handed man was referred to our clinic due to an undiagnosed progressive weakness and numbness in his hands and feet. As a young adult, the patient described an inability to develop muscles in his chest, triceps, and hamstrings. He did not recall previous muscle weakness until the age of 47, after which he noticed that his left index finger subtly began to droop making full extension of this finger difficult. By 55 years of age, progressive hand atrophy and poor finger extension had developed, with a lesser involvement of hand grip. Two years later, he began to notice dorsiflexor weakness and poor sensation in his feet. Since then, muscle atrophy has progressed to his lower leg, with slight atrophy of his thigh. He reports no pain or bladder/bowel involvement. The patient also has no known family history of similarly affected individuals. His mother died of coronary heart failure at the age of 77, and his father died of lung cancer at 74 years. Maternal ancestry was Irish, and paternal ancestry was Swedish.
Before visiting the Individualized Medicine Clinic (Mayo Clinic Florida), he was believed to have a neuropathy that caused muscle atrophy. However, no definitive diagnosis was made. Prior genetic testing for spinal muscular atrophy (SMA1), amyotrophic lateral sclerosis (C9orf72), and limb girdle muscular dystrophy (gene-panel testing) was normal. In addition, previous electromyography (EMG) studies were inconclusive and did not show any changes in muscle activity. The patient was evaluated by a neurosurgeon for foraminal stenosis that was unrelated to his muscle weakness.
A neurological examination determined that his mental status and coordination were normal. However, he displayed severe abnormalities in wrist extension, digit extension, and intrinsic hand muscle weakness (Figs. 1A, B). Trace biceps weakness was also observed. In his lower extremities, tone was slightly reduced, and dorsiflexion and toe extension were absent. He had mildly reduced sensation in the toes to light touch and vibration. Interestingly, he also presented with high arches and hammertoes but does not recall other family members with this predisposition (Figs. 1C–E). He had absent left biceps and left ankle jerk muscle stretch reflexes. Toes were downgoing to plantar stimulation. Casual gait was normal, but he was unable to walk on his heels. Magnetic resonance imaging of the spine showed congenital narrowing of the central canal. A comprehensive blood test was unremarkable. The use of orthotics was recommended should walking become difficult in addition to occupational therapy to assist with his hand weakness.
FIGURE 1.
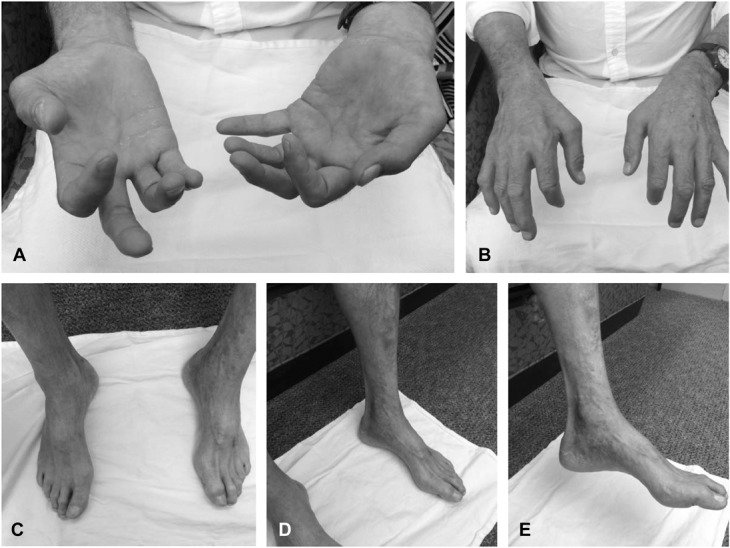
Patient photographs. A, Poor extension of the index finger. B, Progressive hand atrophy and dysfunction of wrist extension. C, Hammertoes. D, Severe atrophy of distal limbs. E, High arches.
