

Supplemental Digital Content is available in the text.
Keywords: axon, brain, clinical trial, gait, stroke
Abstract
Background and Purpose—
One class of poststroke restorative therapy focuses on promoting axon outgrowth by blocking myelin-based inhibitory proteins such as myelin-associated glycoprotein. The purpose of the current study was to extend preclinical and clinical findings of GSK249320, a humanized monoclonal antibody to myelin-associated glycoprotein with disabled Fc region, to explore effects on motor outcomes poststroke.
Methods—
In this phase IIb double-blind, randomized, placebo-controlled study, patients at 30 centers with ischemic stroke 24 to 72 hours prior and gait deficits were randomized to 2 IV infusions of GSK249320 or placebo. Primary outcome measure was change in gait velocity from baseline to day 90.
Results—
A total of 134 subjects were randomized between May 2013 and July 2014. The 2 groups were overall well matched at baseline. The study was stopped at the prespecified interim analysis because the treatment difference met the predefined futility criteria cutoff; change in gait velocity to day 90 was 0.55±0.46 (mean±SD) in the GSK249320 group and 0.56±0.50 for placebo. Secondary end points including upper extremity function were concordant. The 2 IV infusions of GSK249320 were well tolerated. No neutralizing antibodies to GSK249320 were detected.
Conclusions—
GSK249320, within 72 hours of stroke, demonstrated no improvement on gait velocity compared with placebo. Possible reasons include challenges translating findings into humans and no direct evidence that the therapy reached the biological target. The antibody was well tolerated and showed low immunogenicity, findings potentially useful to future studies aiming to use a monoclonal antibody to modify activity in specific biological pathways to improve recovery from stroke.
Clinical Trial Registration—
URL: http://www.clinicaltrials.gov. Unique identifier: NCT01808261.
After an injury from an acute stroke, numerous restorative events evolve within the brain. Targeting these events therapeutically may augment poststroke neural repair and favorably impact long-term outcome.1 Numerous biological targets are under study to develop restorative therapies. One class of therapy focuses on promoting recovery after stroke by blocking myelin-based inhibitory proteins that inhibit axon outgrowth. Three major inhibitors of such growth have been identified, 1 being myelin-associated glycoprotein (MAG). After stroke, MAG levels spontaneously increase in penumbra,2 suggesting that MAG may be a useful target to promote neural repair, an idea bolstered by previous observations that MAG blockade promotes axonal growth.3–5
The main objective of the current study was to determine whether a monoclonal antibody targeting MAG improves stroke recovery in patients with ischemic stroke. The specific therapy under study was GSK249320, an IgG1-type humanized monoclonal antibody to MAG with disabled Fc region. Anti-MAG antibodies have been shown to neutralize MAG-mediated inhibition in preclinical studies6 and to promote regeneration after peripheral nerve injury.7,8 Blocking the action of a related protein, Nogo, 7 days after ischemic stroke in rats improved behavioral recovery by promoting axonal growth.9 The preclinical program for GSK249320 included rodent studies that found that the antibody penetrated the infarct site and had small but significant effects on behavioral outcomes when initiated 24 hours poststroke without affecting infarct volume,10 and primate studies in which IV infusion of GSK249320 beginning 24 hours after experimental ischemic infarct facilitated behavioral recovery.11 GSK249320 was found to be safe in healthy human subjects,12 and a recent randomized, placebo-controlled phase II trial in patients 24 to 72 hours after ischemic stroke also found the antibody to be safe and suggested potential efficacy for improving recovery of gait.13
The current study built on these findings as a phase IIb double-blind, randomized, placebo-controlled, multicenter study. Patients with ischemic stroke 24 to 72 hours prior and deficits in gait were randomized to receive 2 IV infusions of GSK249320 or placebo. The primary outcome measure was change from baseline to day 90 in gait velocity, which is valid, reliable, and sensitive after stroke.14,15 The study was stopped at the interim analysis because there was insufficient evidence to justify continuing the study given that the observed difference between treatment groups met the predefined futility cutoff.
