

Abstract
The development of bitopic ligands directed toward D2-like receptors has proven to be of particular interest to improve the selectivity and/or affinity of these ligands and as an approach to modulate and bias their efficacies. The structural similarity between dopamine D3 receptor (D3R)-selective molecules that display bitopic or allosteric pharmacology and those that are simply competitive antagonists are subtle and intriguing. Herein we synthesized a series of molecules in which the primary and secondary pharmacophores were derived from the D3R-selective antagonists SB269,652 (1) and SB277011A (2) whose structural similarity and pharmacological disparity provided the perfect templates for SAR investigation. Incorporating a trans-cyclopropylmethyl linker between pharmacophores and manipulating linker length resulted in the identification of two bivalent non-competitive D3R-selective antagonists, 18a and 25a, which further delineates SAR associated with allosterism at D3R and provides leads toward novel drug development.
Keywords: dopamine D3 receptor, dopamine D2 receptor, antagonist, allosteric modulator, bitopic ligand, cyclopropyl, primary pharmacophore, secondary pharmacophore, synthon, orthosteric binding site, β-arrestin
INTRODUCTION
The neurotransmitter dopamine signals through a family of five G protein-coupled receptors (GPCRs), subdivided into dopamine D1-like (D1R and D5R) and dopamine D2-like (D2R, D3R and D4R) families, based on sequence similarities and pharmacological actions.1 Although D2-like receptor antagonists have long been prescribed for the treatment of schizophrenia, lack of efficacy and potential extrapyramidal and/or metabolic syndrome-like side effects have precluded successful application to substance abuse disorders.2,3
Since it was cloned and characterized, the D3R has been a target of pharmacotherapeutic interest due to its relatively localized expression within mesolimbic neurocircuitry, including the nucleus accumbens, islands of Calleja and ventral striatum.4–7 As these brain regions are associated with rewarding and motivational characteristics of addictive drugs, the D3R has been an appealing drug development target for substance abuse disorders.8–11
Previous structure-activity relationships (SAR) developed for the D3R have revealed 4-phenylpiperazine as a classical primary pharmacophore (PP), which binds to the orthosteric binding site (OBS) and is primarily responsible for receptor binding affinity and efficacy.12–15 A four-carbon linking chain with or without substituents (e.g. OH or F) or structural rigidity (e.g. trans-cyclohexyl or trans-olefin) can be attached to an extended aryl ring system, generally through an amide group, to create a secondary pharmacophore (SP) which, when suitably designed, results in D3R selectivity.13,16–18
Hybrid molecules that concomitantly engage both an orthosteric and an allosteric site on a receptor have been termed ‘bitopic’ or ‘dualsteric’ ligands. As such, bitopic ligands can be viewed as a specific class of bivalent ligands. The development of bitopic ligands has proven to be of particular interest as a means of improving the selectivity and/or affinity of ligands for GPCRs as well as an approach to modulate and bias their efficacy.19–23
SB269,652 (1) is a potent and D3R-preferential antagonist that has been shown to act at this receptor in a non-competitive or allosteric manner.15,24 Interestingly, this tetrahydroisoquinoline derivative is also an allosteric antagonist at the D2R; a mode of action at this receptor has been proposed whereby 1 engages one protomer of a D2R dimer in a bitopic manner to negatively modulate the binding of dopamine to the second protomer.24–27 Conversely, SB277011A (2) is a competitive D3R-selective antagonist, first described in 2000 and extensively used as an in vivo tool to characterize the D3R and its potential as a target for medication development.28 The only structural differences between these two compounds are the position of the CN group on the tetrahydroisoquinoline ring system (PP) and the replacement of the terminal 2-indole amide (SP) of 1 with a 4-quinoline amide on 2. However, while both molecules display selectivity for the D3R and display high structural similarity, they differ in their competitive versus allosteric pharmacology. Hence, it is of interest to explore the structural determinants of this divergent pharmacological profile.
Recently, another analog of 1, compound 3 was described in which the indole moiety was replaced with a 7-azaindole.26 This simple modification caused ~30-fold increase in binding affinity at the D2R and it also displayed negative cooperativity, suggesting allosteric interactions with the D2R. Another D3R-selective partial agonist, BP1,4979 (4), has recently been evaluated for safety and efficacy in a clinical trial for smoking cessation and has structural similarities, but also differences from compounds 1 and 2; notably a 3-CN-phenyl piperazine, instead of the CN-tetrahydroisoquinolines, and the lack of a terminal aryl amide.29 In comparison, we reported PG622, (5, Fig. 1) as a moderately selective and high affinity D3R weak partial agonist.30 Its PP is the classic 2,3-diCl-phenylpiperazine. This compound is a structural analogue of the D3R antagonist, PG01037 (6, Fig. 1), with the only difference being the trans-olefin in 6 was replaced with a trans-cyclopropylmethyl group. D3R receptor binding affinities of 5 and 6 were virtually identical (~1 nM).30 However, 6 had ~2-fold lower affinity at D2R, resulting in a more D3R-selective compound.
Figure 1.

Selective pharmacological tools for the dopamine D3R.
In the present study we deconstructed compounds 1, 2 and 4 into “synthons” or functional fragments in order to: 1) investigate the binding affinities and functional activities of the PPs at both D2R and D3R; 2) test SP-derivatives of 1 for binding at both D2R and D3R receptors; 3) link these PPs and SPs with a conformationally-restricted trans-cyclopropylmethyl linking chain, as in compound 5, and determine the binding and functional properties of the resulting compounds at both D2R and D3R and finally, 4) synthesize analogues with one additional methylene group in the linking chain in an attempt to improve D3R selectivity, using either 2-indole or 7-azaindole as the SP. We hypothesized that the PPs would bind to both D2R and D3R and their affinities would improve with alkyl homologation. We expected the terminal aryl amide SPs to exhibit very low, if any, affinity for the D2R or D3R, but that linking these PPs and SPs with a trans-cyclopropylmethyl linking chain would result in compounds with high affinity and selectivity for D3R. Moreover, we predicted that adding one additional methylene group to the linking chain would further decrease binding affinity at the D2R resulting in a more D3R-selective ligand. Finally, we wanted to determine if the structural rigidity conferred by the trans-cyclopropyl group on these novel ligands engenders allosteric interactions at D3R, as has been reported for the trans-cyclohexyl linked parent compound 1 at the D2R and D3R.15,25–27
CHEMISTRY
Scheme 1 describes the synthesis of both PP and SP synthons. To synthesize the PPs, 3-aminobenzonitrile (7) was cyclized to N-aryl piperazine (8)31 using bis(2-chloroethyl)amine hydrochloride in diglyme, as a solvent, at 150 °C.32 Piperazine derivative 8 and the 6- or 7-CN-substituted tetrahydroisoquinolines 9a, 9b were N-alkylated to the corresponding 10, 11a and 11b, respectively, in the presence of K2CO3, under reflux conditions. To synthesize the SP of 1, indole-2-carboxylic acid (12a) was reacted with the corresponding amine, in the presence of CDI, as the coupling reagent, to afford the amides 13a, 13b,33 13c34 and 13d.
Scheme 1. Synthesis of synthons a.

aReagents and conditions: (a) bis(2-chloroethyl)amine.HCl, diethyleneglycol monoethylether, 150 °C, 6 h; (b) 1-bromobutane, K2CO3, acetone, reflux, overnight; (c) amine, CDI, THF, 0 °C to RT, overnight.
Scheme 2 outlines the synthetic strategy used for the synthesis of the trans-methyl-aryl carboxamide derivatives 14a–f. 2-(Aminomethyl)-trans-cyclopropyl)methanol (15), was prepared according to a procedure described previously,30,35 and was coupled with 12a or 4-quinoline carboxylic acid (12b) to afford the amides 16a and 16b, respectively, which were oxidized to the corresponding aldehydes 17a and 17b, using a Dess-Martin periodinane reagent in more than 80% yield.36 Compounds 17a and 17b were reductively aminated using NaBH(OAc)3 as the reducing agent, to give compounds 14a–f.
Scheme 2. Synthesis of analogues 14a–f a.

aReagents and conditions: (a) ArCOOH, CDI, THF, 0 °C to RT; overnight (b) Dess-Martin periodinane reagent, CH2Cl2, −78 °C to RT, 3 h ; (c) 2,3-dichlorophenyl piperazine or 3-cyanophenyl piperazine or 6-cyano-1,2,3,4-tetrahydroisoquinoline or 7-cyano-1,2,3,4-tetrahydroisoquinoline, NaBH(OAc)3, AcOH, DCE, RT, overnight.
The synthesis of target compounds 18a and 18b is outlined in Scheme 3. In these compounds, the trans-cyclohexyl linker of 1 and 3 was replaced with a trans-cyclopropylmethyl group, while keeping the same PP and SPs. In Scheme 3, ethyl 2-(hydroxymethyl)cyclopropane-1-carboxylate (19)37 was prepared according to a procedure described previously and was converted to cyclopropylcarbinyl bromide (20)38 using phosphorus tribromide under basic conditions.39 Treatment of the bromide 20 with potassium cyanide in DMSO afforded the nitrile 21,38 which was reduced to the amine 22, using LiAlH4. Compound 22 was coupled to either the indole (12a) or 7-azaindole-2-carboxylic acid (12c) in the presence of the coupling reagent CDI to afford the alcohol amide intermediates, 23a and 23b, respectively. These intermediates were oxidized (24a and 24b) and reductively aminated as described in Scheme 2 to afford the final compounds 18a and 18b.
Scheme 3. Synthesis of analogues 18a and 18b a.

aReagents and conditions: (a) PBr3, K2CO3, ether, −78 °C to RT, overnight; (b) KCN, DMSO, 60 °C, 3 h; (c) LAH, THF, reflux, 3 h; (d) 12a or 12c, CDI, DMF, 0 °C to RT, overnight; (e) Dess-Martin periodinane reagent, CH2Cl2, −78 °C to RT, 3 h ; (f) 9b, NaBH(OAc)3, AcOH, DCE, RT, overnight.
In Scheme 4, two additional analogs of 1 were synthesized in which the trans-cyclopropyl ring of 18a or 18b was moved one methylene group closer to the indole or 7-azaindole ring. By adding a methylene group between the PP and the trans-cyclopropyl ring, the same linker length as in 18a and 18b was retained. To synthesize the target compound 25a, tetrahydropyran (THP) protected ethyl-trans-cyclopropylmethanol 26 was prepared according to a procedure described previously40 and was oxidized to the aldehyde 27. Compound 27 was converted to intermediate 28 in the presence of O-benzylhydroxylamine HCl as a mixture of the cis and trans isomers of the resulting oxime (~1:1) in 59% yield.41 The benzyloxime 28 was reduced in the presence of LiAlH4 to the amine 29 and coupled with 12a to give the amide 30a. The tetrahydropyranyl group was removed under acidic conditions to give the alcohol 31a, which was oxidized to 32a, and reductively aminated to give the target compound 25a, as described in the previous scheme. The same procedure was used to synthesize the 7-azaindole derivative 25b from 29 and 12c, except that the THP group of 30b was removed using pyridinium p-toluenesulfonate (PPTS) to provide 31b.42
Scheme 4. Synthesis of analogues 25a and 25ba.

