

Abstract
A process of regulated necrosis, termed necroptosis, has been recognized as a major contributor to cell death and inflammation occurring under a wide range of pathologic settings. The core event in necroptosis is the formation of the detergent-insoluble “necrosome” complex of homologous Ser/Thr kinases Receptor Interacting Kinase 1 (RIPK1) and Receptor Interacting Kinase 3 (RIPK3), which promotes phosphorylation of a key pro-death effector Mixed Lineage Kinase Domain-like (MLKL) by RIPK3. Core necroptosis mediators are under multiple controls, which have been a subject of intense investigation. Additional, non-necroptotic functions of these factors, primarily in controlling apoptosis and inflammatory responses, have also begun to emerge. This review will provide an overview of the current understanding of the human disease relevance of this pathway, and potential therapeutic strategies, targeting necroptosis mediators in various pathologies.
Necroptosis – what is it and how did we get there?
Many debilitating diseases arise through a disruption of homeostasis, most notably through alterations in the regulation of cell proliferation and cell death [1]. While apoptosis has been long viewed as an exclusive form of regulated cell death, it became clear that this view does not cover all the complexity that occurs pathologically. One of the first definitive observations suggesting existence of the non-apoptotic, yet regulated, forms of cell death emerged from in vitro studies using tumor necrosis factor alpha (TNFα). Early observations had suggested that depending on the cell type, TNFα could induce cell death displaying morphologic features of either apoptosis or pathologic necrosis, which was in part dictated by the activation status of pro-apoptotic caspases [2-4]. These were exciting findings, as it became clear that necrosis can be induced in a regulated manner by the same pro-death signals that were previously associated with regulated apoptotic cell death. Ensuing works showed that kinase activity of a known component of TNF-induced signaling complexes, RIPK1, and inhibition of the activity of the specific initiator caspase, caspase-8, provide conditions for selective initiation of TNF-induced necrosis [5, 6]. These seminal studies provided first insights into the specific factors negotiating this newly defined, non-apoptotic, and regulated form of cell death that manifested with necrotic morphology [7].
Subsequent work by Chan et al. revealed that multiple viral proteins suppress TNF-induced necrosis, while infection with vaccinia virus promoted it, demonstrating that necrosis may be part of physiologic regulation in the context of viral infections, rather than a product of caspase manipulation in vitro [8]. The term necroptosis was first introduced in 2005, when we performed a small molecule screen and identified a highly selective inhibitor of TNF-induced necrosis - necrostatin-1 (Nec-1) [9], which was subsequently found to inhibit RIPK1 [10]. Availability of this inhibitor helped further elucidate the role of necroptosis as a unified regulated mechanism of cell death expressed in multiple cell types as well as in vivo, leading eventually to more than 1,000 references on necroptosis listed on PubMed to date. In this review, we will summarize some of the findings on necroptosis and non-necroptotic regulation by RIPK1 kinase and its partner, RIPK3 kinase, revealing pathologic roles of these exciting druggable targets.
As discussed above, necroptosis is a form of cell death that very closely resembles classic necrosis. Necroptotic cells display enlarged and swollen organelles, early plasma membrane damage with eventual rupture, and the lack of typical nuclear condensation and inter-nucleosomal DNA fragmentation, characteristic for apoptosis. Similar to un-regulated or accidental necrosis, rapid loss of plasma membrane integrity during necroptosis leads to the release of multiple damage (or danger) associated molecular pattern molecules (DAMPs), such as, IL-1α, HMGB1, ATP, uric acid, and HSPs, leading to activation of inflammation [11]. Necroptosis signaling is initiated through a wide range of triggers, including TNFα, FasL, and TRAIL [5], IFNs [12], TLRs [13], and virus-mediated pathways [14]. These and other triggers are further reviewed in [15, 16]. TNFα induced necroptosis is currently best understood mechanistically (Figure 1). Binding of TNFα to TNFR1 leads to the formation of membrane bound Complex I, which is assembled through the recruitment of RIPK1 along with TNF receptor-associated protein with death domain (TRADD) and TNFR-associated factors (TRAF2/TRAF5) [17]. At this point, cellular inhibitors of apoptosis (cIAP1/2) and linear ubiquitin chain assembly complex (LUBAC) are recruited to the complex, and ubiquitinate RIPK1. This ubiquitination of RIPK1 strongly correlates with NFkB activation, although it may not be universally required as no defects in NFkB activation were observed in TNFα-stimulated primary Ripk1-/- hepatocytes and thymocytes, which was in contrast to freshly isolated lung cells [18]. In addition, contribution of RIPK1 to NFkB activation does not require its catalytic kinase activity [19, 20]. The subject of RIPK1 poly-ubiquitination is discussed comprehensively in multiple recent reviews [21-23].
Figure 1.
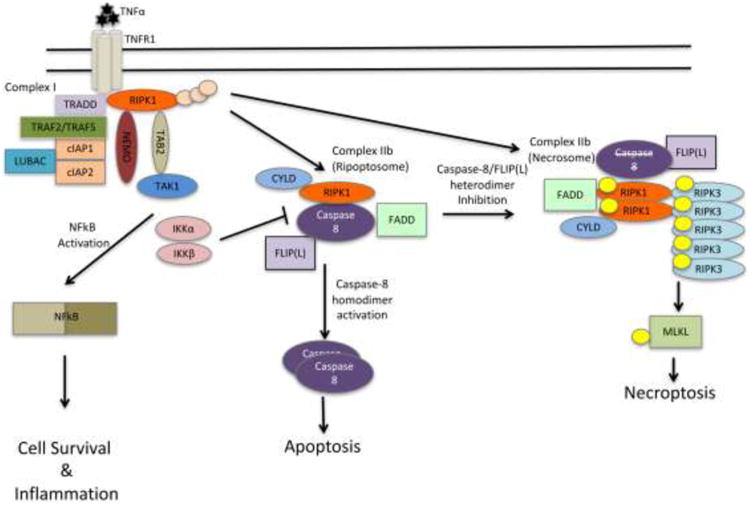
Scheme depicting Complex IIb formation in response to TNFα. Of note, different numbering systems are used in the literature to denote various TNF-induced pro-death complexes. We follow the nomenclature where canonical RIPK1 kinase independent proapototic caspase-8 complex is termed Complex II or Complex IIa, while RIPK1 kinase-dependent complexes are named Complex IIb. Components of ripoptosome and necrosome are indicated putatively as complete composition of these complexes is not yet established. For instance, multiple lines of evidence suggest that RIPK3 may participate in both ripoptosome (as a scaffold) and necrosome (as a kinase). Furthermore, it is also not entirely clear if ripoptosome and necrosome represent largely the same complex, promoting distinct responses in a context-dependent manner, or separate complexes.