Methods
Study Overview
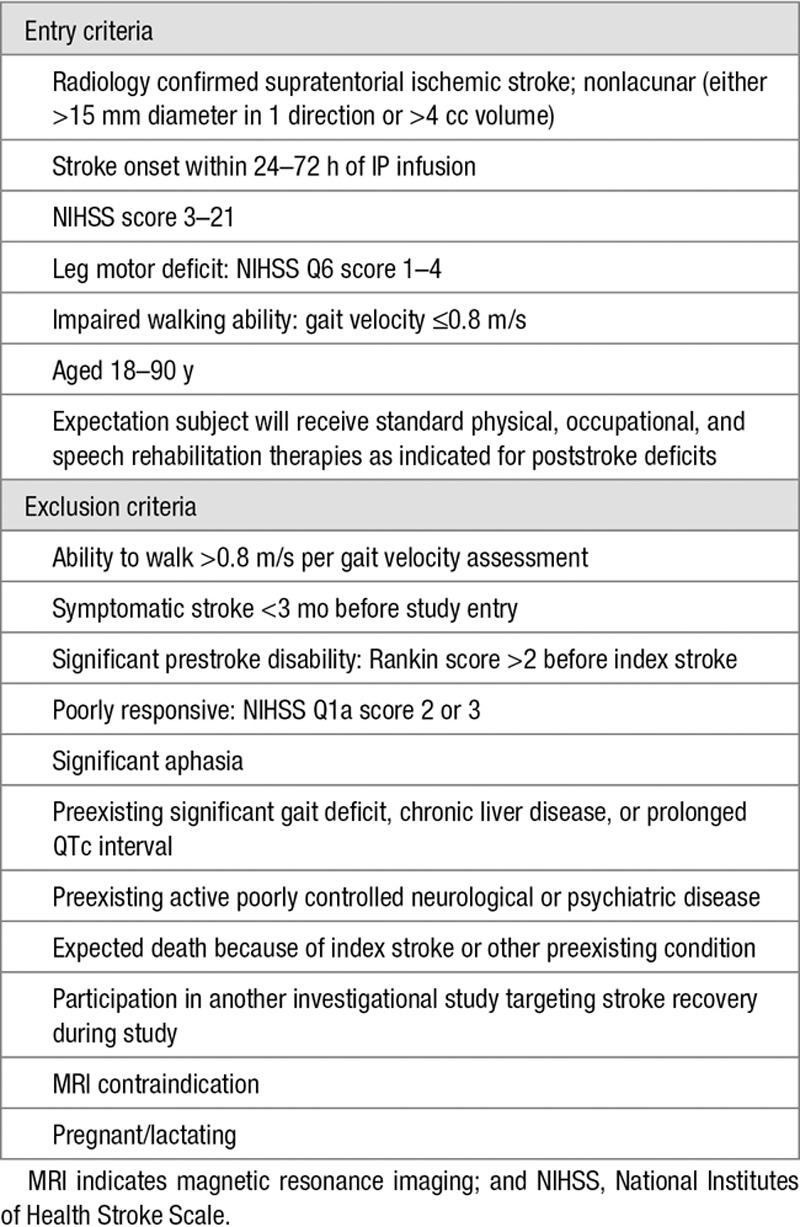
Thirty centers across 4 countries enrolled subjects in the study, between May 2013 and July 2014. The study was approved by each site’s institutional review board. All subjects, or surrogates, gave written informed consent. Participation spanned 6 visits from baseline to day 180. Key entry/exclusion criteria appear in Table 1. See also online-only Data Supplement.
Table 1.
Key Entry and Exclusion Criteria
Randomization
Subjects were centrally randomized to GSK249320 15 mg/kg or placebo in a 1:1 allocation ratio, using permuted blocks, with treatment stratified according to baseline gait velocity (0, >0–<0.4, or 0.4–0.8 m/s). See also online-only Data Supplement.
Study Assessments
At baseline, prior to first infusion and thus <72 hours poststroke, assessments included National Institutes of Health Stroke Scale (NIHSS), modified Rankin Scale, gait velocity, and Box and Blocks (no. blocks transferred during 1 minute). All study assessors were formally trained and certified in each of these outcome measures (see online-only Data Supplement). Patients and assessors were blinded at all times. These were serially evaluated during the remaining 5 visits, as was the amount of rehabilitation (physical and occupational) therapy that patients received. Safety assessments included vital signs, clinical laboratories, ECGs, suicidality, adverse events (AE), serious adverse events, and falls and were monitored by the internal Safety Review Committee. Blood samples were collected at baseline, pre- and post-dosing of IP at visit 2 (day 6), as well as at visits 3 and 6 (day 30 and 180, respectively), or at the time of study withdrawal if applicable, from which free serum MAG levels and GSK249320 levels were measured. See also online-only Data Supplement.
Data Analysis
The primary efficacy end point was the mean change in gait velocity from baseline to day 90. To test the hypothesis that treatment with GSK249320 leads to an improvement of change in gait velocity compared with placebo at day 90, a repeated-measures mixed-effects model was used in a Bayesian framework, including fixed effects for treatment, visit, age, sex, treatment by visit interaction, baseline mean gait velocity by visit interaction, and baseline NIHSS by visit interaction. For additional information, see online-only Data Supplement. At the end of study, a positive signal of efficacy was to be declared if the posterior probability that the true improvement over placebo (GSK249320-placebo) was greater than zero is >95%, and a negative signal of efficacy was to be declared if the posterior probability that the true improvement over placebo is greater than zero is <85%; otherwise the result was to be interpreted as indeterminate. If the true mean gait velocity improvement with GSK249320 is 0.25 m/s over placebo, assuming variance as in the earlier placebo-controlled phase II study of GSK249320,13 enrolling 136 subjects with day 90 data would provide an 85% chance of observing a positive signal of efficacy. Assuming a 16% dropout rate to day 90, enrollment of 162 subjects was planned. Note that a change in gait velocity of 0.1 m/s has been suggested as clinically meaningful in populations with impaired walking speed,16 and an increase of 0.16 m/s is linked to a meaningful improvement in disability.17
One interim and one headline data analysis were planned during the study. The interim analysis was planned for when ≈70 subjects completed the day 90 visit. At that time, the internal Safety Review Committee was to determine whether the estimated treatment effect of GSK249320 was likely to be futile based on a prespecified clinically meaningful treatment effect, that is, if the posterior probability that the true improvement over placebo is greater than zero is <70%. If the data hit the futility threshold, the internal Safety Review Committee would recommend discontinuation of the study.
