

SUMMARY
Sleep remains one of the most mysterious yet ubiquitous animal behaviors. We review current perspectives on the neural systems that regulate sleep/wake states in mammals and the circadian mechanisms that control their timing. We also outline key models for the regulation of rapid eye movement (REM) sleep and non-REM sleep, how mutual inhibition between specific pathways gives rise to these distinct states, and how dysfunction in these circuits can give rise to sleep disorders.
Keywords: REM sleep, non-REM sleep, wake, arousal, circadian, monoamines, orexin, hypocretin, optogenetics, pharmacogenetics, chemogenetics
IN BRIEF
Scammell and colleagues review current perspectives on the neural systems that regulate sleep/wake states in mammals, current models for the control of sleep, and the circadian mechanisms that control the timing of sleep and wakefulness.
INTRODUCTION
From the time of Aristotle until the early 20th century, most philosophers thought that sleep was simply a consequence of reduced sensory input and low levels of brain activity. In fact, leading early anatomists and neurologists such as Purkinje and Lhermitte doubted the existence of specific neural pathways for regulating wakefulness and sleep. Perspectives on the biology of sleep have changed dramatically over the last years, and researchers have now identified many brain systems that selectively regulate the occurrence and timing of wake, rapid eye movement (REM) sleep, and non-REM (NREM) sleep.
Understanding these neural mechanisms is of great scientific interest and clinical importance. Fundamentally, sleep is essential for optimal cognition, immune function, and general health, and sleep disorders are among the most common clinical problems.
This review focuses on the neural networks in mammals that generate sleep/wake states and the circadian mechanisms that control the timing of these states. We emphasize recent research using techniques such as optogenetics and chemogenetics that enables targeting of specific neuronal populations, but we do not delve into sleep in non-mammalian species, sleep-dependent memory consolidation, how arousal affects attention, or the specific circuits in the thalamus and cortex that drive cortical rhythms as these topics have been covered well in other recent papers (Allada and Siegel, 2008; McCormick et al., 2015; Pimentel et al., 2016; Rasch and Born, 2013).
OVERVIEW OF SLEEP AND WAKE STATES
Neurobiologists often define wakefulness as a spectrum of behavioral states during which an animal exhibits voluntary motor activation and is responsive to internal and external stimuli. Consciousness has been little studied in animals, but it is a critical topic in clinical research (Koch et al., 2016). As one enters NREM sleep, consciousness fades, and in the deepest stages, sensory gating can block all but the strongest and most salient stimuli. During REM sleep, vivid, emotional, and story-like dreams are common, and these are accompanied by rapid eye movements and fluctuations in heart rate and respiration.
The electroencephalogram (EEG) and electromyogram (EMG) are excellent biomarkers of sleep/wake states (Fig. 1). During wake, the EEG shows low amplitude, fast frequencies, and the EMG shows variable amounts of muscle activity. During NREM sleep, the EEG is dominated by slower frequencies in the delta (0–4 Hz) and theta (4–7 Hz) ranges, and prolonged periods of wake are usually followed by large amounts of NREM sleep with especially high delta activity. In humans, the EEG during REM sleep shows low amplitude, fast activity much like that of wake with a modest amount of theta activity. In rodents, the cortical EEG during REM sleep shows abundant theta activity that arises from the underlying dorsal hippocampus. Muscle activity is strongly suppressed during REM sleep, preventing the enactment of dreams.
Figure 1. Sleep physiology.

A) Over the night, a typical young adult rapidly enters deep NREM sleep (N3) and then cycles between NREM and REM sleep about every 90 minutes. As homeostatic sleep pressure dissipates across the night, NREM sleep become lighter and REM sleep episodes become longer. B) Features of wake, NREM sleep, and REM sleep.
Sleep research often relies heavily on EEG recordings, but it is important to emphasize that the EEG is simply a biomarker of an underlying state, and changes in the EEG may not always reflect changes in state. For example, EEG delta activity can be high during wake in people with metabolic derangements such as hepatic encephalopathy or the post-ictal state but low during benzodiazepine-induced sleep.
Wake and NREM sleep are often considered discrete states, but arousal levels vary within these states. For example, people rouse easily from light NREM sleep (stage N1), but much stronger stimuli are required to wake from deep NREM sleep (stage N3) when the EEG is dominated by delta activity. In addition, levels of arousal vary during wake and correlate with pupil diameter, eyelid position, and sharp wave ripples in hippocampus (Eldar et al., 2013; McGinley et al., 2015; Reimer et al., 2014).
REGULATION OF WAKE
About 100 years ago, an epidemic of encephalitis lethargica swept through Europe, and affected patients would often sleep more than 20 hours/day for months on end. The Viennese neurologist Constantin von Economo discovered that these patients often had lesions in the midbrain and posterior hypothalamus, and he proposed that these regions contain vital wake-promoting circuitry (von Economo, 1930). Moruzzi and Magoun later showed that electrical stimulation of the reticular formation in anesthetized cats shifts the EEG from the slow activity typical of anesthesia to fast activity similar to that seen during wake (Moruzzi and Magoun, 1949). These observations fit well with the prevailing idea that the reticular formation integrates sensory information and then drives generalized arousal and motor responses. However, over the last few decades, the idea of undifferentiated reticular neurons has faded as researchers established that the major influences on arousal arise from neurochemically distinct systems (Fig. 2).
Figure 2. Wake-promoting pathways.

A) Several neurochemical systems promote arousal and the fast cortical activity typical of wakefulness. Monoaminergic neurons (light green) in the rostral brainstem and caudal hypothalamus directly innervate the cortex as well as many subcortical regions including the hypothalamus and thalamus. These monoaminergic regions include noradrenergic neurons of the locus coeruleus, serotonergic neurons of the dorsal and median raphe nuclei, dopaminergic neurons of the ventral tegmental area, and histaminergic neurons of the tuberomammillary nucleus. Wake-promoting signals also arise from the parabrachial nucleus and cholinergic regions (dark green with hatching), including the pedunculopontine and laterodorsal tegmental nuclei and basal forebrain.
The wake-promoting pathways ascend through the paramedian region of the midbrain and then split into a dorsal pathway into the thalamus and a large ventral pathway that innervates the hypothalamus, basal forebrain (BF), and cortex. The dorsal pathway enables thalamic processing of signals related to sensation, motor responses, and cognition. Photostimulation of GABA or glycine inputs to the thalamus can reduce locomotor activity or even produce complete behavioral arrest, but as soon as the photostimulation ends, motor activity resumes with little effect on the amounts of wake (Giber et al., 2015; Lewis et al., 2015). Similarly, patients with lesions limited just to the thalamus are usually in an awake but unresponsive vegetative state (Posner et al., 2007). In contrast, patients and animals with injury to the ventral pathway often struggle to stay awake and sleep more than the usual amount (Fuller et al., 2011; Ranson, 1939). Increased total sleep has also been reported in patients with injury to the paramedian thalamus (Bassetti et al., 1996), but on close examination, these injuries usually extend into the adjacent posterior hypothalamus and midbrain. Overall, normal wakefulness requires both the dorsal and ventral pathways, and it is most likely that the dorsal pathway to the thalamus is necessary for the content of consciousness by enabling proper thalamocortical signaling, whereas the ventral pathway is essential for the behavioral state of wakefulness.
The thalamus is also required for the generation of some of the rhythmic activity seen in the cortical EEG. Sleep spindles (bursts of 7–15 Hz activity during NREM sleep) are generated by interactions of the reticular nucleus of the thalamus and thalamocortical neurons (Huguenard and McCormick, 2007). The slow waves of NREM sleep probably arise mainly from the cortex, but thalamic circuits may also contribute (Crunelli et al., 2015; Lewis et al., 2015).
In the sections below, we review the specific systems that drive wakefulness, but some caveats should be kept in mind. First, while stimulation of many systems can promote arousal, an awakening may be secondary to engagement of other pathways. As a trivial example, photostimulation of nociceptive C fibers should rapidly rouse an animal from sleep, but the actual awakening is certainly mediated by downstream target neurons. Thus, in gain-of-function experiments, it is essential to fully describe the animal’s behavior in case the arousal arises from circuits mediating pain, fear or other behaviors, and then to control for these influences, with, for example, medications to reduce pain or anxiety. Similarly, the monoaminergic neurons, which are often considered fundamental wake-promoting systems, also influence attention, mood, reward, locomotion, and other behaviors, so one needs to consider whether increases in wake with activation of these systems are a secondary effect of high motivation, movement, etc. Second, while each of the systems described below helps promote wake, some may be more important for promoting wake under specific circumstances that are not yet well understood. Last, we present these arousal systems as discrete entities, but they have many anatomic interconnections and similar targets, and they likely work in concert.
Monoamines
Monoaminergic cell groups that drive arousal produce norepinephrine (NE), serotonin (5HT), dopamine (DA), or histamine, and they diffusely innervate the cerebral cortex, BF, lateral hypothalamus (LH), and many other regions. Their projections to the thalamus generally target the midline, intralaminar, and reticular nuclei of the thalamus through which they diffusely enhance thalamocortical signaling. Monoaminergic neurons also share similar firing patterns, with generally high rates of firing during wake (especially active wake), slow firing during NREM sleep, and a virtual cessation of firing during REM sleep.
The locus coeruleus (LC) is the major source of forebrain NE. Like other monoamine systems, LC neurons project widely throughout the CNS, and receive inputs from other arousal systems, the brainstem, and prefrontal cortex (Luppi et al., 1995). Conditional viral tracing confirms these diverse inputs and also demonstrates that LC neurons have extensive collateral projections (Schwarz et al., 2015). These convergent inputs and highly divergent outputs may enable the LC to function as a broadcast system, producing widespread effects that are likely required for global changes in behavioral state.
LC neurons promote arousal in general and are essential for the high levels of arousal required when responding to salient stimuli and stressors. NE agonists (e.g. α1) increase wake, and photoactivation of the LC rapidly wakes mice from sleep (Carter, 2010). Conversely, drugs that reduce NE release such as α2 agonists are sedating, and photoinhibition of noradrenergic LC neurons promotes transitions into NREM sleep and mildly reduces wake (Carter et al., 2010). LC activity is especially high with stress, novel stimuli, or salient stimuli that indicate reward or threat (Aston-Jones and Cohen, 2005). The importance of the LC in driving arousal with salient stimuli is evident in rats with LC lesions which have much less wakefulness than intact animals when placed in a socially and physically complex environment (Gompf et al., 2010).
Most forebrain 5HT arises from the dorsal raphe (DR) and median raphe nuclei. Like the LC, these nuclei innervate and receive reciprocal inputs from many brain regions that regulate sleep/wake states plus the extended amygdala, insula, and prefrontal cortex (Weissbourd et al., 2014).
Early research suggested that 5HT might promote sleep because large lesions of the raphe nuclei or acute depletion of 5HT causes insomnia in rodents and cats. However, 5HT neurons are essential for thermogenesis, and it now appears that the disruption of sleep with 5HT depletion was a consequence of hypothermia in a cool environment as this insomnia does not occur in a warm environment (Murray et al., 2015).
Instead, moderate quality evidence now demonstrates that 5HT neurons promote wake. 5HT directly excites other wake-promoting neurons, and drugs that increase 5HT tone such as SSRI’s generally increase wakefulness in both humans and rodents. Photoactivation of 5HT neurons doubles the amount of wake and fragments NREM sleep (Ito et al., 2013); some of this effect may be mediated by co-release of glutamate (Liu et al., 2014). These results show that 5HT neurons are sufficient to promote wake, but as these neurons are also implicated in regulating mood, reward, patience, and responding to salient cues, further work is needed to define the specific conditions during which they contribute most to arousal.
Dopamine’s capacity to drive arousal is clear from the potent effects of drugs that increase or decrease DA signaling. Amphetamines and modafinil strongly promote wake, mainly by increasing synaptic concentrations of DA (Wisor et al., 2001), whereas DA antagonists such as antipsychotics are quite sedating.
However, the source of this wake-promoting DA has been a mystery as many brain regions produce DA. Complicating matters further, the substantia nigra and ventral tegmental area (VTA) lack the “wake-on” firing patterns typical of other monoamine neurons, and the modest rise in extracellular DA levels during the active period is mostly driven by changes in dopamine transporter (DAT) activity (Ferris et al., 2014). Consequently, many researchers concluded that the VTA does not regulate sleep/wake states. This now appears incorrect as chemogenetic inhibition of DA VTA neurons reduces wake during baseline conditions and especially under conditions of high motivation such as seeking palatable food or a potential mate (Eban-Rothschild et al., 2016). Interestingly, this reduction in wake is also accompanied by nest building, suggesting engagement of a behavioral pattern tied to sleep. Conversely, photostimulation of these neurons or their terminals in the accumbens nucleus or the central nucleus of the amygdala rapidly wakes mice from sleep. Considered with other research on the VTA, these findings suggest that VTA DA neurons promote wake, especially under conditions of high motivation.
In addition, a small group of DA neurons in the ventral periaqueductal grey just lateral to the DR express Fos during wake, and lesioning these neurons reduces wake about 20% (Lu et al., 2006a). Researchers now need to determine the conditions during which these and other DA pathways are engaged to promote arousal.
The tuberomammillary nucleus (TMN) is the sole neuronal source of histamine in the brain, and during wake, TMN neurons excite neurons in the cortex, thalamus, and other arousal-promoting regions. Centrally acting histamine H1 antagonists are sedating, and mice lacking histamine or the H1 receptor have less wakefulness at the beginning of the dark period and less wake in response to the stress of a novel environment (Parmentier et al., 2016). Conversely, the histamine neurons are likely more excitable in mice lacking GABA receptors on the histamine neurons, and these mice take longer to fall asleep when placed in a novel environment (Zecharia et al., 2012). Recent work suggests that co-release of GABA from the histamine neurons may synergistically promote cortical activation and wake (Yu et al., 2015); other studies saw no evidence of GABA co-release (Williams et al., 2014), possibly due to heterogeneity in the histaminergic population. Histamine likely promotes generalized arousal as histamine levels are consistently high during wake, and chemogenetic stimulation of the TMN increases locomotion (Yu et al., 2014). Still, whether the histaminergic neurons promote motivation, cognition or other specific aspects of arousal remains unknown.
