

SUMMARY
S6K1 has been implicated in a number of key metabolic responses, which contribute to obesity. Critical among these is the control of a transcriptional program required for the commitment of mesenchymal stem cells to the adipocytic lineage. However, in contrast to its role in the cytosol, the functions and targets of nuclear S6K1 are unknown. Here, we show that adipogenic stimuli trigger nuclear translocation of S6K1, leading to H2BS36 phosphorylation and recruitment of EZH2 to H3, which mediates H3K27 trimethylation. This blocks Wnt gene expression, inducing the upregulation of PPARγ and Cebpa and driving increased adipogenesis. Consistent with this finding, white adipose tissue from S6K1-deficient mice exhibits no detectable H2BS36 phosphorylation or H3K27 trimethylation, whereas both responses are highly elevated in obese humans or in mice fed a high-fat diet. These findings define an S6K1-dependent mechanism in early adipogenesis, contributing to the promotion of obesity.
In Brief
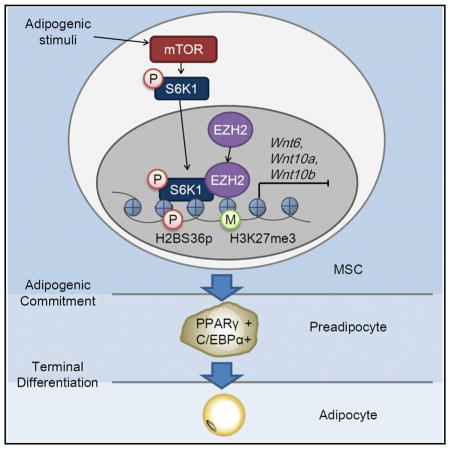
Expansion of adipose mass through adipogenesis is the main cause of obesity. Yi et al. report a role for S6K1 in the transcriptional regulation of an epigenetic network mediated by histone H2B phosphorylation and histone H3 trimethylation. This results in blocking Wnt ligand production and promoting adipogenesis.
INTRODUCTION
The worldwide epidemic in obesity is the chief cause of the dramatic increase in a number of associated pathologies including diabetes, hyperlipidemia, cardiovascular disease, and cancer (Spiegelman and Flier, 2001; Khandekar et al., 2011). It has been suggested that a potential target for therapeutic intervention would be inhibiting the recruitment of adipocyte precursor stem cells to adipose depots (Tang et al., 2008; Rodeheffer et al., 2008). This step is initiated by the commitment of mesenchymal stem cells (MSCs) to the adipocytic lineage, which is regulated by a number of extracellular cues, including growth factors, hormones, and nutrients. These extracellular cues coordinate a complex network of transcriptional factors, including proliferator-activated receptor-γ (PPARγ) and CCAAT/enhancer binding protein-α (C/EBPα) (Rosen and MacDougald, 2006), whose expression is controlled by a family of histone methyltransferases and demethylases acting epigenetically through the Wnt/β-catenin signaling pathway (Ge, 2012). Normally, activated Wnt/β-catenin signaling restrains adipogenesis by suppressing the expression of PPARγ and Cebpa (Rosen and MacDougald, 2006), while promoting differentiation of MSCs into myocytes and osteocytes (Christodoulides et al., 2009). However, the signaling pathway, by which epigenetic modifications mediate adipogenesis is poorly understood.
Previous studies have shown that S6K1, a key downstream effector of the mammalian target of rapamycin (mTOR) signaling pathway (Zoncu et al., 2011), plays a critical role in a number of key catabolic responses, including protein (Ma and Blenis, 2009), nucleotide (Ben-Sahra et al., 2013; Robitaille et al., 2013), and lipid synthesis (Düvel et al., 2010; Owen et al., 2012). In the latter case, de novo lipid biosynthesis is mediated by S6K1-stimulated processing of sterol regulatory element-binding proteins 1 and 2 (SREBP1 and SREBP2) (Düvel et al., 2010; Owen et al., 2012), consistent with S6K1-deficient mice being lean at birth, exhibiting reduced adipocyte cell size and resistance to high-fat diet (HFD)-induced obesity (Um et al., 2004). However, unexpectedly, we previously found that when such mice were maintained on a HFD, their adipocytes increased in size but not in number. The lesion could be traced to an impairment of the ability of mesenchymal stem cells to commit to the adipogenic lineage (Carnevalli et al., 2010). Moreover, the lesion was associated with a failure to upregulate a number of transcription factors critical for adipogenic commitment (Carnevalli et al., 2010). Although no nuclear targets have been identified for S6K1, recent studies have shown that the activated kinase translocates to the nucleus (Rosner and Hengstschläger, 2011; Rosner et al., 2012), raising the possibility that S6K1 may transcriptionally regulate adipogenic commitment through a nuclear mechanism. Here, we demonstrate that in response to an adipogenic stimulus, S6K1 translocates to the nucleus and phosphorylates histone H2B at serine 36, which is required for EZH2 recruitment to target gene promoters and subsequent histone H3 trimethylation at lysine 27. These epigenetic changes suppress the expression of Wnt6, Wnt10a, and Wnt10b and facilitate adipogenic commitment of MSCs. Critically, both phosphorylation of H2B and trimethylation of H3 are absent in adipose from S6K1−/− mice, whereas both responses are highly elevated in obese humans or in mice maintained on a HFD.
