

SUMMARY
Dendritic mislocalization of microtubule associated protein tau is a hallmark of tauopathies, but the role of dendritic tau is unknown. We now report that tau interacts with the RNA-binding protein (RBP) TIA1 in brain tissue, and we present the brain-protein interactome network for TIA1. Analysis of the TIA1 interactome in brain tissue from wild-type (WT) and tau knockout mice demonstrates that tau is required for normal interactions of TIA1 with proteins linked to RNA metabolism, including ribosomal proteins and RBPs. Expression studies show that tau regulates the distribution of TIA1, and tau accelerates stress granule (SG) formation. Conversely, TIA1 knockdown or knockout inhibits tau misfolding and associated toxicity in cultured hippocampal neurons, while overexpressing TIA1 induces tau misfolding and stimulates neurodegeneration. Pharmacological interventions that prevent SG formation also inhibit tau pathophysiology. These studies suggest that the pathophysiology of tauopathy requires an intimate interaction with RNA-binding proteins.
In Brief
Vanderweyde et al. show that the interaction of microtubule associated protein tau with the RNA binding protein (RBP) TIA1 regulates stress granule (SG) formation as well as misfolding and aggregation of tau. TIA1 knockdown prevents tau misfolding and tau-mediated toxicity, which points to RBPs as potential targets for therapy of tauopathies.
INTRODUCTION
RNA-binding proteins (RBPs) are a class of about 800 proteins that function in the nucleus to regulate mRNA maturation, including splicing, RNA helicase activity, RNA polymerase elongation, and nuclear export (Anderson and Kedersha, 2008). RBPs also function in the cytoplasm where they regulate RNA translation, trafficking, sequestration, and degradation. RBP function is strongly regulated by the multiple signaling cascades integrated with RNA translation/protein synthesis, which will be referred to as “translational signaling.” The cytoplasmic actions of RBPs play a particularly crucial role in neurobiology because the large distance between the soma and synapse demands a proportionately large role of RBPs in the trafficking of mRNA transcripts (Liu-Yesucevitz et al., 2011).
Increasing evidence links neurological disease processes to dysfunction of neuronal RBPs, RNA granules, and stress granules (SGs) (Ramaswami et al., 2013; Wolozin, 2012). SGs are a particular type of RNA granule that accumulates during the translational response to stress. RBPs, such as T cell intracellular antigen 1 (TIA1), contain prion-like, poly-glycine-rich domains, which promote their physiological reversible aggregation (Thomas et al., 2011). Nucleation by core RBPs, such as TIA1, is followed by recruitment of secondary RBPs to form a mature SG, which is a key component of stress-induced translational suppression. SGs play a dynamic role in mRNA triage by sorting sequestered mRNAs for re-initiation, storage, or degradation.
Mutations in multiple RBPs cause motor neuron diseases, including amyotrophic lateral sclerosis (ALS) (Li et al., 2013). Many of the mutations in RBPs that are linked to disease appear to increase the tendency of these proteins to aggregate (Johnson et al., 2009; Kwiatkowski et al., 2009). Studies from our lab and others show that the mutations also increase RNA granule formation, leading to SGs that are larger and more abundant, as well as larger and slower transport granules (Alami et al., 2014; Colombrita et al., 2009; Liu-Yesucevitz et al., 2010, 2014). Studies with recombinant FUS and hnRNAPA1 indicate that these proteins exhibit a normal ability to cycle between solution and gel phases, forming liquid droplets. However, mutations in either protein impair the phase transition, leading to formation of stable amyloid-like fibrils (Lin et al., 2015; Molliex et al., 2015; Nott et al., 2015; Patel et al., 2015).
Formation of pathological RNA granules is associated with neuropathology. For instance, TIA1 co-localizes with neuropathology in brain tissue of subjects with Alzheimer’s disease (AD), frontotemporal dementia with parkinsonism (FTDP-17), frontotemporal lobar dementia (FTLD-TDP), ALS, Huntington’s disease, Creutzfeld-Jakob disease, and spinomuscular atrophy, as well as in animal models of these diseases (Liu-Yesucevitz et al., 2010; Thomas et al., 2011; Vanderweyde et al., 2012; Wolozin, 2012). Our previous work suggests that the biology of tau is intimately linked to TIA1, with the proteins accumulating concomitantly with each other over the disease course in brain tissue from subjects with human tauopathies as well as animal models of tauopathies (Vanderweyde et al., 2012).
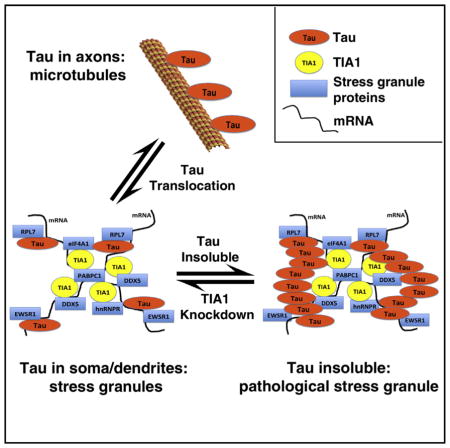
We now report that tau promotes SG formation and modulates the patterns of protein interactions of TIA1, a main SG component. The interaction between tau and TIA1 promotes tau misfolding and assembly at the site of SGs and results in the degeneration of processes and stimulation of apoptotic markers in primary neurons. Reducing TIA1 inhibits tau-misfolding and degeneration in neuronal cultures. These results indicate that tau plays an important role in neuronal RBP biology and suggest that RBPs and SGs contribute to the misfolding of tau. These results also raise the possibility that the pathophysiology of tauopathies, such as AD, is associated with dysfunction of RBP biology.
RESULTS
Tau Increases Somatodendritic Localization of TIA1
The distribution of TIA1 was examined in primary cultures of hippocampal neurons from wild-type (WT) and tau−/− mice to investigate whether tau regulates the distribution of TIA1 (Figure 1A). Primary hippocampal neurons from tau−/− mice were transduced with TIA1-GFP ± AAV1 WT Tau-V5 or P301L Tau-V5. TIA1 exhibited a strong nuclear localization in neurons from tau−/− mice, with few TIA1 granules (Figure 1A). Expressing TIA1 plus tau dramatically increased the amount of somatodendritic TIA1, with the TIA1 exhibiting a strong granular character (Figure 1A); little or no TIA1 was observed in axons (Figure S1A). Quantification shows that P301L tau significantly increased the size of TIA1 SGs compared to WT tau but produced fewer SGs than WT tau (Figures 1A–1C), which is strikingly similar to effects produced in neurons by disease-linked mutations in RBPs, such as TDP-43 (Liu-Yesucevitz et al., 2014).