Repeat EMG at our institution revealed a chronic, mildly active distal greater than proximal myopathy. Nerve conduction studies on the right arm and leg demonstrated diffusely low motor amplitudes with no significant dispersion or conduction block (Table 1). Concentric needle EMG demonstrated increased insertional activity in right T8 paraspinal and pronator teres muscles along with both increased and decreased insertional activity in the first dorsal interosseous (Table 2). There was reduced recruitment; however, motor unit potentials appeared small and polyphasic except in the deltoid. In the right leg, there was increased insertional activity with fibrillation potentials in distal muscles and high and low amplitude, short duration polyphasic motor unit potentials. A left biceps muscle biopsy revealed abnormal variation in fiber size and nonnecrotic fibers containing small vacuoles (Fig. 2A). Rare atrophic muscle fibers were observed overreacting to a nonspecific esterase stain indicative of denervation atrophy (Fig. 2B).
TABLE 1.
Detailed Nerve Conduction Findings
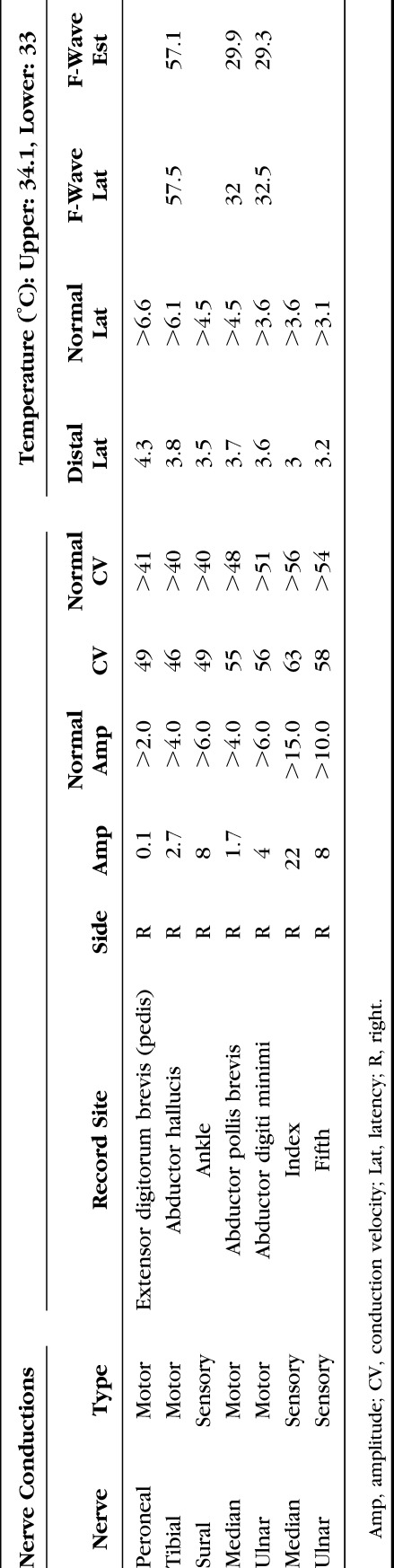
TABLE 2.
Detailed Needle EMG Finding
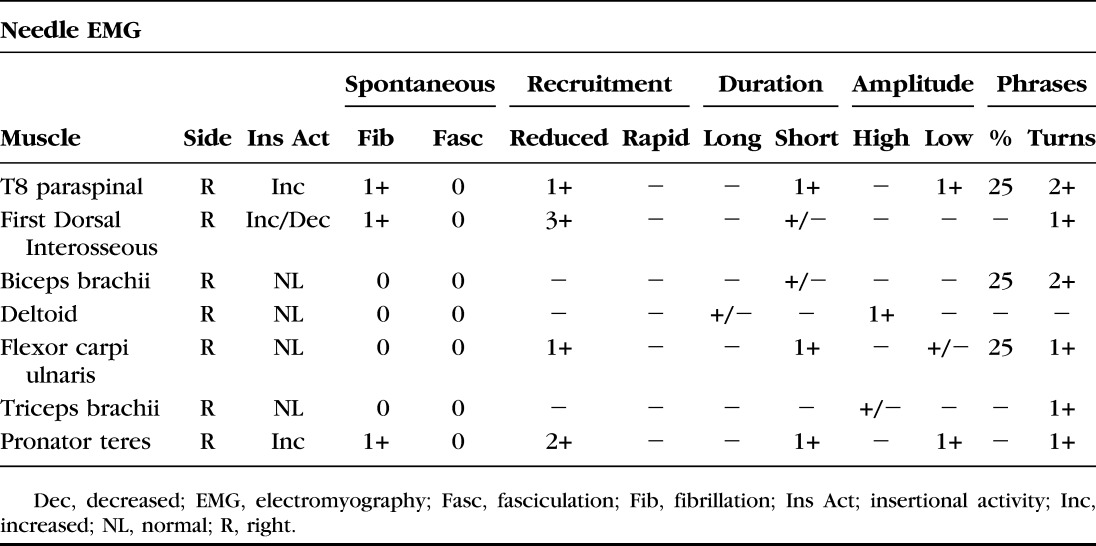
FIGURE 2.