aReagents and conditions: (a) Dess-Martin periodinane reagent, CH2Cl2, −78 °C to RT, 3h; (b) BnONH2.HCl, mol. sieves, THF, RT, overnight; (c) LAH, THF, reflux, 3 h; (d) 12a or 12c, CDI, THF, 0 °C to RT (e) 10% HCl, THF, RT, overnight or PPTS, THF, 55 °C, overnight; (f) Dess-Martin periodinane reagent, THF, −78 °C to RT, 3h; (g) 9b, NaBH(OAc)3, DCE, AcOH, RT, overnight.
PHARMACOLOGICAL RESULTS AND DISCUSSION
Radioligand binding assays
All ligands were evaluated in radioligand binding assays using HEK-293 cells stably transfected with human D2RL and D3R, as described previously.43 The radioligand was the high-affinity D2-like receptor-selective antagonist [3H]N-methylspiperone (NMS). Ki values are displayed in Table 1 for the synthons and Table 2 for the full-length ligands. In addition, cLogP and polar surface area (PSA) values are provided in Table 2 as a relative measure of lipophilicity and predicted brain penetration, respectively.
Table 1.
In vitro pharmacological profile for synthons
| Compd | Structure | D2R | D3R | D2/D3d | D2R | D3R |
|---|---|---|---|---|---|---|
|
|
|
|||||
| Binding | β-Arrestin Recruitment | |||||
|
|
|
|||||
| Kia (nM) ± SEM | pKBb ± SEM (KB, nM) | |||||
| 8 |

|
4890 ±700 | 581 ± 70 | 8.4 | 5.68 ± 0.15 (2090) | 5.42 ± 0.06 (3800) |
| 9a |

|
53800 ±6800 | 5890 ±1700 | 9.1 | <5 (>10,000) | <5 ( >10,000) |
| 9b |

|
11400 ±1800 | 1520 ±36 | 7.5 | 5.37 ± 0.10 (4270) | 5.49 ± 0.22 (3240) |
| 10 |

|
493 ±28 | 34.7 ±0.67 | 14 | 7.12 ± 0.13 (75) | 7.50 ± 0.14 (32) |
| 11a |
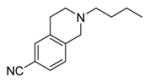
|
8300 ±1100 | 1090 ±20 | 7.7 | 5.78 ± 0.13 (1660) | 5.99 ± 0.09 (1020) |
| 11b |

|
832 ±190 | 149 ±3.0 | 5.6 | 7.00 ± 0.19 (100) | 6.70 ± 0.25 (200) |
| 13a |

|
>30 μM | >50 μM | 0.42 | NAc | NA |
| 13b |

|
>100 μM ±0 | >100 μM ±0 | 0 | NA | NA |
| 13c |

|
>100 μM ±0 | >100 μM ±0 | 0 | NA | NA |
| 13d |

|
13400± 1285 | >100 μM ±0 | 0 | NA | NA |
Estimate of the equilibrium dissociation constant ± standard error of the mean (SEM) determined by inhibition of [3H]N-methylspiperone binding in membranes harvested from HEK 293 cells stably expressing hD2R or hD3R as described in Experimental Methods.
Estimate of the negative logarithm of the equilibrium dissociation constant ± SEM determined in a β-arrestin assay using the DiscoverX PathHunter assay; by fitting IC50 data to equation 5 as described in the Methods.
NA indicates that “no activity” was observed at concentrations up to 100 μM. Values represent the mean obtained from 3 individual experiments.
Table 2.
In vitro pharmacological profile for extended length compounds

| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd | Structure | clogP | PSA | D2R | D3R | D2/D3 | D2R | D3R | |||
|
| |||||||||||
| Radioligand Binding | β-Arrestin Recruitment | ||||||||||
|
| |||||||||||
| Kia (nM) ± SEM | pKB ± SEM (KB, nM) | ||||||||||
| R1 | R2 | m | n | ||||||||
|
|
|||||||||||
| 1 | 4.78 | 68.16 | 485 ± 84 | 3.9 ± 1.6 | 124 | 5.60 ± 0.10c (2510) | 7.72 ± 0.14c (19) | ||||
| 2 | 4.82 | 68.49 | 5160±115 | 11.0±2.63 | 469 | 5.19 ± 0.19b (6490) | 7.55 ± 0.19d (28) | ||||
| 14a |

|

|
1 | 1 | 5.29 | 47.61 | 8.41±0.52 | 0.45± 0.06 | 19 | 6.70 ± 0.23b (200) | 7.13 ± 0.23d (74) |
| 14b |

|

|
1 | 1 | 5.34 | 47.94 | 19.9±1.6 | 1.51±0.18 | 13 | 7.78 ± 0.26b (17) | 7.29 ± 0.12d (51) |
| 14c |

|

|
1 | 1 | 3.75 | 71.73 | 2290±91 | 51.7±9.7 | 44 | 6.68 ± 0.16b (209) | 6.84 ± 0.10d (144) |
| 14d |

|

|
1 | 1 | 3.23 | 68.49 | 9500±3300 | 1040±290 | 9 | 5.76 ± 0.13b (1740) | 5.96 ± 0.19d (1100) |
| 14e |

|

|
1 | 1 | 3.23 | 68.49 | 1440±270 | 256±35 | 6 | 6.96 ± 0.18b (110) | 6.62 ± 0.12d (240) |
| 14f |

|

|
1 | 1 | 3.19 | 68.16 | 695 ±72 | 32.0±4.2 | 22 | 6.56 ± 0.19b (275) | 6.57 ± 0.10d (269) |
| 18a |

|

|
1 | 2 | 3.72 | 68.16 | 182±3.20 | 0.890±0.02 | 204 | 6.88 ± 0.29b (132) | 8.36 ± 0.06e (4) |
| 18b |

|

|
1 | 2 | 3.10 | 80.52 | 14.4±2.5 | 2.47±0.3 | 6 | 8.29 ± 0.13b (5) | 8.44 ± 0.11d (4) |
| 25a |

|

|
2 | 1 | 3.72 | 68.16 | 476±19.4 | 9.81±1.6 | 49 | 5.76 ± 0.08b (1740) | 7.56 ± 0.23e (28) |
| 25b |
|

|
2 | 1 | 3.10 | 80.52 | 190±34 | 23.4±4.4 | 8 | 7.19 ± 0.11b (65) | 7.49 ± 0.11d (32) |
Estimate of the equilibrium dissociation constant ± standard error of the mean (SEM) determined by inhibition of [3H]N-methylspiperone binding in membranes harvested from HEK 293 cells stably expressing hD2R or hD3R as described in the Methods. Estimate of the equilibrium constant for 1 at D2R and D3R determined through application of equation 1.
Estimate of the negative logarithm of the equilibrium dissociation constant ± SEM determined in a β arrestin assay using the DiscoverX PathHunter assay, by; fitting IC50 data to equation 5,
by fitting curve shift data to equation 3,
by fitting curve shift data to equation 5 as described in the methods, or
by fitting curve shift data to equation 4. Values represent the mean obtained from 2–3 individual experiments.
On the basis of SAR and previous modeling studies, we first synthesized the PP of compounds 1, 2 and 4, the SP of 1 and additional alkylated derivatives. The simplest PPs, 8, 9a and 9b (6/7-CN tetrahydroisoquinoline), were nominally D3R-selective (D2/D3 7.5–9.1) based on Ki values in the binding assays (Table 1). Importantly, as the n-butyl linking chain was introduced into these PPs, 10, 11a, and 11b, binding affinities for both D2R and D3R increased (for D3R, Ki range = 34–1090 nM; for D2R, Ki range = 493–8300 nM). Consistent with previous SAR studies, this suggests that hydrophobic interactions between the linker and the receptor contribute to the progressive enhancement of affinity at both D2R and D3R. Although introduction of the n-butyl group enhanced affinity at both receptors, only a modest (5 to 14-fold) D3R selectivity was observed. It is also noteworthy that these PPs have much lower affinities than the previously reported and privileged 2,3-dichlorophenylpiperazine PP, for which the N-n-butyl-substituted synthon had a Ki=1.9 nM.13 As shown previously with other SPs,13,15 13a-d showed no or very weak ability to displace [3H]N-methylspiperone (NMS) binding from either the D2R or D3R. Notably, however, in a separate study, at concentrations over 50 μM, 13b was able to partially inhibit (~20%) [3H]spiperone binding to the D2R, a finding that might be consistent with an allosteric interaction.27
As shown in Table 2, the linking chain between the PPs of compounds 1–4 and the SPs of 1 and 2 was replaced with the conformationally rigid trans-cyclopropylmethyl group of compound 5. We had previously shown that the trans-olefin of 6 could be replaced with a trans-cyclopropyl group without diminishing binding affinity at D3R.30 We were interested to see if the trans-cyclohexyl linker in 1 and 2 could be replaced with the trans-cyclopropylmethyl group, which can often be bioisosteric,44 and retain D3R affinity and selectivity profiles of 1 when the PP and SP were identical. For comparison, we also synthesized compounds 14a and 14b to evaluate the SPs of compounds 1 and 2 with the 2,3-dichlorophenylpiperazine PP. We found that all of the full-length trans-cyclopropyl analogues preferred D3R over D2R and most showed high affinity (nM) D3R binding (Table 2). Analogues 14a (Ki = 0.45 nM) and 14b (K = 1.51 nM) demonstrated the highest D3R affinities among the 14-series that have the same linker, consistent with the higher affinities for their PP compared to the others. Interestingly, compound 14d, with the PP and SP of 2, had the lowest D3R affinity (Ki = 1 μM) suggesting that the trans-cyclopropylmethyl linker is not as well tolerated as the parent compound’s trans-cyclohexyl linker and that the binding mode of these tetrahydroisoquinolines differs from that of the 4-phenylpiperazines. In comparison to its regiosiomer 14e, in which the only structural difference is the position of the CN on the tetrahydroisoquinoline PP, D3R binding affinity was improved by ~4-fold and a more dramatic improvement was seen when the 4-quinoline SP was replaced with the 2-indole in 14f (Ki = 32 nM). Although D2R affinity also increased by ~2-fold, D3R selectivity was still higher with the indole SP. This SAR corresponds with our previously reported 4-phenylpiperazines, wherein the 2-indole analogues are typically more D3R-selective than their quinoline derivatives.45
Generally, the 6/7-CN-substituted tetrahydroisoquinolines 14d–f and the CN-substituted-phenyl piperazine analogue 14c showed lower affinities than the privileged 2,3-diCl-phenylpiperazines and this was particularly significant with the 4-quinoline analogues 14d and 14e. A similar pattern of lower affinity was previously observed at the D2R for the tetrahydroisoquinoline containing compound 1 in comparison to analogues in which this moiety was swapped for 2,3-dichlorophenylpiperazine or 2-methoxyphenylpiperazine moieties.26 In addition, the incorporation of these two privileged D2R structures switched the pharmacological action from that of allosteric (compound 1) to behavior that was best fit by a competitive model.26 However, when an additional methylene group was added to the linking chain in 18a, high affinity and selectivity for the D3R receptor was regained with a Ki = 0.89 nM for D3R and selectivity vs. D2R of >200 (Table 2). The positional isomer, 25a, was synthesized to further compare the effects on D3R affinity and selectivity. Compared to 18a, this analogue showed an 11-fold decrease in binding affinity for D3R and a 2.6-fold decrease in D2R affinity, resulting in lower D3R selectivity (49-fold). When the indole ring of both 18a and 25a was replaced with 7-azaindole a decrease in the binding affinity was observed for D3R with a simultaneous increase for D2R, a change that caused both a significant increase in affinity and negative cooperativity with dopamine in a SAR study of compound 1 derivatives at the D2R.26
Of note, the cLogP value range of 3–5.3 and PSA range of 48–80 for all extended length compounds in Table 2 predict reasonable blood-brain barrier penetration, and thus support future in vivo investigation.
Functional profiling using a β-arrestin recruitment assay
Functional characterization of synthons
Functional profiling of the compounds at the D2R and D3R was achieved using a β-arrestin recruitment assay as previously employed.15,46–49 Each compound was tested dose-dependently in both “agonist mode”, which evaluated the compound’s ability to stimulate β-arrestin recruitment, and “antagonist mode” that evaluated the compound’s ability to inhibit β-arrestin recruitment stimulated by an EC80 concentration of dopamine. The potency of dopamine at the D2R and D3R is different, therefore we used a derivation of the Gaddum/Schild model of competitive antagonism (equation 5 in Experimental Methods) to estimate a value of functional affinity (KB) from these data that can be compared across the receptor subtypes to allow some insight into the functional selectivity of these compounds.
None of the compounds exhibited agonist activity up to 30 μM (data not shown). The simplest PPs, 8, 9a and 9b (6/7-CN tetrahydroisoquinoline), showed nonselective (in agreement with the aforementioned binding) functional antagonism at both D2R and D3R (Table 1). The functional affinity (in “antagonist mode”) of these compounds was low (KB > 2 μM) with 9a having the lowest functional affinity of the series. The addition of the n-butyl linking chain in 10, 11a, and 11b increased functional affinity (KB) for both D2R (75–1660 nM) and D3R (32–1020 nM), with 10 displaying the highest potency. However, in agreement with the binding results, an n-butyl linking chain addition had minimal effects on D3R vs. D2R selectivity (Table 1).
We next examined the abilities of the SPs, 13a–d, to influence dopamine receptor functions. Not surprisingly, given the above binding analysis, the compounds showed no functional inhibition of either the D2R or D3R at concentrations up to 100 μM (Table 1). These findings are in agreement with both the currently presented binding results and previously published data for this SP wherein 13b is reported as CS01-12.15 Notably, however, in a previous study, found that at concentrations over 50 μM, 13b acts as an allosteric antagonist of the D2R with modest negative cooperativity with dopamine.27 Together, these results suggest that the SPs have either no activity at, or very low affinity for, these dopamine receptors when not linked to a PP.
Functional characterization of extended compounds
Following characterization of the PP and SPs independently, we sought to examine, in functional assays, the extended length compounds containing both PP and SPs within the same chemical scaffold (Table 2). Compound 1 has previously been shown to act as a negative allosteric modulator at both the D2R and D3R).15,24,25 This allosteric action has been linked to a bitopic mode of interaction at the D2R.25 The above binding data show that the linkage of a SP to a PP enhances the affinity and selectivity of the extended-length compounds for the D3R and this increase in affinity may be consistent with a bitopic mode of action.22,23 We therefore extended our pharmacological characterization to determine if these novel compounds might display allosteric pharmacology at the D3R. We performed curve shift experiments in which the ability of increasing concentration of our novel compounds to modulate dopamine potency was measured. Increasing concentrations of 1 cause a limited rightward shift in D3R dopamine dose-response curves (Fig. 2A). Analysis of these data with an allosteric ternary complex model (equation 3 in Experimental Methods) allowed estimations of affinity (KB = 19 nM, pKB = 7.72 ± 0.14) and negative cooperativity with dopamine (α = 0.009, Logα = −2.05 ± 0.11) that equates to a maximal 100-fold decrease in dopamine potency (Table 3). Equivalent curve shift experiments using cells expressing the D2R revealed an allosteric mode of action for 1 but with both a lower affinity (KB = 2.5 μM, pKB = 5.60 ± 0.10) and weaker negative cooperativity with dopamine (α = 0.1, Logα = −0.77 ± 0.10). Thus 1 displays 130-fold selectivity towards the D3R (Table 2). In contrast, increasing concentrations of the D2R/D3R competitive antagonist sulpiride produces a limitless shift in the potency of dopamine for D3R (Fig. 2B) and D2R activation (not shown). These data could be fit to a Gaddum/Schild model of competitive antagonism with Schild slopes of unity (equation 5 in Experimental Methods) to derive affinities (KB) of 40 nM (pA2 = 7.40 ± 0.10) and 3 nM (pA2 = 8.55 ± 0.19) at the D3R and D2R respectively. Interestingly, 2, a structural congener of 1 (Fig. 1), exhibits the properties of a competitive antagonist in this curve-shift assay with 230-fold selectivity towards the D3R (Fig. 2C, Table 2). These results suggest that subtle structural features in extended-length D3R selective ligands can engender allosteric behavior.
Figure 2. Curve-shift assays contrasting the competitive vs. non-competitive behavior of known D3R antagonists.