RESULTS AND DISCUSSION
S6K1 Is Required for H3K27 Trimethylation during Adipogenic Commitment
Earlier, we demonstrated that S6K1 plays a key role in the commitment of embryonic stem cells to early adipocyte progenitors, a response apparently regulated at the transcriptional level (Carnevalli et al., 2010). To analyze the potential role of S6K1 in mediating commitment to the adipogenic lineage, we took advantage of 10T1/2 cells, a clonal mouse embryo cell line, which can differentiate into distinct mesodermal progenitor cell lineages, including adipocytes (Huang et al., 2009; Tang and Lane, 2012) (Figure 1A). We initially tested whether S6K1 translocates to the nucleus in 10T1/2 cells treated with the adipogenic stimulus BMP4. The results demonstrated that BMP4 treatment induces the acute re-localization of S6K1 from the cytoplasm to the nucleus within 30 min, such that by 2 hr ~50% of the total cellular S6K1 is nuclear (Figures 1B and S1A). The effect of BMP4 is abolished by pretreatment with rapamycin, an mTORC1 inhibitor, as is the basal level of nuclear S6K1 (Figure 1C). Similarly, serum stimulation of HeLa cells induced a rapid increase in S6K1T389p, a marker of S6K1 activation, which was paralleled by the translocation of S6K1 into the nucleus and suppressed by rapamycin in a time-dependent manner (Figure S1A). Given that site-specific histone methylations play a critical role in gene expression during adipogenesis, we screened for epigenetic modifications of histones following BMP4 treatment of 10T1/2 cells in the absence or presence of the selective S6K1 inhibitor, PF-4708671 (Pearce et al., 2010). Of the modifications analyzed, only H3K27 trimethylation (H3K27me3) was sensitive to PF-4708671 treatment, whereas H3K4 trimethylation (H3K4me3), H3K36 trimethylation (H3K36me3), and H3 acetylation (H3Ac) were unaffected (Figure 1D). Similar results were obtained in HeLa cells (Figure S1B). Moreover, by immunofluorescence S6K1 nuclear translocation preceded that of H3K27me3 (Figure S1C). Consistent with this finding, small interfering RNA (siRNA) depletion of S6K1 abolished H3K27me3 in BMP4-treated or serum-treated 10T1/2 or HeLa cells, respectively (Figures 1E and S1D). Although EZH2, H3K27 methyltransferase, has been shown to be directly phosphorylated and regulated by a number of kinases, including CDK1, CDK2, and AKT (Cha et al., 2005; Chen et al., 2010; Kaneko et al., 2010; Wei et al., 2011), S6K1 did not phosphorylate EZH2 in vitro (Figure S1E), nor did inhibition of S6K1 by PF-4708671 treatment affect the levels of EZH2 or CDK1/2-dependent phosphorylation EZH2T345p and EZH2T487p in BMP4- or serum-treated 10T1/2 or HeLa cells, respectively (Figures 1F and 1G). Moreover, these results did not appear to be due to loss of the S6K1 negative feedback loop to AKT (Um et al., 2004), as AKT activation was unaffected by either S6K1 depletion or PF-4708671 treatment (Figures 1E and 1F). Taken together, these findings suggest an indirect mechanism of S6K1 activation of EZH2-mediated H3K27me3.
Figure 1. S6K1 Is Required for H3K27 Trimethylation during Adipogenic Commitment.

(A) Schematic representation of 10T1/2 cell differentiation into adipocyte.
(B) Immunoblot analysis of cytoplasmic and nuclear extracts from 10T1/2 cells treated with BMP4 for the indicated times, with quantification graphs of S6K1 indicated in the left-hand panel.
(C) Immunoblot analysis of cytoplasmic and nuclear extracts from 10T1/2 cells treated with BMP4 in either the absence or presence of rapamycin (100 nM), which was added for 1 hr prior to the addition of BMP4.
(D) Immunoblot analysis of 10T1/2 cells treated with BMP4 for 4 days in the presence or absence of PF-4708671 (20 μM).
(E) Immunoblot analysis of 10T1/2 cells expressing S6K1 siRNA (#03) in the presence of BMP4 (2 days).
(F) Immunoblot analysis of 10T1/2 cells treated with BMP4 for 1 hr in the presence or absence of PF-4708671 (20 μM).
(G) Immunoblot analysis of HeLa cells stimulated with serum for 15 min after pretreatment with rapamycin (100 nM) or PF-4708671 (20 μM) for 1 hr.
See also Figure S1.