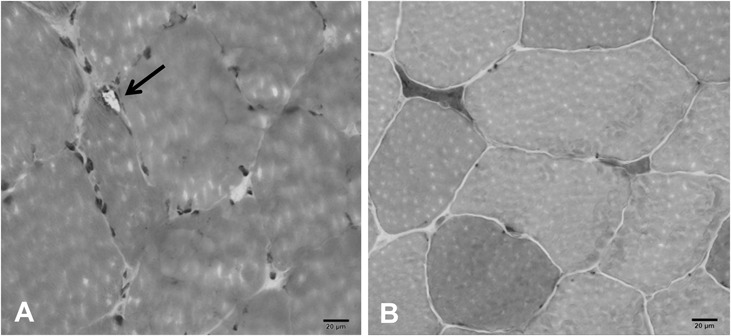
Left biceps brachii muscle biopsy. A, High power hematoxylin and eosin stain revealing a small peripheral vacuole in a nonnecrotic fiber as noted with arrow. B, Atrophic muscle fibers overreacting for nonspecific esterase.
Whole exome sequencing (WES) was performed by a commercial diagnostic laboratory (XomeDx, GeneDx). Briefly, genomic DNA from the patient was sequenced using an Agilent Clinical Research Exome kit to target exonic regions and flanking splice junctions. These regions were sequenced simultaneously by massively parallel next-generation sequencing on an Illumina HiSeq 2000 with 100 base-pair paired-end reads. The sequence was assembled and aligned to reference gene sequences and analyzed using Xome Analyzer. Sanger sequencing was used to confirm potential variants. No reportable secondary findings were identified in the coding regions. No pathogenic variants known to be associated with a disorder of mitochondrial metabolism were identified by sequencing analysis of the entire mitochondrial genome. Analysis of the sequencing data revealed a heterozygous pathogenic variant in TIA-1, c.1150G>A, which in known to cause WDM. All genetic testing was covered by the patient's medical insurance plan.
DISCUSSION
Distal myopathies are rare inherited disorders which cause progressive weakness of voluntary muscles in the hands and feet. To date, over 20 different distal myopathies have been classified by their distinct phenotype and genetic causes.5 Several other distal myopathies have been reported in various families; however, pathogenic variants have yet to be identified.6 With WDM, muscle weakness usually starts in the extensor of the index finger and slowly progresses to the flexor muscle of the wrists and other fingers. As the disease advances, the distal muscles in the feet also become affected, making walking difficult in severe cases.7 Proximal muscles are not affected. After the onset, typically around 50 years of age, symptoms slowly progress but do not affect life expectancy. In severe cases, symptoms may evolve into dysfunction of fine motor movements and walking.8
The TIA-1 gene encodes an RNA-binding protein that is a key component of stress granules.4 Klar et al3 determined that the c.1150G>A missense mutation is associated with increased survival of motor neuron 2 (SMN2) splicing in skeletal muscle and stress markers. Increased number of stress granules in mutated TIA-1 has also been reported. However, the exact mechanism causing mutant TIA-1 to produce myopathy is unknown.