Basal forebrain
The BF is defined by fields of cholinergic neurons extending from the medial septum back to the substantia innominata, and this region also contains neurons producing GABA and glutamate. All these populations directly innervate the cortex, and through local connections, they also influence the activity of neighboring BF neurons (Xu et al., 2015; Zant et al., 2016). As a whole, the BF is thought to play an essential role in producing wake as BF stimulation promotes arousal, and large BF lesions can produce EEG slow waves and even coma (Buzsaki et al., 1988; Fuller et al., 2011). Beyond these global effects, BF neurons project topographically to distinct regions of cortex, and anatomical and neurochemical characteristics of these cortical regions may influence responses to incoming signals from the BF (Coppola, Disney, 16). Thus, the BF is broadly necessary for cortical activation and arousal, and subcircuits within the BF likely affect local cortical circuits.
BF cholinergic neurons promote the fast cortical activity characteristic of wake plus many cortical functions, including attention, memory, sensory processing, and cortical plasticity. Cholinergic BF neurons heavily innervate the cortex, directly and indirectly exciting cortical pyramidal neurons. Cholinergic neurons in the caudal BF also project to the amygdala, and those of the medial septum innervate the dorsal hippocampus and help drive theta activity. The cholinergic neurons fire in association with fast cortical rhythms during wake and REM sleep but much less during NREM sleep (Boucetta et al., 2014; Xu et al., 2015)
Selective manipulations of the cholinergic BF neurons mainly affect cortical activity, with subtler effects on sleep/wake states. For example, selective lesions or inhibition of the cholinergic neurons using optogenetic or chemogenetic techniques mainly increase slow EEG rhythms and reduce awakenings from NREM sleep, but they do not reduce the amount of wake (Chen et al., 2016; Fuller et al., 2011; Shi et al., 2015). Conversely, chemogenetic activation of these neurons suppresses slow EEG activity during NREM sleep and destabilizes NREM sleep for several hours but with little effect on the amount of wake (Anaclet et al., 2015; Chen et al., 2016). Photoactivation similarly suppresses slow waves, but after a few seconds, it also wakes mice from NREM sleep and then prolongs the subsequent period of wake (Irmak and de Lecea, 2014; Xu et al., 2015). These results consistently show that the BF cholinergic neurons promote fast cortical activity, but it is less clear whether these neurons are necessary for wakefulness itself.
Most likely, these divergent results arise from differences in optogenetic and chemogenetic effects. Photostimulation can potently activate neurons, but the resulting firing patterns and release of neurotransmitters may differ from the natural pattern. On the other hand, chemogenetic activation generally produces mild depolarization that should enhance responsiveness to incoming signals, but as the depolarization is small, the behavioral effects may not reflect the neuron’s full potential. Other factors may also contribute such as the number and location of transduced neurons; the number of engineered channels per neuron and their location within the neuron (e.g. soma vs. axon terminals); and the electrophysiological and intracellular signaling characteristics of the target neurons. Ultimately, with these and other gain of function methods, it is important to strive for patterns of neuronal activity similar to those that occur across the range of normal sleep/wake behavior. This is a challenging goal, but it can be approached by combining stimulation with techniques for monitoring the activity of the manipulated neurons such as optrodes and calcium imaging (Eban-Rothschild et al., 2016; Herrera et al., 2016; Weber et al., 2015).
In contrast to the cholinergic neurons, GABAergic BF neurons are clearly capable of promoting wake. These cells promote cortical activation by reducing the activity of inhibitory cortical interneurons, and they may have indirect effects via projections to the midline nuclei of the thalamus and many other subcortical regions that influence arousal. Chemogenetic activation of GABAergic BF neurons dramatically increases wake and fast EEG activity for many hours, whereas inhibition increases NREM sleep for about 3 hours (Anaclet et al., 2015).
However, GABAergic BF neurons are anatomically and functionally heterogeneous. They have been subdivided based on their expression of somatostatin, parvalbumin, calretinin, calbindin, Kv2.2, and other markers, and some are mainly active during wake and REM sleep while others are more active in NREM sleep (Hassani et al., 2009a; Xu et al., 2015). For example, photostimulation of the parvalbumin neurons increases wake and fast EEG activity, but photostimulation of somatostatin neurons mildly increases NREM sleep (Kim et al., 2015; Xu et al., 2015). Future experiments will need to target specific types of BF GABAergic neurons as these GABAergic subpopulations are clearly distinct.
Much less is known about the glutamatergic BF neurons, but they also seem to promote cortical activation. These cells innervate the cortex and arousal-promoting subcortical regions, and most fire at moderate rates in wake and REM sleep (Boucetta et al., 2014; Xu et al., 2015). Photostimulation of these cells consistently wakes mice from NREM sleep, and though chemogenetic stimulation slightly reduces slow EEG rhythms, it does not increase wake (Anaclet et al., 2015; Xu et al., 2015). As with the BF cholinergic neurons, these divergent results may arise from differences in the neural effects of optogenetic and chemogenetic methods.
Pedunculopontine and Laterodorsal Tegmental Nuclei (PPT/LDT)
The PPT and LDT are clusters of cholinergic neurons at the junction of the pons and midbrain, and like the BF, these nuclei also contain separate populations of GABAergic and glutamatergic neurons (Wang and Morales, 2009).
PPT/LDT cholinergic neurons innervate many subcortical regions that influence arousal, but unlike the BF cholinergic neurons, their innervation of the cortex is sparse. These neurons fire fastest during wake, especially active wake and REM sleep (Boucetta et al., 2014), and electrical stimulation of the PPT region induces fast EEG activity, in part through cholinergic excitation of thalamocortical neurons (Steriade et al., 1991). Selective chemogenetic activation of PPT cholinergic neurons strongly suppresses slow EEG activity during NREM sleep (Kroeger et al., in press), and photostimulation of these cells reduces slow wave activity during seizures (Furman et al., 2015). Thus, much like BF cholinergic neurons, PPT/LDT cholinergic neurons can suppress slow cortical activity, but it remains unknown whether they promote wake itself.
PPT/LDT glutamatergic and GABAergic neurons also fire in association with fast EEG frequencies (Boucetta et al., 2014; Cox et al., 2016). Chemogenetic activation of the glutamatergic PPT neurons markedly increases wake for several hours, but the functions of the GABAergic neurons remain unknown (Kroeger et al., in press).
Parabrachial Nucleus (PB)
The PB is essential for relaying sensory signals to the forebrain, and it is also crucial for regulating arousal as injury to the PB region produces coma or persistent vegetative state in both animals and humans (Fischer et al., 2016; Fuller et al., 2011; Parvizi and Damasio, 2003).
Glutamatergic neurons of the medial PB may be especially important for wake as this region heavily innervates the BF, and destruction of medial PB neurons or local deletion of the vesicular glutamate transporter (vGluT2) reduce wake and increase EEG delta power during NREM sleep (Fuller et al., 2011; Kaur et al., 2013). Glutamatergic neurons of the external lateral PB are activated by hypercarbia, and disrupted glutamate signaling in the lateral PB delays arousals to hypercarbia in a mouse model of obstructive sleep apnea (Kaur et al., 2013). Because pain, cold, and nausea also activate neurons in the external lateral PB, this pathway may promote arousal in response to a variety of interoceptive stimuli.
Orexins
Orexin-A and -B (also known as hypocretin-1 and -2) are neuropeptides essential for regulating wake and REM sleep (de Lecea et al., 1998; Sakurai et al., 1998). The orexin neurons are scattered across the LH, and orexins strongly excite all the wake-promoting brain regions discussed above plus the midline thalamus and cortex (Fig. 3). The orexin neuropeptides bind to the OX1R and OX2R receptors and excite target neurons for many minutes. The orexin neurons also produce glutamate and the inhibitory neuropeptide dynorphin, and co-release of glutamate from the orexin neurons can excite target neurons (Schone et al., 2014).
Figure 3. Projections of the orexin neurons.

The orexin neuropeptides are produced by neurons in the lateral hypothalamus and excite neurons in the cortex, midline thalamus, and all wake-promoting brain regions.
The orexin neuropeptides are especially important for maintaining long periods of wake. Orexin neuron activity and extracellular levels of orexin-A are high during the active period, especially during active wake, and ICV injection of orexin-A increases wake and suppresses REM sleep for several hours. Similarly, photoactivation of the orexin neurons rouses mice from sleep (Adamantidis et al., 2007), and chemogenetic activation of the orexin neurons increases wake and strongly suppresses REM sleep (Sasaki et al., 2011). The importance of the orexin system is most obvious in narcolepsy, a common sleep disorder caused by selective loss of the orexin-producing neurons. People with narcolepsy have chronic, often severe sleepiness and dysregulated REM sleep, and animals with disrupted orexin signaling have a very similar phenotype, with wake bouts much shorter than usual and sudden transitions from wake into REM sleep-like states (Chemelli et al., 1999; Lin et al., 1999). This role in maintaining wake is evident in mice with acute loss of the orexin neurons which lose their ability to produce long bouts of wake, but their total amount of wake is only slightly reduced (Branch et al., 2016).
Which are the key pathways through which orexins sustain long periods of wake? Focal restoration of orexin signaling in the TMN and LC regions substantially improves the maintenance of wake (Hasegawa et al., 2014; Mochizuki et al., 2011), suggesting that signaling through these regions is sufficient to improve wake. In addition, photoinhibition of the LC reduces the awakening effect of orexin neuron photostimulation (Carter et al., 2012); this could indicate that the LC is necessary for the wake-promoting effects of orexins, but the interpretation is challenging as this could be an additive effect as LC photoinhibition is sedating (Carter et al., 2010). The orexin neurons project widely, and more work is needed to determine just which projections are necessary for arousal and other effects of orexins.
Orexin signaling also helps maintain arousal when animals are stressed, seeking rewards, or responding to homeostatic challenges (Bonnavion et al., 2015; Mahler et al., 2014). For example, food deprivation markedly increases wake in wild type mice, probably to encourage foraging, but mice lacking the orexin neurons show very little arousal with hunger (Yamanaka et al., 2003), and, in contrast to wild type mice, food deprivation has little antidepressant-like effect in mice lacking orexins (Lutter et al., 2008). In addition, mice lacking orexins show almost no conditioned place preference for drugs such as morphine (Narita et al., 2006). Thus, in addition to maintaining wake under quiet conditions, the orexin neurons enable the high levels of arousal required when defending against homeostatic challenges or when pursuing motivated behaviors.
Lateral Hypothalamus (LH) and Posterior Hypothalamus (PH)
Clinical injury to the LH and PH often causes severe sleepiness; these individuals can sleep 15–20 hours each day, and when woken, they seem apathetic and quickly lapse back to sleep. Loss of the orexin neurons causes sleepiness, but it does not increase the total amount of sleep so researchers have been searching for other wake-promoting systems in these regions. One population of GABAergic neurons in the LH innervates the reticular nucleus of the thalamus, and photoactivation of these LH neurons rapidly wakes mice from NREM sleep whereas photoinhibition increases slow waves and lengthens NREM sleep bouts (Herrera et al., 2016). Another population of GABAergic LH neurons innervates sleep-promoting brain regions, and chemogenetic activation of these neurons promotes wake for several hours (Venner et al., 2016). GABAergic neurons in the LH and PH also have potent effects on seeking food and other rewards, and in future studies, it will be important to determine whether the wake-promoting LH neurons form distinct populations and whether the increases in wake are a consequence of high motivation or hunger.
REGULATION OF NREM SLEEP
NREM Sleep Homeostasis and Somnogens
Prolonged periods of wake are followed by long periods of deep NREM sleep, and this homeostatic response is likely mediated by NREM sleep-promoting substances (somnogens), including adenosine (AD), prostaglandin D2, and cytokines such as interleukin-1 and tumor necrosis factor-α (Krueger et al., 2011). In general, somnogens increase during wake and act as paracrine mediators to promote sleep.
AD is the best understood of these somnogens. In the BF, cortex, and hippocampus, extracellular levels of AD increase across prolonged periods of wake and decline during sleep (Porkka-Heiskanen et al., 1997; Schmitt et al., 2012). Interestingly, astrocytes are a major source of this extracellular AD (Halassa et al., 2009; Schmitt et al., 2012), and AD levels are regulated by AD kinase in astrocytes (Bjorness et al., 2016). Injections into the brain of AD receptor agonists increase NREM sleep, whereas AD antagonists, such as caffeine, promote wake (Radulovacki et al., 1984). AD can promote sleep by directly inhibiting wake-promoting neurons and by activating sleep-promoting neurons (Fredholm et al., 1999). In addition, neurons in the shell of the nucleus accumbens mediate the arousing effects of caffeine, possibly through projections to wake-promoting brain regions (Lazarus et al., 2011).
Somnogens may also contribute to focal increases in cortical slow waves known as local sleep. Intense focal activity in the cortex is often followed by high EEG delta activity within that region. For example, focal EEG slowing is common after focal seizures, but even mild challenges such as learning a new motor task can be followed by increased delta activity during NREM sleep within the corresponding region of supplementary motor cortex (Huber et al., 2004). Similarly, rats kept awake for several hours have increasingly frequent periods of focal slow waves that correlate with worsening performance on a motor task (Vyazovskiy et al., 2011). Possibly, high metabolic demand in local cortical regions increases AD, other somnogens, or ion concentrations that enhance local slow waves (Ding et al., 2016). A fascinating open question is whether somnogens mainly influence global sleep/wake states through a top-down influence of local sleep or from a bottom-up effect on subcortical circuits.
Homeostatic sleep drive may also be encoded by changes in neuronal physiology. The “R2-ExFl2” circuit in Drosophila is not required for sleep per se, but is required for gauging sleep need in response to previous sleep/wake behavior (Liu et al., 2016). Moreover, sleep drive generates reversible synaptic changes at least partially related to increased expression of NMDA receptors. Sleep researchers now need to determine if a similar mechanism contributes to sleep homeostasis in mammals.
Neuronal circuits for NREM sleep
The Preoptic Area (POA)
In his research on encephalitis lethargica, von Economo found that most patients had severe sleepiness with lesions in the posterior hypothalamus, but some had just the opposite - unrelenting insomnia with lesions in the POA, the most rostral part of the hypothalamus (von Economo, 1930). He proposed that the POA contains neurons that promote sleep, and this idea was later confirmed in cats and rats that had much less sleep after lesions of the anterior hypothalamus (McGinty and Sterman, 1968). In addition, extracellular recordings showed that the POA and adjacent BF contain neurons selectively active during NREM and REM sleep (Szymusiak and McGinty, 1986).