Dopamine-mediated β-arrestin recruitment assays were conducted by stimulating the receptor with a concentration response curve of dopamine at the indicated concentrations either with or without various concentrations of test compound. Data were measured as RLU and are expressed as a percentage of the maximum dopamine response seen in the absence of any test compound. Data points represent the mean ± SEM of three independent experiments performed in triplicate. Analysis of data fit is shown in Table 3.
Table 3.
Affinity and cooperativity of allosteric compounds at D3R.
| Compd | pKB ± SEMa (KB, nM) | Logα ± SEMb (α) | Logβ ± SEMc (β) |
|---|---|---|---|
| 1 | 7.72 ± 0.14* (19) | -2.05 ± 0.11 (0.009) | = 0 (1) |
| 18a | 8.36 ± 0.06* (4) | -1.36 ± 0.08 (0.04) | -2.29 ± 0.10 (0.005) |
| 25a | 7.56 ± 0.23* (28) | -1.54 ± 0.19 (0.03) | -1.57 ± 0.34 (0.03) |
Estimate of the negative logarithm of the equilibrium dissociation constanta, negative cooperativity with dopamine affinityb and efficacyc determined in a β-arrestin assay using the DiscoverX PathHunter assay by fitting IC50 data to equation 3 (for 1) or equation 4 (for 18a and 25a) as described in the Methods. Values represent the mean obtained from at least 3 individual experiments.
Values also displayed in Table 2.
We next tested all of the trans-cyclopropylmethyl-linked analogues to determine their mechanism of antagonism at the D3R by performing curve-shift analyses (Figs. 3 and 4). These data were fit with a derivation of the operational model of allosterism (equation 3 in Experimental Methods) or a Gaddum/Schild model of competitive antagonism (equation 4 in Experimental Methods). For each compound, data were analyzed with both models and the best fit was determined by an F-test.
Figure 3. Curve-shift assays indicate the 14-series compounds behave in a competitive manner at the D3R.

Dopamine-mediated β-arrestin recruitment assays were conducted by stimulating the receptor with a concentration response curve of dopamine at the indicated concentrations either with or without various concentrations of test compound. Data were measured as RLU and are expressed as a percentage of the maximum dopamine response seen in the absence of any test compound. Data points represent the mean ± SEM of two to four independent experiments performed in triplicate. Analysis of data fit is shown in Table 3.
Figure 4. Curve-shift assays indicate 18a and 25a behave in a non-competitive manner with dopamine at the D3R.