Figure 1. tau Increases the Somatodendritic Localization of TIA1.

(A) Imaging for V5 (tau), GFP (TIA1), and MAP2 in primary hippocampal tau knockout neurons transduced with TIA1-GFP lentivirus ± AAV1-WT tau-V5 or AAV1-P301L tau-V5. Images indicate tau increases TIA1 movement into processes and SG formation (n = 100/condition, three independent experiments).
(B and C) WT or P301L tau increases TIA1 granules in tau−/− neurons. Granule density (B) and area (C) were determined using ImageJ to quantify TIA1 puncta per neuron for both endogenous TIA1 staining and exogenous TIA1-GFP fluorescence (n = 100/condition).
(D) Live-cell imaging was done on tau−/− primary hippocampal neurons transduced with AAV1-TIA1-mRFP ± AAV9-WT or P301L tau. The number of moving particles per neuron was determined with BitPlane (Imaris) (n = 20/condition, three independent experiments).
(E–G) Scatterplots of TIA1+ granule area versus distance from soma in tau−/− neurons. TIA1 average granule velocity versus granule area for neurons transduced with: (E) TIA1-RFP, (F) WT tau/TIA1-RFP, and (G) P301L tau/TIA1-RFP (n = 20/condition).
(H) Quantification of the net displacement of TIA1-positive granules in both anterograde (+) and retrograde (−) directions (n = 20/condition, three independent experiments).
Scale bars, 10 μm. **p < 0.01, and ***p < 0.001.
The ability of tau to regulate TIA1 RNA granule formation suggests a biological role for tau in RNA granule trafficking, which was investigated using live-cell imaging. Tau−/− neurons (3 days in vitro [DIV] 3) were transduced with AAV1-TIA1-mRFP (a monomeric form of RFP) ± AAV9-WT or P301L tau, and, at DIV 21, the neurons were imaged. Compiled traces showing particle localization and tracks of TIA1 granules show decreased granule movement with tau expression (Figures 1D–1G). Tau inhibited net displacement and velocity of TIA1 granules, with retrograde (−) movement inhibited more than anterograde (+) movement (Figure 1H). Granule size was inversely correlated with granule velocity (Figures 1D–1G), with the trend particularly pronounced with P301L tau where the granule area versus velocity graph shows a distinct inflection at about 1.2 μm2 (Figure 1G). As with SG formation, this effect is strikingly similar to the relationship between size and granule movement among TDP-43 granules (Liu-Yesucevitz et al., 2014; Alami et al., 2014).
tau Increases SG Number and Size
Next, we investigated the effects of tau on SG formation using mouse hippocampal HT22 cells. Stress induces TIA1 to exit the nucleus, aggregate, and bind transcripts to form SGs (Figure 2A); TIA1 SG formation can be prevented by concurrent treatment with protein synthesis inhibitors, such as cycloheximide, that stabilize ribosomes on the mRNA. Expressing human tau (4R0N WT and P301L) increased SG formation in the HT22 cells (Figure 2B). HT22 cells were transfected with human tau (4R0N WT and P301L) and then examined ± arsenite (0.5 mM, 30 min) to induce SGs. Tau overexpression strongly increased RNA granule formation under basal and stressed conditions (Figures 2B, S2A, and S2B). Double labeling of these granules demonstrated that they are bona-fide SGs because they are positive for two SG markers: TIA1 and PABP; in addition, co-treatment with 10 μg/ml cycloheximide prevented formation of the tau/SG complexes (Figure 2B).
Figure 2. tau Promotes Stress Granule Formation.

(A) TIA1 translocates to the cytoplasm with stress, where it nucleates SGs and inhibit synthesis of non-essential proteins.
(B) HT22 cells overexpressing WT tau, P301L tau, or β-gal followed by arsenite treatment (0.5 mM, 30 min) to induce formation of SGs positive for TIA1, PABP, and tau (identified with the Tau13 antibody). Formation of tau+ SGs is reversed with cycloheximide treatment (10 μg/ml, CHX). Quantification is presented in Figure S2B, based on three independent experiments.
(C) Immunoblots and IPs showing levels of Tau13, TIA1, and actin in lysates and immunoprecipitates from HT22 cells transfected with EGFP, WT tau, or P301L ± TIA1-RFP.
(D) Live-cell imaging of TIA1-RFP was done in HT22 cells 24 hr after transfection with EGFP, WT tau-EGFP, or P301L tau-EGFP and co-transfection with TIA1-mRFP. 500 μM arsenite was added to the cells to stimulate SG formation and images were taken every 15 s for 30 min. Representative images shown for 0, 6, and 12 min, with arrows at 6 min indicating the SG formation. High-magnification insets are shown.
(E) Upper panel: Graph of the average number of SGs per cell over time per condition (n = 12 per condition), Lower panel: Graph of the average area of SG formation over time (n = 12).
(F) Graph depicting the mean time to reach 50% maximal SG formation (based on SG numbers per cell) (n = 12/condition).
(G) Overexpressing tau (4R0N, P301L) in HEK293 cells inhibits protein synthesis detected using the SUnSET method and the anti-puromycin antibody, 12D10.
Scale bars, 10 μm. ***p < 0.001.
Immunoprecipitations (IPs) were performed to test whether TIA1 associates with tau biochemically. HT22 cells were transfected with WT tau (4R0N) and TIA1, and the human specific Tau13 antibody was used to IP the complex, followed by immunoblotting for TIA1; IP with anti-TIA1 followed by immunoblotting with Tau13 was also performed, which produced compelling evidence indicating robust associations (Figure 2C).
Next, we applied a live-cell imaging approach to examine the effects of tau on stress granule dynamics. Expressing tau with TIA1-RFP strongly increased the rate of SG formation (Figures 2D–2F). P301L tau accelerated the rate of SGs formation more than WT tau, with increased consolidation into larger SGs (Figures 2D–2F). Thus, tau accelerates SG formation. Taken together, these studies point to an important role of tau in regulating trafficking and assembly of RNA granules, including SGs.