While Complex I promotes cell survival, it is the transition to the secondary cytosolic complex, Complex II, which mediates cell death. This transition requires deubiquitination of Complex I, and association with Fas-associated protein with death domain (FADD) and caspase-8 [17]. Complex II may contain different components and promote cell death in a manner that is either independent or dependent on the kinase activity of RIPK1. Pro-death RIPK1 kinase-dependent versions of this complex are usually denoted as Complex IIb (reviewed in [24]). It is not yet entirely clear which endogenous factors dictate differentiation between formation of Complex II and Complex IIb. Formation of Complex IIb (also called “ripoptosome”) promotes activation of apoptosis, in a caspase-8-dependent manner. Upon inhibition of caspase-8, Complex IIb (in this case referred to as “necrosome”) promotes necroptosis. Notably, inhibition of necroptosis has been shown to be mediated by a heterodimer of caspase-8 with FLIP(L), rather than the apoptosis-mediating homodimers of caspase-8 [25]. Multiple events control necrosome formation and signaling, although the precise order of events is not yet fully understood. RIPK1 undergoes de-ubiquitination by CYLD, which is required for necroptosis [26]. Curiously, CYLD may be the critical target of caspase-8 activity in necrosomes [27, 28]; however, deubiquitination of RIPK1 also appears to be critical for induction of apoptosis by Complex IIb. Another deubiquitinase (DUB) that has been linked to the regulation of necroptosis is A20 (Tnfaip3). Catalytic activity of the N-terminal ovarian tumor (OTU) DUB domain of this protein was shown to remove K63 chains from Lys5 residue of RIPK3, which limited necrosome formation and necroptosis [29]. OTU domain of A20 was previously also shown to mediate removal of K63 chains from RIPK1 in Complex I, while C-terminal zinc finger E3 ligase domain (ZF4) was linked to RIPK1 degradation through K48 poly-ubiquitination [30] and inhibition of TRAIL-induced apoptosis through promoting RIPK1 K63 poly-ubiquitination [31]. However, it has not been established whether these regulatory modalities may be relevant to necroptosis. Notably, mutations of C609/C612 residues in ZF4 domain, which are required for A20 binding to ubiquitinated RIPK1, only marginally promoted necroptosis [29]. Constitutive signaling by Type I interferons appears to be essential, at least in some contexts, as it may provide expression of several critical signaling cascade components [28]. Upon activation, RIPK1 and RIPK3 become phosphorylated on multiple sites either by autophosphorylation or cross-phosphorylation and this is also required for necroptosis [32]. Of note, multiple phospho-specific antibodies against RIPK1 (Ser14/15, Ser166) and RIPK3 (Ser232 (mouse)/Ser227 (human)) have been developed and provide new useful tools to analyze initiation of necroptosis [33-37].
Additional phosphorylation events have been suggested to regulate pro-death activity of RIPK1. This includes negative phosphorylation of RIPK1 on Ser89 by yet to be identified kinase(s) [38], and by IKKα/β on yet to be defined site(s) [39]. The latter appears to represent a new function of the IKK complex, independent of its main role as an activator of NFkB. Notably, the third component of IKK complex poly-Ub binding factor NEMO (IKKγ) as well as upstream activator of this complex TAK1 kinase were also shown to suppress RIPK1-mediated cell death in an NFkB-independent manner [40-43], suggesting tight negative coupling of two major TNFα-dependent pathways (IKK and RIPK1 kinase) at the level of Complex IIb. Furthermore, as NEMO and TAK1 associate with poly-ubiquitinated RIPK1 in Complex I [44-47], this regulation is likely tightly linked to RIPK1 ubiquitination status. However, molecular details of this regulation are not sufficiently clear to determine if IKKs, TAK1 and NEMO may control separate regulatory events or these factors share a mechanism of RIPK1 regulation. Additionally, ubiquitination also plays a direct role in RIPK1 activation. Even though inhibition of ubiquitination promotes formation of Complex IIb, a second wave of linear and K63 RIPK1 ubiquitination was shown to occur in the necrosomes. It involved Lys115 site and required catalytic activity of RIPK1, but neither RIPK3 or MLKL [48]. Mutation of this site reduced RIPK1 phosphorylation and ubiquitination, necrosome formation, and necroptosis in cells treated with TNFα, SMAC mimetic and zVAD.fmk, demonstrating that necrosome-associated Lys115 ubiquitination is an essential early event in the formation of stable necrosomes.
RIPK3 is brought into the complex through interaction between homologous RIPK1 and RIPK3 RIP homotypic interaction motif (RHIM) domains, which promote formation of detergent-insoluble amyloid-like structures [49]. In necrosomes, RIPK3 undergoes phosphorylation on Ser232, which is essential for recruitment of downstream necroptosis effector – pseudokinase MLKL [50, 51]. MLKL is the critical necroptosis effector, as Mlkl-/- cells are completely resistant to necroptosis [52]. Notably, the main function of necrosome formation in necroptosis initiation may be to promote formation of RIPK3 homodimers, as enforced homodimerization of just RIPK3 is sufficient for necroptosis even in the absence of interaction with RIPK1 [53-55]. MLKL is directly phosphorylated by RIPK3 on Thr357/Ser358 sites in the activation loop, inducing a conformational change that releases the pro-necrotic membrane-disrupting N-terminal four-helix bundle [56]. MLKL is a gateway for activation of RIPK3 kinase-dependent necroptosis [57-59]. Plasma membrane translocation of MLKL has been linked to perturbation of calcium and sodium fluxes [60, 61], which is associated with increased osmotic pressure [61]. Furthermore, calcium influx through TRPM7 channel was shown to be an early and essential event in MLKL-dependent necroptosis [60]. MLKL-dependent membrane permeabilization can be induced by enforced dimerization of just the N-terminal bundle and brace domains, and involves interactions with phosphoinositides (especially PI(4,5)P2) [58, 62, 63]. However, the precise mechanism of how this ultimate step in necroptosis execution occurs is still a matter of discussion (reviewed in [64]).
Differential Roles of the Catalytic and Scaffold Activities of RIPK1 and RIPK3
As we will discuss further below, RIPK1 and RIPK3 kinases attracted major interest as potentially drug-targetable mediators of pathologic necrosis and associated inflammation. This stems from initial discoveries suggesting critical and selective roles of these kinases in necroptosis. However, subsequent data revealed new levels of complexity of cell death regulation by these proteins. For example, evidence has revealed contribution of their kinase activities to other modes of cell death, namely apoptosis, as well as direct controls of inflammatory gene expression. On the other hand, kinase-independent functions of these proteins should also be considered when interpreting results obtained in Ripk1-/-and Ripk3-/- animals and cells.
Necrostatin-1 (Nec-1) and its optimized analog Nec-1s (7-N1, 7-Cl-O-Nec-1), are highly selective allosteric Type III RIPK1 inhibitors, which have been used extensively to probe the roles of RIPK1 kinase activity under various settings (summarized recently in [65]). Recently developed kinase dead knock-in RIPK1 mice, harboring D138N (Ripk1D138N/D138N) and K45A (Ripk1K45A/K45A) mutations, provided additional valuable tools for the analyses of the kinase-dependent functions of RIPK1 [36, 66]. Current data, generated using these tools as well as Ripk1-/- mice and cells, clearly indicates that RIPK1 possesses multiple kinase-dependent and independent functions. As mentioned above, an important kinase-independent function of RIPK1 in some, but not all circumstances is the rapid NFkB activation upon formation of Complex I by TNFR1, which involves RIPK1 protein ubiquitination on Lys377 [18, 67, 68]. Another example of kinase-independent activity of RIPK1 is its contribution to caspase-8-dependent apoptosis, which was observed upon expression of a catalytically inactive Ripk3D161N/D161N mutant [51, 69]
Initial analyses using Nec-1 and re-expression of kinase-dead RIPK1 in RIPK1-deficient Jurkat cells suggested that kinase activity may be required exclusively for necroptosis induced by TNF receptor family member, Fas [5, 9]. Importantly, both Ripk1D138N/D138N and Ripk1K45A/K45A mice were viable and fertile and displayed no obvious abnormalities, similar to Ripk3-/- and Mlkl-/- animals, suggesting that inhibition of pathology-associated necroptosis may be a feasible therapeutic strategy. While more recent data further support inhibition of RIPK1 kinase as a therapeutic target, it also suggests that the functions of the kinase activity of RIPK1 in disease are not limited just to necroptosis. Several lines of evidence suggest that kinase activity of RIPK1 can also exert direct cell intrinsic controls on pro-inflammatory gene expression and, thus, promote inflammation independently of the DAMPs released as a result of necroptosis. For example, work by our and other labs suggested that RIPK1 kinase activity in L929 and other cell lines is associated with new synthesis and release of TNFα [70-73]. We examined this regulation in macrophages stimulated with LPS and caspase inhibitors and showed that it is mediated by kinase activities of RIPK1 and RIPK3, but necroptosis and MLKL are dispensable [37]. Rather, the responses were mediated by recruitment of inflammatory effectors, in this case Erk1/2, to necrosome-like RIPK1/RIPK3 aggregates (Fig. 2). In another example, Wong et al. showed that genetic deletion of cIAP1/2 and XIAP led to the greatly enhanced production of inflammatory molecules by myeloid cells, which occurred secondary to the de-regulated expression of TNFα [74]. Recent analyses in mouse models of multiple sclerosis (MS) and amyotrophic lateral sclerosis (ALS) suggested that activation of RIPK1 in microglia promoted inflammatory phenotypes of diseased microglia, rather than necroptosis by virtue of low levels of MLKL expression in this cell type [34, 35]. Other examples of this regulation will be discussed in the subsequent sections of this review. Besides necroptosis and inflammation, RIPK1 kinase activity has also been linked to induction of caspase-8-dependent apoptosis as mentioned earlier. For example, inhibition of TAK1 kinase or depletion of cIAP1/2 has been found to promote caspase-8-dependent apoptosis in the presence of TNFα, which required catalytic activity of RIPK1 [42]. Interestingly, RIPK1 appears to be able to promote apoptosis both in a kinase-independent and kinase-dependent manner [42, 69]. An exact mechanism of how RIPK1 kinase activity facilitates apoptosis is not yet known, but these data may suggest that activation of caspase-8 results from a particular conformation of RIPK1 and kinase activity may aid in this conformational change. Hypothetically, this could occur through autophosphorylation events either promoting conformational changes, or creating high affinity docking site(s) for pro-apoptotic effects, or, alternatively, trans-phosphorylation of proteins in the RIPK1/caspase-8 apoptotic complex, accelerating activation of apoptosis.