Dopamine-mediated β-arrestin recruitment assays were conducted by stimulating the receptor with a concentration response curve of dopamine at the indicated concentrations either with or without various concentrations of test compound. Data were measured as RLU and are expressed as a percentage of the maximum dopamine response seen in the absence of any test compound. 18a and 25a were best fit with a complete operational model of allosterism and agonism, indicating these compounds behave in a non-competitive manner at the D3R (top row). The replacement of the indole ring in 18a and 25a with an azaindole ring as in 18b and 25b resulted in data that were best fit with a competitive Gaddum/Schild model (bottom row). Data points represent the mean ± SEM of three independent experiments performed in triplicate.
The 14-series as described above contains both PP and SPs. As was shown in Fig. 2 for compounds 2 and sulpiride, increasing concentrations of compounds 14a, 14b, 14c, 14d, 14e, and 14f caused a limitless rightward displacement of dopamine concentration response curves with no effect on the Emax. As such, these data, along with those for 2 and sulpiride, were best fit by a competitive Gaddum/Schild model to determine affinity (pA2) and Schild slope (equation 5 in Experimental Methods, Table 2). Consistent with a competitive mode of interaction, the Schild slopes determined for all of the above compounds were not significantly different from unity. These data indicate that this novel series of compounds (14a–f) function as competitive antagonists at the D3R. 14b was found to display the highest functional affinity at both D2R (KB = 17 nM) and D3R (KB = 51 nM), while 14d (KB = 1.1 and 1.7 μM for D3R and D2R, respectively) showed the lowest functional affinity of the 14-series. These findings are consistent with the observed affinities in the binding studies. The regio-isomer of 14d, compound 14e, showed improved functional affinity for the D2R (KB = 110 nM) and D3R (KB = 240 nM) with no appreciable selectivity difference; although the binding analysis revealed a modest (6-foldpreference for D3R. Similar results were obtained for compound 14f suggesting that all of the 14-series compounds were relatively nonselective for D3R vs. D2R.
As noted above, both 18a and 25a contain the same PP and SP, and same absolute cyclopropyl linker length, with the only difference being the length of the methylene chains on either side of the cyclopropyl group. Furthermore, whereas these compounds contain the same PP and SP as 1, they contain a structurally rigid and shorter trans-cyclopropyl linker instead of the trans-cyclohexyl group. Hence, we reasoned that adding an extra methylene group to either side of the trans-cyclopropyl ring system, as compared to the 14-series compounds, might improve D3R selectivity and might also reveal allosterism as observed with compound 1. Indeed, in contrast to the results observed with the compounds from the 14-series, increasing concentrations of 18a caused not only a decrease in dopamine potency but also a decrease in the Emax for dopamine suggesting non-surmountable or non-competitive antagonism (Fig. 4). Accordingly, these data were best fit by an allosteric operational model (equation 4 in Experimental Methods) to derive a value of affinity (KB = 4 nM), negative cooperativity with dopamine affinity (α = 0.04) and negative modulation of dopamine efficacy (β = 0.005, Table 3). Interestingly, compound 25a behaved in a manner similar to 1 causing a limited rightward shift in the dopamine dose-response curve suggesting allostery within its mechanism of antagonism (Fig. 4, Table 3). The more modest decrease in Emax of the dopamine concentration response curve produced by increasing concentrations of 25a was reflected by a value of negative cooperativity with dopamine efficacy that was 6-fold smaller (β = 0.03) than that of 18a whereas it’s negative cooperativity with dopamine affinity was similar (α = 0.03, Fig. 4, Table 3). Thus, compounds 18a and 25a were the only trans-cyclopropyl-linked analogues found to antagonize the D3R in an allosteric mechanism. It should be noted that the addition of a methylene group to the linking chain of 14f to yield compound 18a, caused a 67-fold increase in D3R functional affinity yielding one of the most D3R-selective compounds in this series (Table 2). The positional isomer, 25a, showed reduced D2R and D3R affinities, although D3R-selectivity was retained (Table 2).
Finally, when the indole ring of both 18a and 25a was replaced with 7-azaindole (yielding 18b and 25b respectively), the potency of D2R inhibition was increased (26-fold for both 18b and 25b) with either no effect (18b) or a decrease (25b) in D3R potency resulting in a loss of D3R selectivity (Table 2). In addition to a decrease in subtype selectivity, both 18b and 25b appeared to inhibit dopamine stimulation of the D2R and D3R in a manner similar to the competitive compounds (Fig. 4.)
CONCLUSION
In this study, we designed, synthesized and evaluated a series of novel compounds to extend the SAR for D3R affinity and selectivity over D2R. In addition, we investigated molecular determinants of competitive versus noncompetitive (or negative allosteric) functional antagonism at these receptor subtypes using the PPs and SPs of two D3R-selective antagonists 1 and 2 that are structurally quite similar, but display significant differences in their pharmacological profiles. The synthesis and evaluation of their individual PPs and SPs provided insight into their roles in binding affinity for D3R, however, none of these synthons demonstrated appreciable D3R-selectivity over D2R and the PPs were competitive antagonists in the β-arrestin recruitment assay. However, by linking these PPs and SPs via a trans-cyclopropyl-functionalized linker, D3R selectivities emerged likely due to the bivalent nature of these compounds. Further, extending the linker length by an additional methylene group either before or after the trans-cyclopropylmethyl function engendered allostery within compounds 18a and 25a suggesting that they could be interacting with the receptor in a bitopic fashion. These allosteric effects appear to be enabled by a bivalent compound of sufficient length with the SP correctly oriented in a secondary binding pocket, which is dependent on the nature and conformation of the linker. Such a mode of action may be consistent with the bitopic mechanism proposed for the allosteric action of 1 at the D2R. Although the PP clearly plays a role in D3R (and D2R) affinity, it is the combination of PP, SP and linker between them that determines functional activity. Taken together, the binding and functional data reported herein support the notion that the SPs and linkers within the extended-length compounds can modulate the selectivity of their antagonist activities at D2R and D3R. Furthermore, these data suggest that the specific orientation of the SP within the secondary binding pocket can confer non-competitive or allosteric modes of interaction, but that relatively subtle changes to structure and orientation of the SP can cause a switch between apparently allosteric and competitive antagonism.
EXPERIMENTAL METHODS
General
Reaction conditions and yields were not optimized. Anhydrous solvents were purchased from Aldrich and were used without further purification except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Sigma-Aldrich Co. LLC, Combi-Blocks, TCI America, Acros Organics, Maybridge, and Alfa Aesar. All amine final products were converted into the oxalate salt. Spectroscopic data and yields refer to the free base form of compounds. Teledyne ISCO CombiFlash Rf or glass flash column chromatography was performed using silica gel (EMD Chemicals, Inc.; 230–400 mesh, 60 Å). 1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer at 400 MHz and 100 MHz, respectively. Chemical shifts are reported in parts-per-million (ppm) and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26, CD3OD, 3.31 or DMSO-d6, 2.50) and 13C spectra (CDCl3, 77.2, CD3OD, 49.0 or DMSO-d6, 39.5). Gas chromatography-mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 6890N GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μm film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100 °C) was held for 3 min and then increased to 295 °C at 15 °C/min over 13 min, and finally maintained at 295 °C for 10 min. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and the results agree within ±0.4% of calculated values. cLogP and polar surface area (PSA)values were calculated using ChemDraw Professional Ultra 15.0. Melting point determination was conducted using a Thomas-Hoover melting point apparatus and are uncorrected. On the basis of NMR and combustion data, all final compounds are ≥95% pure.
3-(Piperazin-1-yl)benzonitrile (8)31
The reaction mixture of 3-aminobenzonitrile (7, 0.500 g, 4.23 mmol) and bis(2-chloroethyl)amine hydrochloride (0.831 g, 4.65 mmol) in diethylene glycol monomethyl ether (2.0 mL) was heated at 150 °C for 6 h. The reaction mixture was allowed to come to RT, MeOH (4 mL) was added, followed by dilution with ether (250 mL). The salt was filtered, suspended in CHCl3 (20 mL) and neutralized with 2N NaOH to pH 8–9. The organic layer was collected, concentrated and purified by column chromatography using 3% MeOH/CHCl3 as eluent to provide 0.294 g (37%) of 8.
3-(4-Butylpiperazin-1-yl)benzonitrile (10)
1-Bromobutane (0.263 g, 1.92 mmol) and K2CO3 (0.398g, 2.88 mmol) were added to a solution of 8 (0.180 g, 0.96 mmol) in acetone (15 mL) and stirred at reflux overnight. The reaction mixture was filtered, concentrated and purified by flash chromatography using 4% acetone/CHCl3 as eluent to provide 0.150 g (65%) of 10 as an oil. 1H NMR (400 MHz, CDCl3) δ 7.32–7.28 (m, 1 H), 7.11–7.05 (m, 3H), 3.22 (t, J = 5.2 Hz, 4H), 2.59 (t, J = 5.0 Hz, 4H), 2.39 (t, J = 7.6 Hz, 2H), 1.55–1.47 (m, 2H), 1.33 (sextet, J = 8.0 Hz, 2H), 0.94 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 151.3, 129.8, 122.2, 119.7, 119.3, 118.2, 112.9, 58.3, 52.9, 48.2, 29.0, 20.7, 14.0. The oxalate salt was precipitated from acetone. Anal. (C15H21N3•C2H2O4•0.5H2O) C, H, N.
2-Butyl-1,2,3,4-tetrahydroisoquinoline-6-carbonitrile (11a)
The same procedure was used as described for compound 10 using 9a. The crude product was purified using 15% EtOAc/hexanes as eluent to provide the product as an oil, in 71% yield. 1H NMR (400 MHz, CDCl3) δ 7.38 (s, 1H), 7.37–7.36 (m, 1H), 7.11 (dd, J = 8.0, 0.8 Hz, 1H), 3.65 (s, 2H), 2.91 (t, J = 6.0 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.53–2.50 (m, 2H), 1.59–1.53 (m, 2H), 1.37 (sextet, J = 7.6 Hz, 2H), 0.94 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 140.7, 136.0, 132.4, 129.1, 127.5, 119.1, 109.9, 58.1, 56.1, 50.3, 29.2, 28.9, 20.7, 14.1. GC-MS (EI) m/z 214.1 (M+). The oxalate salt was precipitated from acetone. Anal. (C14H18N2•C2H2O4•0.5H2O) C, H, N. Mp 140–141 °C.
2-Butyl-1,2,3,4-tetrahydroisoquinoline-7-carbonitrile (11b)
The same procedure was used as described for compound 10 using 9b. The crude product was purified using 12% EtOAc/hexanes as eluent to provide the product as an oil, in 58% yield. 1H NMR (400 MHz, CDCl3) δ 7.37 (dd, J = 7.6, 1.6 Hz, 1H), 7.30 (s, 1H), 7.18 (d, J = 8.4 Hz, 1H),3.60 (s, 2H), 2.94 (t, J = 5.6 Hz, 2H), 2.72 (t, J = 5.6 Hz, 2H), 2.53–2.49 (m, 2H), 1.60–1.53 (m, 2H), 1.37 (sextet, J = 7.2 Hz, 2H), 0.94 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 140.4, 136.5, 130.3, 129.5, 129.4, 119.1, 109.3, 58.0, 55.6, 50.2, 29.4, 29.2. GC-MS (EI) m/z 214.2 (M+). The oxalate salt was precipitated from acetone. Anal. (C14H18N2•C2H2O4•0.25H2O) C, H, N. Mp 167–168 °C.
N-(Pentan-2-yl)-1H-indole-2-carboxamide (13a)