The safety population was defined as subjects who received at least 1 infusion of IP. The intent-to-treat (ITT) population was defined as subjects in the safety population who underwent at least 1 postbaseline efficacy assessment, with subjects analyzed according to the treatment to which they were randomized. Intent to treat was the population used for the primary efficacy analysis. The per-protocol (PP) population was defined as all subjects in the intent-to-treat population, who were not protocol violators with regard to inclusion/exclusion criteria, unblinding, IP administration, or gait velocity assessments. Subjects who did not receive both infusions of IP were also excluded from the PP population.
Results
Study Conduct
Across all 4 participating countries, 134 subjects were randomized, including 64 who were enrolled during the 3 months it took for the 70th subject to reach day 90, the futility criteria interim analysis to be completed, and the internal Safety Review Committee to make and communicate the decision to stop the study. Of the 133 who received investigational product, 64 subjects (48%) completed the study, and 69 subjects (52%) withdrew from the study or were lost to follow-up (Figure 1). The primary reason for withdrawal was that the study was terminated at the interim analysis. A total of 100 subjects (75%) were in the study for >90 days. A total of 116 subjects (87%) received both infusions of IP; 1 subject received no IP infusions, 10 subjects received only 1 IP infusion, 2 subjects received an incorrect dose for 1 infusion because of incorrect preparation of the dose, and 3 subjects received less than the full 100 mL volume of IP for at least 1 infusion. Overall, protocol deviations were reported for 109 subjects (81%), most of which were minor and did not require exclusion from the PP population (Table I in the online-only Data Supplement); all protocol deviations were collected, for transparency, regardless as to whether or not they had an impact on outcome. Of the 134 subjects randomized into the study, 133 were included in the safety population (placebo: n=68; GSK249320: n=65), 120 were included in the intent-to-treat population (placebo: n=60; GSK249320: n=60), and 104 were included in the PP population.
Figure 1.
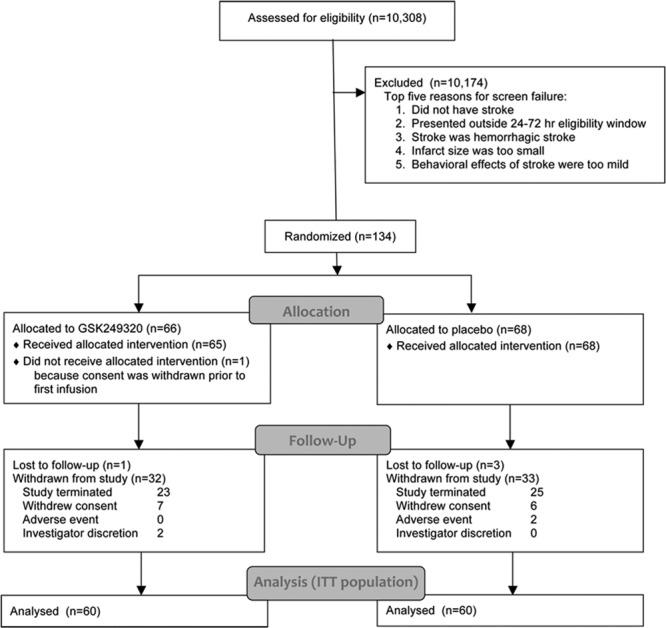
CONSORT (Consolidated Standards of Reporting Trials) diagram. ITT indicates intent to treat.
Subjects
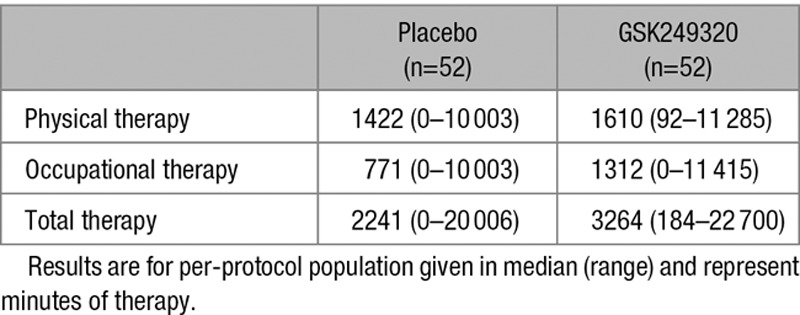
Baseline data (Table 2) were generally balanced across treatment groups. The majority of enrollees (91%) had stroke involving the middle cerebral artery territory. During study participation, the amount of rehabilitation therapy, in minutes, provided to enrollees was substantial and variable, with subjects randomized to GSK249320 receiving a greater amount of therapy (Table 3).
Table 2.