Figure 2.
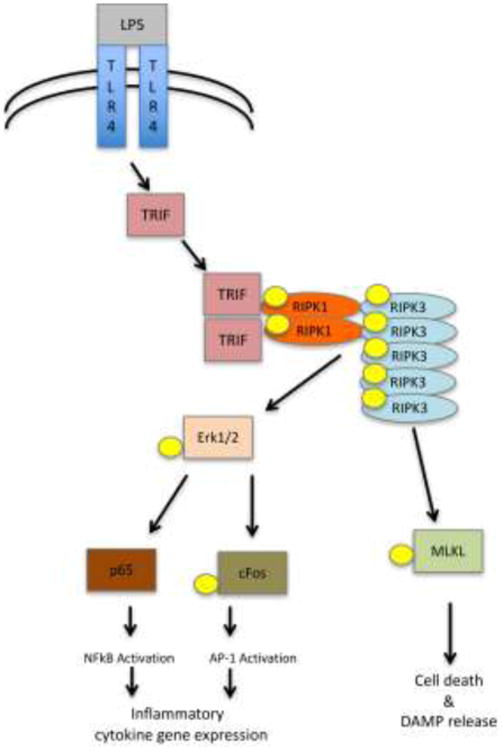
Proposed mechanism for the activation of inflammatory gene expression through RIPK1/RIPK3 kinase activity [37].
Another critical question is whether catalytic activity of RIPK1 is absolutely essential for necroptosis. In a number of instances, upstream signals were shown to induce necroptosis by directly engaging RIPK3 in the absence of RIPK1. In case of virus-induced DAI signaling and in some instances of TLR signaling this was seen in the cells expressing RIPK1. In cases of TNFα and IFN signals this was observed in Ripk1-/- cells [53, 75, 76], highlighting an interesting paradox; inhibition of RIPK1 kinase activity by Nec-1 efficiently prevented necroptosis in cells expressing RIPK1, whereas loss of the entire protein did not prevent necroptosis. One important observation, hinting a possible explanation, is that inhibition of necroptosis by Nec-1 was only observed in the cells expressing RIPK1 [71, 75]. These data suggest that signals normally requiring RIPK1 kinase activity are fundamentally re-wired in the absence of RIPK1 protein to allow RIPK3 activation, resulting in regulation that can by-pass a more upstream RIPK1 step and directly engage RIPK3, as described in the case of DAI [76]. Experiments with Ripk1-/- cells and animals revealed another unexpected and essential function of this protein in guarding against inappropriate activation of both caspase-8-dependent apoptosis and RIPK3-dependent necroptosis in multiple tissues, including skin, liver and intestine [75, 77-81]. These data further suggest that the wiring of both apoptosis and necroptosis is profoundly dependent on the presence of RIPK1 protein, which, in turn, ensures that activation of cell death only occurs upon specific signals through activation of RIPK1 kinase activity. As discussed above, in some instances of apoptosis, RIPK1 may allow cell death in a kinase-independent manner through currently unknown mechanism(s) (e.g. changes to RIPK1 conformation).
Regulation by RIPK3 is similarly complex. In vitro, necroptosis occurs strictly in a RIPK3 kinase dependent manner and is inhibited in both Ripk3-/- cells and by RIPK3 inhibitors. This reflects phosphorylation of MLKL by RIPK3 as a critical step in this regulation. However, we still have limited confirmation for the role(s) of the kinase activity of RIPK3 in vivo due to the lack of available tools. Currently available RIPK3 inhibitors promote apoptosis through the mechanism analogous to the expression of the catalytically inactive RIPK3 mutant gene, Ripk3D161N/D161N [51]. These molecules have not been examined in mice. In contrast, mice expressing the catalytically inactive Ripk3K51A/K51A gene are viable, but display greatly reduced expression of RIPK3 [51]. Thus, data generated using this mutant may reflect both kinase-dependent and kinase-independent functions of RIPK3. In addition, a recent analysis of concanavalin A-induced liver injury in mice [82], which resembles human immune-induced hepatitis, suggested the roles for MLKL and kinase activity of RIPK1, but no role for RIPK3 (as assessed using Ripk3-/- animals). The relevance and mechanistic details of such putative non-canonical RIPK1/MLKL-dependent death remain to be further validated and characterized.
Recently developed Mlkl-/- mice provide a useful selective tool to analyze the role of necroptosis in disease models. To examine the relative roles of known players in necroptosis, a recent study compared Ripk1D138N/D138N and Ripk3-/- mice to Mlkl-/- mice in several models of pathology [83]. For example, in the case of myocardial infarction, loss of any of the three factors was equipotent. However, Mlkl-/- mice were poorly rescued from kidney ischemia-reperfusion and not at all from systemic inflammatory pathology in Tnaip3-/- mice, illustrating that pathologic contributions of RIPK1 kinase activity and/or RIPK3 (acting as a kinase or scaffold) extend beyond necroptosis alone [83].
There are multiple examples of RIPK3 exerting regulation in a kinase-independent manner in vitro. Nogusa et al. examined pathways of Influenza A virus induced death and provided a striking example of RIPK3 acting in a kinase-independent manner to promote clearance of virally infected cells through activation of caspase-8-mediated apoptosis [84]. It has also been shown that RIPK3 can induce inflammatory signaling in a kinase-independent manner through the activation of a non-canonical caspase-8-dependent inflammasome [85]. In a related example of kinase-independent regulation, RIPK3 was found to promote caspase-1 dependent processing of IL-1β, IL-23, and IL-22 in dendritic cells in DSS model of severe colitis [86]. Notably, this was essential for the efficient repair, recovery of the animals, and progression of colorectal carcinogenesis [87]. RIPK3 was also shown to promote activation of canonical NLRP3 inflammasomes and apoptosis, again, acting in a kinase and MLKL-independent manner [88, 89]. This pathway exacerbated interleukin-1 (IL-1)-dependent autoantibody-mediated arthritis.
Overall, the examples we list in this section clearly indicate that the biology of RIPK1 and RIPK3 greatly extends the boundaries of necroptosis and experimental data, generated using Ripk1-/- and Ripk3-/- cells and transgenic animals, should be treated cautiously, given the multitude of the regulatory events that could be affected.
Disease Pathology
Activities of RIPK1 and/or RIPK3 have been implicated in a wide range of disease processes. The following section will highlight some of the established and emerging disease areas that are related to RIPK1 and RIPK3-dependent cell death and/or inflammation triggered by this pathway, which illustrate both the promise as well as complexity of the regulation.