S6K1 Phosphorylates H2BS36 and S6K1-Mediated H2B Phosphorylation Induces Trimethylation of H3K27
Histones are targets of multiple posttranslational modifications, including phosphorylation, leading to subsequent regulation of gene transcription (Tsankova et al., 2007). Although S6K1 had minor effects on H2A and H3 phosphorylation in vitro, it strongly phosphorylated H2B (Figure 2A), a response ablated by PF-4708671 (Figure 2B). As specific histone phosphorylations can regulate posttranslational modifications at other histones (Baek, 2011; Shimada et al., 2008; Yang et al., 2012), we analyzed H2B for S6K1 consensus phosphorylation motifs and found that the N terminus contains two K/RxRxxS motifs at S32 and S36 (Flotow and Thomas, 1992). To determine whether these sites are potential S6K1 phosphorylation sites, two peptides spanning amino acids 27–46, containing the phosphorylation motifs, or amino acids 7–26, as a control, were incubated with S6K1. Analysis of fluorograms showed that only the 27–46 peptide was phosphorylated by S6K1 (Figure 2C). Moreover, peptides covering the same amino acids 27–46 but harboring either S32A or S38A substitution served as substrates for S6K1, but not the same peptide having a S36A substitution (Figure 2D). Consistent with this finding, S6K1 depletion blocked phosphorylation of H2B S36 (H2BS36p) in 10T1/2 and HeLa cells (Figures 2E and S2A, respectively). In contrast, depletion of AKT, which shares the RxRxxS consensus phosphorylation site motif with S6K1, had no effect on H2BS36p in 10T1/2 (Figure S2B), consistent with AKT activation not being affected by either S6K1 depletion or PF-4708671 treatment (Figures 1E and 1F). These results are compatible with both endogenous and ectopically expressed H2B co-immunoprecipitating with S6K1, and no other histones (Figures 2F and S2C–S2E), a response that was enhanced by BMP4 stimulation (Figure 2G). Moreover, BMP4-induced S6K1 activation, measured by S6K1T389p, closely paralleled that of H2BS36p and H3K27me3 (Figure 2H), which were blocked by PF-4708671 treatment (Figure 2I). Finally, a phosphomimetic mutant H2BS36D protected BMP4-induced H3K27me3 from PF-4708671 treatment (Figure 2J). More importantly, H2BS36D, but not H2BS36A, co-immunoprecipitated with EZH2 (Figure 2K). Collectively, the data indicate that S6K1-mediated H2BS36p induces H3K27me3, potentially by recruiting EZH2 to H3 during adipogenic commitment.
Figure 2. S6K1 Phosphorylates H2BS36 and S6K1-Mediated H2B Phosphorylation Induces Trimethylation of H3K27.

(A) In vitro phosphorylation of recombinant H2A, H2B, H3, or H4 with active S6K1 and 32P-ATP.
(B) In vitro phosphorylation of H2B with active S6K1 and 32P-ATP after preincubation of S6K1 with the indicated concentration of PF-4708671 for 30 min.
(C) In vitro phosphorylation of H2B 7–26 and 27–46 peptides with active S6K1 and 32P-ATP.
(D) Peptide sequences containing S6K1 target motifs in H2B or single site mutation (top panel) and in vitro phosphorylation of the indicated peptides with active S6K1 and 32P-ATP (bottom panel).
(E) Immunoblot analysis of 10T1/2 cells expressing S6K1 siRNA in the presence of BMP4 (2 days).
(F) Immunoblot analysis of S6K1 immunoprecipitates (IP) and whole-cell lysates (WCLs) from 10T1/2 cells.
(G) Immunoblot analysis of immunoprecipitates with S6K1 antibody from 10T1/2 cells in the presence or absence of BMP4.
(H) Immunoblot analysis of 10T1/2 cells treated with BMP4 for the indicated time.
(I) Immunoblot analysis of 10T1/2 cells treated with BMP4 in the presence or absence of PF-4708671 (20 μM) for the indicated time.
(J) Immunoblot analysis of 10T1/2 cells transfected with or without the GFP-H2BS36D vector in the presence of BMP4 and PF-4708671 (20 μM) for 24 hr.
(K) Immunoblot analysis of immunoprecipitates with GFP antibody from 10T1/2 cells transfected with GFP-H2B36D or GFP-H2BS36A vectors in the presence of BMP4.
See also Figure S2.
S6K1-Mediated H2BS36 Phosphorylation Suppresses the Expression of Wnt Genes through the Recruitment of EZH2 during Adipogenic Commitment
As Wnt ligands, particularly Wnt6, Wnt10a, and Wnt10b, are known suppressors of adipogenic commitment and their expression is directly repressed by EZH2 during adipogenesis (Ge, 2012; Wang et al., 2010), we assessed the role of S6K1 and EZH2 in this response (Figure S3A). Reverse transcription-quantitative PCR (qRT-PCR) analysis showed that depletion of S6K1 increased Wnt6, Wnt10a, and Wnt10b gene expression during adipogenic commitment (Figure 3A). In contrast, depletion of AKT had no effect on Wnt6, Wnt10a, and Wnt10b gene expression, consistent with it having no impact on H2BS36p 10T1/2 (Figure S3B). Moreover, expression of a dominant-negative S6K1 (S6K1-DN) induced the upregulation of same set of Wnt genes, whereas a constitutively active S6K1 (S6K1-CA) suppressed their expression (Figures 3B and S3C). Importantly, chromatin immunoprecipitation (ChIP) analysis showed that BMP4 treatment led to the recruitment of S6K1 to Wnt6, Wnt10a, and Wnt10b genes promoters, that this was accompanied by increased H2BS36p and that both responses were abrogated by rapamycin treatment (Figures 3C and 3D). Likewise, Wnt gene expression was suppressed in cells expressing gain-of-function H2BS36D variant and induced in cells expressing the loss-of-function H2BS36A variant, as compared to control cells (Figure 3E). In addition, PF-4708671-induced Wnt gene expression was abrogated in H2BS36D-expressing cells (Figure 3F). In parallel, we found that BMP4 treatment resulted in increased EZH2 recruitment and H3K27me3 at the same Wnt promoters, with both responses blocked by rapamycin treatment (Figures 3G and 3H). As with S6K1, H2BS36D-expressing cells exhibited the enriched presence of EZH2 and H3K27me3 at Wnt gene promoters, whereas in H2BS36A-expressing cells both responses were inhibited (Figures 3I and 3J). These differences were not due to alterations in the recruitment of wild-type (WT) H2B, H2BS36D, or H2BS36A to Wnt gene promoters (Figure S3D), nor did their expression alter S6K1 activity (Figures S3E and S3F). It should be noted that the transcription of other EZH2 target genes but not non-EZH2 target genes were also negatively regulated by S6K1-induced H3K27me3 (Figures S3G and S4), further supporting the negative regulation of EZH2 target genes by S6K1-mediated H2BS36p. Together, the results indicate that S6K1-mediated H2BS36p leads to the recruitment of EZH2, H3K27me3 and the suppression of Wnt gene transcription during adipogenic commitment.