Baseline Clinical Measures and Demographics
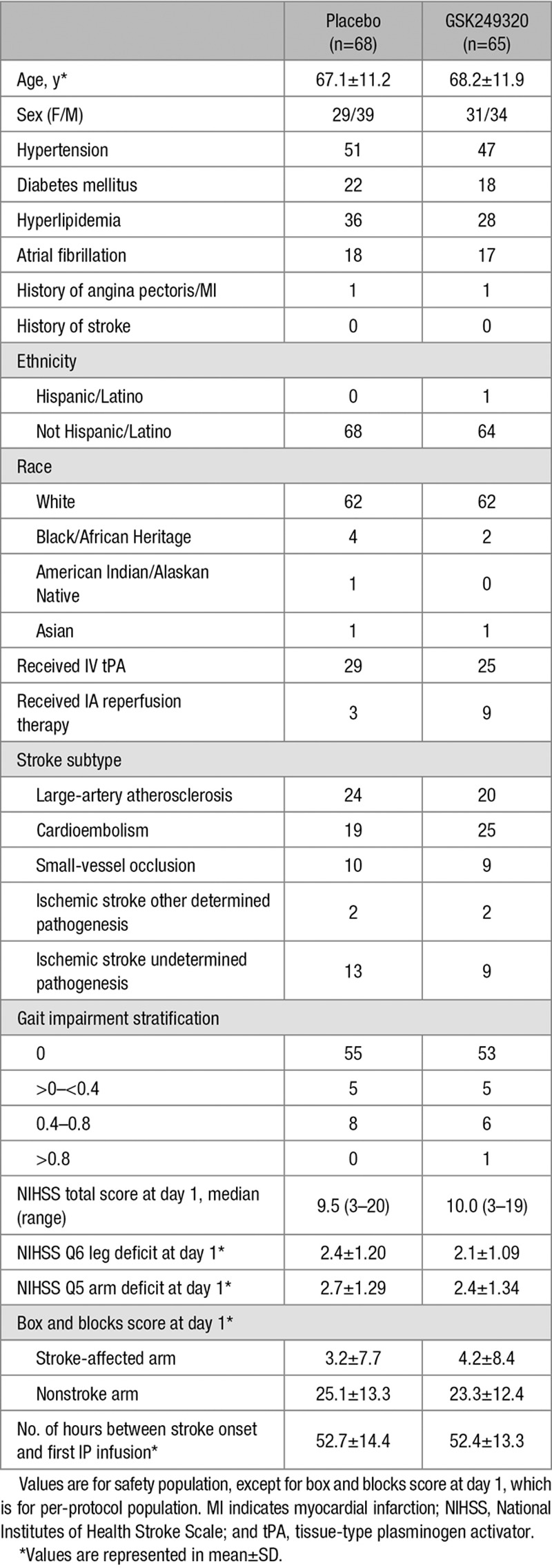
Table 3.
Therapy Provided to Enrollees for the Duration of Study Participation
Analysis of Treatment Efficacy
The study was stopped at the interim analysis because the posterior mean treatment difference was 0.027 at day 90 (95% credible interval, −0.146 to 0.199) and the posterior probability that true treatment difference was greater than zero was 0.621, which was lower than the predefined futility cutoff of 0.70 (Figure 2). Analysis of the PP population and using the final database including subject data for those subjects with an early withdrawal visit because of study termination were concordant (online-only Data Supplement).
Figure 2.
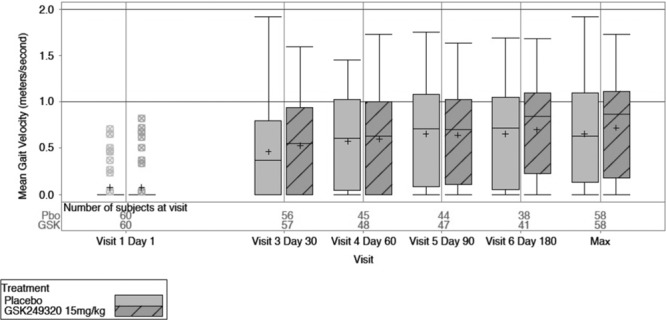
Box-and-whisker plots of gait velocity change over time and maximum value for the 2 treatment arms (intent-to-treat group).
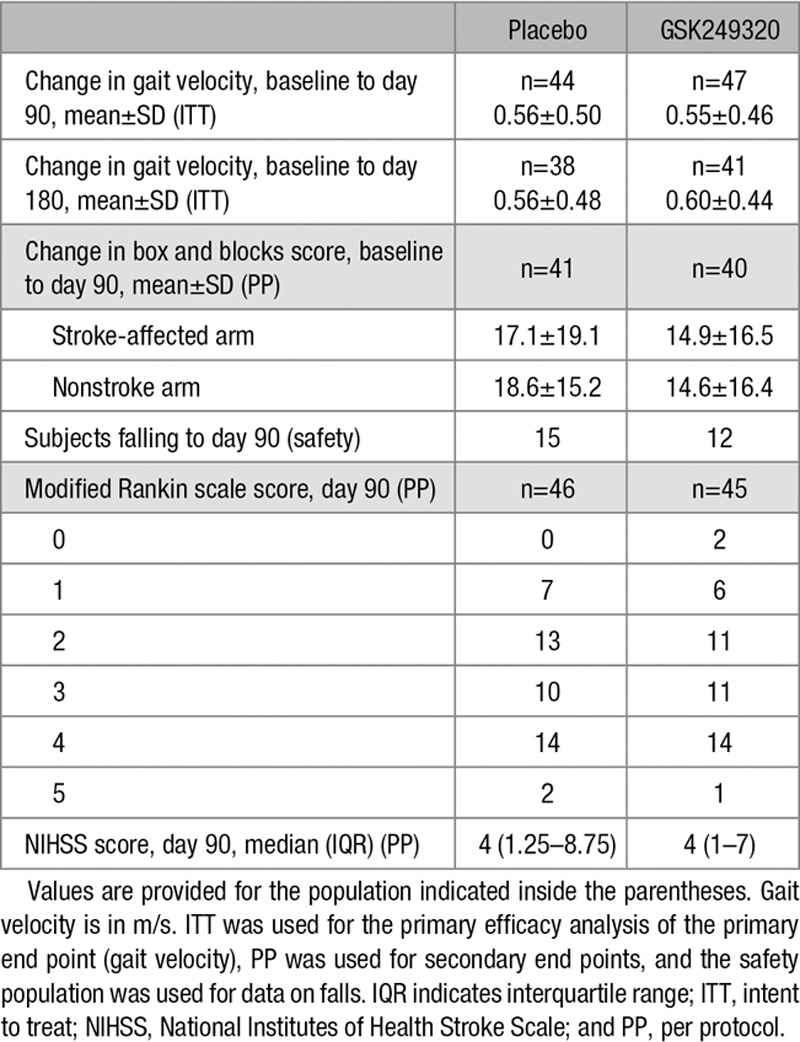
Gait velocity data described the proportion of subjects in each gait impairment category (0, >0–< 0.4, 0.4–0.8, and >0.8 m/s) over time. Most subjects were nonambulatory at baseline and progressed to some level of ambulation by day 180, but a review of summary statistics for the secondary end points (change in gait impairment category, change in box and blocks score, distribution of modified Rankin Scale scores, and total NIHSS score) suggests no obvious differences between treatment groups (Table 4).