Ischemic Injury
Necrosis is well established to be the major form of cell death in ischemic injuries. Necroptosis, representing a regulated form of necrosis dependent on RIPK1-RIPK3-MLKL, has attracted major interest as a potential component of such injuries. Targeting the damaging effects of necroptosis through the use of Nec-1 in middle cerebral artery occlusion (MCAO) mouse model was initially shown to be effective in reducing ischemia-reperfusion injury (IRI) in the brain [90]. Since then, inhibition of IRI injuries by Nec-1 was observed in multiple tissues, including retina, heart and kidney (summarized in [91]). While these studies confirmed the efficacy of RIPK1 kinase inhibition against IRI, they also raised the question of whether the target of the inhibition is always exclusively RIPK1-RIPK3-MLKL-dependent necroptosis.
Several studies showed strong protection from heart IRI injury by Nec-1 in different animal models [92-94]. Multiple reports also demonstrated strong amelioration of the injury in Ripk3-/- mice [83, 95, 96]. However, analysis of cardiomyocyte IRI injury in vitro suggested that RIPK3 promoted both necroptosis and apoptosis. Furthermore, RIPK3-dependent death was shown to proceed through the activation of CamKII kinase, which is well established to play a role in heart IRI, rather than MLKL [96]. CamKII activation, in this case, was a result of a combination of RIPK3-dependent phosphorylation and oxidation, identifying CamKII as a new target of RIPK3 kinase.
Parallel analyses of kidney IRI revealed only a marginal protection in Mlkl-/-animals, even though Ripk3-/- mice were strongly protected and Ripk1D138N/D138N showed some protection [83]. Conversely, Caspase-8-/-;Ripk3-/- animals were completely resistant to IRI, suggesting that caspase-8 dependent apoptosis, rather than necroptosis may be a primary mode of cell death. In the same study, mouse MCAO injury was not significantly reduced in Ripk3-/- mice, suggesting that it may reflect activation of RIPK1 kinase-dependent, but RIPK3-independent cell death. Overall, these data suggest that RIPK1 inhibitors may represent promising agents against IRI injuries, but necroptosis may not be the primary mediator of this form of injury. The role of kinase activity of RIPK3 in IRI is unclear at this point.
Analyses of the role of phosphoglycerate mutase family member 5 (PGAM5) in IRI injury provided interesting further insights into the potential regulation by RIPK1 and RIPK3. PGAM5 was initially identified as a component of the RIPK1-RIPK3-MLKL complex, which anchors this complex to the mitochondria and mediates both necroptosis and passive oxidative stress-induced necrosis [97]. However, necroptosis was not affected in Pgam5-/- mice [98], and, furthermore, these mice displayed increased IRI injury in the heart and brain [99]. PGAM5 is important for the normal mitochondrial homeostasis by promoting mitophagy to rid cells of dysfunctional mitochondria [100]. Thus, the loss of protective mitophagy, counteracting induction of necroptosis, was proposed as a mechanism of increased susceptibility of Pgam5-/- animals to IRI injuries [99]. These data revealed yet another facet of cellular sensitivity to RIPK1 and RIPK3-mediated death.
Acute and Chronic Inflammatory Diseases
Many pathologies involving RIPK1 and RIPK3 are associated with the acute or chronic inflammation as well as disruption of immune homeostasis. Understanding the signaling pathways linking necroptosis mediators to inflammatory and immune regulatory circuits may help uncover potential treatments for debilitating diseases that often have lengthy, difficult, and sometimes unsuccessful treatment options. The contribution of RIPK1 and RIPK3 kinases to inflammatory and autoimmune pathologies has been an area of major focus and has provided many interesting conclusions.
Work by Duprez et al in 2011 suggested that RIPK1/3-mediated necroptosis might be the basis for systemic inflammation and mortality in TNF-induced systemic inflammatory response syndrome (SIRS) [101]. The results, using both Nec-1 and Ripk3-/- animals, indicated efficient protection from hypothermia, death, and reduction in the damage to multiple tissues including liver and heart and/or kidney. In contrast, intestinal injury, primarily occurring through apoptosis, was not affected. Systemic release of multiple DAMPs, such as LDH, HEX, and three different mitochondrial DNA (mtDNA) genes, were also reduced. Although initial inflammatory response was not affected, there was a significant reduction in the sustained inflammation (e.g. levels of IL1 and IL6 at 6 hr after TNF injection), which likely reflects DAMP-mediated responses. Notably, Ripk3-/- mice were protected from sepsis in the cecal ligation and puncture model, suggesting a universal role for RIPK3 in sepsis-like inflammatory conditions.
TNFα plays a critical role in a variety of inflammatory and autoimmune pathologies (summarized in [102]). Findings by Duprez et al. provided a first dramatic demonstration that the systemic toxicity of this cytokine may not be a direct consequence of acute TNFR-mediated pro-inflammatory signaling, but rather reflects widespread necroptotic cell death and DAMP-mediated systemic inflammation. The significant role of necroptosis in SIRS is further supported by recent demonstration that Mlkl-/- mice are also protected from SIRS [83]. However, protection was partial, and Caspase-8-/- provided added benefit to Ripk3-/-, suggesting that overall cell death was a result of both necroptosis and apoptosis.
Even though original observations by Duprez et al. suggested that necroptosis may not be involved in acute intestinal injury induced by injection of TNFα, recent evidence indicates that RIPK1 and RIPK3 may play important roles in intestinal injuries under different settings. Upregulation of RIPK3 and MLKL and reduction of caspase-8 were observed in the biopsies from the children with Inflammatory Bowel Disease (IBD) [103]. In another study, administration of Nec-1 was shown to attenuate disease development, inflammation, and HMGB1 release, as well as, inflammation-driven colorectal tumorigenesis in the DSS model of colitis [104]. Work presented by Vlanis et al. showed that the loss of Nuclear Factor kB (NFkB) essential modulator (NEMO) in intestinal epithelium cells (IEC) led to the disruption of intestinal immune homeostasis, cell death and development of the chronic inflammation [41]. NEMO is a component of the IKK complex, required for NFkB activation. However, comparable injury was not observed in triple knockout mice deficient in all three transactivating subunits of NFkB (p65, c-Rel and RelB), suggesting the phenotype of NEMOIEC-KO was not due to the loss of NFkB activation. Instead, development of intestinal auto-inflammatory disease was dependent on TNFR1 signaling and intestinal microbiota. NEMOIEC-KO mice displayed severe loss of Paneth cells, which was completely prevented by RIPK1D138ND138N or if both caspase-8-dependent apoptosis and RIPK3-dependent necroptosis were inhibited. Overall, the authors proposed a model of intestinal injury in which the loss of NEMO sets in motion a powerful self-amplifying loop, encompassing progressive loss of epithelial barrier integrity due to the RIPK1 kinase-dependent Paneth cell apoptosis and necroptosis, followed by microbiota-dependent inflammation and TNFα production, further potentiating RIPK1 kinase-dependent cell death. NEMO likely restrains RIPK1 activation directly, by binding to poly-ubiquitin chains on RIPK1, rather than through the regulation of NFkB [40]. Similar auto-amplification cascades, involving cooperation between cell death and inflammation, controlled directly by RIPK1 kinase, or mediated by DAMPs, or induced by exposure to new exogenous signals (e.g. exposure to commensal bacteria), may represent a general feature of RIPK-dependent inflammatory pathologies.
Curiously, while NEMO loss in the intestine led to the mixture of necroptosis and apoptosis, loss of this protein in the liver parenchymal cells (LPC) led to the development of steatohepatitis and hepatocellular carcinoma (HCC), which were mediated exclusively by RIPK1 kinase-dependent apoptosis and not necroptosis [105]. However, the reasons for preferential activation of RIPK1-dependent apoptosis vs. necroptosis in different tissues are not yet known. In several animal pathologies, cell death-independent controls of inflammation by RIPK1 and RIPK3 have been shown to primarily contribute to the disease. In one striking example, necroptosis-independent pro-inflammatory signaling by RIPK1 kinase was proposed to promote auto-inflammatory skin disorders, such as neutrophilic dermatosis in Ptnp6spin mice [106]. Development of this pathology was found to be driven by the excessive release of IL-1α, which promoted improper wound healing. Excessive production of IL-1α was found to be mediated by RIPK1 in a Erk and NFkB-dependent manner. Administration of Nec-1 as well as fetal liver transplantation of Ripk1-/- cells protected against this wound-induced inflammation and tissue damage. Notably, no protection was seen in Ripk3-/- mice.