SGs are thought to reflect adaptation of protein synthesis to stress; typically the stress-induced changes in protein synthesis are associated with an overall reduction in protein synthesis. Analysis of cells overexpressing tau (4R0N, P301L) showed a decrease in total protein synthesis (measured by SUnSET assay) in response to tau overexpression in a Tet-inducible HEK WT tau (4R0N) cell line following addition of doxycycline, which is consistent with the data above suggesting that tau promotes SGs and the translational stress response (Figure 2G).
tau Regulates the Interaction of TIA1 with Its Proteome
Proteomic studies revealed a surprising role for tau in regulating the proteins that interact with TIA1. To investigate whether tau exerts control over TIA1 protein interactions, TIA1 was immunoprecipitated from cortical brain tissue of 10-month-old WT (C57BL/6J), tau−/−, and TIA1−/− mice. The specificity of the TIA1 IP was verified by immunoblotting with anti-TIA1 antibody (Figure 3A), and the resulting TIA1 proteome was examined by mass spectrometry. 163 proteins were identified that were present in TIA1 immunoprecipitates from WT or tau−/− brains and absent in the TIA1 immunoprecipitates from TIA1−/− brains (Figure 3C; Table S1). Protein associations identified by proteomics were validated by repeat mass spectrometry on fresh samples, as well as by immunoblot for tau and four proteins that gave strong signals by mass spectrometry: RPL7, EWSR1, hnRNPR, and DDX5 (Figures 3A and 3B). As expected, endogenous mouse tau (MAPT) was abundantly detected in the TIA1 immunoprecipitates (Table S1). Figure 3C summarizes the average protein level for each TIA1-binding protein detected in the WT versus tau−/− mass spectrometry samples by a hierarchically clustered heatmap.
Figure 3. tau Regulates the TIA1-binding Proteome in the Brain.

(A and B) IP of TIA1 from the TIA−/−, C57BL/6 and tau−/− mouse cortical lysates used for the proteomic studies (A), with quantification in (B) (n = 3). TIA1 was evident in the C57BL/6 and tau−/− lysates but absent from the TIA1−/− lysates. Excluding from the proteomic analysis any proteins that were present in the TIA−/− IP eliminates proteins immunoprecipitated non-specifically. Binding of RPL7 and EWSR1 to TIA1 was greatly reduced in the tau−/− cortices, as suggested by the mass spectrometry studies. In contrast, binding of DDX5 to TIA1 was not affected by the absence of tau.
(C) Hierarchical clustering of proteins associated with TIA1 upon IP from WT and tau−/− mice, using the MS1 current channel for semiquantitative measurements.
(D) Network diagram depicting the TIA1-binding proteome in the WT mouse cortex. Edges connecting nodes denote shared functional annotation terms (greater than three) between the connected proteins, as determined by DAVID functional clustering analysis. Proteins highlighted in red were not detected in any of the three tau−/− samples analyzed, indicating that their interaction with TIA1 is tau dependent. Node size is proportional to the degree of replication in the WT samples (n = 3 mice per group). Only edges containing more than three shared annotation clusters are shown. Background circles indicate protein clusters in the TIA1 network including RNA metabolism (orange), cytoskeleton (yellow), vesicles/synaptic function (gray), and mitochondrial (green).
(E and F) Listing of functional categories by GO gene ontology annotation showing statistically significant enrichment (E) or reduction (F) (Benjamini corrected FDR <0.05) in their association with TIA1 by at least 2-fold for either WT or tau−/− cortex as determined using the DAVID bioinformatics resource (Huang et al., 2009). *p < 0.05; #p < 0.1 (trend level).
Next, we analyzed the TIA1-binding proteome using the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resource available via the NIH (Huang et al., 2009). As expected, immunoprecipitating TIA1 from WT mouse brain tissue identified proteins exhibiting statistically significant enrichment (false discovery rate [FDR] <0.05) for annotation terms involving RNA metabolism (Table S2), including ribonucleoprotein (KEYWORDS, FDR = 6.30E-06), RNA-binding (KEYWORDS, FDR = 2.09E-05), and RNA recognition motif RNP-1 (INTERPRO, FDR = 2.15E-03); proteins involved in mitochondrial and vesicular/synaptic function also exhibited significant enrichment (Table S2). We also constructed a network diagram to understand the functional connectivity of TIA1-binding proteins in the brain (Figure 3D). In the network, lines connecting nodes indicate shared functional annotations with other proteins in the TIA1 network; the size of each node corresponds to the degree of replication in the WT samples (n = 3). Note that some proteins listed in the hierarchical table (Figure 3C) are absent from the network (Figure 3D) because they lack known shared functional annotations with the other TIA1-binding proteins. The network of TIA1-binding proteins in WT mouse cortex includes multiple proteins linked to RNA metabolism, including ribosomal proteins (e.g., RPL6, 7, 10A, 13 and 13A, MRPL46, RPS3, and 4X), translational regulatory proteins (EIF4A1, PABPC1, and NACA), small nuclear ribonucleoproteins (SNRNP70 and SNRPB), heterogeneous nuclear ribonucleoproteins (HNRNPF, HNRNPR and HNRNPUL2, EWSR1, and SYNCRIP), and helicases (DDX5 and DDX17) (Figure 3D). Importantly, deletion of tau eliminated the interaction of TIA1 with many proteins in the network, particularly those associated with RNA metabolism and RNA translation (Figure 3D, red circles). This network analysis of the TIA1-binding proteomes from the WT and tau−/− mouse brain highlights the important role that tau plays in regulating proteins that interact with TIA1, with loss of tau abrogating interactions with multiple core TIA1-binding proteins, including EWSR1, RPL6, RPL7, MRPL46, RBM17, and SNRNP70 (Figures 3C–3F). To further validate this finding, we separately analyzed the proteins decreasing (Figure 3E) or increasing (Figure 3F) their association with TIA1 by greater than 2-fold in tau−/− mice compared to WT mice in order to understand how tau affects the protein interactions of TIA1. Proteins decreasing their association with TIA1 were enriched for various functional annotation terms related to RNA metabolism, including ribosomal, ribonucleoprotein, and RNA-binding (Figure 3E). Taken together, these results suggest that the presence of tau protein is required for normal interaction of TIA1 with the RNA metabolism machinery.
RNA-binding Proteins in the TIA1 Proteome Co-localize with tau Pathology In Vivo
The strong role of tau in the TIA1-binding proteome raised the possibility that TIA1-associated RBPs might co-localize with tau pathology in human tauopathies and mouse models of tauopathy. Brain tissue (frontal cortex) from 11-month-old rTg4510 mice were labeled with antibodies for TIA1, EWSR1, DDX5, RPL7, TDP-43, and FUS (Figure 4); all samples were co-labeled with the anti-phospho-tau antibody, PHF1. Proteins present in the TIA1 network (TIA1, EWSR1, DDX5, and RPL7) all co-localized with PHF1-positive tau pathology. The RPL7 reactivity was notable because it tended to localize to the outer rim of tau pathology (Figure 4). No co-localization with PHF1 positive tau pathology was observed for TDP-43 or FUS, which are not part of the TIA1-binding network (Figure 4).
Figure 4. TIA1 Network Members Co-localize with tau Pathology in rTg4510 Mice.