CDI (0.251 g, 1.55 mmol) was added to a solution of indole-2-carboxylic acid (12a, 0.250 g, 1.55 mmol) in THF (10 mL) under an Argon atmosphere. The reaction mixture was stirred for 4 h and cooled to 0–5°C. 2-Aminopentane (0.135 g, 1.55 mmol) was added drop wise in THF (10 mL). The reaction mixture was allowed to come to RT and then stirred overnight. The solvent was evaporated, the crude product was diluted with water (20 mL) and extracted with CHCl3 (2x20 mL). The organic layer was dried, concentrated and purified by flash chromatography using 10% EtOAc/hexanes as eluent to provide 0.321 g (90%) of the desired product. 1H NMR (400 MHz, CDCl3) δ 9.75 (bs, 1H), 7.63 (dd, J = 8.0, 0.8 Hz, 1H), 7.45 (dd, J = 7.6, 0.8 Hz, 1H), 7.28 (dd, J = 7.2, 1.2 Hz, 1H), 7.15–7.11 (m, 1H), 6.82 (m, 1H), 5.96 (bs, 1H), 4.31–4.24 (m, 1H), 1.70–1.38 (m, 4H), 1.38–1.24 (m, 3H), 0.96 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 161.2, 136.5, 131.2, 127.8, 124.4, 121.9, 120.7, 112.2, 101.5, 45.6, 39.4, 21.3, 19.5, 14.1. Anal. (C14H18N2O) C, H, N.
N-(Heptan-4-yl)-1H-indole-2-carboxamide (13d)
The same procedure was used as described for compound 13a using N-heptan-4-ylamine. The crude product was purified using 15% EtOAc/hexanes as eluent to provide the product in 66% yield. 1H NMR (400 MHz, CDCl3) δ 9.75 (bs, 1H), 7.64 (dd, J = 8.0, 0.8 Hz, 1H), 7.45 (dd, J = 8.0, 0.8 Hz, 1H), 7.29–7.25 (m, 1H), 7.15–7.11 (m, 1H), 6.83 (m, 1H), 5.88 (d, J = 9.2 Hz, 1H), 4.24–4.21 (m, 1H), 1.64–1.39 (m, 8H), 0.95 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 161.4, 136.4, 131.2, 127.8, 124.5, 121.9, 120.7, 112.1, 101.4, 49.4, 37.9, 19.4, 14.2. Anal. (C16H22N2O) C, H, N.
N-(2-((4-(2,3-Dichlorophenyl)piperazin-1-yl)methyl)-trans-cyclopropylmethyl)-1H-indole-2-carboxamide (14a)
1-(2,3-Dichlorophenyl)piperazine (0.100 g, 0.43 mmol) and 17a (0.105 g, 0.43 mmol) were mixed in 1,2-dichloroethane (10 mL) and treated with sodium triacetoxyborohydride (0.138 g, 0.64 mmol) and HOAc (0.2 mL) under an atmosphere of Argon. The reaction mixture was stirred at RT for 6 h. The reaction mixture was quenched with 2 N NaOH (10 mL) and extracted with EtOAc (3 x 20 mL). The organic layer was dried over Na2SO4, concentrated and purified by flash chromatography using 2% MeOH/CHCl3 as eluent to provide 0.150 g (76%) of yellow solid product. Mp 215–216° C. 1H NMR (400 MHz, CDCl3) δ 11.53 (s, 1H), 8.57 (t, J = 6.0 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.24–7.09 (m, 4H), 6.96 (dt, J = 7.2, 1.2 Hz, 1H), 6.76 (d, J = 6.8 Hz, 1H), 3.32–3.26 (m, 1H), 3.16–3.09 (m, 1H), 2.79 (bs, 4H), 2.53 (bs, 4H), 2.39 (dd, J = 12.4, 5.2 Hz, 1H), 2.07–2.02 (m, 1H), 0.91–0.83 (m, 2H), 0.49 (quintet, J = 4.8 Hz, 1H), 0.31 (quintet, J = 4.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 161.0, 151.1, 136.3, 132.5, 131.9, 128.3, 127.1, 125.8, 124.1, 123.1, 121.4, 119.6, 119.2, 112.2, 102.2, 61.6, 52.5, 50.8, 42.1, 18.2, 14.7, 9.3. Anal. (C24H26Cl2N4O•0.5H2O) C, H, N.
N-(2-((4-(2,3-Dichlorophenyl)piperazin-1-yl)methyl)-trans-cyclopropylmethyl)quinoline-4-carboxamide (14b)
The same procedure was used as described for compound 14a from 2,3-dichloro, and 17b. The crude product was purified by flash chromatography using 2% MeOH/CHCl3 as eluent to provide the oily product in 96% yield. 1H NMR (400 MHz, CDCl3) δ 8.72 (d, J = 4.4 Hz, 1H), 8.14 (dd, J = 8.4, 0.8 Hz, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.69 (dt, J = 7.2, 1.6 Hz, 1H), 7.53 (dt, J = 7.2, 1.2 Hz, 1H), 7.30 (d, J = 7.2 Hz, 1H), 7.15–7.11 (m, 2H), 7.07 (bt, J = 5.2 Hz, 1H), 6.84 (dd, J = 7.6, 2.8 Hz, 1H), 3.46–3.41 (m, 1H), 3.35–3.30 (m, 1H), 2.98 (bs, 4H), 2.67 (bs, 4H), 2.42–2.37 (m, 1H), 2.32–2.27 (m, 1H), 0.97–0.92 (m, 2H), 0.58 (quintet, J = 5.2 Hz, 1H), 0.45 (quintet, J = 5.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 167.2, 151.1, 149.6, 148.4, 142.1, 133.9, 129.9, 129.6, 127.6, 127.5, 127.3, 125.2, 124.6, 124.4, 118.6, 118.4, 62.3, 53.2, 51.0, 43.9, 17.6, 15.2, 10.2. The oxalate salt was precipitated from MeOH/ether. Anal. (C25H26Cl2N4O•1.5 C2H2O4.C3H6O•1.5H2O) C, H, N. Mp 55–56 °C.
N-(2-((4-(3-Cyanophenyl)piperazin-1-yl)methyl)-trans-cyclopropylmethyl)quinoline-4-carboxamide (14c)
The same procedure was used as described for compound 14a from 8 and 17b. The crude product was purified by flash chromatography using 50% acetone/ CHCl3 as eluent. 1H NMR (400 MHz, CDCl3) δ 8.81 (d, J = 4.0 Hz, 1H), 8.20 (dd, J = 8.4, 1.0 Hz, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.76–7.72 (m, 1H), 7.60–7.56 (m, 1H), 7.36 (d, J = 4.4 Hz, 1H), 7.31–7.27 (m, 1H), 7.07–7.00 (m, 3H), 6.74 (bt, J = 5.6 Hz, 1H), 3.51 (quintet, J = 7.0 Hz, 1H), 3.98–3.33 (m, 1H), 3.14–3.11 (m, 4H), 2.63 (t, J = 5.0 Hz, 4H), 2.42–2.38 (m, 1H), 2.33–2.28 (m, 1H), 1.01–0.94 (m, 2H), 0.64–0.59 (m, 1H), 0.50–0.46 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 167.3, 151.2, 149.8, 148.6, 142.1, 130.1, 130.0, 129.9, 127.7, 125.3, 124.4, 122.5, 119.8, 119.4, 118.4, 118.3, 112.9, 62.2, 52.8, 48.1, 44.0, 17.7, 15.3, 10.3. The oxalate salt was precipitated from acetone. Anal. (C26H27N5O•C2H2O4•2H2O) C, H, N. Mp 125–126 °C.
N-(2-((6-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)methyl)-trans-cyclopropylmethyl)quinoline-4-carboxamide (14d)
The same procedure was used as described for compound 14a from 9a and 17b. The crude product was purified by flash chromatography using 40% acetone/ CHCl3 as eluent to provide yellowish solid in 68% yield. 1H NMR (400 MHz, CDCl3) δ 8.86–8.85 (m, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 7.77–7.31 (m, 1H), 7.59–7.55 (m, 1H), 7.40–7.31 (m, 3H), 7.01 (d, J = 8.0 Hz, 1H), 6.43 (d, J = 4.8 Hz, 1H), 3.76–3.67 (m, 2H), 3.59–3.52 (m, 1H), 3.45–3.39 (m, 1H), 2.88–2.76 (m, 4H), 2.57–2.53 (m, 1H), 2.46–2.41 (m, 1H), 1.09–1.03 (m, 2H), 0.69–0.65 (m, 1H), 0.58–0.53 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 167.4, 149.9, 148.8, 142.1, 140.2, 135.7, 132.6, 130.1, 130.0, 129.2, 127.8, 127.5, 125.3, 124.5, 119.1, 118.5, 110.1, 62.0, 55.9, 50.4, 44.1, 28.8, 17.9, 15.7, 10.4. The oxalate salt was precipitated from EtOH/ ether. Anal. (C25H24N4O•1.25C2H2O4•2H2O) C, H, N. Mp 65–66 °C.
N-(2-((7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)methyl)-trans-cyclopropylmethyl)quinoline-4-carboxamide (14e)
The same procedure was used as described for compound 14a from 9b and 17b. The crude product was purified by flash chromatography using 40% acetone/ CHCl3 as eluent. 1H NMR (400 MHz, CDCl3) δ 8.69 (d, J = 4.4 Hz, 1H), 8.15 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 8.0 Hz, 1H), 7.51 (t, J = 8.0 Hz, 1H), 7.34–7.11 (m, 4H), 7.04–7.00 (m, 1H), 3.65–3.59 (m, 2H), 3.51–3.32 (m, 2H), 2.86–2.75 (m, 4H), 2.50–2.39 (m, 2H), 1.0 (t, J = 6.0 Hz, 2H), 0.65–0.61 (m, 1H), 0.52–0.48 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 167.3, 149.6, 148.4, 142.1, 140.1, 136.1, 130.3, 130.0, 129.9, 129.7, 129.5, 129.5, 127.5, 125.2, 124.3, 119.0, 118.4, 109.2, 61.8, 55.2, 50.1, 43.8, 29.3, 17.7, 15.5, 10.3. The oxalate salt was precipitated from acetone. Anal. (C25H24N4O•1.25 C2H2O4•2H2O) C, H, N. Mp 82–83 °C.
N-(2-((7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)methyl)-trans-cyclopropylmethyl)-1H-indole-2-carboxamide (14f)
The same procedure was used as described for compound 14a from 9b and 17a. The crude product was purified by flash chromatography using 25% acetone/ CHCl3 as eluent to provide the oily product in 86% yield. 1H NMR (400 MHz, CDCl3) δ 10.15 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.43 (dd, J = 9.6, 1.0 Hz, 1H), 7.33 (dd, J = 8.0, 1.6 Hz, 1H), 7.28–7.17 (m, 2H), 7.14–7.09 (m, 2H), 6.85 (s, 1H), 6.73 (bt, J = 5.6 Hz, 1H), 3.67 (s, 2H), 3.52 (quintet, J = 6.0 Hz, 1H), 3.37 (quintet, J = 6.0 Hz, 1H), 2.91–2.75 (m, 4H), 2.62–2.56 (m, 1H), 2.36–2.31 (m, 1H), 1.05–0.99 (m, 2H), 0.64–0.60 (m, 1H), 0.52–0.50 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 161.8, 140.1, 136.5, 136.0, 130.7, 130.3, 129.6, 129.5, 127.6, 124.4, 121.9, 120.6, 119.0, 112.1, 109.3, 102.2, 61.9, 55.3, 50.1, 43.5, 29.2, 18.2, 15.4, 9.9. The oxalate salt was precipitated from acetone. Anal. (C24H24N4O• C2H2O4) C, H, N. Mp 132–133 °C.
N-(2-Hydroxymethyl-trans-cyclopropylmethyl)-1H-indole-2-carboxamide (16a)
CDI (0.184 g, 1.13 mmol) was added to a solution of 12a (0.183 g, 1.13 mmol) in THF (10 mL) under an Argon atmosphere. The reaction mixture was stirred for 4 h and cooled to 0–5°C. trans-(2-(Aminomethyl)cyclopropyl)methanol (15)30,35 (0.115 g, 1.13 mmol) was added drop wise after dissolving in THF (10 mL). The reaction mixture was allowed to come to RT and then stirred overnight. The solvent was evaporated, the crude product was diluted with water (20 mL) and extracted with CHCl3 (2x20 mL). The organic layer was dried, concentrated and purified by flash chromatography using 25% acetone/ CHCl3 as eluent to provide 0.161 g (63%) of the desired product. 1H NMR (400 MHz, CD3OD) δ 8.46 (s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.22 (t, J = 8.0 Hz, 1H), 7.10–7.05 (m, 2H), 3.52–3.48 (m, 1H), 3.39–3.26 (m, 3H), 1.05 (m, 2H), 0.60–0.56 (m, 1H), 0.55–0.49 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 164.1, 138.3, 132.3, 129.0, 125.0, 122.7, 121.1, 113.0, 104.4, 66.4, 4.19, 20.8, 17.8, 9.1. GC-MS (EI) m/z 244.1 (M+).
N-(2-Hydroxymethyl-trans-cyclopropylmethyl)quinoline-4-carboxamide (16b)
The same procedure was used as described for 16a using 4-quinoline carboxylic acid (12b). The crude product was purified by flash chromatography using 30% acetone/ CHCl3 as eluent to provide the desired product in 75% yield. 1H NMR (400 MHz, (CDCl3) δ 8.57 (bs, 1H), 7.95 (dd, J = 13.0, 8.4 Hz, 2H), 7.69 (t, J = 5.2 Hz, 1H), 7.64–7.60 (m, 1H), 7.46–7.42 (m, 1H), 7.11 (d, J = 4.4 Hz, 1H),3.53–3.48 (m, 1H), 4.25 (bs, 1H), 3.93–3.33 (m, 1H), 2.98–2.90 (m, 2H), 0.86–0.77 (m, 2H), 0.38–0.30 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 167.4, 149.4, 148.0, 142.1, 130.0, 129.2, 127.5, 125.2, 124.2, 118.4, 65.5, 43.8, 20.1, 16.6, 8.4.
N-(2-Formyl-trans-cyclopropylmethyl)-1H-indole-2-carboxamide (17a)
Dess-Martin periodinane (0.338 g, 0.79 mmol) was added in fractions to a solution of 16a (0.130 g, 0.53 mmol) in THF (15 mL) and stirred for 30 min at −78° C, under Argon. The reaction mixture was allowed to come to RT and stirred for another 3h. The reaction mixture was concentrated and purified by flash chromatography using 10% acetone/ CHCl3 as eluent to provide 0.110 g (86%) of the desired product. 1H NMR (400 MHz, (CD3)2SO) δ 11.57 (s, 1H), 8.90 (d, J = 2.4 Hz, 1H), 8.63 (t, J = 6.0 Hz, 1H), 7.61 (dd, J = 8.0, 0.8 Hz, 1H), 7.42 (dd, J = 8.0, 0.8 Hz, 1H), 7.19–7.15 (m, 1H), 7.11–7.10 (m, 1H), 7.05–7.01 (m, 1H), 3.43–3.37 (m, 1H), 3.29–3.22 (m, 1H), 1.85–1.78 (m, 2H), 1.31–1.26 (m, 1H), 1.15–1.10 (m, 1H); 13C NMR (100 MHz, (CD3)2SO) δ 201.2, 161.2, 136.4, 131.5, 127.0, 123.3, 121.4, 119.7, 112.3, 102.5, 40.5, 28.6, 21.1, 12.1. GC-MS (EI) m/z 242.1 (M+).
N-(2-Formyl-trans-cyclopropylmethyl)quinoline-4-carboxamide (17b)
The same procedure was used as described for 17a from 16b. The crude product was purified by flash chromatography using 25% acetone/ CHCl3 as eluent to provide the desired product in 87% yield. 1H NMR (400 MHz, (CD3)2SO) δ 8.96 (d, J = 4.4 Hz, 1H), 8.92 (d, J = 5.6 Hz, 1H), 8.06 (dd, J = 14, 8.0 Hz, 2H), 7.84–7.78 (m, 1H), 7.68–7.62 (m, 1H), 7.51 (d, J = 4.4 Hz, 1H), 3.50–3.44 (m, 1H), 3.30–3.25 (m, 2H), 1.88–1.80 (m, 2H), 1.33–1.29 (m, 1H), 1.17–1.12 (m, 1H); 13C NMR (100 MHz, (CD3)2SO) δ 201.1, 166.6, 150.2, 147.9, 142.0, 129.8, 129.4, 127.3, 125.3, 124.1, 119.0, 40.8, 28.5, 20.9, 12.2. GC-MS (EI) m/z 254.1 (M+).
N-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)methyl-trans-cyclopropylethyl)-1H-indole-2-carboxamide (18a)
The same procedure was used as described for compound 14a from 9b and 24a. The crude product was purified by flash chromatography using 25% acetone/ CHCl3 as eluent to provide the oily product in 62% yield. 1H NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 7.52 (dd, J = 8.0, 0.8 Hz, 1H), 7.43 (dd, J = 8.4, 0.4 Hz, 1H), 7.33 (dd, J = 8.0, 2.0 Hz, 1H), 7.28–7.24 (m, 2H),7.13–7.09 (m, 2H), 6.75 (dd, J = 2.4, 0.8 Hz, 1H), 6.71 (bt, J = 5.6 Hz, 1H), 3.74–3.61 (m, 3H), 3.58–3.50 (m, 1H), 2.93 (t, J = 6.0 Hz, 2H), 2.85–2.82 (m, 1H), 2.77–2.71 (m, 1H), 2.63–2.49 (m, 1H), 2.41–2.17 (m, 1H), 1.72–1.55 (m, 2H), 0.88–0.80 (m, 1H), 0.73–0.65 (m, 1H), 0.50–0.44 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 161.8, 140.1, 136.4, 136.0, 130.8, 130.3, 129.6, 129.5, 127.5, 124.3, 121.8, 120.6, 119.1, 112.0, 109.4, 101.9, 62.5, 55.6, 50.1, 39.6, 33.3, 29.2, 15.8, 15.7, 11.4. The oxalate salt was precipitated from acetone. Anal. (C25H26N4O•1.5C2H2O4) C, H, N. Mp 185–186 °C.
N-(2-(2-((7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)methyl-trans-cyclopropyl)ethyl)-1H-pyrrolo[2,3-b]pyridine-2-carboxamide (18b)