IL-1α is also the cause for inflammation that drives atherosclerotic progression as an antibody against this cytokine reduced development of the atherosclerotic plaques in ApoE-/- mice [107]. Two independent studies [33, 107] demonstrated that Ripk3-/- significantly attenuated plaque formation by controlling systemic inflammation, including IL1α production. However, in this case, systemic inflammatory responses were proposed to reflect macrophage necroptosis in plaques, rather than direct control of gene expression by RIPK3. The contribution of RIPK1 to this disease has not yet been reported.
Loss of SHARPIN, a component of linear ubiquitination LUBAC complex, in Sharpincpdm/cpdm mice leads to robust dermatitis and multi-organ inflammatory pathology, which is dependent on TNFR1-mediated cell death [108]. Strikingly, all aspects of this pathology were prevented in Ripk1K45/K45A kinase dead knock-in animals [36]. Further, in line with our earlier discussion of tissue specific roles of RIPK1-dependent apoptosis and necroptosis, Rickard et al. showed that dermatitis was specifically attenuated in Sharpincpdm/cpdm;Caspase-8-/+ mice, while deficiency in RIPK3 or MLKL attenuated splenomegaly and liver inflammation [108]. Combined loss of caspase-8 and RIPK3 markedly delayed development of pathology in Sharpincpdm/cpdm;Casp8+/-;Ripk3-/- including sparing of Peyer's patches in the gut, which was not prevented by single loss of RIPK3 or caspase-8. Notably, human keratinocyte HaCaT cells expressing catalytically inactive mutant of another LUBAC component HOIP protein displayed similar hypersensitivity to TNF-induced cell death. These data may hold new hope for the patients with deficiencies in LUBAC components HOIP and HOIL1, which lead to the development of the auto-inflammatory pathology [109, 110]. Notably, complete loss of caspase-8 in Sharpincpdm/cpdm;Casp8-/−;Ripk3−/− mice displayed perinatal mortality [108]. This is curious, as similar mortality was not observed in Sharpincpdm/cpdm;Ripk1K45A/K45A mice [36]. It is tempting to speculate that this model may unexpectedly reveal the consequences of unbalanced activation of RIPK1 in the absence of RIPK3/caspase-8, but this possibility has not yet been addressed.
Axonal Degeneration
In addition to the above examples, necroptosis mediators were also implicated in the sustained inflammation in multiple other disease states. Multiple Sclerosis (MS) is a neuroinflammatory disease characterized by the progressive demyelination of axons in the central nervous system. Intriguingly, Ofengeim et al. observed that active white matter lesions of MS patients displayed defective caspase-8 activation and an elevation in TNFα [35]. The authors further showed that these conditions provide a prime setting for the activation of RIPK1 and RIPK3 kinases and necroptosis. Consistently, elevated levels of phosphorylated and/or total RIPK1, RIPK3 and MLKL were further found in lesioned tissue. In in vivo experiments, administration of optimized Nec-1 analog, 7N-1 (Nec-1s), was shown to preserve oligodendrocytes, reduce the loss of myelin, and reduce neuroinflammation in the cuprizone model of MS. Notably, oligodendrocytes were found to be highly sensitive to TNFα-induced necroptosis in vitro. Oligodendrocyte degeneration is a prominent event in the cuprizone model, and Ripk3-/- mice were protected comparably to 7N-1 in this model. In contrast, autoimmune/allergic encephalomyelitis (EAE) model of MS involves neuroinflammation mediated by infiltrating monocytes and activated microglia. The latter express high levels of RIPK1, but low levels of MLKL and are resistant to necroptosis. Mice were strongly protected by 7N-1 in the EAE model, but weaker protection was observed in Ripk3-/- mice, consistent with RIPK1 kinase-dependent inflammatory signaling in microglia cells playing a significant role. Overall, these data suggested that RIPK1 kinase plays a primary role in the development of MS-like demyelination through both RIPK3/MLKL-dependent necroptosis of oligodendrocytes and microglia/monocyte-dependent inflammation.
Similar findings were recently made in another disease, involving axonal degeneration - amyotrophic lateral sclerosis (ALS). The initial work by Re et al. showed that human adult primary sporadic and familial ALS (sALS and fALS) astrocytes triggered death of human embryonic stem cell-derived motor neurons (MNs) in vitro [111]. This new model reproduced a key feature of the disease: ability of diseased astrocytes, but not the control cells to induce death in MNs. Furthermore, both sALS and fALS astrocytes induced necroptosis, which was blocked by Nec-1 and necrosulfonamide, a small molecule inhibitor of human MLKL [57], suggesting that necroptosis may be a new player in this devastating disease. Recently published analysis by Ito et al. examined the consequences of the loss-of-function mutations in the Optineurin (Optn) gene, which are associated with both sALS and fALS [34]. The authors reported that the loss of Optn (Optn-/-) in the oligodendrocytes and myeloid cells led to the axonal loss and dysmyelination in the spinal cords, resembling pathology in ALS. Levels of RIPK1, RIPK3 and MLKL were increased in the spinal cords of Optn-/- mice as well as in the white matter lesions in the spinal cords of ALS patients. In part, these changes were explained by the direct association of OPTN and RIPK1, which promoted the proteosomal degradation of the latter. Optn-/-oligodendrocytes showed significantly greater sensitivity to TNF-induced death, which was blocked by Nec-1s and RIPK1D138N/D138N. Optn-/- microglia showed increased phosphorylation of RIPK1 and increased expression of a number of inflammatory genes suggestive of M1-like polarization. Importantly, the axonal pathology of Optn–/– mice was completely rescued in Optn–/–;Ripk1D138N/D138N and the Optn–/–;Ripk3–/– double-mutant mice as well as by the administration of Nec-1s. Notably, Ripk3–/– and Nec-1s also rescued axonal pathology in the SOD1G93A fALS model. Overall, these data, again, provided a striking demonstration of a cooperative contribution of both necroptosis and inflammatory signaling downstream from RIPK1 kinase in the development of axonal degeneration.
Autoinflammatory and autoimmune pathology in the absence of necroptosis
Extensive analyses by Alvrez-Diaz et al. and Zhang et al. examined the roles of RIPK3 and MLKL in autoimmune diseases. Both groups observed that while Mlkl-/- rescued embryonic lethality due to activation of necroptosis in Caspase-8-/- and Fadd-/- mice, these mice rapidly succumbed to extremely severe systemic autoimmune disease, lymphadenopathy, and thrombocytopenia [112, 113]. Interestingly, while the development of autoimmunity may be hypothesized to result from a deficiency of both apoptosis and necroptosis in these mice; however, cell death-deficient Ripk3-/-;Caspase-8-/- and Ripk3-/-; Fadd-/- mice developed much milder autoimmune disease. One potential clue to understanding this discrepancy comes from the observation that deficiency in MLKL, which has been previously shown to mediate necroptosis exclusively, led to the increased systemic inflammation, and this was not seen in RIPK3-deficient mice [113]. This may reflect our recent observation that caspase-8 inhibition in macrophages promoted inflammatory gene expression, which was blocked in Ripk3-/-, but not Mlkl-/- cells [37]. Further, this raises the possibility that the combination of intact pro-inflammatory signaling by RIPK1/RIPK3 and deficient apoptosis and necroptosis provides an optimal environment for the development of autoimmune disease. Another interesting example of RIPK1/3-dependent inflammatory pathology was revealed in the experiments with mice deficient in the deubiquitinase A20 (Tnaip3). Tnaip3-/- mice develop severe inflammation and cachexia, die prematurely, and show hypersensitivity to LPS and TNFα [114]. Haploinsufficiency in A20 in humans leads to early onset auto-inflammatory disease [115]. It has been proposed that this phenotype is dictated by the critical role of A20 in terminating TNFα-induced cell death and NFkB activation, mediated by K63 ubiquitination of multiple signaling intermediates (TRAF6, NEMO, RIPK1) [30, 114, 115]. However, recent data suggest that pathology may arise primarily from the activation of the RIPK1/RIPK3 axis, although the exact mechanism remains to be fully uncovered. Work by Onizawa et al. identified a new ubiquitination site on Lys5 of RIPK3, which was negatively regulated by A20, and was essential for RIPK1/RIPK3 complex formation and necroptosis [29]. However, Mlkl-/- mice were not protected from the disease induced by the loss of A20, unlike RIPK1D138N/D138N or Ripk3-/- animals [83], again indicating that non-necroptotic regulation plays a primary role under these conditions.