Researchers have now established that the ventrolateral preoptic area (VLPO) and median preoptic nucleus (MnPO) contain neurons essential for promoting NREM sleep (Fig. 4). Single unit recordings and analysis of Fos expression show that NREM sleep-active neurons are concentrated in the VLPO and MnPO (Alam et al., 2014; Sherin et al., 1996), and lesions of these nuclei produce large and long-lasting reductions in sleep (John and Kumar, 1998; Lu et al., 2000). VLPO and MnPO sleep-active neurons are GABAergic, and VLPO neurons also produce the neuropeptide galanin. Both cell groups strongly innervate and inhibit arousal-promoting brain regions, including the cholinergic neurons of the BF, orexin neurons, TMN, DR, median raphe, PB, and LC (Sherin et al., 1998; Uschakov et al., 2007). Photostimulation of GABAergic projections from the POA directly inhibits the orexin neurons (Saito et al., 2013). Conversely, the VLPO and possibly the MnPO are innervated by arousal-promoting brain regions (Chou et al., 2002), and acetylcholine, NE, and 5HT all directly inhibit VLPO neurons; histamine suppresses VLPO activity via local GABAergic interneurons (Williams et al., 2014), and orexins may have similar effects.
Figure 4. NREM sleep-promoting pathways.

GABAergic neurons in the ventrolateral preoptic area and median preoptic nucleus promote sleep by inhibiting wake-promoting neurons in the caudal hypothalamus and brainstem. The basal forebrain also contains sleep-active neurons that may promote sleep via projections within the BF and direct projections to the cortex. GABAergic neurons of the parafacial zone (PZ) may promote sleep by inhibiting the parabrachial nucleus. The cortex contains scattered NREM sleep-active neurons that contain both GABA and neuronal nitric oxide synthase (nNOS). Blue circles with hatching denote NREM sleep-promoting nuclei.
In addition to promoting NREM sleep, VLPO and MnPO neurons may help mediate the homeostatic response to sleep deprivation. With rising sleep pressure, neurons in both regions fire faster during the sleep deprivation period and during the subsequent sleep (Alam et al., 2014). This surge in VLPO activity can be blocked by an AD antagonist, and in vitro electrophysiological studies show that AD excites VLPO neurons directly and indirectly by pre-synaptically inhibiting GABAergic afferents (Chamberlin et al., 2003; Gallopin et al., 2005).
The VLPO and MnPO are the most-studied NREM sleep-promoting brain regions, but the precise identity and function of these neurons is only partially understood. One challenge is that while these nuclei contain many sleep-active neurons, they are intermixed with wake-active neurons and the precise neurochemical identities of these populations are unknown. In addition, the VLPO may contain two types of sleep-active neurons: one population is excited by AD and may promote sleep in response to homeostatic sleep pressure, and the other population does not respond to AD but is inhibited by acetylcholine, NE and 5HT, suggesting it may help consolidate sleep once the arousal systems are inactive (Gallopin et al., 2005).
A recent study suggests that sleep-promoting preoptic neurons may be more extensive than previously suspected. A tet-tagging method enabled expression of an excitatory DREADD receptor in sleep-active neurons, and chemogenetic activation of these cells produced abundant EEG slow waves and reduced body temperature. Combining these approaches with in vitro electrophysiological recordings should help establish whether this broader population is active during spontaneous sleep (Zhang et al., 2015).
Basal Forebrain
Though most neurons in the BF are wake-active, others are mainly active in NREM sleep (Hassani et al., 2009a). These neurons are mostly GABAergic, and they usually begin to fire a few seconds before NREM sleep onset and then fire at their maximal rate throughout NREM sleep (Hassani et al., 2009a). Some of these sleep-active neurons innervate the cortex, so they may promote NREM sleep via direct inhibition of cortical neurons (Manns et al., 2000).
The identity of these NREM sleep-active BF neurons is unclear, but at least some appear to be GABAergic neurons that produce somatostatin. Some somatostatin BF neurons are selectively active in NREM sleep, and optogenetic stimulation of this population mildly increases NREM sleep, perhaps via inhibition of other BF neurons (Kim et al., 2015; Xu et al., 2015). Furthermore, in vitro optogenetic studies show that somatostatin BF neurons directly inhibit putative wake-active BF neurons producing acetylcholine, parvalbumin/GABA, and glutamate. Thus, BF somatostatin neurons may promote NREM sleep by inhibiting local, wake-active BF neurons (Xu et al., 2015).
Parafacial Zone (PZ)
Most research on NREM sleep circuitry has focused on the POA and BF, but early transection studies suggested that the caudal brainstem also contains neurons that promote NREM sleep (Batini et al., 1958). Using a combination of chemogenetic techniques and lesions, researchers recently identified a cluster of NREM sleep-active neurons in the parafacial zone (PZ), a region just dorsal and lateral to the facial nerve in the rostral medulla. These GABAergic/glycinergic neurons express Fos during NREM sleep, and cell specific lesions or disruption of GABA/glycinergic transmission in the PZ region increases wake (Anaclet et al., 2014). Chemogenetic activation of these neurons rapidly induces sustained periods of NREM sleep with high EEG delta power similar to that seen after sleep deprivation. Conversely, chemogenetic inhibition of PZ GABAergic neurons strongly decreases NREM sleep, even during times of high sleep drive, indicating that these neurons are necessary for NREM sleep. PZ GABAergic neurons directly inhibit PB neurons, including those that project to the BF. These experiments suggest that the PZ could promote NREM sleep by inhibiting wake-promoting neurons in the PB, but as this has not yet been tested in vivo, the PZ may also promote sleep via inhibition of additional wake-promoting systems.
Cortical Sleep-Active nNOS Neurons
Though most cortical neurons are wake-active, one population is especially active during NREM sleep. These cells produce neuronal nitric oxide synthase (nNOS) and are a small subset of the broader population of GABAergic cortical interneurons. Fos expression in cortical nNOS neurons correlates with the amounts of NREM sleep and slow wave activity during NREM sleep (Gerashchenko et al., 2008; Morairty et al., 2013). The nNOS neurons are thought to respond to homeostatic sleep drive and synchronize slow cortical rhythms via long-range, intracortical projections and release of GABA and nitric oxide (NO). In support of this idea, mice constitutively lacking nNOS have shorter NREM sleep bouts, less total NREM sleep, and a blunted homeostatic response to sleep deprivation (Morairty et al., 2013). The cortical nNOS neurons are quite intriguing as they may drive local or widespread increases in slow waves, and future studies will need to examine their firing patterns in relation to sleep and sleep pressure and test whether selective loss of function in these neurons alters NREM sleep or slow waves.
Models of NREM sleep control
NREM sleep clearly relies on neurons in the POA, BF, brainstem, and possibly the cortex, but how these generate transitions into NREM sleep and how they sustain NREM sleep is still being established. The VLPO inhibits many wake-promoting brain regions, and these regions in turn inhibit the VLPO. One model proposes that this mutual inhibition generates patterns of activity akin to an electrical flip-flop switch (Saper et al., 2010). Specifically, inhibition of wake-promoting neurons during sleep lessens any inhibition of the VLPO by the wake-promoting systems, enabling high levels of activity in the VLPO; just the opposite likely occurs during wake, and this dynamic interaction should enable rapid transitions between states and the generation of full sleep and wake states with little time spent in intermediate drowsy states. The NREM sleep-promoting neurons of the BF and brainstem may have similar relations with wake-active neurons, but this has not yet been studied. The cortical nNOS neurons probably interact mainly with other cortical neurons, but as these too are GABAergic, they may also form a mutually inhibitory circuit with wake-active cortical neurons that influences patterns of cortical activity across states.
REGULATION OF REM SLEEP
Neural circuits in the pons are required for REM sleep. Early studies showed that when in REM sleep, animals with a knife cut at the caudal edge of the pons show the usual low amplitude, fast EEG activity but lack muscle atonia, whereas transections at the rostral edge of the pons preserve the atonia of REM sleep but eliminate the fast EEG activity (Jouvet, 1962; Siegel et al., 1984). This focused attention on the pons, and in the late 70’s, many researchers thought REM sleep was controlled by reciprocal connections between REM sleep-promoting, cholinergic LDT/PPT neurons and REM sleep-suppressing, monoaminergic neurons (Hobson et al., 1975). Over the last several years, another REM sleep model has taken hold in which glutamatergic neurons in the sublaterodorsal nucleus of the pons play a central role in generating REM sleep (Fig. 5).
Figure 5. REM sleep-promoting pathways.

The sublaterodorsal nucleus (SLD) plays a crucial role in regulating REM sleep. Glutamatergic neurons of the SLD produce the muscle paralysis of REM sleep by exciting GABAergic/glycinergic neurons in the ventromedial medulla and spinal cord that hyperpolarize motor neurons. Cholinergic neurons of the pedunculopontine and laterodorsal tegmental nuclei also promote REM sleep and may help drive the fast EEG activity typical of REM sleep. During wake and NREM sleep, the SLD is inhibited by GABAergic neurons of the ventrolateral periaqueductal grey and adjacent lateral pontine tegmentum as well as monoaminergic neurons of the locus coeruleus and raphe nuclei. During REM sleep, the ventrolateral periaqueductal grey is likely inhibited by GABAergic neurons of the SLD and medulla. REM sleep-promoting nuclei are shown in blue with hatching; REM sleep-suppressing nuclei are shown in green. DPGi, dorsal paragigantocellular reticular nucleus; LPGi, lateral paragigantocellular nucleus; GiV, ventral gigantocellular reticular nucleus; GiA, alpha gigantocellular reticular nucleus.
Pedunculopontine and Laterodorsal Tegmental Nuclei (PPT/LDT)
Historically, PPT/LDT cholinergic neurons were considered key elements of the pontine REM sleep generator, but more recently, these neurons are seen as modulators rather than central elements (discussed below). Acetylcholine levels in the dorsal pons are high during REM sleep just as in wake, and microinjection of the cholinergic agonist carbachol into the laterodorsal pons produces a long-lasting REM sleep-like state in cats and to a lesser degree in rodents (Kubin, 2001). Juxtacellular recordings have shown that the cholinergic neurons mainly fire during REM sleep and wake (Boucetta et al., 2014). In fact, these neurons begin firing just before REM sleep or wake, suggesting they may help promote transitions into these state. In line with this idea, photostimulation of PPT/LDT cholinergic neurons promotes transitions from NREM to REM sleep (Van Dort et al., 2015).
Recent studies have begun to shed light on the non-cholinergic neurons of the PPT/LDT (Boucetta et al., 2014; Cox et al., 2016). Most glutamatergic neurons in this region begin firing just before and continue during REM sleep, and many also fire during wake. The GABAergic neurons in the PPT/LDT region seem mainly wake-active, but some may be more active during REM sleep than in NREM sleep. Defining how these three PPT/LDT populations work together to regulate REM sleep remains an important goal for future research.
Sublaterodorsal Nucleus (SLD)
Seminal research in the 1960’s and 1970’s identified another pontine region that is crucial for generating muscle atonia during REM sleep. This region lies just ventral to the caudal LDT and LC and is named the sublaterodorsal nucleus (SLD) or subcoeruleus.
Considerable research indicates that glutamatergic neurons of the SLD are essential for generating the muscle atonia of REM sleep. SLD neurons are active during REM sleep as demonstrated by c-Fos immunoreactivity and by single unit recordings (Lu et al., 2006b; Sakai et al., 2001). The SLD neurons fire tonically throughout REM sleep, and pharmacological activation of the SLD region rapidly produces a long-lasting REM sleep-like state characterized by a low voltage EEG, prominent EEG theta activity, and continuous muscle atonia (Boissard et al., 2002). Most importantly, animals with focal lesions of the SLD or deletion of glutamate signaling in the SLD often have a state much like REM sleep but without atonia, during which they twitch, jump, and sometimes exhibit complex motor behaviors. SLD lesions that extend into adjacent regions can also shorten REM sleep bouts and reduce the total amount of REM sleep (Krenzer et al., 2011; Lu et al., 2006b). Thus, the SLD is necessary for the atonia of REM sleep, and it likely works with nearby regions to generate the REM sleep state.
During REM sleep, SLD neurons are probably activated by cholinergic neurons of the PPT/LDT. Cholinergic PPT/LDT neurons innervate the SLD, and the SLD is the most sensitive region for the promotion of REM sleep by carbachol (Kubin, 2001). In vivo and in vitro recordings show that carbachol excites REM sleep-active and spinally-projecting SLD neurons (Sakai et al., 2001; Weng et al., 2014). However, this cholinergic input may not be necessary as blocking cholinergic signaling in the SLD does not alter the amount of REM sleep or the duration of REM sleep bouts (Grace et al., 2014).
Researchers continue to debate just how the SLD generates muscle atonia. One model proposes that the SLD excites neurons in the ventromedial medulla (VMM) that inhibit spinal motor neurons. These glycinergic/GABAergic VMM neurons, as discussed below, are located just rostral to the inferior olive in the ventral gigantocellular reticular (GiV) and the alpha gigantocellular reticular (GiA) nuclei (Luppi et al., 2012). In support of this idea, single unit recordings demonstrate that the VMM contains neurons that fire fastest in REM sleep, slower in NREM sleep, and very little during wake, correlating with the amount of muscle atonia (Chase et al., 1984). Furthermore, VMM lesions partially disrupt the atonia of REM sleep (Holmes and Jones, 1994; Schenkel and Siegel, 1989). Another model proposes that the SLD neurons bypass the medulla and directly excite spinal interneurons that use glycine and/or GABA to inhibit motor neurons (Lu et al., 2006b). In line with this idea, focal disruption of glycinergic/GABAergic transmission in the spinal ventral horn produces phasic movements during REM sleep (Krenzer et al., 2011). These models are not mutually exclusive, and additional work is needed to establish the relative contributions of these two descending pathways in the control of REM sleep atonia.
REM Sleep-Suppressing Neurons in the Pons
REM sleep has many similarities to wake; cortical activity is desynchronized, cortical metabolism is high, and mental activity is complex. In addition, both people and animals can rapidly transition from REM sleep to wakefulness, but what prevents transitions from wake into REM sleep?
Several neuronal populations in the pons are thought to suppress REM sleep during wake, including the monoaminergic neurons of the LC and DRN. These neurons are very active during wake but nearly silent during REM sleep. They project to the SLD where local application of NE inhibits REM sleep-active neurons and suppresses REM sleep (Crochet and Sakai, 1999). In addition, NE and 5HT inhibit cholinergic neurons of the PPT/LDT (Luebke et al., 1992; Williams and Reiner, 1993), and antidepressants that increase monoamine levels often suppress REM sleep. Surprisingly, photoinhibition of LC neurons did not alter REM sleep (Carter et al., 2010), but this approach only partially reduced NE levels and may not have been sufficient to test whether silencing the LC neurons permits entry into REM sleep.