Table 4.
Study Outcomes
Analysis of Safety
The 2 IV infusions of GSK249320 were well tolerated as evidenced by an AE rate comparable to placebo, the majority of AEs having been reported as mild or moderate in severity, and the low withdrawal rate because of AEs (Table II in the online-only Data Supplement). No clinically important safety trends were observed post-dosing with GSK249320. There was no difference in the proportion of subjects having a fall, or in the number of falls, between treatment groups. The overall incidence of events common to stroke was comparable across the treatment groups (Table III in the online-only Data Supplement). AEs were reported in 57 subjects (84%) in the placebo group and in 49 subjects (75%) in the GSK249320 group. The most common AEs were constipation, nausea, and headache. No AE reports suggested peripheral neuropathy, infusion site reaction, or hypersensitivity reaction with GSK249320. Withdrawal from the study because of an AE occurred in 2 subjects in the placebo group and in no subjects in the GSK249320 group.
Sixteen subjects (24%) in the placebo group experienced serious adverse events, compared with 9 subjects (14%) in the GSK249320 group. Five subjects (7%) died in the placebo group. Two subjects (3%) died in the GSK249320 group: respiratory failure in a 90-year-old subject 4 days after first infusion and cardiorespiratory arrest in a 76-year-old subject 22 days after first infusion, both considered unrelated to IP infusions.
Immunogenicity
Five subjects had preexisting antibodies at low titers that were not related to treatment. Six of the 64 subjects who received GSK249320 developed antidrug antibodies. Eight of the 68 subjects in the placebo treatment group had antidrug antibodies against GSK249320 that were also not related to treatment. No neutralizing antibodies were detected.
GSK249320 Reduced Free Serum MAG Levels
Before administration of IP, soluble, free MAG plasma levels were similar between placebo and GSK249320 groups (33.0±42.0 versus 30.0±30.7 pg/mL, mean±SD). A progressive slow decline in free MAG level was seen after day 6 for placebo subjects, whereas subjects receiving GSK249320 exhibited an abrupt decline in free MAG level between day 1 and day 6 that was maintained until at least day 30: median inhibition of free MAG in plasma was 97.5% after the first infusion of GSK249320 on day 1 and was maintained after the second infusion on day 6 at 97% until at least day 30, with free MAG levels in GSK249320-treated subjects resuming to levels similar to placebo group subjects at day 180 (Figure I in the online-only Data Supplement). The median GSK249320 concentration at the end of the second IP infusion, which can be considered the maximum concentration, was 494.5 μg/mL, and the mean half-life of GSK249320 was 23.7±5.2 days (Figure II in the online-only Data Supplement).
Discussion
The current study hypothesized that GSK249320, administered as 2 IV infusions beginning 24 to 72 hours poststroke and spaced 5±2 days apart, would improve gait recovery over 90 days in subjects with ischemic stroke and leg weakness with impaired walking ability. The data do not support this, and the study was stopped at interim analysis because observed difference between treatment groups met the predefined futility threshold.
The primary outcome measure was gait velocity, a choice that in retrospect had both advantages and disadvantages. Gait velocity has an established record as a valid, reliable assessment sensitive to treatment effects.14,15 Another advantage is that it measures function (ie, disability and activities limitations), rather than impairment, and can be directly linked with participation level (ie, handicap).15,16,18,19 As a modality-specific outcome measure, gait velocity has potential advantages compared with global outcomes for understanding recovery such as granularity of assessment.20 Furthermore, reduced gait velocity is common after stroke, gait improvements after stroke are linked to better quality of life, and in some studies gait recovery is ranked as the top priority by patients with hemiplegia after stroke.16,21,22 The value of gait velocity as primary end point was also based in part on its direct link with entry criteria (Table 1), which required slow gait for study entry. However, at baseline, >80% of subjects were entirely unable to ambulate at all (gait velocity=0 m/s), masking accurate understanding of within-subject gait recovery. This produced a floor effect such that several different degrees of neural abnormality were scored identically, although the study did make the key distinction between patients with gait velocity=0 m/s and patients in whom gait velocity could not be assessed. Another potential disadvantage of gait as the primary end point is that it is a complex behavior influenced by activity at multiple nervous system levels. Many patients with severe hemiparesis learn to walk on their spasticity, further complicating interpretation of changes in gait velocity after stroke. Putting it in perspective, the current placebo group mean gait velocity change from baseline to day 90 (0.56 m/s) was >3-fold greater compared with placebo group of the previous phase II GSK249320 trial (0.18 m/s),13 a difference possibly because of play of chance but that reduced ability of the current study to detect a treatment group difference. Level of impairment also differed between studies, with median placebo group baseline total NIHSS score of 7 in the previous trial compared with 9.5 herein.