The same procedure was used as described for compound 14a from 9b and 24b. The crude product was purified by flash chromatography using 50% acetone/ CHCl3 as eluent to provide the product in 41% yield. 1H NMR of oxalate salt (400 MHz, CD3OD) δ 8.32 (dd, J = 4.4, 1.6 Hz, 1H), 8.07 (dd, J = 8.0, 1.2 Hz, 1H), 7.63–7.56 (m, 3H), 7.44–7.39 (m, 2H), 7.16 (dd, J = 8.0, 4.8 Hz, 1H), 7.07 (s, 1H), 4.55–4.46 (m, 2H), 4.31 (s, 1H), 3.65–3.58 (m, 3H), 3.53–3.42 (m, 2H), 3.23–3.10 (m, 2H), 1.83–1.75 (m, 1H), 1.55–1.46 (m, 1H), 1.26–1.11 (m, 1H), 0.96 (m, 1H), 0.71–0.67 (m, 2H); 13C NMR of free base (100 MHz, CD3OD) δ 161.5, 147.7, 145.2, 140.4, 139.9, 135.5, 131.7, 130.9, 120.4, 118.8, 116.7, 109.2, 102.2, 62.3, 55.1, 49.9, 39.6, 33.3, 28.6, 15.6, 15.5, 11.1. The oxalate salt was precipitated from acetone; mp 178–179 °C. Anal. (C24H25N5O• 2C2H2O4) C, H, N.
Ethyl trans-2-(bromomethyl)cyclopropane-1-carboxylate (20)38
A solution of PBr3 (1.178 g, 4.35 mmol) in ether (20 mL) was added dropwise to a mixture of ethyl trans-2-(hydroxymethyl)cyclopropane-1-carboxylate (19)37 (1.697 g, 11.77 mmol) and K2CO3 (3.248 g, 23.54 mmol) in ether (50 mL) at −78 °C under an atmosphere of Argon. The reaction mixture was allowed to come to RT, stirred overnight and filtered. The ether layer was washed with saturated NaHCO3 solution, dried, concentrated and purified by flash chromatography using 1% EtOAc/ hexanes as eluent to provide 0.708 g (29%) of 20.
trans-(2-(Aminoethyl)cyclopropyl)methanol (22)
A suspension of LiAlH4 (0.239 g, 6.31 mmol) in THF (30 mL) was stirred at RT for 30 min, cooled to 0 °C, and a solution of 2138 (0.483 g, 3.15 mmol) in THF (10 mL) was added slowly. The reaction mixture was brought to RT and then heated and stirred at reflux for 3 h. Upon cooling to 0 °C, the reaction mixture was carefully quenched by dropwise addition of water (1.0 mL). The reaction mixture was stirred further for 15 min, filtered and the residue was further washed with EtOAc (10 mL). The combined organic layer was concentrated, diluted with water and extracted in CHCl3 to give the 22 as an oil, 0.363 g (89%), which was used without further purification in the next step.
N-(2-Hydroxymethyl-trans-cyclopropylethyl)-1H-indole-2-carboxamide (23a)
The same procedure was used as described for 16a from 12a and 22. The crude product was purified by prep. TLC using 60% EtOAc/ hexanes as eluent to provide the volatile product in 57%. 1H NMR (400 MHz, CDCl3) 9.58 δ (s, 1H), 7.61 (dd, J = 8.4, 0.8 Hz, 1H), 7.40 (dd, J = 8.4, 0.8 Hz, 1H), 7.27–7.23 (m, 1H), 7.12–7.08 (m, 1H), 7.06 (bt, J = 5.2 Hz, 1H), 6.97 (dd, J = 2.0, 0.8 Hz, 1H),3.98–3.88 (m, 2H), 3.38–3.32 (m, 1H), 3.19 (bs, 1H), 1.99–1.93 (m, 2H), 1.12–1.03 (m, 1H), 0.97–0.90 (m, 1H), 0.71–0.65 (m, 1H), 0.43–0.34 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1, 136.3, 130.9, 127.6, 124.4, 121.9, 120.5, 112.0, 102.5, 67.1, 39.8, 34.0, 20.9, 16.0, 9.0. GC-MS (EI) m/z 258.1 (M+).
N-(2-(2-(Hydroxymethyl-trans-cyclopropyl)ethyl)-1H-pyrrolo[2,3-b]pyridine-2-carboxamide (23b)
The same procedure was used as described for the 16a from 12c and 22. The crude product was used in the next step without further purification.
N-(2-Formyl-trans-cyclopropylethyl)-1H-indole-2-carboxamide (24a)
The same procedure was used as described for the 17a using 23a. The crude product was purified by flash chromatography using 15% acetone/ CHCl3 as eluent to provide the desired product in 54% yield. 1H NMR (400 MHz, CDCl3) δ 9.54 (s, 1H), 9.08 (d, J = 5.2 Hz, 1H), 7.64 (dd, J = 8.4, 0.8 Hz, 1H), 7.44 (dd, J = 8.4, 0.8 Hz, 1H), 7.29 (dd, J = 7.0, 0.8 Hz, 1H), 7.15–7.11 (m, 1H),6.85 (m, 1H), 6.46 (bt, J = 5.2 Hz, 1H), 3.59 (dd, J = 12.8, 7.0 Hz, 2H), 1.76–1.68 (m, 3H), 1.58–1.53 (m, 1H), 1.37–1.32 (m, 1H), 1.02–0.98 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 200.7, 161.8, 136.3, 130.5, 127.6, 124.6, 121.9, 120.7, 112.0, 102.0, 39.3, 32.8, 29.9, 20.2, 14.6.
N-(2-(2-Formyl-trans-cyclopropyl)ethyl)-1H-pyrrolo[2,3-b]pyridine-2-carboxamide (24b)
The same procedure was used as described for the 17a using 23b. The crude product was purified by flash chromatography using 15% acetone/ CHCl3 as eluent. 1H NMR (400 MHz, CD3OD + CDCl3, 1:1) δ 8.93 (d, J = 5.2 Hz, 1H), 8.34 (d, J = 4.0 Hz, 1H), 7.90 (dd, J = 8.0, 1.6 Hz, 1H), 7.07–6.97 (m, 3H), 6.93 (s, 1H), 3.47–3.43 (m, 2H), 1.66–1.55 (m, 3H), 1.50–1.46 (m, 1H), 1.26–1.22 (m, 1H), 0.94–0.89 (m, 1H); 13C NMR (100 MHz, CD3OD + CDCl3, 1:1) δ 201.4, 161.6, 147.8, 145.3, 131.5, 130.9, 120.3, 116.7, 102.1, 39.1, 32.4, 29.9, 20.4, 14.7.
N-((2-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)-trans-cyclopropyl)methyl)-1H-indole-2-carboxamide (25a)
The same procedure was used as described for compound 14a from 9b and 32a. The crude product was purified by flash chromatography using 22% acetone/ CHCl3 as eluent to provide the desired product in 69% yield. 1H NMR (400 MHz, CD3OD + CDCl3, 2:1) δ 7.88 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.48–7.40 (m, 3H), 7.32 (t, J = 8.0 Hz, 1H), 7.26 (s, 1H), 3.77 (s, 2H), 3.68 (dd, J = 13.6, 6.4 Hz, 1H), 3.33 (dd, J = 13.6, 8.0 Hz, 1H), 3.15 (t, J = 6.0 Hz, 2H), 3.02–2.88 (m, 2H), 2.86–2.77 (m, 2H), 1.89 (sextet, J = 7.2 Hz, 1H), 1.61 (sextet, J = 7.2 Hz, 1H), 1.16–1.14 (m, 1H), 0.98–0.97 (m, 1H), 0.77–0.72 (m, 1H), 0.65–0.62 (m, 1H); 13C NMR (100 MHz, CD3OD + CDCl3, 2:1) δ 162.5, 140.0, 136.7, 135.5, 130.9, 130.3, 129.7, 129.5, 127.5, 124.0, 121.6, 120.1, 118.7, 111.9, 109.1, 103.4, 57.7, 55.2, 49.8, 43.4, 30.5, 28.5, 18.4, 15.6, 10.4. Anal. (C25H26N4O•C2H2O4•0.25H2O) C, H, N. Mp 182–183°C.
N-((2-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)-trans-cyclopropyl)methyl)-1H-pyrrolo[2,3-b]pyridine-2-carboxamide (25b)
The same procedure was used as described for compound 14a from 9b and 32b. The crude product was purified by flash chromatography using 50% acetone/ CHCl3 as eluent to provide the desired product in 44% yield. 1H NMR of oxalate salt (400 MHz, CD3OD) δ 8.33 (d, J = 3.6 Hz, 1H), 8.08 (d, J = 7.2 Hz, 1H), 7.62 (m, 2H), 7.53 (m, 1H), 7.42 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.17 (dd, J = 8.0, 5.2 Hz, 1H), 7.06 (s, 1H), 4.44–4.40 (m, 4H), 3.57–3.45 (m, 5H), 3.41–3.16 (m, 1H), 1.90 (m, 1H), 1.70 (m, 1H), 1.01 (s, 1H), 0.84 (s, 1H), 0.62 (m, 1H), 0.52 (m, 1H); 13C NMR of free base (100 MHz, CDCl3) δ 161.4, 148.4, 145.7, 140.7, 140.3, 136.1, 131.9, 130.5, 129.4, 120.1, 119.1, 116.7, 109.2, 100.3, 57.9, 55.8, 49.7, 44.1, 30.9, 29.3, 18.2, 15.9, 10.7. The oxalate salt was precipitated from acetone; mp 189–190 °C. Anal. (C24H25N5O•2.5C2H2O4•1H2O) C, H, N.
2-(2-((Tetrahydro-2H-pyran-2-yl)oxy)ethyl)-trans-cyclopropane-1-carbaldehyde (27)
The same procedure was used as described for compound 17a from 2640. The crude aldehyde was purified by flash chromatography using 15% EtOAc/hexanes as eluent to provide 0.159 g (80%) of the desired product.1H NMR (400 MHz, CDCl3) δ 8.93 (m, 1H), 4.50 (s, 1H), 3.75–3.70 (m, 2H), 3.42–3.36 (m, 2H), 1.74–1.64 (m, 1H), 1.62–1.43 (m, 9H), 1.25–1.22 (m, 1H), 0.92–0.89 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 200.8, 98.7, 66.4, 62.1, 32.7, 30.6, 30.0, 25.3, 19.8, 19.4, 14.3. GC-MS (EI) m/z 197.1 (M+).
2-(2-((Tetrahydro-2H-pyran-2-yl)oxy)ethyl)-trans-cyclopropane-1-carbaldehyde O-benzyl oxime (28)
A mixture of 27 (1.210 g, 6.10 mmol), O-benzylhydroxylamine hydrochloride (1.948 g, 12.21 mmol), and molecular sieves 4A (1.000 g) in THF (20 mL) was stirred at RT overnight under Argon. The reaction mixture was filtered through a Florisil bed, and the filtrate was concentrated and purified by flash chromatography using 10% EtOAc/ hexanes as eluent to provide 1.09 g (59%) of an oily material - a mixture of trans and cis isomers (~1:1). 1H NMR (400 MHz, CDCl3) δ 7.40–7.28 (m, 5H), 6.98 (d, J = 8.4 Hz, 0.5H) trans, 6.00 (dd, J = 8.8, 1.2 Hz, 0.5H) cis, 5.10 (s, 1H), 5.02 (s, 1H), 4.60–4.57 (m, 1H), 3.87–3.78 (m, 2H), 3.51–3.42 (m, 2H), 2.10–2.07 (m, 0.5H), 1.84–1.78 (m, 1H), 1.73–1.49 (m, 7H), 1.44–1.40 (m, 0.5H), 1.14–1.06 (m, 1H), 0.83–0.78 (m, 1.5H), 0.74–0.70 (m, 0.5H); 13C NMR (100 MHz, CDCl3) δ 154.6, 154.2, 138.2, 137.6, 128.4, 128.2, 127.9, 127.8, 127.7, 98.8, 98.7, 75.7, 75.6, 66.8, 62.3, 62.2, 33.4, 30.7, 25.5, 19.5, 18.1, 17.8, 17.3, 15.1, 15.0, 13.2, 13.1, 12.5, 12.4. ESI 304.1909 (M+H)+, 326.1725 (M+Na)+.
(2-(2-((Tetrahydro-2H-pyran-2-yl)oxy)ethyl)-trans-cyclopropyl)methanamine (29)
A suspension of LiAlH4 (0.067 g, 1.78 mmol) in THF (10 mL) was stirred at RT for 30 min, cooled to 0 °C, and a solution of 28 (0.180 g, 0.59 mmol) in THF (10 mL) was added slowly. The reaction mixture was brought to RT and then heated and stirred at reflux for 3 h. It was cooled to 0 °C and carefully quenched by dropwise addition of water (1.0 mL). The reaction mixture was stirred further for 15 min, filtered and the residue was further washed with EtOAc (10 mL). The combined organic layer was concentrated, diluted with water and extracted in CHCl3 to give the title compound as an oil 0.093 g (79%), which was used without further purification in the next step. 1H NMR (400 MHz, CDCl3) δ 4.54–4.52 (m, 1H), 3.85–3.73 (m, 2H), 3.48–3.36 (m, 2H), 2–80–2.56 (bs, 2H), 2.46–2.34 (m, 1H), 1.81–1.72 (m, 1H), 1.69–1.62 (m, 2H), 1.55–1.46 (m, 6H), 0.61–0.52 (m, 2H), 0.34–0.24 (m, 2H); GC-MS (EI) m/z 198.2 (M+).
N-((2-(2-((Tetrahydro-2H-pyran-2-yl)oxy)ethyl)-trans-cyclopropyl)methyl)-1H-indole-2-carboxamide (30a)
The same procedure was used as described for the 16a from 12a and 29. The crude product was purified by flash chromatography using 20% EtOAc/ hexanes as eluent to provide 0.359 g (41%) of the product as an oil. 1H NMR (400 MHz, CDCl3) δ 10.73 (s, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 7.6 Hz, 1H), 7.07–6.96 (m, 3H), 4.53 (d, J = 18.8 Hz, 1H), 3.86–3.74 (m, 2H), 3.53–3.43 (m, 3H), 3.23–3.18 (m, 1H), 1.74–1.32 (m, 8H), 0.88–0.87 (m, 1H), 0.75 (m, 1H), 0.46–0.44 (m, 1H), 0.35–0.33 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 162.0, 136.8, 131.8, 127.6, 124.0, 121.7, 120.2, 112.3, 102.5, 99.2, 68.6, 62.6, 44.1, 33.6, 30.8, 27.7, 25.4, 22.0, 20.0, 19.8, 17.9, 15.9, 10.5. GC-MS (EI) m/z 342.2 (M+).
N-((2-(2-((Tetrahydro-2H-pyran-2-yl)oxy)ethyl)-trans-cyclopropyl)methyl)-1H-pyrrolo[2,3-b]pyridine-2-carboxamide (30b)
The same procedure was used as described for the 16a from 12c and 29. The crude product was purified by flash chromatography using 15% acetone/ CHCl3 as eluent to provide 0.348 g (40%) of the product as oil. 1H NMR (400 MHz, CDCl3) δ 10.63 (bs, 1H), 8.51 (d, J = 3.2 Hz, 1H), 7.97 (dd, J = 8.4, 1.6 Hz, 1H), 7.12 (dd, J = 8.4, 5.6 Hz, 1H), 6.94–6.88 (m, 1H), 6.60–6.57 (m, 1H), 4.62–4.56 (m, 1H), 3.95–3.75 (m, 2H), 3.68–3.58 (m, 2H), 3.53–3.50 (m, 1H), 3.16–3.06 (m, 1H), 1.90–1.24 (m, 8H), 0.90–0.83 (m, 1H), 0.80–0.74 (m, 1H), 0.51–0.49 (m, 1H), 0.46–0.40 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 160.9, 148.1, 146.0, 130.3, 120.2, 116.9, 100.6, 99.5, 67.8, 63.1, 44.3, 33.5, 30.9, 25.3, 19.9, 17.7, 15.3, 10.4.
N-((2-(2-Hydroxyethyl)-trans-cyclopropyl)methyl)-1H-indole-2-carboxamide (31a)
Aqueous HCl solution (10%, 6 mL) was added to a solution of 30a in THF (4 mL) and stirred overnight. The crude product was extracted with EtOAc, concentrated and purified by flash chromatography using 40% EtOAc/hexanes as eluent to provide 0.178 g (67%) of a white solid product. 1H NMR (400 MHz, CD3OD + CDCl3, 1:1) δ 10.41 (s, 1H), 7.99 (s, 1H), 7.59 (d, J = 7.6 Hz, 1H), 7.4 (d, J = 9.2 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 7.07–7.03 (m, 2H), 3.63 (t, J = 6.4 Hz, 2H),3.51 (dd, J = 13.6, 6.0 Hz, 1H), 2.99 (dd, J = 14.0, 8.0 Hz, 1H), 1.67–1.59 (m, 1H), 1.29–1.21 (m, 1H), 0.86–0.79 (m, 1H), 0.75–0.67 (m, 1H), 0.47–0.43 (m, 1H), 0.37–0.32 (m, 1H); 13C NMR (100 MHz, CD3OD + CDCl3, 1:1) δ 162.6, 136.6, 130.8, 127.5, 124.1, 121.7, 120.1, 111.9, 103.6, 62.0, 43.9, 36.0, 17.6, 14.8, 9.8. GC-MS (EI) m/z 258.1 (M+).
N-((2-(2-Hydroxyethyl)-trans-cyclopropyl)methyl)-1H-pyrrolo[2,3-b]pyridine-2-carboxamide (31b)