The pons contains another population of REM sleep-suppressing neurons in the ventrolateral periaqueductal grey matter (vlPAG) and the lateral pontine tegmentum (LPT), a region also known as the deep mesencephalic reticular nucleus (Boissard et al., 2003). Based on the expression of Fos, the vlPAG/LPT neurons are probably silent during REM sleep and active during wake and NREM sleep (Sapin et al., 2009), but whether these neurons fire in a state-dependent fashion is still unknown. The vlPAG/LPT sends GABAergic projections to the SLD, and lesions or pharmacological inactivation of the vlPAG/LPT region increase REM sleep (Lu et al., 2006b; Sapin et al., 2009). Similarly, inhibition of vlPAG neurons by optogenetic activation of the GABAergic afferent input from the VMM increases REM sleep and triggers transitions from NREM to REM sleep (Weber et al., 2015), whereas chemogenetic activation or inhibition of GABAergic vlPAG/LPT neurons reduces or increases REM sleep, respectively (Hayashi et al., 2015; Weber et al., 2015). Considered together, these results strongly suggest that the vlPAG/LPT inhibits REM sleep via GABAergic projections to the SLD.
During REM sleep, the vlPAG/LPT is thought to be held in check by GABAergic neurons. In addition to the glutamatergic, REM sleep-promoting neurons described above, the SLD also contains REM sleep-active, GABAergic neurons that innervate the vlPAG/LPT (Lu et al., 2006b; Maloney et al., 2000), and additional REM sleep-suppressing neurons may reside in the vlPAG itself (Sapin et al., 2009). Thus, the vlPAG/LPT and SLD may form a mutually inhibitory circuit that regulates REM sleep, much like the wake-NREM sleep flip-flop model introduced above.
Medullary Reticular Formation
In the VMM, neurons in the GiV and GiA nuclei are essential for REM sleep atonia (Luppi et al., 2012). These glycinergic and probably GABAergic neurons are active during REM sleep; they receive input from the SLD; they project to spinal and brainstem motor neurons; and, when stimulated, they produce glycinergic IPSPs in motor neurons (Boissard et al., 2002; Soja et al., 1987). Consistent with this model, glutamate levels in the VMM increase as animals enter REM sleep, and blockade of glutamatergic signaling onto these neurons results in REM sleep without muscle atonia, suggesting that glutamatergic input, probably from the SLD, activates the VMM neurons to produce muscle atonia (Lai and Siegel, 1988). In addition, a glutamatergic cell group in the caudal GiV just above the inferior olive appears to drive REM sleep atonia (Vetrivelan et al., 2009).
Other medullary neurons also may promote REM sleep by inhibiting REM sleep-suppressing neurons of the pons such as the LC, DRN and vlPAG/LPT. These neurons reside in the dorsal paragigantocellular reticular (DPGi) and lateral paragigantocellular (LPGi) nuclei, regions lateral and dorsal to the GiV and GiA. In support of this model, REM sleep-active, GABAergic neurons of the DPGi and LPGi project to the LC and vlPAG/LPT, and during REM sleep, release of GABA suppresses activity of the monoaminergic neurons and vlPAG/LPT neurons (Goutagny et al., 2008). Remarkably, photostimulation of neurons in the LPGi and adjacent regions or their terminals in the vlPAG can prolong REM sleep or trigger transitions into REM sleep, whereas chemogenetic inhibition of these neurons decreases REM sleep (Weber et al., 2015). These findings were unexpected as medullary neurons were thought simply to generate atonia, and additional experiments will be necessary to determine whether REM sleep is mainly driven by the pons, medulla, or the two regions together.
Hypothalamic Control of REM Sleep
For decades, REM sleep research focused on the brainstem, but researchers have recently identified neurons in the POA, LH, and PH that help generate and regulate REM sleep. These regions contain many neurons that are maximally active during both NREM and REM sleep, but some are predominantly active in REM sleep. Though the neurochemical identity of most of these neurons remains unknown, two specific groups have been identified.
One cluster of REM sleep-active neurons is located just dorsal and medial to the VLPO, in a region referred to as the extended VLPO. These GABAergic/galaninergic neurons are active during REM sleep, and lesions of the extended VLPO reduce REM sleep (Lu et al., 2002). These cells innervate the DR, LC, and vlPAG/LPT, suggesting that the extended VLPO may promote REM sleep by inhibiting brainstem neurons that suppress REM sleep.
Another group of REM sleep-promoting neurons is scattered across the LH and PH and produces the neuropeptide melanin-concentrating hormone (MCH). These cells innervate the SLD and many other regions, and they fire maximally during REM sleep (Hassani et al., 2009b). Several studies show that photoactivation or chemoactivation of the MCH neurons increases REM sleep (Jego et al., 2013; Tsunematsu et al., 2014; Vetrivelan et al., 2016). Whether the MCH neurons promote NREM sleep is debated as this has been reported by only one group (Konadhode et al., 2013). Other studies have shown that chronic loss of the MCH neurons disrupts NREM sleep, but compensatory changes may contribute (Tsunematsu et al., 2014; Varin et al., 2016). Most evidence suggests that while MCH neurons can promote REM sleep, they may not be necessary for spontaneous REM sleep as ablation or photoinhibition of MCH neurons has only minor effects on REM sleep, and MCH receptor null mice exhibit only minor changes in REM sleep (Adamantidis et al., 2008; Jego et al., 2013; Tsunematsu et al., 2014; Varin et al., 2016).
MCH neurons may also affect target neurons via co-release of GABA and/or glutamate. MCH neurons contain GAD65 and GAD67, but they also contain the vesicular glutamate transporter 2 (vGlut2) (Chee et al., 2015; Jego et al., 2013; Jennings et al., 2015). One optogenetic study demonstrated release of GABA from MCH neurons while another showed glutamate release but no signs of GABA release (Chee et al., 2015; Jego et al., 2013). The cause of this discrepancy is unclear, but as the target regions differed, it may reflect heterogeneity of the MCH population. In addition to their projections to the SLD, MCH neurons densely innervate the REM sleep-suppressing neurons of the LC, DRN, and vlPAG/LPT. Possibly, release of glutamate from MCH neurons directly activates pontine REM sleep-promoting neurons and release of MCH and GABA inhibits REM sleep-suppressing neurons.
The orexin neurons of the lateral hypothalamus are intermixed with the MCH neurons but have completely opposite effects on REM sleep. ICV injection of orexin-A reduces REM sleep for many hours (Mieda et al., 2011), and photoactivation of the orexin neurons awakens mice from REM sleep (Adamantidis et al., 2007). Similarly, chemogenetic activation of the orexin neurons robustly suppresses REM sleep (Sasaki et al., 2011), whereas orexin antagonists increase REM sleep and decrease REM sleep latency. Chronic loss of the orexin neurons in rodents and people with narcolepsy results in very poor regulation of REM sleep, with REM sleep at any time of day and fragments of REM sleep such as muscle atonia and dream-like hallucinations mixing into wake. The precise regions through which orexins inhibit REM sleep are still being mapped, but the orexin neurons heavily innervate REM sleep suppressing region such as the LC, DR, vlPAG/LPT, and focal rescue of orexin signaling in 5HT DR neurons reduces episodes of atonia during wake (Hasegawa et al., 2014). As the orexin neurons are mainly active during wake, it is likely that orexins tonically suppress REM sleep during wake.
Models of REM Sleep Control
In 1975, Hobson and McCarley proposed that REM sleep is regulated by reciprocal interactions between cholinergic, REM sleep-promoting PPT/LDT neurons and monoaminergic, REM sleep-suppressing neurons (Hobson et al., 1975). Over the last decade, work from a number of research groups has given rise to a new model in which glutamatergic neurons of the SLD are now a key central element, and the cholinergic and monoaminergic neurons are considered modulatory. The SLD region is thought to provide ascending projections for EEG activation and descending projections that trigger muscle atonia. During REM sleep, SLD neurons inhibit REM sleep-suppressing neurons of the vlPAG/LPT, but during wake and NREM sleep, the SLD is reciprocally inhibited by the vlPAG/LPT. Lu and colleagues proposed that this mutual inhibition forms another mutually inhibitory circuit that helps generate the distinct state of REM sleep (Lu et al., 2006b). Pharmacological and recent optogenetic studies support this model (Boissard et al., 2002; Weber et al., 2015).
This dynamic interaction between the SLD and vlPAG/LPT is modulated by several brain regions. Acetylcholine from the PPT/LDT, ascending projections from the medulla, and MCH likely bias the system towards REM sleep, whereas monoamines and orexins do the opposite.
Still, the model is incomplete; much has been learned about the descending pathways that control REM muscle atonia, but the ascending pathways that control cortical activity and dreaming during REM sleep remain poorly understood.
CIRCADIAN REGULATION OF SLEEP AND WAKE
The transitions between sleep and wake states are regulated over short time scales of seconds to hours, but what mechanisms promote sleep in the night and banish it from the day? Unlike the homeostatic control of sleep, circadian-controlled processes are driven by an autonomously rhythmic system. The rotation of the Earth every 24 hours exposes most organisms to daily oscillations in light and ambient temperature, and the circadian system (for circa diem, ‘about a day’) is a biological clock mechanism that synchronizes the internal state of the organism with these predictable environmental changes. In mammals, circadian timekeeping is organized by the suprachiasmatic nucleus of the hypothalamus (SCN), which is both necessary and sufficient for orchestrating circadian timing of sleep/wake behavior, metabolism, and physiology in synchrony with changes in the light/dark cycle (Marcheva et al., 2013; Welsh et al., 2010).
The Suprachiasmatic Nucleus is the Central Pacemaker
The SCN is a pair of compact nuclei in the most ventral and medial part of the hypothalamus, directly abutting the third ventricle and just above the optic chiasm from which it receives a major retinal input via the retinohypothalamic tract (RHT) (Morin, 2013). The SCN is composed of tightly apposed, neurochemically heterogeneous neurons, which are subdivided into a ventrolateral ‘core’ and a dorsomedial ‘shell’ (Bedont and Blackshaw, 2015; Welsh et al., 2010). While most neurons in the SCN are GABAergic, the core contains neuronal groups that also co-express either vasoactive intestinal peptide (VIP), calretinin, gastrin related peptide (GRP), or neurotensin; those in the shell express arginine vasopressin (AVP), angiotensin II, prokineticin-2, and met-enkephalin.
The SCN is perhaps unique in the entire mammalian brain as its neurons form a coupled, intercellular network capable of self-sustained, circadian oscillations of both neuronal activity and gene expression, even in the absence of external stimuli (Colwell, 2011; Welsh et al., 2010). How this network enables circadian timekeeping is an area of intense investigation. Intercellular communication between the ‘core’ and ‘shell’ regions is essential for the generation of SCN network synchrony (Buhr et al., 2010). VIP neurons in the core may act as a prime coupling signal for the influence of other neuropeptides, including AVP and GRP (Aton et al., 2005; Maywood et al., 2011; Mieda et al., 2015). Indeed, VIP appears essential for SCN network function as its loss desynchronizes the SCN, and exogenous application of VIP produces phase shifts (Maywood et al., 2011). While VIP is thus critically important, other signals within the SCN are certainly involved as even VIP knockout tissues exhibit some residual circadian rhythms. Recently, neuromedin S (Nms) has been identified in many SCN neurons, and while molecular oscillations in Nms neurons are necessary for the maintenance of circadian behavior in constant darkness, Nms peptide itself is not (Lee et al., 2015).
Most neurons of the SCN are GABAergic, and while the coupling of their oscillatory activity is essential to SCN function, the role of GABA in establishing and maintaining coupling remains controversial. The available evidence suggests that the effects of GABA in the SCN depend on its network state. For example, GABA has been proposed to have either excitatory or inhibitory actions, promote synchrony or desynchrony, and have region-specific effects on the ventral compared to dorsal SCN (Albus et al., 2005; Aton et al., 2006; Mohawk and Takahashi, 2011; Wagner et al., 1997). Recent evidence suggests that tonic (as opposed to pulsatile) release of GABA enables network function (DeWoskin et al., 2015). To date, most studies have simply blocked GABAA receptors in the SCN region, and in the future, genetic ablation of all GABA transmission will be valuable to assess the full spectrum of GABA’s role in maintenance and transmission of SCN synchrony.
The SCN most likely regulates activity in other brain regions through both its anatomical connections (reviewed below) and the release of secreted factors, but only a few of these such as TGFα and prokineticin-2 have been characterized in detail (Hatcher et al., 2008; Southey et al., 2014). In SCN-ablated animals, transplantation of the SCN in semi-permeable membranes partially rescues circadian rhythms, suggesting a role for secreted factors (Silver et al., 1996), but whether these act locally within the SCN or distally on other neurons remains unclear.
Another, likely crucial factor is SCN-mediated control of the circadian rhythm of body temperature. The rhythm of core body temperature is one of the most strongly circadian variables in endothermic animals and is a potent timing signal for the brain and peripheral tissues. Importantly, the SCN’s circadian timekeeping is largely impervious to changes in temperature, so it can regulate the timing of temperature oscillations in other cells and tissues without affecting its own timekeeping (Buhr et al., 2010).
Timing signals such as variations in temperature, nutrients, or steroid hormones synchronize peripheral tissues by engaging a conserved, ubiquitous molecular oscillator comprised of a negative feedback loop of transcription-translation and post-translational modifications (Fig. 6)(Buhr and Takahashi, 2013). The transcription factors BMAL1 and CLOCK heterodimerize in the nucleus and bind E box elements in the promoters of thousands of clock controlled genes (CCGs). Among the CCGs are the Period gene products PER1-3 and the cryptochromes, CRY1, 2, which curb the transcriptional activity of BMAL1 and CLOCK, closing the loop. Post-translational modification and subsequent degradation of PERs and CRYs disinhibits BMAL1/CLOCK activity, and the cycle is renewed roughly every 24 hours. In addition to this main loop, Bmal1 transcription is directly inhibited by other feedback proteins, including REV-ERBα and DEC1/2. In addition, circadian oscillations exist beyond transcription and translational, and post-translational pathways are crucial for various aspects of cellular timing (Lipton et al., 2015; O’Neill et al., 2013). These findings support the notion that circadian timing is a fundamental property of cellular function.