Figure 3. S6K1-Mediated H2BS36 Phosphorylation Suppresses the Expression of Wnt Genes through the Recruitment of EZH2 during Adipogenic Commitment.

(A) The mRNA levels of Wnt genes in 10T1/2 cells expressing S6K1 siRNA during commitment.
(B) The mRNA levels of Wnt genes in 10T1/2 cells expressing WT, constitutively active, and dominant-negative S6K1 vectors during commitment.
(C and D) 10T1/2 cells treated with BMP4 or co-treated with BMP4 and rapamycin (100 nM) for 24 hr, followed by ChIP-qPCR analyses for S6K1 (C) and H2BS36p
(D) antibodies in the promoter regions of the Wnt genes.
(E) The mRNA levels of Wnt genes in 10T1/2 cells expressing GFP-WT H2B, GFP-H2BS36D, or GFP-H2BS36A vectors during commitment.
(F) The mRNA levels of Wnt genes in 10T1/2 cells expressing the GFP-H2BS36D vector in the presence or absence of PF-4708671 (20 μM) during commitment.
(G and H) 10T1/2 cells treated with BMP4 or co-treated with BMP4 and rapamycin (100 nM) for 24 hr, followed by ChIP-qPCR analyses for EZH2 (G) and H3K27me3 (H) antibodies in the promoter regions of the Wnt genes.
(I and J) 10T1/2 cells transfected with GFP-WT H2B, GFP-H2BS36D, or GFP-H2BS36A vectors and incubated with BMP4 for 24 hr, followed by ChIP-qPCR analyses for EZH2 (I) and H3K27me3 (J) antibodies in the promoter region of the Wnt genes.
In (A)–(J), data are presented as the mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001. See also Figures S3 and S4.
S6K1-Mediated H2BS36 Phosphorylation Is Required for Adipogenic Commitment and Correlates with BMI
Critical for driving the adipogenic response is the suppression of Wnt genes, which triggers the upregulation of transcription factors PPARγ and C/EBPα (Cawthorn et al., 2012; Christodoulides et al., 2009). We found that S6K1 depletion during commitment reduced PPARγ and Cebpa mRNA levels (Figure 4A). Consistent with this result, ectopic expression of S6K1-DN repressed PPARγ and Cebpa expression, whereas transfection with S6K1-CA upregulated the transcription of these same genes (Figure 4B). Likewise, expression of the H2BS36D mutant during commitment increased PPARγ and Cebpa mRNA levels, whereas H2BS36A expression produced the opposite effect (Figure 4C). Critically, the expression of the PPARγ and the Cebpa genes in cells transfected with H2BS36D were resistant to suppression by PF-4708671 treatment (Figure 4D). Moreover, H2B36D, but not H2B36A, promoted the differentiation of the progenitor cells into adipocytes, as determined by the expression of adipocyte marker genes Fabp4, Adipsin, Adipoq, and Apoe (Figure 4E). Consistent with S6K1 phosphorylation of H2B regulating this response, the differentiation of H2B36D-expressing progenitor cells was resistant to PF-4708671 treatment (Figure 4F). If S6K1-dependent H2BS36p and H3K27me3 are playing a critical role in adipogenic commitment, their levels S6K1−/− would be expected to be reduced in the fat pads of mice. To test this, we isolated epididymal white adipose tissue (eWAT) from WT- and S6K1-deficient mice. As we previously reported (Carnevalli et al., 2010; Um et al., 2004), eWAT from S6K1-deficient mice was reduced in mass as compared to eWAT from WT mice (Figure 4G). Moreover, the mRNA levels of Wnt6, Wnt10a, and Wnt10b were elevated in S6K1-deficient mice as compared to WT mice (Figure 4H). Consistent with these findings, the mRNA levels of white adipocyte markers (Adipsin, Fabp4, and PPARγ) were significantly reduced in S6K1-deficient mice (Figure 4I). Critically, H2BS36p and H3K27me3 were completely absent in eWAT from S6K1−/− mice (Figure 4J), underscoring their potential key role in S6K1-mediated adipogenic commitment.
Figure 4. S6K1-Mediated H2BS36 Phosphorylation Is Required for Adipogenic Commitment and Correlates with BMI.