Other study design features may also be important for understanding results. Choice of patient population influences how hypotheses are tested. Patients with small-vessel infarcts, operationally <15 mm maximum diameter or 4 cc volume,23 were excluded given their comparatively favorable prognosis.24,25 Study entry required total NIHSS score of 3 to 21 and leg motor score of 1 to 4. This enrolled subjects with milder strokes, who might be expected to have a favorable prognosis regardless of treatment arm. The amount of IP infused could also be important. Median GSK249320 concentration at the end of the second infusion (maximum concentration) was lower herein as compared with subjects receiving the same dose in the previous study20 in which the second infusion was administered 9±1 days apart (median 494.5 versus 723.0 μg/mL); conceivably infusing a higher amount of antibody might have increased its effect size.
It is useful to revisit assumptions that supported current study design. The antibody showed a favorable preclinical and clinical profile. It was well characterized and the progression of therapy development conformed to published recommendations.17 Preclinical studies in rodents10 and primates11 suggested efficacy. The antibody was found to be safe in 37 healthy subjects, who received a single IV infusion ≤25 mg/kg,12 and in a phase II study of 42 patients 24 to 72 hours after ischemic stroke, among whom 25 subjects received 2 IV infusions ≤15 mg/kg13; significant benefit compared with placebo was found over time for gait velocity, an end point well aligned with preclinical behavioral end points.
Other issues relevant to current results pertain to translation from animals to humans. Behavioral recovery26,27 and neural plasticity28–30 after stroke are accelerated in rodents compared with humans. On the basis of this, time of first infusion in animals (24 hours poststroke) was extended to 72 hours in humans, but this may not have been an appropriate extrapolation. The same concern might extend to presence of MAG, the biological target: in rats with experimental stroke, MAG levels start to increase by 3 days poststroke and peak at 2 weeks poststroke,2 but it is uncertain whether this is true in humans. White matter constitutes 14% of rodent versus 50% of human brain volume31,32; axons might be more difficult for a large antibody to access in humans. Other limitations of animal models may also pertain, including that animal models incompletely capture the complex psychosocial issues that patients face after stroke, such as depression, caregiver support, and financial stressors.33
Direct evidence that substantial quantities of the therapy reached the biological target was not available. Indirect evidence of target binding in the current study was suggested by the substantial reduction in free MAG plasma levels with GSK249320 treatment. The half-life of GSK249320 in the current study was 23.7±5.2 days, similar to the value of 21 days found in healthy control subjects and typical of a monoclonal antibody.12 Neutralizing antibodies were not detected and so did not contribute to current findings.
The experience of translating therapies targeting acute ischemic stroke has provided several lessons,34 and in many cases, these inform translation of restorative stroke therapies to clinical trials. Examples include stepwise translation from preclinical to clinical studies, the need to standardize performance of assessments, careful selection of study sample size to insure adequate study power, and centralized data management. However, neuroprotection differs in many ways from restoration—restorative trials are not simply delayed neuroprotection trials. On the contrary, trials targeting brain restoration must address unique aspects of study design issues33 within the context of topics such as end point selection, target population identification, and intervention timing because the optimal approach in these and other areas often does not directly extend from neuroprotection trials to restorative trials.1,35
This proof-of-concept study for GSK249320, a monoclonal antibody GSK249320 administered IV and initiated within 72 hours of stroke onset, demonstrated no improvement on gait outcomes compared with placebo. As mentioned above, many possible reasons might have contributed to these findings, including using an end point with too large a floor effect at baseline, enrolling patients with too severe a level deficit, using too low an antibody dose, interspecies differences in pharmacokinetics, lack of direct evidence that the therapy reached the biological target, or simply that GSK249320 does not work in human stroke. In the current study, the antibody was well tolerated and showed low immunogenicity, findings that may prove useful to future studies aiming to use a monoclonal antibody to modify activity in specific biological targets to promote improved stroke recovery.
Sources of Funding
S.C. Cramer is supported by NIH K24 HD074722. This study was sponsored by GlaxoSmithKline.
Disclosures
S.C. Cramer has served as a consultant for GlaxoSmithKline, Roche, Dart Neuroscience, MicroTransponder, and RAND Corporation. L.A. Enney and M. Simeoni are employees of GlaxoSmithKline and own shares in the company. C.K. Russell and T.R. Thompson are former employees of GlaxoSmithKline and own shares in the company.
Supplementary Material
Footnotes
Presented in part at the International Stroke Conference of the American Heart Association, Houston, TX, February 22–24, 2017.
The online-only Data Supplement is available with this article at http://stroke.ahajournals.org/lookup/suppl/doi:10.1161/STROKEAHA.116.014517/-/DC1.