Figure 6. Circadian timekeeping in cells is orchestrated by transcriptional-translational and post-translational feedback loops.
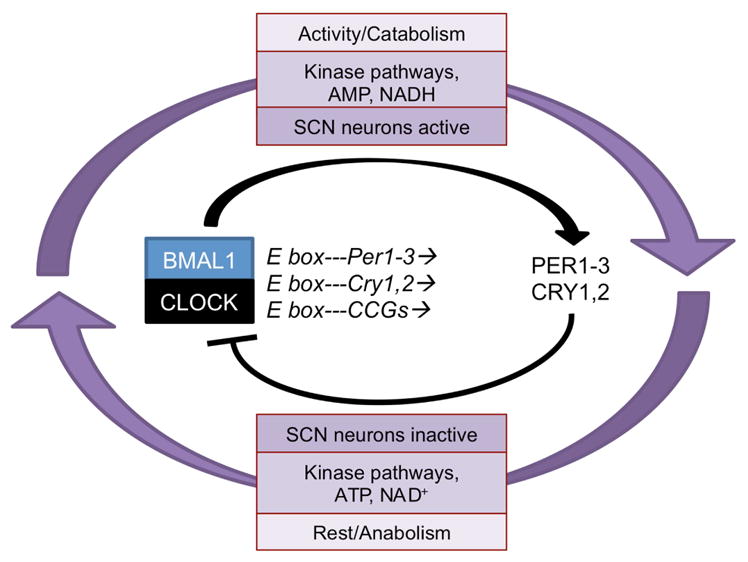
The transcription factors BMAL1 and CLOCK form heterodimers that activate the transcription of E box-containing clock-controlled genes (CCGs) including the Period (Per) and Cryptochrome (Cry) genes. Per and Cry gene products subsequently inhibit the activity of BMAL1/CLOCK. Additionally, circadian oscillations in kinase pathways, energy metabolism (AMP/ATP ratio), and redox state (NAD+/NADH ratio) link global control of cellular physiology and metabolism to the molecular clock.
Remarkably, the clock’s molecular oscillations are coupled with circadian oscillations in SCN neuronal activity. Multiunit electrode recordings in freely behaving rats demonstrate strong circadian oscillations in SCN electrical activity, with the highest firing rates in the light period (Deboer et al., 2003). Interestingly, in both diurnal and nocturnal animals, SCN firing rates are highest in the light period, suggesting that regions downstream of the SCN determine the behavioral responses to SCN signals. Thus, the SCN somehow integrates molecular timing with rhythms of neuronal firing on circadian timescales.
Circadian Timing of Sleep and Wake
The circadian system synchronizes sleep/wake cycles with the light/dark cycle. SCN ablation in rodents reduces the diurnal peaks and troughs of wake and sleep but does not increase sleep over a 24 hour period, supporting a primary role of the circadian system in sleep timing not quantity (Mistlberger, 2005). In one experiment, SCN ablation in squirrel monkeys produced more sleep, but these experiments relied on electrolytic lesions that may have injured wake-promoting neurons in the nearby basal forebrain (Edgar et al., 1993).
The nature of the interactions between the circadian clock and sleep/wake behavior are far from clear. Several studies in both mice and humans suggest that the clock may regulate sleep quality (Franken et al., 2006; He et al., 2009; Naylor et al., 2000), whereas others point to more targeted roles on just the timing of sleep. For example, in advanced sleep phase syndrome, the sleep period begins much earlier than usual, and some of these individuals have mutations in Per2 or casein kinase 1, one of PER2’s phosphorylating kinases (Toh et al., 2001; Xu et al., 2005).
The interaction of sleep and circadian rhythms has been examined using “forced desynchrony’ protocols in which people are placed in non-24 hour light/dark cycles. Very long (28 hour) or very short (22 hour) light/dark cycles decouple circadian influences from sleep/wake behavior. These studies show that sleep timing is strongly phase-locked to intrinsic circadian clock-controlled temperature rhythms (Czeisler et al., 1980a; Czeisler et al., 1980b). These studies have also suggested that the circadian clock promotes wake during the day even as homeostatic sleep drive builds (Dijk and Czeisler, 1995). This wake-promoting influence turns off just before habitual bedtime, and the SCN then seems to promote sleep through the night, even as homeostatic sleep drive dissipates in the early morning hours.
While SCN neuronal firing is clearly circadian, it also varies with sleep state. At any time of day, SCN activity is relatively high during REM sleep and plummets during NREM sleep, especially when slow EEG activity is prominent such as during the intense rebound NREM sleep after sleep deprivation (Deboer et al., 2007; Deboer et al., 2003). These data show that sleep state affects SCN activity, but how this occurs and whether it affects the circadian regulation of sleep are unknown.
SCN Outflow: Circadian Influences on Sleep/Wake Systems
How does the SCN promote wake at some times and sleep at others? The SCN mainly projects to other hypothalamic nuclei, with especially heavy projections to the subparaventricular zone (SPZ; a region just above the SCN), and moderate projections to the paraventricular nucleus (PVN), dorsomedial nucleus (DMH), ventromedial nucleus (VMH), LH, and medial preoptic area (Fig. 7)(Deurveilher and Semba, 2005; Morin, 2013).
Figure 7. Neural pathways that regulate the circadian timing of sleep and other rhythms.

The suprachiasmatic nucleus is the master circadian pacemaker and sits just above the optic chiasm. Neuronal rhythms generated by the SCN are synchronized with the daily light-dark cycle by direct inputs from the retina via the retinohypothalamic tract (RHT). The RHT also has small projections that may influence the activity of sleep-promoting neurons in the preoptic area. Most output signals of the SCN are relayed through the subparaventricular zone (SPZ) and then to the dorsomedial nucleus of the hypothalamus (DMH). The DMH regulates the timing of wakefulness via excitatory projections to the orexin neurons and locus coeruleus and inhibitory projections to the preoptic area. Signals from the SPZ and DMH also regulate the circadian rhythms of heart rate, blood pressure, body temperature, locomotor activity, and feeding. Circadian signals to the paraventricular nucleus of the hypothalamus regulate the daily melatonin rhythm.
The SPZ receives a major concentration of the SCN’s efferent connections. In nocturnal rodents, SCN firing rates are highest in the light period, but SPZ firing rates are highest in the dark period. Indeed, the timing of SPZ neuron firing parallels that of wake-promoting brain regions such as the BF (Kubota et al., 1981; Miyamoto et al., 2012), and Fos expression in the SPZ is high in the dark period of nocturnal rodents and high in the light period of diurnal rodents (Nunez et al., 1999). Cell-specific lesions of the ventral SPZ in rats severely reduce circadian rhythms of wake, NREM and REM sleep (Lu et al., 2001), suggesting that the SPZ is a critical circadian relay that promotes arousal during the active period and sleep during the rest period.
The ventral SPZ in particular is thought to regulate sleep/wake rhythms primarily via its heavy projections to the DMH, as cell-specific lesions of the DMH also eliminate circadian rhythms of sleep/wake behavior (Chou et al., 2003). Glutamatergic neurons of the DMH strongly innervate wake-promoting brain regions, including orexin neurons, TMN, LC, VTA, DR and LDT, whereas GABAergic DMH neurons innervate sleep-promoting regions including the VLPO and MnPO (Saper et al., 2010; Vujovic et al., 2015). Thus, the DMH likely integrates circadian signals to actively promote wake at certain times and sleep at others. Direct projections from the SCN to the VLPO and MnPO may also play a role, but their functional importance has not been tested (Chou et al., 2002; Deurveilher and Semba, 2005).
Synchronizing the Clock with the Environment
One of the major functions of the circadian clock is the integration of external and internal timing cues. This requires entrainment – the synchronization of the autonomous oscillator with an external timing cue, or zeitgeber. The most potent circadian zeitgeber is light. The retinohypothalamic tract is the major source of light information to the SCN (Freedman et al., 1999; Lucas et al., 2001). The retina contains intrinsically photosensitive retinal ganglion cells (ipRGCs) containing the specialized photopigment melanopsin. Unlike the rod/cone system, the ipRGCs and melanopsin are required for photoentrainment (Berson et al., 2010; Gooley et al., 2001; Hattar et al., 2003). Melanopsin has specialized physiological properties that enable integration over relatively long times and wavelengths – specializations well suited for detecting slow variations in light as the sun rises and sets (Emanuel and Do, 2015). Interestingly, the melanopsin system is most sensitive to relatively short wavelengths, paralleling the particularly strong effect of blue light in entrainment.
Another important physiological timing cue is the hormone melatonin which is released from the pineal gland during the dark period. Interestingly, melatonin release is driven by the circadian clock, and melatonin acts on the SCN, forming yet another feedback loop within the timekeeping network. Melatonin secretion is regulated by a multi-synaptic pathway connecting the SCN to the PVN and then to sympathetic fibers innervating the pineal gland, but how light suppresses melatonin secretion remains unclear. The circadian phase-shifting effects of melatonin inversely mirror those of light, suggesting that melatonin acts as an internal timing signal (Scheer and Czeisler, 2005). Though widely touted as a sedative, melatonin’s sleep-promoting effects are weak, and it is mainly a chronotherapeutic, helping regularize sleep onset by entraining circadian rhythms (Fisher and Sugden, 2010; Mendelson and Bergmann, 2001).
Sleep Rhythms and Society
In our post-industrial society, air travel across time zones and exposure to artificial light, including cell phones and other personal electronic devices, have created myriad opportunities for people to ignore or suppress natural circadian cues (Wright et al., 2013). For example, evening use of personal electronic devices, which are rich in blue light, delays circadian rhythms and sleep onset, suppresses melatonin, and increases morning sleepiness (Chang et al., 2015). Circadian rhythm disruption frequently leads to disrupted sleep, and both have been linked to metabolic disease, cancer risk, neurological disease, and mood disorders (Maury et al., 2010; Wulff et al., 2010). The control of sleep/wake behavior cannot be divorced from circadian timing, and a deeper understanding of the relationship between rhythms of cellular metabolism and complex behaviors such as sleep will likely be a boon to human health.
CONCLUSIONS
Neuroscientists have made much progress defining the key circuits that regulate sleep/wake behavior, but this is just a start as many fundamental questions remain unanswered. In this review, we have emphasized brainstem and hypothalamic pathways that drive wake or sleep and how these likely influence cortical activity in a bottom-up fashion, but might cortical activity have a top-down influence? For example, high levels of cortical activity promote arousal, and patients with insomnia have high levels of activity in certain cortical regions, even during sleep (Nofzinger et al., 2004). On the other hand, might focal reductions in cortical activity during wake trigger transitions into sleep? Is the deep and long rebound sleep seen after sleep deprivation driven by cortical or subcortical mechanisms?
We have portrayed these pathways in a simple fashion, but these circuits are not static. For example, SCN neurons exhibit circadian rhythms in sodium and potassium currents that affect their firing (Colwell, 2011; Flourakis et al., 2015), and sleep deprivation alters glutamatergic and noradrenergic inputs to the orexin neurons (Rao et al., 2007; Uschakov et al., 2011). In addition, neuronal excitability is likely altered by variations in ion concentrations across sleep/wake states (Ding et al., 2016), and research in mammals and Drosophila suggest that sleep homeostasis is influenced by changes in specific ion channels (Liu et al., 2016; Pimentel et al., 2016; Tononi and Cirelli, 2014). An important direction for future research will be determining whether variations in neuronal excitability drive the essential characteristics of sleep/wake behavior.
Ultimately, sleep and wake states arise from dynamic interactions throughout the sleep/wake circuitry, but how do neurons acting on a millisecond time scale give rise to emergent sleep/wake behaviors that last for minutes to hours? Might short-acting neurotransmitters like GABA and glutamate drive rapid changes in state, whereas longer-acting neuromodulators like orexins and MCH influence behavioral states for longer periods? Studying these network interactions will require new tools such as simultaneously imaging the activity of many neurons in multiple brain regions and sophisticated computational analysis.
With a better understanding of the anatomy, physiology, and dynamics of these circuits, we should gain novel insights into the bigger mysteries of how sleep is controlled, how it benefits the brain, and how it can be improved.
Acknowledgments
The authors thank Dr. Clifford Saper and Dr. Mark Andermann for their thoughtful comments. Figures 2, 3, 4, 5 and 7 © 2016 Natalie Koscal. This work was supported by NIH grants HL095491, DE022912, NS091126, and HD071026.