Interestingly, myopathies can mimic a variety of disorders making them difficult to diagnose in some patients.6 Our patient developed reduced sensation to touch and vibration in his feet suggestive of a peripheral neuropathy. In fact, sensory symptoms are atypical for WDM adding to the complexity of this diagnosis.8 He also had very high arches and hammertoes uncharacteristic of WDM and often seen in patients with peripheral neuropathy such as Charcot–Marie–Tooth disease.9
On the other hand, EMG findings were suggestive of a distal myopathy, with little involvement of proximal muscles. Occasionally, EMG findings in patients with WDM can display a myopathic and neuropathic pattern.6 A muscle biopsy from our patient indicated a slight myopathy and mild denervation atrophy (Fig. 2). WDM pathology in muscle fibers usually affects fiber diameter resulting in atrophic fibers with rimmed vacuoles.
To conclude, WES of our patient's DNA revealed a pathogenic variant in TIA-1 which causes WDM and explains most of his symptoms. Previous genetic tests performed on our patient were unremarkable due to the small subset of genes tested. WES and targeted next generation sequencing panels are powerful clinical tools when investigating patients with confounding clinical presentations. Many of the clinical tests and observations of our patient displayed mixed myopathic and neuropathic findings confounding the diagnosis. In fact, certain diseases such as Welander distal myopathy and valosin-containing protein–mutated distal myopathy can be clinically indistinguishable in some cases.5 Therefore, caution should be exercised when diagnosing and treating patients without establishing the molecular genetic cause, as valosin-containing protein–related disorders are associated with frontotemporal dementia and amyotrophic lateral sclerosis, which have a poor prognosis and can have life-altering effects on patients.10,11 The identification of possible genetic causes of disease is very important for determining future treatments and family planning of affected individuals. Our report highlights the use of careful clinical electrodiagnostic tests and the need to consider genetic screening of patients as an early diagnostic tool. This can prevent delayed diagnosis and reduce the amount of unnecessary tests and examinations, as with our patient.
ACKNOWLEDGMENTS
The authors thank the patient and his family for their permission to publish this article and the Mayo Clinic Center for Individualized Medicine for supporting this work.
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Welander L. Myopathia distalis tarda hereditaria; 249 examined cases in 72 pedigrees. Acta med Scand Suppl. 1951;265:1–124. [PubMed] [Google Scholar]
- 2.Hackman P, Sarparanta J, Lehtinen S, et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol. 2013;73:500–509. [DOI] [PubMed] [Google Scholar]
- 3.Klar J, Sobol M, Melberg A, et al. Welander distal myopathy caused by an ancient founder mutation in TIA1 associated with perturbed splicing. Hum Mutat. 2013;34:572–577. [DOI] [PubMed] [Google Scholar]
- 4.Waris S, Wilce MC, Wilce JA. RNA recognition and stress granule formation by TIA proteins. Int J Mol Sci. 2014;15:23377–23388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Udd B. Distal myopathies–new genetic entities expand diagnostic challenge. Neuromuscul Disord. 2012;22:5–12. [DOI] [PubMed] [Google Scholar]
- 6.Dimachkie MM, Barohn RJ. Distal myopathies. Neurol Clin. 2014;32:817–842. x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindberg C, Borg K, Edstrom L, et al. Inclusion body myositis and Welander distal myopathy: a clinical, neurophysiological and morphological comparison. J Neurol Sci. 1991;103:76–81. [DOI] [PubMed] [Google Scholar]
- 8.Borg K, Tome FM, Edstrom L. Intranuclear and cytoplasmic filamentous inclusions in distal myopathy (Welander). Acta Neuropathol. 1991;82:102–106. [DOI] [PubMed] [Google Scholar]
- 9.Pareyson D, Saveri P, Piscosquito G. Charcot-marie-tooth disease and related hereditary neuropathies: from gene function to associated phenotypes. Curr Mol Med. 2014. [DOI] [PubMed] [Google Scholar]
- 10.Kazamel M, et al. Clinical spectrum of valosin containing protein (VCP)-opathy. Muscle Nerve. 2015. [DOI] [PubMed] [Google Scholar]
- 11.Palmio J, et al. Distinct distal myopathy phenotype caused by VCP gene mutation in a Finnish family. Neuromuscul Disord. 2011;21:551–555. [DOI] [PubMed] [Google Scholar]