References
- 1.Cramer SC. Repairing the human brain after stroke. II. Restorative therapies. Ann Neurol. 2008;63:549–560. doi: 10.1002/ana.21412. doi: 10.1002/ana.21412. [DOI] [PubMed] [Google Scholar]
- 2.Li S, Carmichael ST. Growth-associated gene and protein expression in the region of axonal sprouting in the aged brain after stroke. Neurobiol Dis. 2006;23:362–373. doi: 10.1016/j.nbd.2006.03.011. doi: 10.1016/j.nbd.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 4.Domeniconi M, Filbin MT. Overcoming inhibitors in myelin to promote axonal regeneration. J Neurol Sci. 2005;233:43–47. doi: 10.1016/j.jns.2005.03.023. doi: 10.1016/j.jns.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 5.Walmsley AR, Mir AK. Targeting the Nogo-A signalling pathway to promote recovery following acute CNS injury. Curr Pharm Des. 2007;13:2470–2484. doi: 10.2174/138161207781368611. [DOI] [PubMed] [Google Scholar]
- 6.Irving EA, Vinson M, Rosin C, Roberts JC, Chapman DM, Facci L, et al. Identification of neuroprotective properties of anti-MAG antibody: a novel approach for the treatment of stroke? J Cereb Blood Flow Metab. 2005;25:98–107. doi: 10.1038/sj.jcbfm.9600011. doi: 10.1038/sj.jcbfm.9600011. [DOI] [PubMed] [Google Scholar]
- 7.Torigoe K, Lundborg G. Selective inhibition of early axonal regeneration by myelin-associated glycoprotein. Exp Neurol. 1998;150:254–262. doi: 10.1006/exnr.1997.6775. doi: 10.1006/exnr.1997.6775. [DOI] [PubMed] [Google Scholar]
- 8.Mears S, Schachner M, Brushart TM. Antibodies to myelin-associated glycoprotein accelerate preferential motor reinnervation. J Peripher Nerv Syst. 2003;8:91–99. doi: 10.1046/j.1529-8027.2003.03012.x. [DOI] [PubMed] [Google Scholar]
- 9.Lee JK, Kim JE, Sivula M, Strittmatter SM. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J Neurosci. 2004;24:6209–6217. doi: 10.1523/JNEUROSCI.1643-04.2004. doi: 10.1523/JNEUROSCI.1643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cash D, Easton AC, Mesquita M, Beech J, Williams S, Lloyd A, et al. GSK249320, a monoclonal antibody against the axon outgrowth inhibition molecule myelin-associated glycoprotein, improves outcome of rodents with experimental stroke. J Neurol Exp Neurosci. 2016;2:28–33. [PMC free article] [PubMed] [Google Scholar]
- 11.Barbay S, Plautz EJ, Zoubina E, Frost SB, Cramer SC, Nudo RJ. Effects of postinfarct myelin-associated glycoprotein antibody treatment on motor recovery and motor map plasticity in squirrel monkeys. Stroke. 2015;46:1620–1625. doi: 10.1161/STROKEAHA.114.008088. doi: 10.1161/STROKEAHA.114.008088. [DOI] [PubMed] [Google Scholar]
- 12.Abila B, Cunningham E, Simeoni M. First-time-in-human study with GSK249320, a myelin-associated glycoprotein inhibitor, in healthy volunteers. Clin Pharmacol Ther. 2013;93:163–169. doi: 10.1038/clpt.2012.227. doi: 10.1038/clpt.2012.227. [DOI] [PubMed] [Google Scholar]
- 13.Cramer SC, Abila B, Scott NE, Simeoni M, Enney LA MAG111539 Study Investigators. Safety, pharmacokinetics, and pharmacodynamics of escalating repeat doses of GSK249320 in patients with stroke. Stroke. 2013;44:1337–1342. doi: 10.1161/STROKEAHA.111.674366. doi: 10.1161/STROKEAHA.111.674366. [DOI] [PubMed] [Google Scholar]
- 14.Richards C, Malouin F, Dumas F, Tardif D. Gait velocity as an outcome measure of locomotor recovery after stroke. In: Craik R, Oates C, editors. In: Gait Analysis: Theory and Application. St. Louis: Mosby; 1995. pp. 355–364. [Google Scholar]
- 15.Duncan PW, Sullivan KJ, Behrman AL, Azen SP, Wu SS, Nadeau SE, et al. LEAPS Investigative Team. Body-weight-supported treadmill rehabilitation after stroke. N Engl J Med. 2011;364:2026–2036. doi: 10.1056/NEJMoa1010790. doi: 10.1056/NEJMoa1010790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fritz S, Lusardi M. White paper: “walking speed: the sixth vital sign.”. J Geriatr Phys Ther. 2009;32:46–49. [PubMed] [Google Scholar]
- 17.Tilson JK, Sullivan KJ, Cen SY, Rose DK, Koradia CH, Azen SP, et al. Locomotor Experience Applied Post Stroke (LEAPS) Investigative Team. Meaningful gait speed improvement during the first 60 days poststroke: minimal clinically important difference. Phys Ther. 2010;90:196–208. doi: 10.2522/ptj.20090079. doi: 10.2522/ptj.20090079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winstein CJ, Stein J, Arena R, Bates B, Cherney LR, Cramer SC, et al. American Heart Association Stroke Council, Council on Cardiovascular and Stroke Nursing, Council on Clinical Cardiology, and Council on Quality of Care and Outcomes Research. Guidelines for Adult Stroke Rehabilitation and Recovery: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2016;47:e98–e169. doi: 10.1161/STR.0000000000000098. doi: 10.1161/STR.0000000000000098. [DOI] [PubMed] [Google Scholar]