Footnotes
Conflicts of Interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamantidis A, Salvert D, Goutagny R, Lakaye B, Gervasoni D, Grisar T, Luppi PH, Fort P. Sleep architecture of the melanin-concentrating hormone receptor 1-knockout mice. Eur J Neurosci. 2008;27:1793–1800. doi: 10.1111/j.1460-9568.2008.06129.x. [DOI] [PubMed] [Google Scholar]
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam MA, Kumar S, McGinty D, Alam MN, Szymusiak R. Neuronal activity in the preoptic hypothalamus during sleep deprivation and recovery sleep. J Neurophysiol. 2014;111:287–299. doi: 10.1152/jn.00504.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albus H, Vansteensel MJ, Michel S, Block GD, Meijer JH. A GABAergic mechanism is necessary for coupling dissociable ventral and dorsal regional oscillators within the circadian clock. Curr Biol. 2005;15:886–893. doi: 10.1016/j.cub.2005.03.051. [DOI] [PubMed] [Google Scholar]
- Allada R, Siegel JM. Unearthing the phylogenetic roots of sleep. Curr Biol. 2008;18:R670–R679. doi: 10.1016/j.cub.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaclet C, Ferrari L, Arrigoni E, Bass CE, Saper CB, Lu J, Fuller PM. The GABAergic parafacial zone is a medullary slow wave sleep-promoting center. Nat Neurosci. 2014;17:1217–1224. doi: 10.1038/nn.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaclet C, Pedersen NP, Ferrari LL, Venner A, Bass CE, Arrigoni E, Fuller PM. Basal forebrain control of wakefulness and cortical rhythms. Nat Commun. 2015;6:8744. doi: 10.1038/ncomms9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- Aton SJ, Colwell CS, Harmar AJ, Waschek J, Herzog ED. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat Neurosci. 2005;8:476–483. doi: 10.1038/nn1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aton SJ, Huettner JE, Straume M, Herzog ED. GABA and Gi/o differentially control circadian rhythms and synchrony in clock neurons. Proc Natl Acad Sci U S A. 2006;103:19188–19193. doi: 10.1073/pnas.0607466103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetti C, Mathis J, Gugger M, Lovblad KO, Hess CW. Hypersomnia following paramedian thalamic stroke: a report of 12 patients. Ann Neurol. 1996;39:471–480. doi: 10.1002/ana.410390409. [DOI] [PubMed] [Google Scholar]
- Batini C, Moruzzi G, Palestini M, Rossi GF, Zanchetti A. Presistent patterns of wakefulness in the pretrigeminal midpontine preparation. Science. 1958;128:30–32. doi: 10.1126/science.128.3314.30-a. [DOI] [PubMed] [Google Scholar]
- Bedont JL, Blackshaw S. Constructing the suprachiasmatic nucleus: a watchmaker’s perspective on the central clockworks. Front Syst Neurosci. 2015;9:74. doi: 10.3389/fnsys.2015.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson DM, Castrucci AM, Provencio I. Morphology and mosaics of melanopsin-expressing retinal ganglion cell types in mice. J Comp Neurol. 2010;518:2405–2422. doi: 10.1002/cne.22381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorness TE, Dale N, Mettlach G, Sonneborn A, Sahin B, Fienberg AA, Yanagisawa M, Bibb JA, Greene RW. An Adenosine-Mediated Glial-Neuronal Circuit for Homeostatic Sleep. J Neurosci. 2016;36:3709–3721. doi: 10.1523/JNEUROSCI.3906-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissard R, Fort P, Gervasoni D, Barbagli B, Luppi PH. Localization of the GABAergic and non-GABAergic neurons projecting to the sublaterodorsal nucleus and potentially gating paradoxical sleep onset. Eur J Neurosci. 2003;18:1627–1639. doi: 10.1046/j.1460-9568.2003.02861.x. [DOI] [PubMed] [Google Scholar]
- Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16:1959–1973. doi: 10.1046/j.1460-9568.2002.02257.x. [DOI] [PubMed] [Google Scholar]
- Bonnavion P, Jackson AC, Carter ME, de Lecea L. Antagonistic interplay between hypocretin and leptin in the lateral hypothalamus regulates stress responses. Nat Commun. 2015;6:6266. doi: 10.1038/ncomms7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucetta S, Cisse Y, Mainville L, Morales M, Jones BE. Discharge profiles across the sleep-waking cycle of identified cholinergic, GABAergic, and glutamatergic neurons in the pontomesencephalic tegmentum of the rat. J Neurosci. 2014;34:4708–4727. doi: 10.1523/JNEUROSCI.2617-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch AF, Navidi W, Tabuchi S, Terao A, Yamanaka A, Scammell TE, Diniz Behn C. Progressive Loss of the Orexin Neurons Reveals Dual Effects on Wakefulness. Sleep. 2016;39:369–377. doi: 10.5665/sleep.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol. 2013:3–27. doi: 10.1007/978-3-642-25950-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr ED, Yoo SH, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Science. 2010;330:379–385. doi: 10.1126/science.1195262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Bickford RG, Ponomareff G, Thal LJ, Mandel R, Gage FH. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J Neurosci. 1988;8:4007–4026. doi: 10.1523/JNEUROSCI.08-11-04007.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ME, Brill J, Bonnavion P, Huguenard JR, Huerta R, de Lecea L. Mechanism for Hypocretin-mediated sleep-to-wake transitions. Proc Natl Acad Sci U S A. 2012;109:E2635–2644. doi: 10.1073/pnas.1202526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K, de Lecea L. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci. 2010;13:1526–1533. doi: 10.1038/nn.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin NL, Arrigoni E, Chou TC, Scammell TE, Greene RW, Saper CB. Effects of adenosine on gabaergic synaptic inputs to identified ventrolateral preoptic neurons. Neuroscience. 2003;119:913–918. doi: 10.1016/s0306-4522(03)00246-x. [DOI] [PubMed] [Google Scholar]
- Chang AM, Aeschbach D, Duffy JF, Czeisler CA. Evening use of light-emitting eReaders negatively affects sleep, circadian timing, and next-morning alertness. Proc Natl Acad Sci U S A. 2015;112:1232–1237. doi: 10.1073/pnas.1418490112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase MH, Enomoto S, Hiraba K, Katoh M, Nakamura Y, Sahara Y, Taira M. Role of medullary reticular neurons in the inhibition of trigeminal motoneurons during active sleep. Exp Neurol. 1984;84:364–373. doi: 10.1016/0014-4886(84)90233-4. [DOI] [PubMed] [Google Scholar]
- Chee MJ, Arrigoni E, Maratos-Flier E. Melanin-concentrating hormone neurons release glutamate for feedforward inhibition of the lateral septum. J Neurosci. 2015;35:3644–3651. doi: 10.1523/JNEUROSCI.4187-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Chen L, Yin D, Wang TX, Guo W, Dong H, Xu Q, Luo YJ, Cherasse Y, Lazarus M, Qiu ZL, et al. Basal Forebrain Cholinergic Neurons Primarily Contribute to Inhibition of Electroencephalogram Delta Activity, Rather Than Inducing Behavioral Wakefulness in Mice. Neuropsychopharmacology. 2016;41:2133–2146. doi: 10.1038/npp.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Bjorkum AA, Gaus SE, Lu J, Scammell TE, Saper CB. Afferents to the ventrolateral preoptic nucleus. J Neurosci. 2002;22:977–990. doi: 10.1523/JNEUROSCI.22-03-00977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J. Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J Neurosci. 2003;23:10691–10702. doi: 10.1523/JNEUROSCI.23-33-10691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS. Linking neural activity and molecular oscillations in the SCN. Nat Rev Neurosci. 2011;12:553–569. doi: 10.1038/nrn3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Pinto L, Dan Y. Calcium imaging of sleep-wake related neuronal activity in the dorsal pons. Nat Commun. 2016;7:10763. doi: 10.1038/ncomms10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochet S, Sakai K. Effects of microdialysis application of monoamines on the EEG and behavioural states in the cat mesopontine tegmentum. Eur J Neurosci. 1999;11:3738–3752. doi: 10.1046/j.1460-9568.1999.00760.x. [DOI] [PubMed] [Google Scholar]
- Crunelli V, David F, Lorincz ML, Hughes SW. The thalamocortical network as a single slow wave-generating unit. Curr Opin Neurobiol. 2015;31:72–80. doi: 10.1016/j.conb.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Czeisler CA, Weitzman E, Moore-Ede MC, Zimmerman JC, Knauer RS. Human sleep: its duration and organization depend on its circadian phase. Science. 1980a;210:1264–1267. doi: 10.1126/science.7434029. [DOI] [PubMed] [Google Scholar]
- Czeisler CA, Zimmerman JC, Ronda JM, Moore-Ede MC, Weitzman ED. Timing of REM sleep is coupled to the circadian rhythm of body temperature in man. Sleep. 1980b;2:329–346. [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FSn, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deboer T, Detari L, Meijer JH. Long term effects of sleep deprivation on the mammalian circadian pacemaker. Sleep. 2007;30:257–262. doi: 10.1093/sleep/30.3.257. [DOI] [PubMed] [Google Scholar]
- Deboer T, Vansteensel MJ, Detari L, Meijer JH. Sleep states alter activity of suprachiasmatic nucleus neurons. Nat Neurosci. 2003;6:1086–1090. doi: 10.1038/nn1122. [DOI] [PubMed] [Google Scholar]
- Deurveilher S, Semba K. Indirect projections from the suprachiasmatic nucleus to major arousal-promoting cell groups in rat: Implications for the circadian control of behavioural state. Neuroscience. 2005;130:165–183. doi: 10.1016/j.neuroscience.2004.08.030. [DOI] [PubMed] [Google Scholar]
- DeWoskin D, Myung J, Belle MD, Piggins HD, Takumi T, Forger DB. Distinct roles for GABA across multiple timescales in mammalian circadian timekeeping. Proc Natl Acad Sci U S A. 2015;112:E3911–3919. doi: 10.1073/pnas.1420753112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15:3526–3538. doi: 10.1523/JNEUROSCI.15-05-03526.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, O’Donnell J, Xu Q, Kang N, Goldman N, Nedergaard M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science. 2016;352:550–555. doi: 10.1126/science.aad4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR, de Lecea L. VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat Neurosci. 2016;19:1356–1366. doi: 10.1038/nn.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar DM, Dement WC, Fuller CA. Effect of SCN lesions on sleep in squirrel monkeys: evidence for opponent processes in sleep-wake regulation. J Neurosci. 1993;13:1065–1079. doi: 10.1523/JNEUROSCI.13-03-01065.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar E, Cohen JD, Niv Y. The effects of neural gain on attention and learning. Nat Neurosci. 2013;16:1146–1153. doi: 10.1038/nn.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel AJ, Do MT. Melanopsin tristability for sustained and broadband phototransduction. Neuron. 2015;85:1043–1055. doi: 10.1016/j.neuron.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris MJ, Espana RA, Locke JL, Konstantopoulos JK, Rose JH, Chen R, Jones SR. Dopamine transporters govern diurnal variation in extracellular dopamine tone. Proc Natl Acad Sci U S A. 2014;111:E2751–2759. doi: 10.1073/pnas.1407935111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer DB, Boes AD, Demertzi A, Evrard HC, Laureys S, Edlow BL, Liu H, Saper CB, Pascual-Leone A, Fox MD, et al. A human brain network derived from coma-causing brainstem lesions. Neurology. 2016;87:2427–2434. doi: 10.1212/WNL.0000000000003404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SP, Sugden D. Endogenous melatonin is not obligatory for the regulation of the rat sleep-wake cycle. Sleep. 2010;33:833–840. doi: 10.1093/sleep/33.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flourakis M, Kula-Eversole E, Hutchison AL, Han TH, Aranda K, Moose DL, White KP, Dinner AR, Lear BC, Ren D, et al. A Conserved Bicycle Model for Circadian Clock Control of Membrane Excitability. Cell. 2015;162:836–848. doi: 10.1016/j.cell.2015.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken P, Dudley CA, Estill SJ, Barakat M, Thomason R, O’Hara BF, McKnight SL. NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: genotype and sex interactions. Proc Natl Acad Sci U S A. 2006;103:7118–7123. doi: 10.1073/pnas.0602006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. 1999;51:83–133. [PubMed] [Google Scholar]
- Freedman MS, Lucas RJ, Soni B, von Schantz M, Munoz M, David-Gray Z, Foster R. Regulation of mammalian circadian behavior by non-rod, non-cone, ocular photoreceptors. Science. 1999;284:502–504. doi: 10.1126/science.284.5413.502. [DOI] [PubMed] [Google Scholar]
- Fuller PM, Sherman D, Pedersen NP, Saper CB, Lu J. Reassessment of the structural basis of the ascending arousal system. J Comp Neurol. 2011;519:933–956. doi: 10.1002/cne.22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman M, Zhan Q, McCafferty C, Lerner BA, Motelow JE, Meng J, Ma C, Buchanan GF, Witten IB, Deisseroth K, et al. Optogenetic stimulation of cholinergic brainstem neurons during focal limbic seizures: Effects on cortical physiology. Epilepsia. 2015;56:e198–202. doi: 10.1111/epi.13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallopin T, Luppi PH, Cauli B, Urade Y, Rossier J, Hayaishi O, Lambolez B, Fort P. The endogenous somnogen adenosine excites a subset of sleep-promoting neurons via A2A receptors in the ventrolateral preoptic nucleus. Neuroscience. 2005;134:1377–1390. doi: 10.1016/j.neuroscience.2005.05.045. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Wisor JP, Burns D, Reh RK, Shiromani PJ, Sakurai T, de la Iglesia HO, Kilduff TS. Identification of a population of sleep-active cerebral cortex neurons. Proc Natl Acad Sci U S A. 2008;105:10227–10232. doi: 10.1073/pnas.0803125105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giber K, Diana MA, Plattner VM, Dugue GP, Bokor H, Rousseau CV, Magloczky Z, Havas L, Hangya B, Wildner H, et al. A subcortical inhibitory signal for behavioral arrest in the thalamus. Nat Neurosci. 2015;18:562–568. doi: 10.1038/nn.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompf HS, Mathai C, Fuller PM, Wood DA, Pedersen NP, Saper CB, Lu J. Locus ceruleus and anterior cingulate cortex sustain wakefulness in a novel environment. J Neurosci. 2010;30:14543–14551. doi: 10.1523/JNEUROSCI.3037-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooley JJ, Lu J, Chou TC, Scammell TE, Saper CB. Melanopsin in cells of origin of the retinohypothalamic tract. Nat Neurosci. 2001;4:1165. doi: 10.1038/nn768. [DOI] [PubMed] [Google Scholar]