(A) Immunohistochemistry for TIA1, EWSR1, DDX5, RPL7, and TDP-43 and FUS (green), PHF1 (red), and DAPI (blue) in frontal cortex of 11-month-old rTg4510 mice.
(B and C) Quantification of co-localization demonstrates strong localization of TIA1 network members with PHF1 reactivity (n = 5 mice).
Scale bars, 10 μm. *p < 0.05, **p < 0.01, and ***p < 0.001.
TIA1 and tau Interact Biochemically
Studies using primary neuronal cultures confirmed the interactions between tau and TIA1. Hippocampal neurons from tau−/− mice were grown in culture and transduced with AAV9-WT or P301L tau ± AAV1-TIA1-mRFP (or mRFP) (Figure S4A). Immunoblotting demonstrated that TIA1 regulates tau levels. TIA1 overexpression reduced levels of total tau, while TIA1 knockout increased levels of total tau (Figures S4A and S4B). The mechanism of regulation appeared to have limited dependence on the proteasomal or autophagic systems, because neither MG132 nor chloroquine prevented tau reductions upon TIA1 expression (Figure S4C). IP with TIA1 indicated that total tau (Tau13, recognizes total human tau) or phospho-tau (PHF1, recognizing phospho-tau S396/404) bound exogenous and endogenous TIA1 (Figure S4A). Pre-treating the lysate with RNase A, or use of a TIA1 construct lacking the three RNA recognition motifs abolished the association, indicating that tau associated with TIA1 through an RNA intermediate (Figure S4D, data for RNase A treatment shown). The role of RNA intermediates supports the hypothesis that tau participates in RNA granule biology. Tau could also interact with RNA directly as suggested by prior studies (Kampers et al., 1996; Wang et al., 2006).
TIA1 Modulates the Misfolding, Stability, and Insolubility of Granular tau
The association of tau with TIA1 complexes raised the possibility that TIA1 might also modulate tau misfolding. TIA1 forms RNA granules, which provide an environment with abundant aggregated proteins as well as RNA, a known promoter of tau misfolding. To test this hypothesis, the relationship between TIA1 levels and tau misfolding was examined. The response of tau misfolding to knockdown of endogenous TIA1 was examined first. TIA1 knockdown was validated by immunoblot (Figure 5A). Next, primary hippocampal neurons (DIV 2) were transduced with AAV1 WT human tau, then transduced with AAV9 shTIA1 or scrambled (shScr) on DIV 4 to knock down TIA1, and aged to DIV 21. Analysis of MC1 reactivity at DIV 21 demonstrated that TIA1 knockdown elicited a robust decrease in MC1 levels (Figures 5B and 5C).
Figure 5. TIA1 Regulates tau Misfolding and Stimulates Consolidation of Misfolded tau into SGs.

(A) Validation of TIA1 knockdown by shTIA1. Immunoblots showing high levels of total tau (Tau13 antibody) in lysates from HT22 cells transfected with EGFP, WT tau, P301L tau co-transfected with shControl (shCtrl) or shTIA1.
(B) Immunocytochemistry for of hippocampal neurons transduced with TIA1 or control short hairpin RNA (shRNA) (using co-transduction with AAV-GFP to identify transduced neurons) and stained for misfolded tau (MC1, red) and MAP2 (blue). Neurons transfected with TIA1 shRNA show reduced MC1 reactivity.
(C) Quantification of MC1 levels in neurons transfected with TIA1 or control shRNA (n = 50 neurons per condition, three independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars, 10 μm.
(D) Immunocytochemistry for MC1 tau and MAP2 in primary tau−/− hippocampal neurons transduced with AAV9-WT or P301L tau co-transduced with EGFP or TIA1-GFP lentivirus. Scale bar, 10 μm. High-magnification inset of dendritic process shows MC1 staining; scale bar, 4 μm.
(E) Quantification of MC1 granule count per neuron (n = 100 neurons/condition, three independent experiments).
(F) Quantification of average MC1 granule area (n = 100/condition, three independent experiments).
(G) Live-cell imaging of photoconvertable WT tau (PC-Tau). Following photo-conversion of PC-Tau, neurons were imaged for up to 6 hr. Representative images are shown for the 0- and 6-hr time points showing stabilization of tau in granules in cells co-expressing TIA1.
(H) Quantification of PC-Tau from neurons at varying time points after photo-conversion (n = 20 per condition, three independent experiments).
Scale bar, 10 μm. *p < 0.05, and ***p < 0.001.
Overexpressing TIA1 exhibited a robust reciprocal response, increasing levels of MC1 reactivity. Tau−/− hippocampal neurons were transduced with WT or P301L tau AAV9 and TIA1-GFP or GFP lentivirus, and immunolabeled for MC1 tau and MAP2. Imaging demonstrated that neurons co-expressing tau and TIA1 displayed abundant MC1+ granules in processes that were not apparent in neurons expressing tau alone, and that co-expressing TIA1 increased the size of the granules (Figures 5D–5F).
The ability of TIA1 to promote tau granules led us to hypothesize that TIA1 increased the stability of tau in granules. To test this hypothesis, we generated a photo-convertible tau (PC-Tau, WT 4R0N) construct that stably converts from cyan to green, transfected cortical neurons (DIV 5) with the PC-Tau ± TIA1, performed the photo-activation at DIV 21 and then imaged for 6 hr. Neurons transfected with PC-Tau plus mCherry control exhibited green tau fluorescence present both diffusely and in granules. The tau fluorescence decreased steadily over 6 hr down to a level of approximately 40% of the original fluorescence levels (Figures 5G and 5H). In contrast, cortical neurons transfected with PC-Tau + TIA1 exhibited greatly reduced decay rates, with PC-Tau fluorescence remaining above 80% at 6 hr (Figures 5G and 5H). Expressing tau with TIA1 also caused a higher proportion of the tau to localize to granules, consistent with Figures 5D–5F. These data indicate that TIA1 stabilizes tau in granules.
The stabilization of tau in granules combined with the presence of misfolded, MC1+ tau in SGs raised the possibility that TIA1 increases formation of insoluble tau in granules. To test this hypothesis, HT22 cells were transfected with WT tau or P301L tau ± TIA1-RFP (Figure S5A). TIA1 was immunoprecipitated; the TIA1 bound and unbound fractions were biochemically fractionated into sarkosyl soluble and insoluble fractions. Overexpressing TIA1 promoted the clearance of tau in all fractions except the insoluble-TIA-bound (Figure S5A) where the amount of insoluble tau (WT and P301L) associated with TIA1 was increased (Figures S5A and S5B). Thus, our findings suggest that association with TIA1 promotes formation of insoluble tau, which is thought to be a key step in forming neurofibrillary tangles.