(A) The mRNA levels of PPARγ and Cebpa genes in 10T1/2 cells expressing S6K1 siRNA during commitment.
(B) The mRNA levels of PPARγ and Cebpa genes in 10T1/2 cells expressing WT, constitutively active, and dominant-negative S6K1 vectors during commitment.
(C) The mRNA levels of PPARγ and Cebpa genes in 10T1/2 cells expressing GFP-WT H2B, GFP-H2BS36D, or GFP-H2BS36A vectors during commitment.
(D) The mRNA levels of PPARγ and Cebpa genes in 10T1/2 cells expressing GFP-H2BS36D vector in the presence or absence of PF-4708671 (20 μM) during commitment.
(E) The mRNA levels of adipocytic marker genes in fully differentiated adipocytes from 10T1/2 cells expressing GFP-WT H2B, GFP-H2BS36D, or GFP-H2BS36A vectors during commitment.
(F) The mRNA levels of adipocytic marker genes in fully differentiated adipocytes from 10T1/2 cells expressing GFP-H2BS36D vector in the presence or absence of PF-4708671 (20 μM) during commitment.
(G) Epididymal white adipose tissues (eWAT) from WT and S6K1-knockout (S6K1−/−) mice.
(H) The mRNA levels of Wnt genes in eWAT from WT and S6K1−/− mice.
(I) The mRNA levels of adipogenic marker genes in eWAT from WT and S6K1−/− mice.
(J) Immunoblot analysis of eWAT from WT and S6K1−/− mice.
(K) Immunoblot analysis of human abdominal subcutaneous WAT from normal or overweight specimens.
(L) Semiquantitative analysis of pS6K1, H3K27me3, and H2BS36p level in individual WAT samples.
(M) Immunoblot analysis of eWAT from WT mice fed normal chow diet (NCD) or high-fat diet (HFD) for 10 weeks.
(N) Mechanisms of metabolic regulation by S6K1 in the nucleus (1) and the cytoplasm (2).
In (A)–(F), (H), and (I), data are presented as the mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001.
We have previously demonstrated that S6K1 phosphorylation is greatly elevated in adipose tissue of mice maintained on HFD versus a normal chow diet (NCD) (Um et al., 2004). Given this finding, we asked whether S6K1T389p, H2BS36p, and H3K27me3 were associated with obesity in humans. We were able to obtain six human biopsies of abdominal subcutaneous adipose tissues, which we stratified based on BMI into either a normal group, BMI <23, or overweight group, BMI >23. Immunoblot analysis showed that S6K1T389p, H2BS36p and H3K27me3 appeared to be highly elevated in adipose tissues from overweight individuals, as compared to adipose tissues of patients of normal weight (Figure 4K). Quantification of the immunoblot analyses showed that the level of H2BS36p and S6K1T389p appeared strongly correlating with BMI (Figure 4L). To determine whether our observations, in the limited number of human samples, were responsible for the development of obesity, we analyzed eWAT from mice maintained on HFD versus NCD, for 10 weeks. The results show, as for S6K1T389p, that both H2BS36p and H3K27me3 were elevated in mice maintained on a HFD versus those maintained on a NCD (Figure 4M). Taken together, these data suggest that hyperactivation of S6K1 is a major player in driving the obese phenotype through H2BS36p and H3K27me3.
Our studies have uncovered a molecular mechanism by which S6K1 regulates early adipogenesis through the direct regulation of gene transcription by the multistep histone modifications of H2BS36p and H3K27me3 (Figure 4N). These findings also raise the question of whether inhibition of S6K1 would offer a potential therapeutic avenue for the treatment of obesity. We recently advocated such an approach for the treatment of insulin resistance (Um et al., 2015), as S6K1−/− mice remain exquisitely insulin sensitive even on a HFD (Um et al., 2004). Interest in such an approach had been questioned, given that transplant patients maintained on rapamycin exhibit impaired glucose tolerance and post-transplant type 2 diabetes (T2D), attributed to inhibition of mTORC1 (Johnston et al., 2008). However, it has recently been demonstrated that the long-term effects of chronic rapamycin treatment on insulin resistance are due to the disruption and loss of mTORC2, not to the inhibition of mTORC1 (Lamming et al., 2012). Moreover, liver-specific depletion of S6K1 protects against both HFD-induced hepatic steatosis and systemic whole-body insulin resistance (Bae et al., 2012). Thus, direct inhibitors of S6K1, which would bypass mTORC2 inhibition, may serve as a potential therapeutic route not only to suppress obesity, but to treat insulin resistance, the hallmark of T2D.