- Goutagny R, Luppi PH, Salvert D, Lapray D, Gervasoni D, Fort P. Role of the dorsal paragigantocellular reticular nucleus in paradoxical (rapid eye movement) sleep generation: a combined electrophysiological and anatomical study in the rat. Neuroscience. 2008;152:849–857. doi: 10.1016/j.neuroscience.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Grace KP, Vanstone LE, Horner RL. Endogenous cholinergic input to the pontine REM sleep generator is not required for REM sleep to occur. J Neurosci. 2014;34:14198–14209. doi: 10.1523/JNEUROSCI.0274-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa E, Yanagisawa M, Sakurai T, Mieda M. Orexin neurons suppress narcolepsy via 2 distinct efferent pathways. J Clin Invest. 2014;124:604–616. doi: 10.1172/JCI71017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Henny P, Jones BE. Discharge profiles of identified GABAergic in comparison to cholinergic and putative glutamatergic basal forebrain neurons across the sleep-wake cycle. J Neurosci. 2009a;29:11828–11840. doi: 10.1523/JNEUROSCI.1259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci U S A. 2009b;106:2418–2422. doi: 10.1073/pnas.0811400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher NG, Atkins N, Jr, Annangudi SP, Forbes AJ, Kelleher NL, Gillette MU, Sweedler JV. Mass spectrometry-based discovery of circadian peptides. Proc Natl Acad Sci U S A. 2008;105:12527–12532. doi: 10.1073/pnas.0804340105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattar S, Lucas RJ, Mrosovsky N, Thompson S, Douglas RH, Hankins MW, Lem J, Biel M, Hofmann F, Foster RG, et al. Melanopsin and rod-cone photoreceptive systems account for all major accessory visual functions in mice. Nature. 2003;424:76–81. doi: 10.1038/nature01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Kashiwagi M, Yasuda K, Ando R, Kanuka M, Sakai K, Itohara S. Cells of a common developmental origin regulate REM/non-REM sleep and wakefulness in mice. Science. 2015;350:957–961. doi: 10.1126/science.aad1023. [DOI] [PubMed] [Google Scholar]
- He Y, Jones CR, Fujiki N, Xu Y, Guo B, Holder JL, Jr, Rossner MJ, Nishino S, Fu YH. The transcriptional repressor DEC2 regulates sleep length in mammals. Science. 2009;325:866–870. doi: 10.1126/science.1174443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera CG, Cadavieco MC, Jego S, Ponomarenko A, Korotkova T, Adamantidis A. Hypothalamic feedforward inhibition of thalamocortical network controls arousal and consciousness. Nat Neurosci. 2016;19:290–298. doi: 10.1038/nn.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–58. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- Holmes CJ, Jones BE. Importance of cholinergic, GABAergic, serotonergic and other neurons in the medial medullary reticular formation for sleep-wake states studied by cytotoxic lesions in the cat. Neuroscience. 1994;62:1179–1200. doi: 10.1016/0306-4522(94)90352-2. [DOI] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Irmak SO, de Lecea L. Basal forebrain cholinergic modulation of sleep transitions. Sleep. 2014;37:1941–1951. doi: 10.5665/sleep.4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Yanase M, Yamashita A, Kitabatake C, Hamada A, Suhara Y, Narita M, Ikegami D, Sakai H, Yamazaki M, et al. Analysis of sleep disorders under pain using an optogenetic tool: possible involvement of the activation of dorsal raphe nucleus-serotonergic neurons. Mol Brain. 2013;6:59. doi: 10.1186/1756-6606-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego S, Glasgow SD, Herrera CG, Ekstrand M, Reed SJ, Boyce R, Friedman J, Burdakov D, Adamantidis AR. Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat Neurosci. 2013;16:1637–1643. doi: 10.1038/nn.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JH, Ung RL, Resendez SL, Stamatakis AM, Taylor JG, Huang J, Veleta K, Kantak PA, Aita M, Shilling-Scrivo K, et al. Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell. 2015;160:516–527. doi: 10.1016/j.cell.2014.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John J, Kumar VM. Effect of NMDA lesion of the medial preoptic neurons on sleep and other functions. Sleep. 1998;21:587–598. doi: 10.1093/sleep/21.6.587. [DOI] [PubMed] [Google Scholar]
- Jouvet M. Research on the neural structures and responsible mechanisms in different phases of physiological sleep. Arch Ital Biol. 1962;100:125–206. [PubMed] [Google Scholar]
- Kaur S, Pedersen NP, Yokota S, Hur EE, Fuller PM, Lazarus M, Chamberlin NL, Saper CB. Glutamatergic signaling from the parabrachial nucleus plays a critical role in hypercapnic arousal. J Neurosci. 2013;33:7627–7640. doi: 10.1523/JNEUROSCI.0173-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Thankachan S, McKenna JT, McNally JM, Yang C, Choi JH, Chen L, Kocsis B, Deisseroth K, Strecker RE, et al. Cortically projecting basal forebrain parvalbumin neurons regulate cortical gamma band oscillations. Proc Natl Acad Sci U S A. 2015;112:3535–3540. doi: 10.1073/pnas.1413625112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C, Massimini M, Boly M, Tononi G. Neural correlates of consciousness: progress and problems. Nat Rev Neurosci. 2016;17:307–321. doi: 10.1038/nrn.2016.22. [DOI] [PubMed] [Google Scholar]
- Konadhode RR, Pelluru D, Blanco-Centurion C, Zayachkivsky A, Liu M, Uhde T, Glen WB, Jr, van den Pol AN, Mulholland PJ, Shiromani PJ. Optogenetic stimulation of MCH neurons increases sleep. J Neurosci. 2013;33:10257–10263. doi: 10.1523/JNEUROSCI.1225-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenzer M, Anaclet C, Vetrivelan R, Wang N, Vong L, Lowell BB, Fuller PM, Lu J. Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One. 2011;6:e24998. doi: 10.1371/journal.pone.0024998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger D, Ferrari L, Petit G, Mahoney C, Fuller P, Arrigoni E, Scammell T. Cholinergic, glutamatergic, and GABAergic neurons of the PPT have distinct effects on sleep/wake behavior in mice. Journal of Neuroscience. doi: 10.1523/JNEUROSCI.1405-16.2016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Clinton JM, Winters BD, Zielinski MR, Taishi P, Jewett KA, Davis CJ. Involvement of cytokines in slow wave sleep. Prog Brain Res. 2011;193:39–47. doi: 10.1016/B978-0-444-53839-0.00003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubin L. Carbachol models of REM sleep: recent developments and new directions. Arch Ital Biol. 2001;139:147–168. [PubMed] [Google Scholar]
- Kubota A, Inouye ST, Kawamura H. Reversal of multiunit activity within and outside the suprachiasmatic nucleus in the rat. Neurosci Lett. 1981;27:303–308. doi: 10.1016/0304-3940(81)90447-x. [DOI] [PubMed] [Google Scholar]
- Lai YY, Siegel JM. Medullary regions mediating atonia. J Neurosci. 1988;8:4790–4796. doi: 10.1523/JNEUROSCI.08-12-04790.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M, Shen HY, Cherasse Y, Qu WM, Huang ZL, Bass CE, Winsky-Sommerer R, Semba K, Fredholm BB, Boison D, et al. Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. J Neurosci. 2011;31:10067–10075. doi: 10.1523/JNEUROSCI.6730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IT, Chang AS, Manandhar M, Shan Y, Fan J, Izumo M, Ikeda Y, Motoike T, Dixon S, Seinfeld JE, et al. Neuromedin s-producing neurons act as essential pacemakers in the suprachiasmatic nucleus to couple clock neurons and dictate circadian rhythms. Neuron. 2015;85:1086–1102. doi: 10.1016/j.neuron.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LD, Voigts J, Flores FJ, Schmitt LI, Wilson MA, Halassa MM, Brown EN. Thalamic reticular nucleus induces fast and local modulation of arousal state. Elife. 2015;4:e08760. doi: 10.7554/eLife.08760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, Guttler T, Davis F, Asara JM, Sahin M. The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell. 2015;161:1138–1151. doi: 10.1016/j.cell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu Q, Tabuchi M, Wu MN. Sleep Drive Is Encoded by Neural Plastic Changes in a Dedicated Circuit. Cell. 2016;165:1347–1360. doi: 10.1016/j.cell.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhou J, Li Y, Hu F, Lu Y, Ma M, Feng Q, Zhang JE, Wang D, Zeng J, et al. Dorsal raphe neurons signal reward through 5-HT and glutamate. Neuron. 2014;81:1360–1374. doi: 10.1016/j.neuron.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Bjorkum AA, Xu M, Gaus SE, Shiromani PJ, Saper CB. Selective activation of the extended ventrolateral preoptic nucleus during rapid eye movement sleep. J Neurosci. 2002;22:4568–4576. doi: 10.1523/JNEUROSCI.22-11-04568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Greco MA, Shiromani P, Saper CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000;20:3830–3842. doi: 10.1523/JNEUROSCI.20-10-03830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci. 2006a;26:193–202. doi: 10.1523/JNEUROSCI.2244-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006b;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Lu J, Zhang YH, Chou TC, Gaus SE, Elmquist JK, Shiromani P, Saper CB. Contrasting effects of ibotenate lesions of the paraventricular nucleus and subparaventricular zone on sleep-wake cycle and temperature regulation. J Neurosci. 2001;21:4864–4874. doi: 10.1523/JNEUROSCI.21-13-04864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas RJ, Freedman MS, Lupi D, Munoz M, David-Gray ZK, Foster RG. Identifying the photoreceptive inputs to the mammalian circadian system using transgenic and retinally degenerate mice. Behav Brain Res. 2001;125:97–102. doi: 10.1016/s0166-4328(01)00274-1. [DOI] [PubMed] [Google Scholar]
- Luebke JI, Greene RW, Semba K, Kamondi A, McCarley RW, Reiner PB. Serotonin hyperpolarizes cholinergic low-threshold burst neurons in the rat laterodorsal tegmental nucleus in vitro. Proc Natl Acad Sci U S A. 1992;89:743–747. doi: 10.1073/pnas.89.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luppi PH, Aston-Jones G, Akaoka H, Chouvet G, Jouvet M. Afferent projections to the rat locus coeruleus demonstrated by retrograde and anterograde tracing with cholera-toxin B subunit and Phaseolus vulgaris leucoagglutinin. Neuroscience. 1995;65:119–160. doi: 10.1016/0306-4522(94)00481-j. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O, Sapin E, Peyron C, Gervasoni D, Leger L, Fort P. Brainstem mechanisms of paradoxical (REM) sleep generation. Pflugers Arch. 2012;463:43–52. doi: 10.1007/s00424-011-1054-y. [DOI] [PubMed] [Google Scholar]
- Lutter M, Krishnan V, Russo SJ, Jung S, McClung CA, Nestler EJ. Orexin signaling mediates the antidepressant-like effect of calorie restriction. J Neurosci. 2008;28:3071–3075. doi: 10.1523/JNEUROSCI.5584-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Moorman DE, Smith RJ, James MH, Aston-Jones G. Motivational activation: a unifying hypothesis of orexin/hypocretin function. Nat Neurosci. 2014;17:1298–1303. doi: 10.1038/nn.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney KJ, Mainville L, Jones BE. c-Fos expression in GABAergic, serotonergic, and other neurons of the pontomedullary reticular formation and raphe after paradoxical sleep deprivation and recovery. J Neurosci. 2000;20:4669–4679. doi: 10.1523/JNEUROSCI.20-12-04669.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns ID, Alonso A, Jones BE. Discharge properties of juxtacellularly labeled and immunohistochemically identified cholinergic basal forebrain neurons recorded in association with the electroencephalogram in anesthetized rats. J Neurosci. 2000;20:1505–1518. doi: 10.1523/JNEUROSCI.20-04-01505.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Peek CB, Affinati A, Maury E, Bass J. Circadian clocks and metabolism. Handb Exp Pharmacol. 2013:127–155. doi: 10.1007/978-3-642-25950-0_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury E, Ramsey KM, Bass J. Circadian rhythms and metabolic syndrome: from experimental genetics to human disease. Circ Res. 2010;106:447–462. doi: 10.1161/CIRCRESAHA.109.208355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, Chesham JE, O’Brien JA, Hastings MH. A diversity of paracrine signals sustains molecular circadian cycling in suprachiasmatic nucleus circuits. Proc Natl Acad Sci U S A. 2011;108:14306–14311. doi: 10.1073/pnas.1101767108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, McGinley MJ, Salkoff DB. Brain state dependent activity in the cortex and thalamus. Curr Opin Neurobiol. 2015;31:133–140. doi: 10.1016/j.conb.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinley MJ, Vinck M, Reimer J, Batista-Brito R, Zagha E, Cadwell CR, Tolias AS, Cardin JA, McCormick DA. Waking State: Rapid Variations Modulate Neural and Behavioral Responses. Neuron. 2015;87:1143–1161. doi: 10.1016/j.neuron.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty DJ, Sterman MB. Sleep suppression after basal forebrain lesions in the cat. Science. 1968;160:1253–1255. doi: 10.1126/science.160.3833.1253. [DOI] [PubMed] [Google Scholar]
- Mendelson WB, Bergmann BM. Effects of pinealectomy on baseline sleep and response to sleep deprivation. Sleep. 2001;24:369–373. doi: 10.1093/sleep/24.4.369. [DOI] [PubMed] [Google Scholar]
- Mieda M, Hasegawa E, Kisanuki YY, Sinton CM, Yanagisawa M, Sakurai T. Differential roles of orexin receptor-1 and -2 in the regulation of non-REM and REM sleep. J Neurosci. 2011;31:6518–6526. doi: 10.1523/JNEUROSCI.6506-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieda M, Ono D, Hasegawa E, Okamoto H, Honma K, Honma S, Sakurai T. Cellular clocks in AVP neurons of the SCN are critical for interneuronal coupling regulating circadian behavior rhythm. Neuron. 2015;85:1103–1116. doi: 10.1016/j.neuron.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Mistlberger RE. Circadian regulation of sleep in mammals: role of the suprachiasmatic nucleus. Brain Res Brain Res Rev. 2005;49:429–454. doi: 10.1016/j.brainresrev.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Miyamoto H, Nakamaru-Ogiso E, Hamada K, Hensch TK. Serotonergic integration of circadian clock and ultradian sleep-wake cycles. J Neurosci. 2012;32:14794–14803. doi: 10.1523/JNEUROSCI.0793-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T, Arrigoni E, Marcus JN, Clark EL, Yamamoto M, Honer M, Borroni E, Lowell BB, Elmquist JK, Scammell TE. Orexin receptor 2 expression in the posterior hypothalamus rescues sleepiness in narcoleptic mice. Proc Natl Acad Sci U S A. 2011;108:4471–4476. doi: 10.1073/pnas.1012456108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk JA, Takahashi JS. Cell autonomy and synchrony of suprachiasmatic nucleus circadian oscillators. Trends Neurosci. 2011;34:349–358. doi: 10.1016/j.tins.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morairty SR, Dittrich L, Pasumarthi RK, Valladao D, Heiss JE, Gerashchenko D, Kilduff TS. A role for cortical nNOS/NK1 neurons in coupling homeostatic sleep drive to EEG slow wave activity. Proc Natl Acad Sci U S A. 2013;110:20272–20277. doi: 10.1073/pnas.1314762110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin LP. Neuroanatomy of the extended circadian rhythm system. Exp Neurol. 2013;243:4–20. doi: 10.1016/j.expneurol.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moruzzi G, Magoun H. Brain stem reticular formation and activation of the EEG. Electroenceph Clin Neurophysiol. 1949;1:455. [PubMed] [Google Scholar]