- 19.Perry J, Garrett M, Gronley JK, Mulroy SJ. Classification of walking handicap in the stroke population. Stroke. 1995;26:982–989. doi: 10.1161/01.str.26.6.982. [DOI] [PubMed] [Google Scholar]
- 20.Cramer SC, Koroshetz WJ, Finklestein SP. The case for modality-specific outcome measures in clinical trials of stroke recovery-promoting agents. Stroke. 2007;38:1393–1395. doi: 10.1161/01.STR.0000260087.67462.80. doi: 10.1161/01.STR.0000260087.67462.80. [DOI] [PubMed] [Google Scholar]
- 21.Salbach NM, Mayo NE, Higgins J, Ahmed S, Finch LE, Richards CL. Responsiveness and predictability of gait speed and other disability measures in acute stroke. Arch Phys Med Rehabil. 2001;82:1204–1212. doi: 10.1053/apmr.2001.24907. doi: 10.1053/apmr.2001.24907. [DOI] [PubMed] [Google Scholar]
- 22.Bohannon R, Andrews A, Smith M. Rehabilitation goals of patients with hemiplegia. Int J Rehab Research. 1988;11:181–183.. [Google Scholar]
- 23.Fisher CM. Lacunar strokes and infarcts: a review. Neurology. 1982;32:871–876. doi: 10.1212/wnl.32.8.871. [DOI] [PubMed] [Google Scholar]
- 24.Pullicino P, Nelson RF, Kendall BE, Marshall J. Small deep infarcts diagnosed on computed tomography. Neurology. 1980;30:1090–1096. doi: 10.1212/wnl.30.10.1090. [DOI] [PubMed] [Google Scholar]
- 25.Sprigg N, Gray LJ, Bath PM, Lindenstrøm E, Boysen G, De Deyn PP, et al. TAIST Investigators. Early recovery and functional outcome are related with causal stroke subtype: data from the tinzaparin in acute ischemic stroke trial. J Stroke Cerebrovasc Dis. 2007;16:180–184. doi: 10.1016/j.jstrokecerebrovasdis.2007.02.003. doi: 10.1016/j.jstrokecerebrovasdis.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Kawamata T, Dietrich WD, Schallert T, Gotts JE, Cocke RR, Benowitz LI, et al. Intracisternal basic fibroblast growth factor enhances functional recovery and up-regulates the expression of a molecular marker of neuronal sprouting following focal cerebral infarction. Proc Natl Acad Sci USA. 1997;94:8179–8184. doi: 10.1073/pnas.94.15.8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duncan PW, Goldstein LB, Matchar D, Divine GW, Feussner J. Measurement of motor recovery after stroke. Outcome assessment and sample size requirements. Stroke. 1992;23:1084–1089. doi: 10.1161/01.str.23.8.1084. [DOI] [PubMed] [Google Scholar]
- 28.Dijkhuizen RM, Ren J, Mandeville JB, Wu O, Ozdag FM, Moskowitz MA, et al. Functional magnetic resonance imaging of reorganization in rat brain after stroke. Proc Natl Acad Sci USA. 2001;98:12766–12771. doi: 10.1073/pnas.231235598. doi: 10.1073/pnas.231235598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calautti C, Leroy F, Guincestre JY, Baron JC. Dynamics of motor network overactivation after striatocapsular stroke: a longitudinal PET study using a fixed-performance paradigm. Stroke. 2001;32:2534–2542. doi: 10.1161/hs1101.097401. [DOI] [PubMed] [Google Scholar]
- 30.Tombari D, Loubinoux I, Pariente J, Gerdelat A, Albucher JF, Tardy J, et al. A longitudinal fMRI study: in recovering and then in clinically stable sub-cortical stroke patients. Neuroimage. 2004;23:827–839. doi: 10.1016/j.neuroimage.2004.07.058. doi: 10.1016/j.neuroimage.2004.07.058. [DOI] [PubMed] [Google Scholar]
- 31.Goldberg MP, Ransom BR. New light on white matter. Stroke. 2003;34:330–332. doi: 10.1161/01.str.0000054048.22626.b9. [DOI] [PubMed] [Google Scholar]
- 32.Cramer SC. Clinical issues in animal models of stroke and rehabilitation. ILAR J. 2003;44:83–84. doi: 10.1093/ilar.44.2.83. [DOI] [PubMed] [Google Scholar]
- 33.Cramer SC. Drugs to Enhance Motor Recovery After Stroke. Stroke. 2015;46:2998–3005. doi: 10.1161/STROKEAHA.115.007433. doi: 10.1161/STROKEAHA.115.007433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. STAIR Group. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dirnagl U. Thomas Willis lecture: is translational stroke research broken, and if so, how can we fix it? Stroke. 2016;47:2148–2153. doi: 10.1161/STROKEAHA.116.013244. doi: 10.1161/STROKEAHA.116.013244. [DOI] [PubMed] [Google Scholar]