EXPERIMENTAL PROCEDURES
DNA Constructs and Antibodies
The DNA constructs used in this study were pRK5-myc-S6K1, pRK5-myc-S6K1-CA, pRK5-myc-S6K1-DN, and pCDNA-EGFP-H2B. The mutant constructs for H2B were generated using site-directed mutagenesis (pCDNA-EGFP-H2B S36D and pCDNA-EGPF-H2B S36A). Anti-p70 S6K1 (Cell Signaling Technology; #9202, Santa Cruz Biotechnology; SC-8418), anti-phospho (T389) p70 S6K1 (Cell Signaling Technology; #9234), anti-S6 (Cell Signaling Technology; #2217), anti-phospho (S235/236) S6 (Cell Signaling Technology; #4856), anti-GFP (Santa Cruz Biotechnology; SC-9996), anti-histone H3 (Santa Cruz Biotechnology; SC-10809), anti-histone H3 Lys27 trimethylation (Millipore; 07-449), anti-histone H2B (Abcam; ab18977) anti-histone H2B Ser36 phosphorylation (ECM Biosciences; HP4331), anti-tubulin (Santa Cruz Biotechnology; SC-32293), anti-actin (Millipore; mab1501), anti-KMT6/EZH2 (Abcam; ab3748), anti-Lamin A/C (Cell Signaling Technology; #2032), and anti-PolII (Santa Cruz Biotechnology; SC-900) antibodies were used in this study.
Cell Culture and Differentiation
HeLa cells were purchased from the American Type Culture Collection (ATCC) and C3H10T1/2 (10T1/2) cells were a generous gift from Jong Sun Kang (Sungkyunkwan University). The cells were grown and maintained in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). For adipogenic commitment to pre-adipocytes, 10T1/2 cells maintained in DMEM with 10% FBS and 1% P/S were treated for 4 days with 10 μg/mL BMP4. For terminal differentiation of pre-adipocytes to adipocytes, the cells were incubated in DMEM with 10% FBS, 1% P/S, 0.5 mM 3-isobuyl-1-methylxanthine (IBMX), 1 μM dexamethasone, and 1 μg/mL insulin.
Inhibition and Knockdown of S6K1
Cells were treated with rapamycin (Calbiochem, 100 nM) and PF-4708671 (Tocris, 20 μM) to inhibit S6K1 activity. For S6K1 knockdown, cells were transfected with siRNA targeting S6K1 using Lipofectamine 2000 reagent (Life Technologies) according to the manufacturer’s protocol. The siRNA sequences targeting S6K1 are as follows: #03 forward, 5′-GGACCAGCCAGAA GAUGCAGGCUCU-3′; #03 reverse, 5′-AGAGCCUGCAUCUUCUGGCUGG UCC-3′; #04 forward, 5′-CACCCUUUCAUUGUGGACCUGAUUU-3′; and #04 reverse, 5′-AAAUCAGGUCCACAAUGAAAGGGUG-3′. To generate stable cell lines, lentiviral short hairpin RNA (shRNA) for S6K1 (5′-GCGACATCTTTCT CAACCTTA-3′) from Open Biosystems was used. The lentivirus was obtained from 293FT cells according to the manufacturer’s protocol. HeLa cells were seeded onto plates and infected with the S6K1 shRNA-containing lentivirus. Cells expressing S6K1 shRNA were selected using puromycin (2 μg/mL).
In Vitro Kinase Assay
S6K1 kinase assays were performed according to the manufacturer’s protocol (SignalChem; #R21-10H). Recombinant histones (1 μg) (BioLab) or peptides (1 μg) were incubated for 1 hr at 30°C in kinase buffer containing 5 mM MgCl2 and 0.3 mM [γ-32P] ATP (0.5 Ci/mL). After the reaction, proteins or peptides were subjected to SDS-PAGE. The gel was stained with Coomassie blue R-150 to visual protein bands and then subjected to fluorography for 24 hr at 70 °C on sensitized AGFA 100 NIF (non-interleaved films).
Immunoblotting and Immunoprecipitation
For immunoblotting, each sample was subjected to SDS-PAGE. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes using semi-dry transfer (Bio-Rad). The membranes were incubated overnight with the indicated primary antibodies, followed by incubation with horseradish-peroxidase-conjugated secondary antibodies for 1 hr (Abcam). The signals were detected using chemiluminescence reagents (Intron). For immunoprecipitation, the cells were lysed with immunoprecipitation (IP) lysis buffer (HEPES 40 mM [pH 7.4] containing 120 mM NaCl, 1 mM EDTA, 50 mM NaF, 1.5 mM Na3VO4, 10 mM β-glycerophosphate, 0.3% CHAPSO, and protease inhibitors). The lysates were centrifuged for 20 min at 13,000 rpm at 4°C. The specific antibodies were incubated with the supernatants overnight at 4°C, followed by incubation with anti-rabbit Ig-IP beads (Trueblot) for 1 hr at 4°C. The beads were spun down for 1 min at 2,000 rpm and washed with IP wash buffer (IP lysis buffer without CHAPSO). The proteins were eluted by boiling for 5 min in Laemmli buffer (Bio-Rad) and subjected to immunoblotting.
Nuclear Fractionation
Cytoplasmic and nuclear extracts were prepared as previously described (Park et al., 2012). In brief, cells were suspended in buffer A (10 mM HEPES containing 1.5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 1 mM DTT, 0.5 μg/mL leupeptin, 1 mM PMSF, 1 μM pepstatin A, and 0.05% NP-40), and cytoplasmic extracts were separated by centrifugation at 4°C at 3,000 rpm for 10 min. The remained pellet was resuspended in buffer B (20 mM HEPES containing 1.5 mM MgCl2, 420 mM KCl, 25% glycerol, 0.2 mM EDTA, 1 mM DTT, 0.5 μg/mL leupeptin, 1 mM PMSF, and 1 μM pepstatin A) and incubated on ice for 30 min. Nuclear extracts were separated by centrifugation at 4°C at 13,000 rpm for 20 min.