A solution of 30b (0.268 g, 0.78 mmol) and pyridinium p-toluenesulfonate (PPTS) (0.019 g, 0.08 mmol) in EtOH (5 mL) was stirred at 55 °C overnight. The solvent was evaporated and the crude product was purified by column chromatography, using 45% acetone/ CHCl3 as eluent to provide 0.142 g (70%) of white solid product. 1H NMR (400 MHz, CD3OD + CDCl3, 1:1) δ 8.29 (d, J = 4.0 Hz, 1H), 8.13 (t, J = 5.2 Hz, 1H), 7.96 (dd, J = 7.6, 1.6 Hz, 1H), 7.08–7.01 (m, 2H), 3.64 (t, J = 6.4 Hz, 2H), 3.56–3.50 (m, 1H), 3.01–2.95 (m, 1H), 1.67–1.59 (m, 1H), 1.28–1.19 (m, 1H), 0.85–0.80 (m, 1H), 0.73–0.68 (m, 1H), 0.47–0.43 (m, 1H), 0.37–0.32 (m, 1H); 13C NMR (100 MHz, CD3OD + CDCl3, 1:1) δ 161.6, 147.7, 145.1, 131.7, 131.0, 120.5, 116.6, 102.6, 62.1, 44.1, 35.9, 17.7, 14.8, 9.9.
N-((2-(2-Oxoethyl)-trans-cyclopropyl)methyl)-1H-indole-2-carboxamide (32a)
The same procedure was used as described for 17a from 31a. The crude product was purified by flash chromatography using 5% acetone/ CHCl3 as eluent to provide the desired product in 79% yield. 1H NMR (400 MHz, acetone-d6) δ 10.77 (bs, 1H), 9.79 (s, 1H), 7.96 (s, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.24–7.16 (m, 2H), 7.07 (t, J = 7.6 Hz, 1H),3.62–3.56 (m, 1H), 3.12–3.06 (m, 1H), 2.62 (dd, J = 18.0, 5.6 Hz, 1H), 2.30 (d, J = 18.0, 8.0 Hz, 1H), 0.99–0.93 (m, 2H), 0.64–0.60 (m, 1H), 0.49–0.44 (m, 1H); 13C NMR (100 MHz, acetone-d6) δ 203.3, 162.0, 137.6, 132.9, 128.8, 124.4, 122.4, 120.8, 113.0, 102.6, 48.0, 44.1, 11.5, 10.5.
N-((2-(2-Oxoethyl)-trans-cyclopropyl)methyl)-1H-pyrrolo[2,3-b]pyridine-2-carboxamide (32b)
The same procedure was used as described for 17a from 31b. The crude product was purified by flash chromatography using 30% acetone/ CHCl3 as eluent to provide the desired product in 69% yield. 1H NMR (400 MHz, (CD3)2SO) δ 12.01 (s, 1H), 8.48–8.46 (m, 1H), 8.28 (d, J = 4.4 Hz, 1H), 8.04–7.91 (m, 2H), 7.09–7.06 (m, 2H), 3.17–3.09 (m, 2H), 1.52–1.27 (m, 2H), 0.85–0.77 (m, 1H), 0.72–0.64 (m, 1H), 0.39–0.37 (m, 1H), 0.28–0.26 (m, 1H); 13C NMR (100 MHz, (CD3)2SO) δ 203.6, 168.1, 148.7, 145.6, 134.9, 126.8, 120.8, 116.8, 100.5, 47.1, 43.1, 18.3, 13.7, 10.4.
Radioligand binding assays
Binding at dopamine D2-like receptors was determined using previously described methods.43 Membranes were prepared from HEK293 cells expressing human D2R, D3R or D4R, grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37°C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using pre-mixed Earle’s Balanced Salt Solution (EBSS) with 5 mM EDTA (Life Technologies) and centrifuged at 3000 rpm for 10 min at 21 °C. The supernatant was removed and the pellet was resuspended in 10 mL hypotonic lysis buffer (5 mM MgCl2 · 6 H2O, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 20,000 rpm for 30 min at 4 °C. The pellet was then resuspended in fresh EBSS buffer made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration and membranes were diluted to 500 μg/mL and stored in a −80 °C freezer for later use.
Radioligand competition binding experiments were conducted using thawed membranes. Test compounds were freshly dissolved in 30% DMSO and 70% H2O to a stock concentration of 100 μM. To assist the solubilization of free-base compounds, 10 μl of glacial acetic acid was added along with the DMSO. Each test compound was then diluted into 13 half-log serial dilutions using 30% DMSO vehicle; final test concentrations ranged from 10 μM to 10 pM. Previously frozen membranes were diluted in fresh EBSS to a 100 μg/mL (for hD2R or hD3R) or 200 μg/mL (hD4R) stock for binding. Radioligand competition experiments were conducted in glass tubes containing 300 μl fresh EBSS buffer with 0.2 mM sodium metabisulfite, 50 μl of diluted test compound, 100 μl of membranes (10 μg total protein for hD2R or hD3R, 20 μg total protein for hD4R), and 50 μl of [3H]N-methylspiperone (0.4 nM final concentration; Perkin Elmer). Nonspecific binding was determined using 10 μM (+)-butaclamol (Sigma-Aldrich, St. Louis, MO) and total binding was determined with 30% DMSO vehicle. All compound dilutions were tested in triplicate and the reaction incubated for one hour at room temperature. The reaction was terminated by filtration through Whatman GF/B filters, presoaked for one hour in 0.5% polyethylenimine, using a Brandel R48 filtering manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 mL of ice cold EBSS buffer and transferred to scintillation vials. 3 mL CytoScint liquid scintillation cocktail (MP Biomedicals, Solon, OH) was added and vials were counted using a Perkin Elmer Tri-Carb 2910 TR liquid scintillation counter (Waltham, MA). IC50 values for each compound were determined from dose-response curves and Ki values were calculated using the Cheng-Prusoff equation; these analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA).50 Reported Ki values were determined from least three independent experiments.
β-Arrestin Recruitment Assays
Measurement of β-arrestin recruitment was conducted with minor modifications as previously published by our laboratory using the DiscoverX PathHunter technology (DiscoverX Inc., Fremont, CA).46,49,51,52 The amino-terminal fragment of β-galactosidase is fused to the DAR while the carboxyl-terminal fragment of the enzyme is fused to β-arrestin-2. Upon agonist activation, β-arrestin is recruited to the receptor resulting in the formation of an active β-galactosidase, which can be detected using a luminescent substrate. Briefly, CHO-K1 cells expressing either the D2R long isoform or the D3R were maintained in Ham’s F12 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 800 μg /ml G418, and 300 μg/ml hygromycin; at 37°C, 5% CO2, and 90% humidity. Cells were seeded in Cell Plating Media 2 (DiscoverX) at a density of 2,625 cells/well in 384-well black, clear-bottom plates. Test compounds were diluted in PBS in the presence of 0.2 μM sodium metabisulfite for agonist mode assays and in an identical assay buffer with an EC80 concentration of dopamine (1 μM for D2R assays, and 30 nM for D3R assays) for antagonist mode assays. For curve shift experiments, concentration response curves of dopamine were generated with or without the indicated concentration of test compound. Cells were preincubated with the compounds at 37°C for 90 min followed by the addition of DiscoverX reagent followed by a 30 min incubation at room temperature. Luminescence was determined on a Hamamatsu FDSS μCell plate reader. Data were collected as RLUs and subsequently normalized to a percentage of the control luminescence seen with a maximum concentration of dopamine for agonist mode assays and as a percentage of the EC80 dopamine concentration for antagonist mode assays.
Data analysis of functional experiments
Data analysis
GraphPad Prism 6.0b (San Diego, CA) was used for all statistical analysis, nonlinear regression, and simulations.
Radioligand binding data
Competition-binding curves between [3H]raclopride and 1 could be fit to the allosteric ternary complex model using the following equation:53
| (1) |
Where Y is percentage (vehicle control) binding; [A] and [B] are the concentrations of [3H]raclopride and 1, respectively; KA and KB are the equilibrium dissociation constants of [3H]raclopride and 1, respectively; α is the cooperativity between 1 and [3H]raclopride. Values of α >1 denote positive cooperativity; values <1 (but >0) denote negative cooperativity, and values = 1 denote neutral cooperativity.
Analysis of functional data
All concentration response (C/R) data were fitted to the following modified four-parameter Hill equation to derive potency estimates:54
| (2) |
Where E is the effect of the system, nH is the Hillslope, and EC50 is the concentration of agonist [A] that gives the midpoint response between basal and maximal effect of dopamine or other agonists (Emax), which are the lower and upper asymptotes of the response, respectively.
Functional data describing the interaction between 1 and dopamine were globally analyzed according to the allosteric ternary complex model.
| (3) |
Where Em is the maximum possible cellular response, [A] and [B] are the concentrations of orthosteric and allosteric ligands, respectively, and KB is the equilibrium dissociation constant of the allosteric ligand, αβ is a composite cooperativity parameter between the orthosteric and allosteric ligand that includes effects upon orthosteric ligand affinity and efficacy and nH is the Hill slope of the orthosteric agonist concentration-response curve. Values of α and/or β greater than 1 denote allosteric potentiation, whereas values less than 1 (but greater than 0) denote allosteric inhibition.
Functional data describing the interaction between 25a & 18a and dopamine at the D3R were analyzed using a complete operational model of allosterism and agonism according to equation 355:
| (4) |
Where Em is the maximum possible cellular response, [A] and [B] are the concentrations of orthosteric and allosteric ligands, respectively, KA and KB are the equilibrium dissociation constant of the orthosteric and allosteric ligands, respectively, and τB (constrained to 0.001) are operational measures of orthosteric and allosteric ligand efficacy (which incorporate both signal efficiency and receptor density), respectively, α is the binding cooperativity parameter between the orthosteric and allosteric ligand, and β denotes the magnitude of the allosteric effect of the modulator on the efficacy of the orthosteric agonist. KA was constrained to a value determined in a radioligand binding assay (0.3 μM).
A logistic equation of competitive agonist-antagonist interaction was globally fitted to data from functional experiments measuring the interaction between dopamine and test compounds which caused an unlimited rightward displacement of a dopamine dose-response curve and no decrease in Emax within the range of concentrations used (2, 14f, 14a, 14c, 14e, 14d, 18b, 25b):54
| (5) |
Where s represents the Schild slope for the test compound and KB is the equilibrium dissociation constant of the test compound, nH is the Hillslope, and EC50 is the concentration of agonist [A] that gives the midpoint response between basal and maximal effect of dopamine or other agonists (Emax), which are the lower and upper asymptotes of the response, respectively. For all cases where a competitive mode of interaction was preferred then the Schild slope did not differ significantly from unity as determined by an extra-sum-of-squares F-test, P > 0.05. For each of the novel compounds the two equations (models) were then compared for their fit using an extra-sum-of-squares F test, whereby the simpler model was selected unless the P value was less than 0.05.
Supplementary Material
Acknowledgments
Support for this research was provided by the Intramural Research Programs of the National Institute on Drug Abuse (VK, TMK, AB, ME and AHN) and the National Institute of Neurological Disorders and Stroke (AEM, CDS RBF, and DRS) JRL is a RD Wright Biomedical Career Development Fellow (NHMRC).
ABBREVIATIONS USED
- DA
dopamine
- SAR
structure activity relationship
- TM
transmembrane
- D2R
D2 dopamine receptor
- D3R
D3 dopamine receptor
- D4R
D4 dopamine receptor
- OBS
orthosteric binding site
- PPTS
pyridinium p-toluenesulfonate
- SBP
secondary binding pocket
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
ASSOCIATED CONTENT
Supporting information. Elemental analysis results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 2.Lieberman JA, Bymaster FP, Meltzer HY, Deutch AY, Duncan GE, Marx CE, Aprille JR, Dwyer DS, Li XM, Mahadik SP, Duman RS, Porter JH, Modica-Napolitano JS, Newton SS, Csernansky JG. Antipsychotic drugs: comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol Rev. 2008;60:358–403. doi: 10.1124/pr.107.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez Y, Pérez-Mañá C, Torrens M, Farré M. Antipsychotic drugs in cocaine dependence: a systematic review and meta-analysis. J Subst Abuse Treat. 2013;45:1–10. doi: 10.1016/j.jsat.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 4.Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 5.Searle G, Beaver JD, Comley RA, Bani M, Tziortzi A, Slifstein M, Mugnaini M, Griffante C, Wilson AA, Merlo-Pich E, Houle S, Gunn R, Rabiner EA, Laruelle M. Imaging dopamine D3 receptors in the human brain with positron emission tomography, [11C]PHNO, and a selective D3 receptor antagonist. Biol Psychiatry. 2010;68:392–399. doi: 10.1016/j.biopsych.2010.04.038. [DOI] [PubMed] [Google Scholar]