- Murray NM, Buchanan GF, Richerson GB. Insomnia Caused by Serotonin Depletion is Due to Hypothermia. Sleep. 2015;38:1985–1993. doi: 10.5665/sleep.5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nagumo Y, Hashimoto S, Khotib J, Miyatake M, Sakurai T, Yanagisawa M, Nakamachi T, Shioda S, Suzuki T. Direct involvement of orexinergic systems in the activation of the mesolimbic dopamine pathway and related behaviors induced by morphine. J Neurosci. 2006;26:398–405. doi: 10.1523/JNEUROSCI.2761-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor E, Bergmann BM, Krauski K, Zee PC, Takahashi JS, Vitaterna MH, Turek FW. The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci. 2000;20:8138–8143. doi: 10.1523/JNEUROSCI.20-21-08138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, Kupfer DJ. Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry. 2004;161:2126–2128. doi: 10.1176/appi.ajp.161.11.2126. [DOI] [PubMed] [Google Scholar]
- Nunez AA, Bult A, McElhinny TL, Smale L. Daily rhythms of Fos expression in hypothalamic targets of the suprachiasmatic nucleus in diurnal and nocturnal rodents. J Biol Rhythms. 1999;14:300–306. doi: 10.1177/074873099129000713. [DOI] [PubMed] [Google Scholar]
- O’Neill JS, Maywood ES, Hastings MH. Cellular mechanisms of circadian pacemaking: beyond transcriptional loops. Handb Exp Pharmacol. 2013:67–103. doi: 10.1007/978-3-642-25950-0_4. [DOI] [PubMed] [Google Scholar]
- Parmentier R, Zhao Y, Perier M, Akaoka H, Lintunen M, Hou Y, Panula P, Watanabe T, Franco P, Lin JS. Role of histamine H1-receptor on behavioral states and wake maintenance during deficiency of a brain activating system: A study using a knockout mouse model. Neuropharmacology. 2016;106:20–34. doi: 10.1016/j.neuropharm.2015.12.014. [DOI] [PubMed] [Google Scholar]
- Parvizi J, Damasio AR. Neuroanatomical correlates of brainstem coma. Brain. 2003;126:1524–1536. doi: 10.1093/brain/awg166. [DOI] [PubMed] [Google Scholar]
- Pimentel D, Donlea JM, Talbot CB, Song SM, Thurston AJ, Miesenbock G. Operation of a homeostatic sleep switch. Nature. 2016;536:333–337. doi: 10.1038/nature19055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner J, Saper C, Schiff N, Plum F. Plum and Posner’s Diagnosis of Stupor and Coma. New York: Oxford University Press; 2007. [Google Scholar]
- Radulovacki M, Virus RM, Djuricic NM, Green RD. Adenosine analogs and sleep in rats. J Pharmacol Exp Ther. 1984;228:268–274. [PubMed] [Google Scholar]
- Ranson S. Somnolence caused by hypothalamic lesions in the monkey. Archives of Neurology and Psychiatry. 1939;41:1–23. [Google Scholar]
- Rao Y, Liu ZW, Borok E, Rabenstein RL, Shanabrough M, Lu M, Picciotto MR, Horvath TL, Gao XB. Prolonged wakefulness induces experience-dependent synaptic plasticity in mouse hypocretin/orexin neurons. J Clin Invest. 2007;117:4022–4033. doi: 10.1172/JCI32829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasch B, Born J. About sleep’s role in memory. Physiol Rev. 2013;93:681–766. doi: 10.1152/physrev.00032.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer J, Froudarakis E, Cadwell CR, Yatsenko D, Denfield GH, Tolias AS. Pupil fluctuations track fast switching of cortical states during quiet wakefulness. Neuron. 2014;84:355–362. doi: 10.1016/j.neuron.2014.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito YC, Tsujino N, Hasegawa E, Akashi K, Abe M, Mieda M, Sakimura K, Sakurai T. GABAergic neurons in the preoptic area send direct inhibitory projections to orexin neurons. Front Neural Circuits. 2013;7:192. doi: 10.3389/fncir.2013.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Crochet S, Onoe H. Pontine structures and mechanisms involved in the generation of paradoxical (REM) sleep. Arch Ital Biol. 2001;139:93–107. [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–1042. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapin E, Lapray D, Berod A, Goutagny R, Leger L, Ravassard P, Clement O, Hanriot L, Fort P, Luppi PH. Localization of the brainstem GABAergic neurons controlling paradoxical (REM) sleep. PLoS One. 2009;4:e4272. doi: 10.1371/journal.pone.0004272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Suzuki M, Mieda M, Tsujino N, Roth B, Sakurai T. Pharmacogenetic modulation of orexin neurons alters sleep/wakefulness states in mice. PLoS One. 2011;6:e20360. doi: 10.1371/journal.pone.0020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer FA, Czeisler CA. Melatonin, sleep, and circadian rhythms. Sleep Med Rev. 2005;9:5–9. doi: 10.1016/j.smrv.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Schenkel E, Siegel JM. REM sleep without atonia after lesions of the medial medulla. Neurosci Lett. 1989;98:159–165. doi: 10.1016/0304-3940(89)90503-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt LI, Sims RE, Dale N, Haydon PG. Wakefulness affects synaptic and network activity by increasing extracellular astrocyte-derived adenosine. J Neurosci. 2012;32:4417–4425. doi: 10.1523/JNEUROSCI.5689-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schone C, Apergis-Schoute J, Sakurai T, Adamantidis A, Burdakov D. Coreleased orexin and glutamate evoke nonredundant spike outputs and computations in histamine neurons. Cell Rep. 2014;7:697–704. doi: 10.1016/j.celrep.2014.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, et al. Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature. 2015;524:88–92. doi: 10.1038/nature14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Shi YF, Han Y, Su YT, Yang JH, Yu YQ. Silencing of Cholinergic Basal Forebrain Neurons Using Archaerhodopsin Prolongs Slow-Wave Sleep in Mice. PLoS One. 2015;10:e0130130. doi: 10.1371/journal.pone.0130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Tomaszewski KS. REM sleep signs rostral to chronic transections at the pontomedullary junction. Neurosci Lett. 1984;45:241–246. doi: 10.1016/0304-3940(84)90233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver R, LeSaute rJ, Tresco P, Lehman M. A diffusible coupling signal from the transplanted suprachiasmatic nucleus controlling circadian locomotor rhythms. Nature. 1996;382:810–813. doi: 10.1038/382810a0. [DOI] [PubMed] [Google Scholar]
- Soja PJ, Morales FR, Baranyi A, Chase MH. Effect of inhibitory amino acid antagonists on IPSPs induced in lumbar motoneurons upon stimulation of the nucleus reticularis gigantocellularis during active sleep. Brain Res. 1987;423:353–358. doi: 10.1016/0006-8993(87)90862-6. [DOI] [PubMed] [Google Scholar]
- Southey BR, Lee JE, Zamdborg L, Atkins N, Jr, Mitchell JW, Li M, Gillette MU, Kelleher NL, Sweedler JV. Comparing label-free quantitative peptidomics approaches to characterize diurnal variation of peptides in the rat suprachiasmatic nucleus. Anal Chem. 2014;86:443–452. doi: 10.1021/ac4023378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Dossi RC, Pare D, Oakson G. Fast oscillations (20–40 Hz) in thalamocortical systems and their potentiation by mesopontine cholinergic nuclei in the cat. Proc Natl Acad Sci U S A. 1991;88:4396–4400. doi: 10.1073/pnas.88.10.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymusiak R, McGinty D. Sleep-related neuronal discharge in the basal forebrain of cats. Brain Res. 1986;370:82–92. doi: 10.1016/0006-8993(86)91107-8. [DOI] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291:1040–1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu T, Ueno T, Tabuchi S, Inutsuka A, Tanaka KF, Hasuwa H, Kilduff TS, Terao A, Yamanaka A. Optogenetic manipulation of activity and temporally controlled cell-specific ablation reveal a role for MCH neurons in sleep/wake regulation. J Neurosci. 2014;34:6896–6909. doi: 10.1523/JNEUROSCI.5344-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uschakov A, Gong H, McGinty D, Szymusiak R. Efferent projections from the median preoptic nucleus to sleep- and arousal-regulatory nuclei in the rat brain. Neuroscience. 2007;150:104–120. doi: 10.1016/j.neuroscience.2007.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uschakov A, Grivel J, Cvetkovic-Lopes V, Bayer L, Bernheim L, Jones BE, Muhlethaler M, Serafin M. Sleep-deprivation regulates alpha-2 adrenergic responses of rat hypocretin/orexin neurons. PLoS One. 2011;6:e16672. doi: 10.1371/journal.pone.0016672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dort CJ, Zachs DP, Kenny JD, Zheng S, Goldblum RR, Gelwan NA, Ramos DM, Nolan MA, Wang K, Weng FJ, et al. Optogenetic activation of cholinergic neurons in the PPT or LDT induces REM sleep. Proc Natl Acad Sci U S A. 2015;112:584–589. doi: 10.1073/pnas.1423136112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varin C, Arthaud S, Salvert D, Gay N, Libourel PA, Luppi PH, Leger L, Fort P. Sleep architecture and homeostasis in mice with partial ablation of melanin-concentrating hormone neurons. Behav Brain Res. 2016;298:100–110. doi: 10.1016/j.bbr.2015.10.051. [DOI] [PubMed] [Google Scholar]
- Venner A, Anaclet C, Broadhurst RY, Saper CB, Fuller PM. A Novel Population of Wake-Promoting GABAergic Neurons in the Ventral Lateral Hypothalamus. Curr Biol. 2016 doi: 10.1016/j.cub.2016.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivelan R, Fuller PM, Tong Q, Lu J. Medullary circuitry regulating rapid eye movement sleep and motor atonia. J Neurosci. 2009;29:9361–9369. doi: 10.1523/JNEUROSCI.0737-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivelan R, Kong D, Ferrari LL, Arrigoni E, Madara JC, Bandaru SS, Lowell BB, Lu J, Saper CB. Melanin-concentrating hormone neurons specifically promote rapid eye movement sleep in mice. Neuroscience. 2016;336:102–113. doi: 10.1016/j.neuroscience.2016.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Economo C. Sleep as a problem of localization. Journal of Nervous and Mental Disorders. 1930;71:249–259. [Google Scholar]
- Vujovic N, Gooley JJ, Jhou TC, Saper CB. Projections from the subparaventricular zone define four channels of output from the circadian timing system. J Comp Neurol. 2015;523:2714–2737. doi: 10.1002/cne.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature. 2011;472:443–447. doi: 10.1038/nature10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Castel M, Gainer H, Yarom Y. GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature. 1997;387:598–603. doi: 10.1038/42468. [DOI] [PubMed] [Google Scholar]
- Wang HL, Morales M. Pedunculopontine and laterodorsal tegmental nuclei contain distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur J Neurosci. 2009;29:340–358. doi: 10.1111/j.1460-9568.2008.06576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Chung S, Beier KT, Xu M, Luo L, Dan Y. Control of REM sleep by ventral medulla GABAergic neurons. Nature. 2015;526:435–438. doi: 10.1038/nature14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbourd B, Ren J, DeLoach KE, Guenthner CJ, Miyamichi K, Luo L. Presynaptic partners of dorsal raphe serotonergic and GABAergic neurons. Neuron. 2014;83:645–662. doi: 10.1016/j.neuron.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DK, Takahashi JS, Kay SA. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol. 2010;72:551–577. doi: 10.1146/annurev-physiol-021909-135919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng FJ, Williams RH, Hawryluk JM, Lu J, Scammell TE, Saper CB, Arrigoni E. Carbachol excites sublaterodorsal nucleus neurons projecting to the spinal cord. J Physiol. 2014;592:1601–1617. doi: 10.1113/jphysiol.2013.261800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Reiner PB. Noradrenaline hyperpolarizes identified rat mesopontine cholinergic neurons in vitro. J Neurosci. 1993;13:3878–3883. doi: 10.1523/JNEUROSCI.13-09-03878.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RH, Chee MJ, Kroeger D, Ferrari LL, Maratos-Flier E, Scammell TE, Arrigoni E. Optogenetic-mediated release of histamine reveals distal and autoregulatory mechanisms for controlling arousal. J Neurosci. 2014;34:6023–6029. doi: 10.1523/JNEUROSCI.4838-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. Dopaminergic role in stimulant-induced wakefulness. J Neurosci. 2001;21:1787–1794. doi: 10.1523/JNEUROSCI.21-05-01787.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KP, Jr, McHill AW, Birks BR, Griffin BR, Rusterholz T, Chinoy ED. Entrainment of the human circadian clock to the natural light-dark cycle. Curr Biol. 2013;23:1554–1558. doi: 10.1016/j.cub.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff K, Gatti S, Wettstein JG, Foster RG. Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci. 2010;11:589–599. doi: 10.1038/nrn2868. [DOI] [PubMed] [Google Scholar]
- Xu M, Chung S, Zhang S, Zhong P, Ma C, Chang WC, Weissbourd B, Sakai N, Luo L, Nishino S, et al. Basal forebrain circuit for sleep-wake control. Nat Neurosci. 2015;18:1641–1647. doi: 10.1038/nn.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptacek LJ, Fu YH. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434:640–644. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, et al. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Yu X, Ye Z, Houston CM, Zecharia AY, Ma Y, Zhang Z, Uygun DS, Parker S, Vyssotski AL, Yustos R, et al. Wakefulness Is Governed by GABA and Histamine Cotransmission. Neuron. 2015;87:164–178. doi: 10.1016/j.neuron.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Zecharia A, Zhang Z, Yang Q, Yustos R, Jager P, Vyssotski AL, Maywood ES, Chesham JE, Ma Y, et al. Circadian factor BMAL1 in histaminergic neurons regulates sleep architecture. Curr Biol. 2014;24:2838–2844. doi: 10.1016/j.cub.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zant JC, Kim T, Prokai L, Szarka S, McNally J, McKenna JT, Shukla C, Yang C, Kalinchuk AV, McCarley RW, et al. Cholinergic Neurons in the Basal Forebrain Promote Wakefulness by Actions on Neighboring Non-Cholinergic Neurons: An Opto-Dialysis Study. J Neurosci. 2016;36:2057–2067. doi: 10.1523/JNEUROSCI.3318-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecharia AY, Yu X, Gotz T, Ye Z, Carr DR, Wulff P, Bettler B, Vyssotski AL, Brickley SG, Franks NP, et al. GABAergic inhibition of histaminergic neurons regulates active waking but not the sleep-wake switch or propofol-induced loss of consciousness. J Neurosci. 2012;32:13062–13075. doi: 10.1523/JNEUROSCI.2931-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ferretti V, Guntan I, Moro A, Steinberg EA, Ye Z, Zecharia AY, Yu X, Vyssotski AL, Brickley SG, et al. Neuronal ensembles sufficient for recovery sleep and the sedative actions of alpha2 adrenergic agonists. Nat Neurosci. 2015;18:553–561. doi: 10.1038/nn.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]