Translational Inhibitors and Kinase Inhibitors Modulate SGs and tau Granules in Dendrites
The interaction between tau and TIA1 points to compelling approaches for modulating formation of tau granules in neurons. SGs are regulated by translational inhibition. Cycloheximide prevents elongation, which leaves ribosomes stalled on mRNA and inhibits SG formation, while puromycin causes premature translational termination leading to release of the 60S ribosomal subunit from the mRNA, promoting SG formation. Analysis of tau granules in neuronal cell lines demonstrated that cycloheximide prevented formation of tau granules (Figures 2A, S2A, and S2B), while puromycin stimulated formation of tau granules (data not shown). We hypothesized that tau granules might be regulated in a similar manner in neuronal dendrites.
Primary cultures of tau−/− hippocampal neurons were transduced with AAV9-WT tau or P301L tau ± AAV1-mRFP or TIA1-mRFP (Figure 6). Neurons expressing WT or P301L tau and mRFP control exhibited tau that was spread relatively diffusely along processes, with only weak granules evident (Figure 6A). However, co-transducing WT or P301L tau with TIA1 resulted in processes with large granules positive for tau and TIA1, and little to no diffuse tau (Figure 6A, arrows). Comparison of the effects of the two different translational inhibitors, puromycin and cycloheximide, highlighted the role of translational signaling. Neurons were treated at DIV 21, immunolabeled for Tau13 and MAP2, and imaged (Figures 6B and 6C). Treatment with puromycin yielded larger and more abundant tau granules that were particularly accentuated by TIA1/tau overexpression (Figure 6B). Conversely, treatment with cycloheximide (10 μg/ml) yielded dendritic tau (and TIA1) that was spread diffusely with only small, less defined granules apparent (Figure 6C). Thus, the localization and granule formation of tau and TIA1 are both modulated by translational signaling.
Figure 6. Tau Granules Are Regulated by Translational Inhibitors.

(A–C) Immunocytochemistry for tau13 antibody and MAP2 in primary cultures of tau/hippocampal neurons transduced with AAV1-RFP or AAV1-TIA1-mRFP ± AAV9-WT or P301L tau. (A–C) No treatment (A), puromycin (B), cycloheximide (C). The columns on the right show images at high magnification (arrows identify tau granules).
(D) In HT22 cells following transfection with TIA1 and WT tau and treatment with the p38 kinase inhibitor (SB203580, 20 μM).
(E) Quantification of inhibition of SGs and tau granules in HT22 cells following treatment for 24 hr with one of five different kinase inhibitors: GSK3β (XXVI, 20 μM), CDK2/5 (alkylbenzyldimethyammonium chloride, 5 μM), p38 MAPK (SB203580, 20 μM), MARK/Par-1 (39621, 20 μM), and Fyn (PP2, 20 nM); 100 cells per condition, based on three independent experiments.
(F) Inhibition of tau-induced TIA1+ SGs in HT22 cells following transfection with TIA1 and WT tau and treatment for 24 hr with the PKR inhibitor (C16, 1 μM) or PERK inhibitor (GSK2606414, 50 nM).
(G) Quantification of granule count in cells treated with kinase inhibitors. *p < 0.05, **p < 0.01, ***p < 0.001; 100 cells per condition, based on three independent experiments.
Scale bars, low-magnification: 10 μm; inset, 2 μm.
The role of tau in SG biology in neurons also suggests that kinases that regulate tau dynamics might also regulate tau-mediated SG formation in neurons. For instance, proline-directed kinases are known to phosphorylate tau, which leads to dissociation of tau from microtubules and increases the propensity of tau to aggregate (Lee et al., 2011; Matenia and Mandelkow, 2009). Chemical inhibitors of GSK3β, CDK5, p38, MARK, and Fyn all significantly inhibited formation of granules positive for TIA1 and phospho-tau (P-S396/404, PHF1) (Figures 6D and 6E). The strongest SG inhibition was observed with the p38 inhibitor, which is known to act downstream of each of these kinases (Roux and Blenis, 2004).
Use of phospho-mimetic tau constructs demonstrated a direct role for tau phosphorylation in modulating SG formation. Transfections were performed using phospho-mimetic (PMIM) or phospho-null (PNULL) tau constructs in which 14 sites exhibiting increased phosphorylation in AD were replaced with either aspartate or alanine (Hoover et al., 2010). HT22 cells were transfected with P301L, P301L PMIM, or P301L PNULL tau ± TIA1-RFP. Cells were treated ± 25 μM salubrinal and SG were imaged for endogenous TIA1 (Figures S6A and S6B) or transfected TIA1-RFP (Figures S6B and S6C). Imaging showed more SGs in the presence of phospho-mimetic tau, and fewer SGs in the presence of phospho-null tau (Figures S6A–S6C). Salubrinal treatment increased the number of SGs in all conditions; salubrinal inhibits the PP2A adaptor protein, GADD34, which increases eIF2α phosphorylation and reduces mRNA translation (Boyce et al., 2005). These data point to direct phosphorylation of tau as a modulator of SG and tau granule formation in neurons.
The eukaryotic translation initiation factor eIF2α is regulated by phosphorylation. We investigated whether inhibiting PKR or PERK, two kinases that phosphorylate eIF2α, might also inhibit tau mediated SG formation. SGs were induced by transfection with TIA1 ± WT tau. After 24 hr, the cells were treated with inhibitors of PKR (C16, 1 μM, Sigma) or PERK (GSK2606414, 50 nM, EMD/Millipore), and then SG number was quantified after 24 hr. PKR or PERK antagonists strongly inhibited tau-mediated SG formation (Figures 6F and 6G). Thus, inhibiting translationally directed kinases also decreases tau-mediated SG formation.
TIA1 Modulates the Pathophysiology of tau
The results above identify a functional interaction between TIA1 and tau, with tau promoting formation of TIA1+ SGs, and TIA enhancing both tau catabolism and tau consolidation. Further studies suggest that the functional interactions between TIA1 and tau also extend to neurodegeneration, where our results show that TIA1 knockout inhibits tau-mediated degeneration, while TIA1 overexpression increases tau-mediated degeneration. Effects on degeneration were investigated by transducing hippocampal neurons (DIV 3) from tau−/− mice with AAV9-WT or P301L tau ± AAV1-TIA1-mRFP or mRFP and imaging for MAP2 at DIV 21 (Figures 7A and 7B). Next, hippocampal neurons (DIV 3) from TIA1−/− mice with AAV9-WT or P301L tau ± AAV1-TIA1-mRFP or mRFP and imaging for MAP2 at DIV 21 (Figures 7A and 7C). Co-expressing TIA1 with tau significantly decreased dendritic length but had no effect independent of tau (Figure 7B). P301L tau also caused toxicity on its own; neurons transduced with P301L tau exhibited significant dendritic shortening compared to neurons transduced with WT tau. However, TIA1 knockout prevented this toxicity (Figure 7C). Hippocampal neurons (DIV 3) from TIA1−/− mice transduced with AAV9-P301L tau exhibited neurite lengths similar to neurons transduced with AAV9-WT tau (Figure 7C). These data indicate that TIA1 expression is necessary for dendrite shortening associated with expression of P301L tau (Figure 7B, right panel).