- 6.Cho DI, Zheng M, Kim KM. Current perspectives on the selective regulation of dopamine D2 and D3 receptors. Arch Pharm Sci Res. 2010;33:1521–1538. doi: 10.1007/s12272-010-1005-8. [DOI] [PubMed] [Google Scholar]
- 7.Gurevich EV, Joyce JN. Distribution of dopamine D3 receptor expressing neurons in the human forebrain: comparison with D2 receptor expressing neurons. Neuropsychopharmacology. 1999;20:60–80. doi: 10.1016/S0893-133X(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 8.Heidbreder CA, Newman AH. Current perspectives on selective dopamine D3 receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann N Y Acad Sci. 2010;1187:4–34. doi: 10.1111/j.1749-6632.2009.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Micheli F. Recent advances in the development of dopamine D3 receptor antagonists: a medicinal chemistry perspective. ChemMedChem. 2011;6:1152–1162. doi: 10.1002/cmdc.201000538. [DOI] [PubMed] [Google Scholar]
- 10.Newman AH, Blaylock BL, Nader MA, Bergman J, Sibley DR, Skolnick P. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol. 2012;84:882–890. doi: 10.1016/j.bcp.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Micheli F, Heidbreder C. Dopamine D3 receptor antagonists: a patent review (2007 – 2012) Expert Opin Ther Pat. 2013;23:363–381. doi: 10.1517/13543776.2013.757593. [DOI] [PubMed] [Google Scholar]
- 12.Ehrlich K, Gotz A, Bollinger S, Tschammer N, Bettinetti L, Harterich S, Hubner H, Lanig H, Gmeiner P. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J Med Chem. 2009;52:4923–4935. doi: 10.1021/jm900690y. [DOI] [PubMed] [Google Scholar]
- 13.Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, Javitch JA, Shi L. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J Med Chem. 2012;55:6689–6899. doi: 10.1021/jm300482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keck TM, Burzynski C, Shi L, Newman AH. Beyond small molecule SAR – using the dopamine D3 receptor crystal structure to guide drug design. Adv Pharmacol. 2014;69:267–300. doi: 10.1016/B978-0-12-420118-7.00007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furman CA, Roof RA, Moritz AE, Miller BN, Doyle TB, Free B, Banala AK, Paul NM, Kumar V, Sibley CD, Newman AH, Sibley DR. Investigation of the binding and functional properties of extended length D3 dopamine receptor-selective antagonists. Eur Neuropsychopharmacol. 2015;25:1448–1461. doi: 10.1016/j.euroneuro.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar V, Banala AK, Garcia EG, Cao J, Keck TM, Bonifazi A, Deschamps JR, Newman AH. Chiral resolution and serendipitous fluorination reaction for the selective dopamine D3 receptor antagonist BAK2–66. ACS Med Chem Lett. 2014;5:647–651. doi: 10.1021/ml500006v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keck TM, John WS, Czoty PW, Nader MA, Newman AH. Identifying medication targets for psychostimulant addiction: unraveling the dopamine D3 receptor hypothesis. J Med Chem. 2015;58:5361–5380. doi: 10.1021/jm501512b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gentry PR, Sexton PM, Christopoulos A. novel allosteric modulators of G protein-coupled receptors. J Biol Chem. 2015;290:19478–19488. doi: 10.1074/jbc.R115.662759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shonberg J, Kling RC, Gmeiner P, Löber S. GPCR crystal structures: medicinal chemistry in the pocket. Bioorg Med Chem. 2015;23:3880–3906. doi: 10.1016/j.bmc.2014.12.034. [DOI] [PubMed] [Google Scholar]
- 21.Shonberg J, Lopez L, Scammells PJ, Christopoulos A, Capuano B, Lane JR. Biased agonism at G protein-coupled receptors: the promise and the challenges-a medicinal chemistry perspective. Med Res Rev. 2014;34:1286–1330. doi: 10.1002/med.21318. [DOI] [PubMed] [Google Scholar]
- 22.Lane JR, Sexton PM, Christopoulos A. Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol Sci. 2013;34:59–66. doi: 10.1016/j.tips.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Mohr K, Tränkle C, Kostenis E, Barocelli E, De Amici M, Holzgrabe U. Rational design of dualsteric GPCR ligands: quests and promise. Br J Pharmacol. 2010;159:997–1008. doi: 10.1111/j.1476-5381.2009.00601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silvano E, Millan MJ, Mannoury lCC, Han Y, Duan L, Griffin SA, Luedtke RR, Aloisi G, Rossi M, Zazzeroni F, Javitch JA, Maggio R. The tetrahydroisoquinoline derivative non-competitive69,652 is an allosteric antagonist at dopamine D3 and D2 receptors. Mol Pharmacol. 2010;78:925–934. doi: 10.1124/mol.110.065755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lane JR, Donthamsetti P, Shonberg J, Draper-Joyce CJ, Dentry S, Michino M, Shi L, López L, Scammells PJ, Capuano B, Sexton PM, Javitch JA, Christopoulos A. A new mechanism of allostery in a G protein-coupled receptor dimer. Nat Chem Biol. 2014;10:745–752. doi: 10.1038/nchembio.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shonberg J, Draper-Joyce C, Mistry SN, Christopoulos A, Scammells PJ, Lane JR, Capuano B. Structure-activity study of N-((trans)-4-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)cyclohexyl)-1H-indole-2-carboxamide (SB269652), a bitopic ligand that acts as a negative allosteric modulator of the dopamine D2 receptor. J Med Chem. 2015;58:5287–5307. doi: 10.1021/acs.jmedchem.5b00581. [DOI] [PubMed] [Google Scholar]
- 27.Mistry SN, Shonberg J, Draper-Joyce CJ, Herenbrink CK, Michino M, Shi L, Christopoulos A, Capuano B, Scammells PJ, Lane JR. Discovery of a novel class of negative allosteric modulator of the dopamine D2 receptor through fragmentation of a bitopic ligand. J Med Chem. 2015;58:6819–6843. doi: 10.1021/acs.jmedchem.5b00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stemp G, Ashmeade T, Branch CL, Hadley MS, Hunter AJ, Johnson CN, Nash DJ, Thewlis KM, Vong AKK, Austin NE, Jeffrey P, Avenell KY, Boyfield I, Hagan JJ, Middlemiss DN, Reavill C, Riley GJ, Routledge C, Wood M. Design and synthesis of trans-N-[4-[2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011): a potent and selective dopamine D3 receptor antagonist with high oral bioavailability and CNS penetration in the rat. J Med Chem. 2000;43:1878–1885. doi: 10.1021/jm000090i. [DOI] [PubMed] [Google Scholar]
- 29. [August 25, 2016]; https://clinicaltrials.gov/ct2/show/NCT01785147.
- 30.Grundt P, Prevatt KM, Cao J, Taylor J, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR, Newman AH. Heterocyclic analogues of N-(4-(4-(2, 3-dichlorophenyl)piperazin-1-yl)-butyl)-aryl-carboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J Med Chem. 2007;50:4135–4146. doi: 10.1021/jm0704200. [DOI] [PubMed] [Google Scholar]
- 31.Belfield AJ, Brown GR, Foubister AJ, Ratcliffe PD. Synthesis of meta-substituted aniline derivatives by nucleophilic substitution. Tetrahedron. 1999;55:13285–13300. [Google Scholar]
- 32.Liu KG, Robichaud AJ. A general and convenient synthesis of N-aryl piperazines. Tetrahedron Lett. 2005;46:7921–7922. [Google Scholar]
- 33.Alvarez GL, Correa ME, Cruces VMA, Elorriaga RC, Fernandez-Alvarez E, Nieto LO. Preparation of acetylenic and allenic derivatives of 2-indolylmethylamines and preliminary results of their study as selective inhibitors for the monoamine oxidases A and B. An Quim Ser C. 1986;82:129–134. [Google Scholar]
- 34.Baytas SN, Inceler N, Orhan Didem D, Ozkan S. Synthesis characterization and antioxidant and antimicrobial properties of new ester and amide derivatives of indole-2-carboxylic acid. FABAD J Pharm Sci. 2013;36:53–61. [Google Scholar]
- 35.Ashton WT, Meurer LC, Cantone CL, Field AK, Hannah J, Karkas JD, Liou R, Patel GF, Perry HC, Wagner AF, Walton E, Tolman RL. Synthesis and antiherpetic activity of (+/-)-9-[[(Z)-2-(hydroxymethyl)cyclopropyl]methyl]guanine and related compounds. J Med Chem. 1988;31:2304–2315. doi: 10.1021/jm00120a010. [DOI] [PubMed] [Google Scholar]
- 36.Dess DB, Martin JC. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J Org Chem. 1983;48:4155–4156. [Google Scholar]
- 37.Baldwin JE, Chang GEC. Stereochemistry of the thermal isomerizations of (2S,3R)-2-methoxymethyl-2,3-dideuterio-1-(dideuteriomethylene)cyclopropane. Tetrahedron. 1982;38:825–835. [Google Scholar]
- 38.Kennewell PD, Matharu SS, Taylor JB, Westwood R. Synthesis of y-aminobutyric acid analogues of restricted conformation. Part 1. The 2-aminocycloalkylacetic acids. J Chem Soc, Perkin Trans I. 1982:2553–2562. [Google Scholar]
- 39.Li RT, Sato Y. D-homoannulation of 5alpha-pregnane-3beta,20beta-diol 3-acetate with phosphorus pentabromide. J Org Chem. 1968;33:3632–3635. doi: 10.1021/jo01273a061. [DOI] [PubMed] [Google Scholar]
- 40.Csuk R, Kern A. Synthesis of spacered cyclopropyl nucleoside analogues as potential antiviral agents. Tetrahedron. 1999;55:8409–8422. [Google Scholar]
- 41.Kazuta Y, Matsuda A, Shuto S. Development of versatile cis- and trans-dicarbon-substituted chiral cyclopropane units: synthesis of (1S,2R)- and (1R,2R)-2-aminomethyl-1-(1H-imidazol-4-yl)cyclopropanes and their enantiomers as conformationally restricted analogues of histamine. J Org Chem. 2002;67:1669–1677. doi: 10.1021/jo010852x. [DOI] [PubMed] [Google Scholar]
- 42.Miyashita M, Yoshikoshi A, Grieco PA. Pyridinium p-toluenesulfonate. A mild and efficient catalyst for the tetrahydropyranylation of alcohols. J Org Chem. 1977;42:3772–3774. [Google Scholar]
- 43.Chen J, Levant B, Jiang C, Keck TM, Newman AH, Wang S. Tranylcypromine substituted cis-hydroxycyclobutylnaphthamides as potent and selective dopamine D(3) receptor antagonists. J Med Chem. 2014;57:4962–4968. doi: 10.1021/jm401798r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Talele TT. The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules. J Med Chem. 2016;59:8712–8756. doi: 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- 45.Boateng CA, Bakare OM, Zhan J, Banala AK, Burzynski C, Pommier E, Keck TM, Donthamsetti P, Javitch JA, Rais R, Slusher BS, Xi ZX, Newman AH. High affinity dopamine D3 receptor (D3R)-selective antagonists attenuate heroin self-administration in wildtype but not D3R knockout mice. J Med Chem. 2015;58:6195–6213. doi: 10.1021/acs.jmedchem.5b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bergman J, Roof RA, Furman CA, Conroy JL, Mello NK, Sibley DR, Skolnick P. Modification of cocaine self-administration by buspirone (buspar®): potential involvement of D3 and D4 dopamine receptors. Int J Neuropsychopharmacol. 2013;16:445–458. doi: 10.1017/S1461145712000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chun LS, Free RB, Doyle TB, Huang XP, Rankin ML, Sibley DR. D1-D2 dopamine receptor synergy promotes calcium signaling via multiple mechanisms. Mol Pharmacol. 2013;84:190–200. doi: 10.1124/mol.113.085175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao J, Free RB, Barnaeva E, Conroy JL, Doyle T, Miller B, Bryant-Genevier M, Taylor MK, Hu X, Dulcey AE, Southall N, Ferrer M, Titus S, Zheng W, Sibley DR, Marugan JJ. Discovery, optimization, and characterization of novel D2 dopamine receptor selective antagonists. J Med Chem. 2014;57:3450–3463. doi: 10.1021/jm500126s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meade JA, Free RB, Miller NR, Chun LS, Doyle TB, Moritz AE, Conroy JL, Watts VJ, Sibley DR. (−)-Stepholidine is a potent pan-dopamine receptor antagonist of both G protein- and β-arrestin-mediated signaling. Psychopharmacology (Berl) 2015;232:917–930. doi: 10.1007/s00213-014-3726-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–1308. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 51.Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J Med Chem. 2011;54:3581–3594. doi: 10.1021/jm200288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Free RB, Chun LS, Moritz AE, Miller BN, Doyle TB, Conroy JL, Padron A, Meade JA, Xiao J, Hu X, Dulcey AE, Han Y, Duan L, Titus S, Bryant-Genevier M, Barnaeva E, Ferrer M, Javitch JA, Beuming T, Shi L, Southall NT, Marugan JJ, Sibley DR. Discovery and characterization of a G protein-biased agonist that inhibits β-arrestin recruitment to the D2 dopamine receptor. Mol Pharmacol. 2014;86:96–105. doi: 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 54.Motulsky HJ, Christopoulos A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical guide to Curve Fitting. 4. Oxford university Press; San Diego: 2003. [Google Scholar]
- 55.Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.