Part of the Tnaip3-/- inflammatory phenotype, such as formation of foci of neutrophilic inflammation in the liver, and development of dermatitis and/or panniculitis, were reduced only in Ripk3-/- mice and not in RIPK1D138N/D138N mice, indicating that both RIPK1-dependent and exclusively RIPK3-dependent events cooperate to promote the pathology. While not yet examined, the latter are reminiscent of the kinase-independent inflammatory regulation by RIPK3, reported by Francis Chan's laboratory [116]. Overall, loss of A20, which normally restricts activation of RIPK1 and RIPK3, appears to primarily promote non-necroptotic cell death and systemic inflammation, which is partially controlled by the kinase activity of RIPK1, and, in part, by the RIPK1-independent signaling through RIPK3.
Overall, the complexity of the data in this section illustrates the main theme of our review. Even though RIPK1 and RIPK3 kinases initially emerged as specific mediators of necroptosis, it is now clear that the regulation is much more complex. RIPK1 and RIPK3 promote at least three distinct responses: necroptosis, apoptosis, and cell death-independent inflammation, and the loss of homeostasis of any of these pathways may contribute to the development of pathology. In some cases, a particular pathway may be a dominant disease driver. For instance, limited levels of active caspase-8 were linked to activation of necroptosis or low expression of MLKL was tied to inflammatory signaling by RIPK1 and RIPK3 as observed in MS and ALS models [34, 35]. However, it seems likely that additional intrinsic factors controlling differential activation of RIPK1/RIPK3-dependent responses may exist. From a therapeutic prospective, kinase activity of RIPK1 appears to be positioned at the apex of the regulation, and multiple lines of evidence suggest that it represents a promising therapeutic target. It is less clear at this point to which extent kinase activity of RIPK3 plays a role and if it can be safely targeted by small molecule inhibitors.
Emerging roles of RIPK1 and RIPK3 in cancer
As described above, activation of RIPK1 and RIPK3 signaling has been shown to mediate and exacerbate injury both systemically and in a tissue-specific manner. Recent evidence summarized in this section also suggests that activity of these kinases may participate in tumorigenesis; however, as in other pathologies, RIPK1 and RIPK3 play complex roles. Thus far, research into the roles of necroptosis in cancer revealed three different, putatively therapeutically exploitable angles: a) induction of necroptosis can be used to eliminate cancer cells that show resistance to treatment; b) promoting necroptosis can enhance surveillance and protective immunity against cancer, and c) conversely, inhibition of necroptosis can lead to a reduction in the cancer-promoting inflammation.
Activation of necroptosis in cancer cells
Consistent across many cancers is the development of resistance to drug-induced apoptosis. Initial work using a necroptosis activator shikonin in MCF-7 and HEK293 cancer cell lines provided confirmation that induction of RIPK-dependent necroptosis could provide an avenue to eliminate cancer cells, which acquired resistance to apoptosis [117]. Apoptosis-resistant cells expressing anti-apoptotic Bcl-2 family members Bcl-2 and Bcl-xl were found to remain sensitive to shikonin-induced cell death, which was prevented by Nec-1. Furthermore, shikonin retained efficacy in cells expressing high levels of P-glycoprotein, which is one of the central mechanisms in acquiring multi-drug resistance by cancer cells [118]. Subsequent work demonstrated the ability of shikonin to induce necroptosis in cell lines representing a variety of cancer cell types, including glioma, breast cancer, osteosarcoma and multiple myeloma [119-122]. Interestingly, inhibition of necroptosis in various cell types has been shown to promote reversal of cell death phenotype and activation of apoptosis, suggesting that unlike many other in vitro experimental systems where apoptosis is induced first and is reverted to necroptosis in the presence of caspase inhibitors, shikonin preferentially promotes necroptosis even in apoptosis-competent cells. Oxidative stress was shown to contribute to the activation of necroptosis by shikonin [120], but the more precise mechanism(s) is not yet known.
Combined inhibition of cIAP1/2 and XIAP by SMAC mimetic inhibitors unleashes multiple RIPK1/RIPK3-dependent mechanisms. This includes synthesis of TNFα, and the generation of an auto-feedback loop promoting apoptotic cell death and expression of multiple cytokines and chemokines [74, 123, 124]. These responses required kinase activity of RIPK1 and RIPK3, acting in a kinase and MLKL-independent manner [74, 125]. SMAC mimetics were also shown to promote IL-1β processing by both NLRP3 and caspase-8 inflammasomes, which was abolished in Ripk3-/- cells [89]. Conversely, addition of zVAD.fmk converted RIPK1 kinase-dependent apoptosis into RIPK3 kinase-dependent necroptosis [125]. Notably, not all cancer cells produce TNFα in response to SMAC mimetics. In this case, SMAC mimetics lack toxicity as single agents, but still promote killing by exogenous TNFα or TRAIL [126-128].
The sum of these observations set the stage for the use of SMAC mimetics to promote RIPK1/3-dependent death as a new anti-cancer therapeutic strategy. In particular, the work by Simone Fulda's laboratory showed that SMAC mimetic BV6 efficiently promoted elimination of cancer cells in combination with multiple anti-cancer agents, including DNA damaging chemotherapeutic agents, ionizing radiation, TRAIL, interferon-alpha, HDAC inhibitors and others [129-140]. Oftentimes this was achieved in the cells that are otherwise highly refractory to chemo- or radio-therapies. Importantly, these and other works have revealed several noteworthy implications discussed below.
First, resistance of cancer cells to apoptosis could be efficiently circumvented by specific activation of necroptosis by the combination of SMAC mimetics with caspase inhibitors. Recent data reported by Brumatti et al. showed that acute myelogenous leukemia (AML) cells display a range of sensitivities to cIAP1/2-selective SMAC mimetic birinapant, depending on the oncogenic mutations present in the cells [141]. However, efficient killing through necroptosis was achieved in all cases when birinapant was combined with a clinical pan-caspase inhibitor Emricasan (IDN-6556). Strikingly, even the cells selected in culture to be resistant to birinapant alone, remained highly sensitive to the combination with caspase inhibitors. In addition, primary AML samples, including those from patients with relapsed or secondary disease, showed good response to the combination. Efficiency of Emricasan in promoting necroptosis was linked to its high affinity against necroptosis-inhibiting caspase-8/FLIP(L) heterodimers. Importantly, birinapant/emricasan combination was well tolerated in vivo and efficiently eliminated AML cells, suggesting that it may offer new hope for AML patients who have failed standard chemotherapy. Similarly, BV6 was shown to efficiently kill apoptosis-resistant pancreatic cancer cells by necroptosis when combined with zVAD.fmk [142]. In contrast, analysis of refractory or relapsed acute lymphoblastic leukemia (ALL) patient samples identified a subset which was highly sensitive to brinipant alone [143]. In this case, specific activation of necroptosis by addition of caspase inhibitor was not necessary, as cells were efficiently eliminated by brinipant alone inducing a mixture of RIPK1 kinase-dependent apoptosis and necroptosis. Overall, these data indicate that SMAC mimetics may provide a new therapeutic agent against the tumors that have developed resistance to standard chemotherapies. Furthermore, ex vivo profiling of patient samples may provide a powerful approach for selecting the optimal SMAC mimetic-based treatment regimen.