Figure 7. TIA1 Modulates the Pathophysiology of tau.

(A–C) Images (A) and quantification (B and C) of dendrite traces of hippocampal neurons (primary culture, DIV 21) using MAP2 labeling of WT and tau−/− mice (B) or TIA1−/− mice (C), transduced with AAV1-TIA1-mRFP or mRFP ± AAV9-WT or P301L tau (n = 30/condition, four independent experiments).
(D) Immunoblots showing levels of synaptophysin, PSD-95, caspase-3, and cleaved caspase-3 in WT primary cortical neurons transduced with AAV1-TIA1-mRFP or mRFP ± AAV9-WT or P301L tau.
(E) Luminescent quantification of caspase cleavage (Caspase-Glo 3/7 Assay kit, Promega). Comparison of the amount of caspase cleavage in tau−/− primary hippocampal neurons (DIV 21) transduced with AAV1-TIA1-mRFP or mRFP ± AAV9-WT or P301L treated with 25 μM salubrinal (n = 3 wells/condition, four independent experiments).
(F) Colorimetric quantification of DNA fragmentation to measure apoptosis (TiterTACS Colorimetric Apoptosis Detection kit, Trevigen). Tau−/− primary hippocampal neurons (DIV 21) transduced with AAV1-TIA1-mRFP or mRFP ± AAV9-WT or P301L tau under basal conditions and treatments with 25 μM salubrinal (n = 3 wells/condition, four independent experiments).
Scale bar, 10 μm. **p < 0.01, and ***p < 0.001.
Additional studies suggest that the modulation of dendritic length by TIA1 and tau is sensitive to translational signaling. Tau−/− and TIA1−/− primary hippocampal neurons were transduced with AAV9-WT tau or P301L tau ± AAV1-TIA1-mRFP or mRFP, and at DIV 21 treated with translation inhibitors puromycin (5 μg/ml) or cycloheximide (10 μg/ml). Translation inhibition with cycloheximide did not affect dendrite length, while treatment with puromycin, which induces SGs, potentiated the decrease in dendritic length associated with TIA1/tau overexpression (Figures S7A and S7B).
Induction of toxicity was also apparent using biochemical assays. We examined levels of synaptic and apoptotic markers by immunoblot in WT primary cortical neurons transduced with AAV1-mRFP or TIA1-mRFP ± AAV9-WT tau or P301L tau. Markers examined included synaptophysin, PSD-95, caspase-3, and cleaved caspase-3. The data indicate a striking loss in the pre-synaptic marker synaptophysin in neurons co-transduced with tau and TIA1, indicating a corresponding loss of axonal terminals (Figure 7D). Levels of cleaved caspase-3 were also elevated in TIA1 and tau co-transduced neurons indicating enhanced toxicity (Figure 7E), which was potentiated by concurrent treatment with 25 μM salubrinal (Figure 7E). Interestingly, changes in the post-synaptic marker PSD-95 levels were not prominent (Figure 7D). Analysis of DNA fragmentation also showed that TIA1 increased apoptosis in tau expressing neurons (Figure 7F). These data suggest that the interaction of TIA1 with tau can promote neurodegeneration under conditions where SG formation is enhanced, such as occurs with TIA1 overexpression or exposure to SG inducers.
DISCUSSION
Tau is classically considered to function as a microtubule binding protein that plays an important role in axonal trafficking; however, in tauopathies tau accumulates in the somatodendritic compartment where it forms protein aggregates. The cellular logic behind somatodendritic accumulation is poorly understood. Our results suggest that the shift in tau localization to the somatodendritic compartment occurs to facilitate formation of SGs, which are RNA/protein complexes that are part of the translational stress response. SGs normally accumulate in the soma and dendrites as small insoluble macromolecular complexes in response to stress. In neurodegenerative diseases SGs become very large, and associate with pathological proteins, such as tau (in AD) and TDP-43 (in amyotrophic lateral sclerosis) (Liu-Yesucevitz et al., 2010; Vanderweyde et al., 2012). In moderation, this stress response is likely beneficial, but an over-active SG response causes a deleterious, degenerative response, such as that caused by overexpressing tau and either co-expressing with TIA1 or treating with puromycin.
TIA1 is known to be a protein involved in nuclear splicing, but recent studies also show that it is one of the core proteins that nucleates cytoplasmic SGs (Anderson and Kedersha, 2008). The network of proteins that associate with TIA1 in the brain includes 14 proteins that are very strongly linked from a functional perspective. This group of proteins includes RBPs typically associated with the spliceosome (SNRPB, snRNP70, DDX5, and RBM17), RBPs associated with mRNA transport (HNRNPR and EWSR1), and multiple ribosomal proteins (e.g., RPL6, 7 10A, 13, 13A, RPS 3, 4X). The prominence of ribosomal proteins highlights the important role of RNA translation in this network. Loss of tau abrogates TIA1-binding to five out of 14 proteins in this core network, RPL6, RPL7, EWSR1, SNRNP70, and RBM17, which points to a role for tau in this translational and transport machinery (Figure 3D). TIA1 shows reduced dendritic localization in tau−/− neurons (Figures 1A and 1B), which suggests altered interactions with trafficking proteins, but the mechanism for this altered localization remains to be determined. The interaction between tau and TIA1 parallels a recent study demonstrating that TIA1 interacts with tubulin to regulate microtubules in yeast (Li et al., 2014). In addition, the interaction of TIA1 with proteins, such as SIRT2 and clathrin (CLTB), have not been reported previously and might point to regulatory interactions unique to the brain. The prominence of members of the U1 SNRNP family (SNRNP70, SNRPB, and RBM17) in the TIA1 network is striking (Figures 3C and 3D). Previous work identified strong accumulation of cytoplasmic SNRNP70 aggregates in the AD brain (Bai et al., 2013). The presence of these TIA1-binding proteins whose binding is tau dependent points to interactions that might be more prominent in neurons or glia than in somatic cells.