Quantitative Real-Time PCR
Total RNA was extracted using Easy-Blue reagent (iNtRON). 1 μg of total RNA was reverse transcribed into cDNA using a Reverse Transcription kit (Promega). Quantitative real-time PCR was performed using KAPATM SYBR FAST qPCR (KAPABIOSYSTEMS) with a CFX96TM or Chromo4TM real-time PCR detector (Bio-Rad). Relative levels of mRNA were normalized to the values of GAPDH (HeLa cells) of β-actin (10T1/2 cells and mouse eWAT) mRNA for each reaction. The qPCR primer sequences used are as follows: β-actin forward, 5′-ACGGCCAGGTCATCACTATTG-3′; β-actin reverse, 5′-TGGA TGCCACAGGATTCCA-3′; Wnt6 forward, 5′-GCGGAGACGATGTGGACTTC-3′; Wnt6 reverse, 5′-ATGCACGGATATCTCCACGG-3′; Wnt10a forward, 5′-CCACTCCGACCTGGTCTACTTTG-3′; Wnt10a reverse, 5′-TGCTGCTCT TATTGCACAGGC-3′; Wnt10b forward, 5′-GCTGACTGACTCGCCCACCG-3′; Wnt10b reverse, 5′-AAGCACACGGTGTTGGCCGT-3′; PPARγ forward, 5′-GCATGGTGCCTTCGCTGA-3′; PPARγ reverse, 5′-TGGCATCTCTGTGT CAACCATG-3′; Cebpa forward, 5′-CTCCCAGAGGACCAATGAAA-3′; Cebpa reverse, 5′-AAGTCTTAGCCGGAGGAAGC-3′; Adipsin forward, 5′-CATG CTCGGCCCTACATG-3′; Adipsin reverse, 5′-CACAGAGTCGTCATCCGTCAC-3′; Fabp4 forward, 5′-AAGGTGAAGAGCATCATAACCCT-3′; Fabp4 reverse, 5′-TCACGCCTTTCATAACACATTCC-3′; GAPDH forward, 5′-GAGT CAACGGATTTGGTCGT-3′; GAPDH reverse, 5′-TTGATTTTGGAGGGATCTCG-3′; DAB2IP forward, 5′-TGGACGATGTGCTCTATGCC-3′; DAB2IP reverse, 5′-GGATGGTGATGGTTTGGTAG-3′; HOXC4 forward, 5′-TCCC TCCCACTGTTAAGGAC-3′; HOXC4 reverse, 5′-GAAATTCACCCAAGCCAGAC-3′; HOXA9 forward, 5′-TTGGAGGAAATGAATGCTGA-3′; HOXA9 reverse, 5′-TGGTCAGTAGGCCTTGAGGT-3′; HOXA11 forward, 5′-CCATT GAATCTCCTTTGCCT-3′; HOXA11 reverse, 5′-CACACACGGTGGGTAAGAAC-3′.
Chromatin Immunoprecipitation and Real-Time PCR
Chromatin immunoprecipitation was performed as previously described (Lee et al., 2012). In brief, a small portion of the cross-linked, sheared chromatin solution was reserved as the input DNA, and the remainder was subjected to immunoprecipitation overnight at 4°C using antibodies. After immunoprecipitation, the recovered chromatin fragments were subjected to qPCR using primer pairs specific for the target gene promoter. The primer sequences are available upon request.
Mice
Mice were maintained on a 12-hr light-dark cycle. Eight- to 10-week-old male mice were used for experiments. To induce obesity, mice were fed with normal chow diet or high-fat diet purchased from Daehan biolink for 10 weeks. All animal studies were performed according to the guidelines of the Sungkyunkwan University Institutional Animal Care and Use Committee (SKKUIACUC).
Human Adipose Tissue
Human abdominal subcutaneous adipose tissue was obtained from adults with diverse BMI during liposuction or abdominal plastic surgeries. All protocols and procedures were approved by the institutional review board (IRB) at the Catholic Medical Center (CMC).
Statistical Analysis
Statistical significance was analyzed using Student’s t test (two-tailed) and assessed based on a p value.
Highlights.
S6K1 directly phosphorylates histone H2B at S36 during adipogenic commitment
S6K1-mediated H2BS36 phosphorylation promotes H3K27 trimethylation by EZH2
These epigenetic changes suppress Wnt gene expression and drive adipogenesis
H2BS36p and H3K27me3 are elevated in adipose tissue from obese humans and mice
Acknowledgments
We thank Jong Sun Kang (Sungkyunkwan University) for the cell line and Kai Ge (NIH) for the primer sequences. We would also like to thank Dr. Alex Vaquero for his critical reading of the manuscript. This research was supported by Medical Research Center programs to J.-W.H through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2012R1A5A2A28671860). G.T. and S.C.K. are supported by Instituto de Salud Carlos III (ISCIII) grants (IIS10/00015 and II12/00002, respectively). S.C.K. is supported by the Spanish Ministry of Economy and Competitive (BFU2012-38867) grant. G.T. is supported by the Spanish Ministry of Science and Innovation (SAF2014-52162-P), the CIG European Commission (PCIG10-GA-2011-304160), the NIH/NCI National Cancer Institute (R01-CA158768) and the Asociacion Espanola Contra el Cancer (AECC GCB14-2035), ISCIII-RTICC (RD12/0036/0049), AGAUR (SGR 870), and Ministerio de Economia y Competitividad, ISCIII (PIE13/00022) grants. The Spanish Ministry grant to G.T. and S.C.K. are co-funded by FEDER (a way to build Europe) funds.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.03.011.