Second, some tumor cell lines display resistance to necroptosis, which is, in part, explained by the reduction of RIPK3 expression through genomic hypermethylation [144]. Thus, combinations of SMAC mimetics with hypomethylating agents may present a good strategy in these cases. Indeed, combination of BV6 with demethylating agents 5-azacytidine or 5-aza-2′-deoxycytidine resulted in a highly synergistic activation of apoptosis in ALL and AML cells, which was not observed in normal peripheral lymphocytes [145, 146]. Cell death was promoted through an autocrine/paracrine TNFα loop described above and inhibition of caspases led to the alternative activation of necroptosis.
Third, the use of SMAC mimetics may not be without complications. Although most cytokine responses to cIAP1/2 and XIAP loss in myeloid cells were mediated by autocrine TNFα signaling, production of CCL2/MCP-1 was induced separately [74]. The possibility of inducing this response should be considered with caution as BV6-induced production of CCL2 has been recently shown to promote migration and invasion of glioblastoma cells and, to modify tumor microenvironment, promoting migration of astroglial cells [147]. Furthermore, while SMAC mimetics generally showed preferential toxicity towards cancer cells, killing of normal cells cannot be completely discounted and may negatively impact therapy. For example, BV6 was shown to increase sensitivity of multiple hematologic and solid cancer cells to lysis by cytokine-induced killer (CIK) cells. However, this beneficial effect was reduced by some toxicity of BV6 towards CIK cells [148].
Tumorigenic activities of RIPK1 and RIPK3
Activation of RIPK1/RIPK3 signaling may not always be a beneficial anti-cancer strategy as these kinases were also shown to promote tumorigenesis in some contexts. One interesting recent example comes from the analysis of RIPK1/RIPK3 signaling in pancreatic ductal adenocarcinoma (PDA) cells [149]. RIPK1 and RIPK3 were found to be highly expressed in PDA cells and their expression was further induced by gemcitabine treatment that triggered MLKL-dependent necroptosis. However, counterintuitively, expression of RIPK1 and RIPK3 was tumor-promoting in this case. Expression of chemokine CXCL1 was high in both human PDA specimens and in mouse p48Cre;KrasG12D (KC) PDA model and was further enhanced by gemcitabine. High CXCL1 expression was mitigated by Ripk3-/- in vivo and RIPK1 and RIPK3 kinase inhibitors in vitro, suggesting a key role of necrosomes in driving CXCL1 expression. Most importantly, loss of Ripk3 or administration of Nec-1s significantly reduced tumorigenesis in the KC model. Mechanistically, this was linked to an increased RIPK1/RIPK3-dependent production of SAP130 protein in PDA cells, which promoted cellular immune suppression by tumor infiltrating macrophages, recruited in a CXCL1-dependent manner and expressing SAP130 receptor Mincle. Overall, this work provided a clear demonstration of tumor promoting activity of RIPK1/RIPK3-mediated inflammation through myeloid-dependent immune suppression. Notably, a number of tumor promoting cytokines and chemokines beyond CXCL1, including CXCL2, GM-CSF and IL6, are induced by necrosomes [37]. This may indicate that tumors that are known to be promoted by these inflammatory mediators may be generally targeted by RIPK1 inhibitors. It is encouraging that another recent study showed that CRISPR-mediated genetic deletion of RIPK1, RIPK3 or MLKL in murine or human breast cancer cell lines reduced xenograft growth in vivo [150]. This, again, was putatively linked to the defects in the levels of a number of tumor-promoting cytokines and chemokines, including IL6, IL8, LCN2, CCL2/MCP1, and CCL5.
Another example of tumorigenesis promoted by RIPK1/RIPK3 comes from the analysis of mice lacking tumor suppressor Hace1 [151]. Loss of Hace1, which acts as a E3 ubiquitin ligase of TRAF2 in TNFR Complex I, results in the loss of both NFkB activation and caspase-8-dependent apoptosis, but does not affect necroptosis. Furthermore, hace1-/- mice displayed greatly exacerbated development of colitis in response to DSS and propensity to develop colon cancer upon challenge with DSS and the genotoxic drug azoxymethane. Overall, deficiency in Hace1 led to greatly increased epithelial cell death, tissue destruction and severe sustained inflammation, which provided the necessary microenvironment for accelerated tumorigenesis that was completely reversed in Ripk3-/- and TNFR1-/- animals. As mentioned earlier, inhibition of RIPK1 may be a promising strategy in attenuating colitis-associated tumorigenesis [104]. The roles of RIPK3 in intestinal tumorigenesis are more complex. It may contribute to the initial injury as seen in hace1-/- mice [151], but it also promotes repair through kinase-independent regulation of IL-1β, IL-23, and IL-22 cascade in severe colitis [86, 87]. It should be noted that there is currently no direct evidence that kinase activity of RIPK3 contributes to any of this regulation.
RIPK1, RIPK3 and anti-tumor immunity
The work by Yatim et al. showed that necroptotic cells produce signals promoting cross-priming of CD8+ T cells by dendritic cells [152]. Using activation of necroptosis by dimerization of RIPK3, the authors showed that this regulation required two sets of signals: DAMPs released from the dead cells, and active RIPK1-NFkB dependent pro-inflammatory transcription in the dying cells. These data provided striking example of two distinct necrosome-dependent responses cooperating to induce functional regulation of cellular immune responses to necroptotic cells. Intriguing work presented this year showed further promise in vaccination against tumors using necroptotic murine colon carcinoma cells [153]. The authors showed that DAMPs, such as ATP and HMGB1, were released by necroptotic cancer cells, and promoted activation of anti-tumor cellular immunity. Vaccination induced dendritic cell maturation, cross-priming of CD8a+ T-cells, and IFN-γ production, all of which enhanced the immune response to the cancer cells.
Another recent study suggested that RIPK3-dependent signaling may be essential for promoting IL-12 production by dendritic cells, which is critical for promoting anti-tumor immunity [154]. In this work, cervical carcinoma C4-I cells, expressing high levels of RIPK3, were found to undergo necroptosis-like death in response to frequently used immune adjuvant polyinosinic:polycytidylic acid (PolyIC). This was shown to promote release of IL-1α, which led to greatly enhanced production of IL-12 by dendritic cells. The authors concluded that RIPK1 was not essential in this regulation because siRNA mediated silencing of RIPK1 failed to abate the response. However, as discussed before, loss of RIPK1 (e.g. in R/pk1-/- mice) may produce new regulation that is not necessarily a reflection of the endogenous role of this protein. Furthermore, these data are reminiscent of an earlier report, examining the role of necroptosis in adjuvant activity of PolyIC [155]. In that case, a small fraction of dendritic cells was found to undergo necroptosis mediated by cathepsin D/IPS-1/RIPK1 complex. HMGB1 released by the dying cells was found to drastically promote IFNJβ synthesis by the non-necroptotic DCs, enhancing cellular immune responses.
Overall, these reports reveal complex and often non-cell autonomous roles of RIPK1 and RIPK3 in tumorigenesis. It is clear that modulating these responses may result in powerful anti-cancer effects. In some cases, promoting RIPK1-dependent cell death or, even, specifically RIPK3-dependent necroptosis may be highly beneficial, while in other cases reducing necrosome-dependent inflammation, driving immune suppression by tumor-infiltrating myeloid cells, may be highly beneficial. In yet another axis of regulation, anti-tumor functions of dendritic cells may be promoted by necroptosis in tumor cells. Furthermore, while these responses have been, thus far, examined separately, the possibility remains that inhibition of RIPK1 may simultaneously promote both pro- and anti- tumor responses. Thus, developing new approaches to selectively target particular downstream effector events may be important.