The TIA1-binding proteome might differ between neurons and most peripheral cells because neurons must manage RNA biology in dendrites and synapses. RNA must be transported to the synapse, where RNA translation is tightly linked to synaptic activity through activity-dependent translation. This means that RBPs exert a much larger footprint on cellular activity outside of the nucleus in neurons (and possibly glia) than in somatic cells. The prominence of ribosomal proteins in the brain TIA1 network combined with the presence of RBPs important for RNA transport, such as HNRNPR, SYNCRIP, and EWSR1, emphasizes an important role for tau in regulating RNA transport and translation during stress. Under basal conditions, tau is present in dendrites only at low levels but localizes to the somatodendritic compartment during stress (Frandemiche et al., 2014; Hoover et al., 2010; Zempel et al., 2013; Zempel and Mandelkow, 2014). Tau might function in this context to slow RNA granule transport and regulate the interaction of TIA1 with other SG proteins, which would facilitate SG formation and the translational stress response.
The immunohistochemical studies of the TIA1 proteome components complement existing studies to highlight an important role for the TIA1 proteome network in the pathophysiology of AD and other tauopathies. Each of the four RBPs examined that were part of the TIA1 protein interactome co-localized with tau pathology. One of these proteins, RPL7, is a tau-dependent TIA1-interacting protein that has also been observed to be associated with tau in human pathological samples (Minjarez et al., 2013). Another tau-dependent TIA1-interacting protein is EWSR1, which is genetically linked to amyotrophic lateral sclerosis (Couthouis et al., 2012). These intersecting pieces of evidence suggest a model in which the accumulation of aggregated RNA-binding proteins in tauopathies might result from shared hyperactive SG pathways that also leads to the accumulation of aggregated tau.
The intersection of SGs with tau biology becomes particularly important when considering therapeutic implications. Protein aggregation in neurodegenerative disease has been classically considered to result from dysfunctional protein misfolding. However, SGs and other RNA granules exhibit protein aggregation that occurs as part of a normal physiological pathway, exhibiting inherent abilities to cycle between the liquid and solid states (Lin et al., 2015; Molliex et al., 2015; Nott et al., 2015; Patel et al., 2015; Vanderweyde et al., 2012). In the cell, TIA1 functions as a core component of SGs, promoting their nucleation. The studies above take advantage of the importance of TIA1 for nucleation of SGs and of tau granules. We show that deletion or knockdown of TIA1 in cultured neurons reduces the ability of cells to form SGs, inhibits pathological tau misfolding, and prevents tau-mediated degeneration. Thus, our work identifies TIA1 knockdown as a potentially important approach to inhibit tau misfolding and tau-mediated degeneration.
A large number of biochemical pathways also have the ability to disperse SGs and RNA granules. Each of these pathways are potential targets of drug discovery. We show that proline-directed kinases, which are known to regulate the association of tau with microtubules, also regulate the tau-mediated SG pathway. Attention to the SG pathway highlights compelling approaches to regulation. For instance, drug discovery efforts built around inhibiting SG formation have been successfully used to identify novel agents that prevent aggregation of TDP-43, which might be an effective treatment for ALS (Boyd et al., 2014; Kim et al., 2014). Translational inhibitors provide an additional mechanism for regulation. The translational inhibitors, puromycin or cycloheximide, reciprocally induce or prevent tau-mediated SG formation and also modulate the degeneration associated with overexpression of TIA1 with tau. Puromycin and cycloheximide are admittedly toxic, but we show that kinases regulating eIF2α phosphorylation, including PKR and PERK, regulate tau-mediated SG formation. These kinases might be particularly effective for tauopathies, such as AD, because they appear to inhibit disease processes at multiple levels, including preventing toxicity associated with β-amyloid (Ohno, 2014). Thus, the role of tau in RNA granule biology highlights the potential role of reversible protein aggregation in the pathophysiology of tauopathies and presents a corresponding wide range of avenues for pharmacotherapy of AD and other tauopathies.
EXPERIMENTAL PROCEDURES
Mice
Use of all animals was approved by the Boston University Institutional and Animal Care and Use Committee. Tau−/− and TIA1−/− mice as described previously (Dawson et al., 2001; Piecyk et al., 2000).
Cell Culture
HT22 cells were transfected using Lipofectamine (Invitrogen), incubated 24 hr, treated after 24 hr (25 μM salubrinal, 5 μg/ml puromycin, 10 μg/ml cycloheximide), and fixed in 4% paraformaldehyde (PFA) at 48 hr. Primary mouse P0 hippocampal cultures were grown for 21 days in Neurobasal medium supplemented with B-27 (Invitrogen).
AAV Transduction
At DIV 2, neurons were transduced with AAV9 vectors at MOI 20 (mRFP, TIA1 shTIA1-GFP, or shControl-GFP). At DIV 7, neurons were transduced with AAV9-WT or P301L tau virus (MOI 20).
Immunocytochemistry was performed as described previously (Vanderweyde et al., 2012). Primary antibodies used were Tau: CP-13, PHF1, MC1 (1:150 each), and Tau13 (1:5,000); SGs were TIA1 (1:400, Santa Cruz Biotechnology); and neurons were MAP2 (1:1,000, Aves).
Image Analysis
SG density and dendritic processes were quantified using ImageJ (using ImageJ plug-ins NeuronJ, tracing MAP2 positive processes for dendritic measurements). Granule movement and formation was quantified using Bitplane Imaris Track software (Imaris).
Photo-conversion
Photo-convertable WT human tau (PC-Tau) was generated by sub-cloning human 4N0R tau into the pPS-CFP2-C mammalian expression vector (Evrogen catalog no. FP801). Primary cortical cultures (E16) were transfected at DIV 5 and aged to DIV 18–23 prior to photo-conversion using a diode 405-nm laser on a Zeiss LSM-710 Duo Scan microscope.
Biochemical Fractionation
HT22 cells were lysed in RIPA buffer with 1 × Halt protease inhibitor cocktail (Thermo Scientific), 1× phosphatase inhibitor cocktail (PhosSTOP, Roche), sonicated, and centrifuged for 1 hr at 100,000 × g at 4°C, and the supernatants were collected in RIPA buffer. The pellets were sonicated and suspended in urea buffer.
Sarkosyl Insoluble and Soluble tau Fractions
Supernatant (100 μl with 100 μg protein) in RIPA buffer plus 1% sarkosyl detergent was rotated at room temperature for 1 hr and then centrifuged 1 hr (100,000 × g at room temperature). The supernatant and the sarkosyl pellet were suspended in sample buffer containing 100 mM DTT.