AUTHOR CONTRIBUTIONS
S.A.Y., S.H.U., J.C.L., and J.-W.H. conceived and designed the experiments. S.A.Y., J.H.Y., M.G.L., Y.J.J., and S.-J.L. performed experiments. S.Y.B., E.K.P., K.H.N., J.W.P., J.S.Y., and G.-U.B. provided experimental assistance and conceptual advice. J.W.R. offered human adipose tissue samples. S.A.Y., S.C.K., G.T., and J.-W.H. helped guide these studies and wrote the manuscript. All authors read and commented on the manuscript.
References
- Bae EJ, Xu J, Oh DY, Bandyopadhyay G, Lagakos WS, Keshwani M, Olefsky JM. Liver-specific p70 S6 kinase depletion protects against hepatic steatosis and systemic insulin resistance. J Biol Chem. 2012;287:18769–18780. doi: 10.1074/jbc.M112.365544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek SH. When signaling kinases meet histones and histone modifiers in the nucleus. Mol Cell. 2011;42:274–284. doi: 10.1016/j.molcel.2011.03.022. [DOI] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevalli LS, Masuda K, Frigerio F, Le Bacquer O, Um SH, Gandin V, Topisirovic I, Sonenberg N, Thomas G, Kozma SC. S6K1 plays a critical role in early adipocyte differentiation. Dev Cell. 2010;18:763–774. doi: 10.1016/j.devcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthorn WP, Bree AJ, Yao Y, Du B, Hemati N, Martinez-Santibañez G, MacDougald OA. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone. 2012;50:477–489. doi: 10.1016/j.bone.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol. 2010;12:1108–1114. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulides C, Lagathu C, Sethi JK, Vidal-Puig A. Adipogenesis and WNT signalling. Trends Endocrinol Metab. 2009;20:16–24. doi: 10.1016/j.tem.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotow H, Thomas G. Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J Biol Chem. 1992;267:3074–3078. [PubMed] [Google Scholar]
- Ge K. Epigenetic regulation of adipogenesis by histone methylation. Biochim Biophys Acta. 2012;1819:727–732. doi: 10.1016/j.bbagrm.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Song TJ, Li X, Hu L, He Q, Liu M, Lane MD, Tang QQ. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci USA. 2009;106:12670–12675. doi: 10.1073/pnas.0906266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston O, Rose CL, Webster AC, Gill JS. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol. 2008;19:1411–1418. doi: 10.1681/ASN.2007111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer. 2011;11:886–895. doi: 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Kang SU, Jeon Y, Park JW, You JS, Ha SW, Bae N, Lubec G, Kwon SH, Lee JS, et al. Protein L-isoaspartyl methyltransferase regulates p53 activity. Nat Commun. 2012;3:927. doi: 10.1038/ncomms1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE, Goldstein JL, Brown MS. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci USA. 2012;109:16184–16189. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Lee JC, Ha SW, Bang SY, Park EK, Yi SA, Lee MG, Kim DS, Nam KH, Yoo JH, et al. Requirement of protein l-isoaspartyl O-methyltransferase for transcriptional activation of trefoil factor 1 (TFF1) gene by estrogen receptor alpha. Biochem Biophys Res Commun. 2012;420:223–229. doi: 10.1016/j.bbrc.2012.02.072. [DOI] [PubMed] [Google Scholar]
- Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, Alessi DR. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) Biochem J. 2010;431:245–255. doi: 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339:1320–1323. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135:240–249. doi: 10.1016/j.cell.2008.09.036. [DOI] [PubMed] [Google Scholar]
- Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- Rosner M, Hengstschläger M. Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR. Oncogene. 2011;30:4509–4522. doi: 10.1038/onc.2011.165. [DOI] [PubMed] [Google Scholar]
- Rosner M, Schipany K, Hengstschläger M. p70 S6K1 nuclear localization depends on its mTOR-mediated phosphorylation at T389, but not on its kinase activity towards S6. Amino Acids. 2012;42:2251–2256. doi: 10.1007/s00726-011-0965-4. [DOI] [PubMed] [Google Scholar]
- Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–232. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Tang QQ, Lane MD. Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem. 2012;81:715–736. doi: 10.1146/annurev-biochem-052110-115718. [DOI] [PubMed] [Google Scholar]
- Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, Graff JM. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322:583–586. doi: 10.1126/science.1156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Um SH, Sticker-Jantscheff M, Chau GC, Vintersten K, Mueller M, Gangloff YG, Adams RH, Spetz JF, Elghazi L, Pfluger PT, et al. S6K1 controls pancreatic β cell size independently of intrauterine growth restriction. J Clin Invest. 2015;125:2736–2747. doi: 10.1172/JCI77030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jin Q, Lee JE, Su IH, Ge K. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc Natl Acad Sci USA. 2010;107:7317–7322. doi: 10.1073/pnas.1000031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC, Hung MC. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]