Immunoblotting
Immunoblots were performed using 15-well- 4%–12% Bis-Tris gels (Invitrogen) for electrophoresis, 1 hr blocking in 5% milk, 4°C overnight antibody incubation (1:7,500 Tau13, 1:500 PHF1, 1:500 TIA1 (Santa Cruz), 1:500, synaptophysin (Santa Cruz), 1:1,000 PSD-95 (NeuroMab), 1:1,000 caspase-3 or cleaved caspase 3 (Cell Signaling Technology), or 1:10,000 actin (Millipore) in PBS. Secondary antibodies (Jackson) were incubated in 5% milk for 1 hr at room temperature. Developing used SuperSignal West Pico Substrate (Thermo Fisher Scientific).
IP
Lysates (100–300 μg) were pre-cleared with rec-Protein G-Sepharose 4B Conjugate beads (Invitrogen) and then immunoprecipitated using ON 4°C with 0.5 μl PHF1 antibody, 1 μl Tau-5 antibody (Abcam), 1 μL MC1 antibody, or 0.5 μL of TIA1 antibody (Santa Cruz) or 1:200 HA antibody (Covance), followed by addition of 50 μl protein G rec-Protein G-Sepharose 4B conjugate beads and incubation for 1 hr at 4°C. The beads were spun down and washed, boiled in SDS sample buffer. and blotted.
Measurement of protein synthesis followed the SUnSET protocol using a 30-min puromycin treatment (Schmidt et al., 2009).
Proteomics
Quantitative proteomic analysis was performed using the total ion current (TIC) for proteins identified by LC-MS/MS normalized to the TIC level of TIA1 detected in each sample. The 163 proteins identified as unique in the WT and tau KO conditions were submitted to the DAVID Functional Clustering tool (Huang et al., 2009). Twelve resulting clusters with enrichment FDR <0.05 were identified, and each of the 163 proteins was associated with the cluster(s) based on its membership in the clustered gene sets. A network was induced between proteins by counting the number of clusters shared between pairs of proteins and visualized in the program Gephi 0.8.2.
Cell Death Studies
Caspase 3/7 cleavage was quantified with the Caspase-Glo 3/7 Assay kit, Promega. Apoptosis detected DNA fragmentation (TiterTACS Colorimetric Apoptosis Detection kit, Trevigen).
Supplementary Material
Highlights.
Tau is required for normal interactions of RNA binding proteins in brain tissue
Tau promotes stress granules, while TIA1 promotes tau misfolding and insolubility
TIA1 knockdown or knockout inhibits tau pathology and toxicity
TIA1 and tau act synergistically to modulate degeneration of cultured neurons
Acknowledgments
Paul Anderson and Nancy Kedersha (BWH) kindly provided the TIA1−/− mouse line and advice. Antibodies were kindly provided by Peter Davies (Einstein, MC1 and PHF1), and Lester Binder and Nicholas Kanaan (Michigan State University, Tau5 and Tau13). Adam Labadorf (Boston University) provided initial bioinformatics. Funding: B.W.: NIH ES020395, AG050471, NS089544 (B.W./L.P.), Alzheimer Association, BrightFocus Foundation, CurePSP Foundation, and the CureAlzheimer Foundation, L.P.: ES20395, AG16574-17JP2, and NS089544, H.L.: NIH CA196631, J.F.A.: NIH NS091329, MD009205, DOD 11811993; T.V.: NIH AG042213.
Footnotes
ACCESSION NUMBERS
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2014) partner repository with the dataset identifier PXD003922 and http://dx.doi.org/10.6019/PXD003922.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.04.045.
AUTHOR CONTRIBUTIONS
T.V., D.J.A., and K.Y.-K. helped design, perform, and analyze the experiments. T.V. and D.J.A. co-wrote the manuscript. P.E.A.A., A.A.F., C.C., K.J.-W., and A.C. performed experiments or produced essential reagents. E.L.d.R. and H.L. performed bioinformatic analysis. J.F.A., J.D.L., P.I., M.S., and L.P. provided advice on experiment design or analysis and provided reagents and/or manuscript editing. B.W. conceived of the project, helped design the experiments, analyzed the data, and co-wrote the manuscript.
References
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ, Wang X, Xia Q, Duong DM, Street C, et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013;110:16562–16567. doi: 10.1073/pnas.1310249110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Boyd JD, Lee-Armandt JP, Feiler MS, Zaarur N, Liu M, Kraemer B, Concannon JB, Ebata A, Wolozin B, Glicksman MA. A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J Biomol Screen. 2014;19:44–56. doi: 10.1177/1087057113501553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111:1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, Lanteé F, Buisson A. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci. 2014;34:6084–6097. doi: 10.1523/JNEUROSCI.4261-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lee VM, Brunden KR, Hutton M, Trojanowski JQ. Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Cold Spring Harb Perspect Med. 2011;1:a006437. doi: 10.1101/cshperspect.a006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Rayman JB, Kandel ER, Derkatch IL. Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol Cell. 2014;55:305–318. doi: 10.1016/j.molcel.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Protter DS, Rosen MK, Parker R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol Cell. 2015;60:208–219. doi: 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE. 2010;5:e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST, Wolozin B. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31:16086–16093. doi: 10.1523/JNEUROSCI.4105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF, Kobrin K, Murphy GJ, Petrucelli L, Wolozin B. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci. 2014;34:4167–4174. doi: 10.1523/JNEUROSCI.2350-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matenia D, Mandelkow EM. The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci. 2009;34:332–342. doi: 10.1016/j.tibs.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Minjarez B, Valero Rustarazo ML, Sanchez del Pino MM, González-Robles A, Sosa-Melgarejo JA, Luna-Muñoz J, Mena R, Luna-Arias JP. Identification of polypeptides in neurofibrillary tangles and total homogenates of brains with Alzheimer’s disease by tandem mass spectrometry. J Alzheimers Dis. 2013;34:239–262. doi: 10.3233/JAD-121480. [DOI] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, Baldwin AJ. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell. 2015;57:936–947. doi: 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M. Roles of eIF2α kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci. 2014;7:22. doi: 10.3389/fnmol.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J. 2000;19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Thomas MG, Loschi M, Desbats MA, Boccaccio GL. RNA granules: the good, the bad, and the ugly. Cell Signal. 2011;23:324–334. doi: 10.1016/j.cellsig.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, Duff K, Wolozin B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 2012;32:8270–8283. doi: 10.1523/JNEUROSCI.1592-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang D, Zhao J, Qu M, Zhou X, He H, He R. The proline-rich domain and the microtubule binding domain of protein tau acting as RNA-binding domains. Protein Pept Lett. 2006;13:679–685. doi: 10.2174/092986606777790566. [DOI] [PubMed] [Google Scholar]
- Wolozin B. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener. 2012;7:56. doi: 10.1186/1750-1326-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014;37:721–732. doi: 10.1016/j.tins.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, Mandelkow EM. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013;32:2920–2937. doi: 10.1038/emboj.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.