New inhibitors of RIPK1 and RIPK3
A great amount of work has been done since the first specific inhibitor of necroptosis, Nec-1 [9], was found to target RIPK1 [10]. A growing understanding of signaling of RIPK1 and RIPK3 has been accompanied by the development of new classes of inhibitors. We will not provide a comprehensive overview of RIPK1, RIPK3 and MLKL inhibitors, which are shown in Figs. 3 and 4 and we have already reviewed recently [65]. Rather, we will focus on some of the interesting new findings, which broaden our understanding of the therapeutic targeting of these factors.
Figure 3.
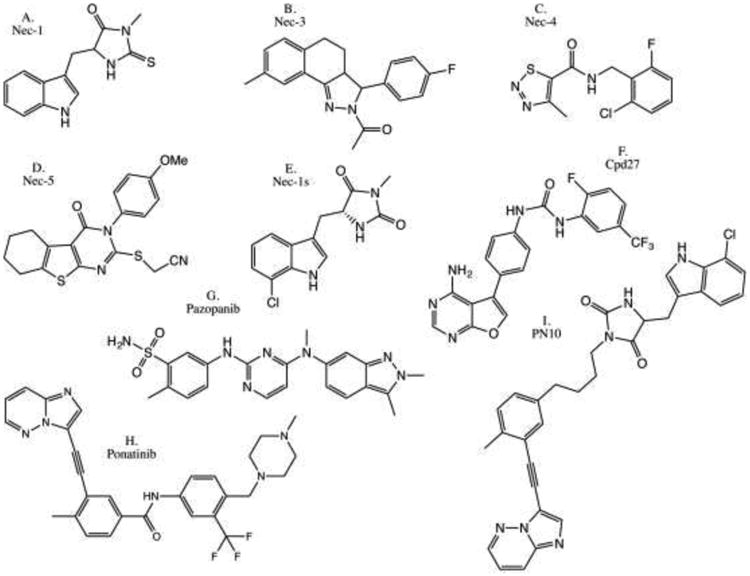
Examples of previously reported RIPK1 kinase inhibitors. a) Necrostatin-1 [90]. b) Necrostatin-3 [10, 171]. c) Necrostatin-4 [10, 172, 173]. d) Necrostatin-5 [10, 174]. e) Optimized Necrostatin-1s [10, 90, 175]. f) Compound 27 [176]. g) Pazopanib [177]. h) Dual RIPK1/RIPK3 inhibitor Ponatinib [125, 177]. i) RIPK1-specific ponatinib/Nec-1s hybrid PN10 [125].
Figure 4.
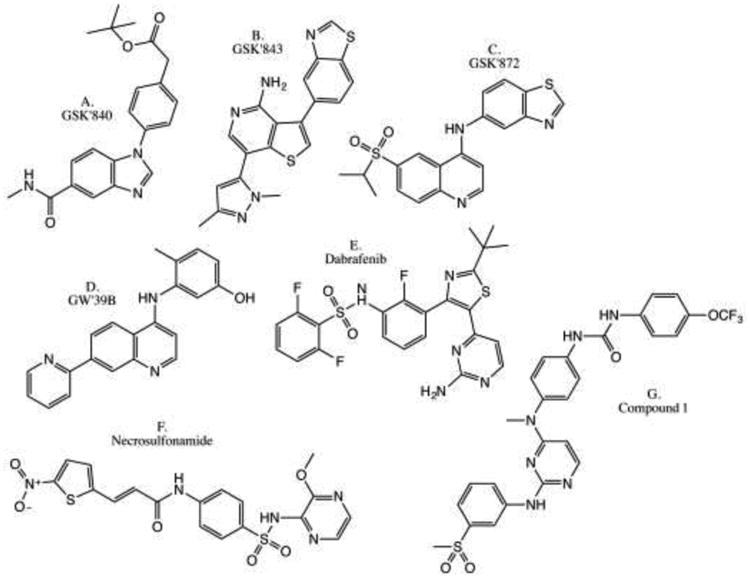
Previously described RIPK3 and MLKL inhibitors. a) GSK'840 [51]. b) GSK'843 [51, 178]. c) GSK'872 [51, 178]. d) GW'39B [179]. e) Dabrafenib [180]. f) Necrosulfonamide [57]. g) Compound 1 [181].
Aside from previously reported classes of inhibitors, Zhou et al. recently introduced new 1-benzyl-1H-pyrazole-based inhibitors of RIPK1 [156]. Structure-activity relationship (SAR) analysis led to development of optimized compound 4b (Fig. 5a). This molecule displayed a Kd value of 78 nM towards RIPK1 and an EC50 value of 160 nM in inhibiting necroptosis, which is comparable to Nec-1s (EC50=210 nM) [157]. Notably, daily injection of 10 mg/kg of 4b for 5 days provided efficient protection against L-arginine-induced pancreatic injury. Another inhibitor was reported by Berger et al. at GlaxoSmithKline [158] (Fig. 5b). The inhibitor GSK'963 was identified in an in vitro enzymatic screen of a collection of ∼2 million compounds. This molecule showed potent, low nanomolar inhibition of necroptosis in the cells and complete protection from TNF/zVAD-induced shock in vivo at 2 mg/kg dose [158]. A screen of DNA-encoded chemical library at GlaxoSmithKline yielded a structurally distinct low nanomolar and mono-specific benzo[b][1,4]oxazepin-4-one based inhibitor of human RIPK1 (Compound 14, GSK'481), which displayed a particularly promising developability profile [159] (Fig. 5c). Notably, structural analysis indicated that this inhibitor and Nec-1s, which is also highly specific for RIPK1 [71, 157], target the same allosteric αC-Glu-out/DLG-out back pocket [125, 160] that, thus far, has only been reported for RIPK1. Another unusual property of these molecules was a high degree of selectivity (>200 fold) towards human and primate RIPK1 versus rodent homologs. This translated into low μM EC50 in mouse cells in vitro, and a relatively high (50 mg/kg, po delivery) dose of optimized inhibitor of this class (Compound 31) that was required to protect mice from TNF/zVAD-induced shock. This was explained by an increased flexibility of the activation loop of human kinase, which was necessary to accommodate the major conformational changes in the activation segment, the C-helix, and the glycine-rich loop upon compound binding. We have previously reported that the DLG motif of RIPK1 (as opposed to DFG present in the majority of the kinases) was also essential in providing flexibility to the allosteric pocket of RIPK1 [125]. These data reveal that the allosteric back pocket of RIPK1 possesses unusual flexibility, which allows development of small molecule inhibitors with unprecedented specificity. Overall, these recent reports identify several exciting new classes of efficient RIPK1 inhibitors, adding to the repertoire of powerful pharmacologic tools to study RIPK1 kinase biology in vitro and in vivo.
Figure 5.
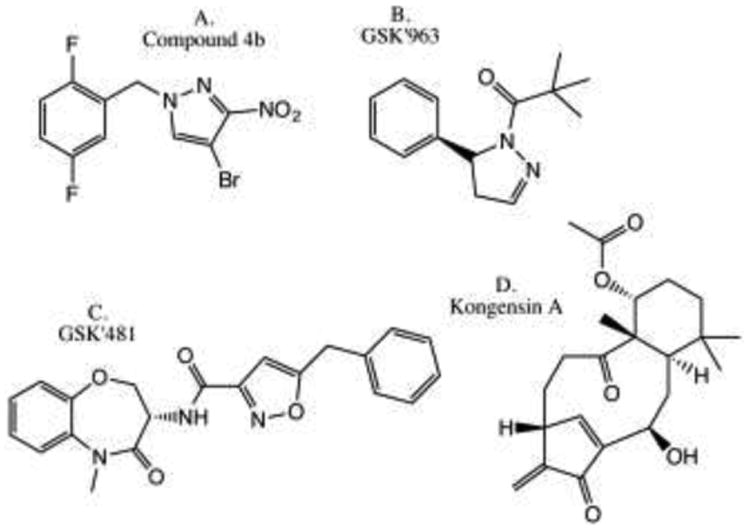
Recently developed inhibitors of RIPK1 and RIPK3. a) Compound 4b [156]. b) GSK'963 [158]. c) GSK'481 [159]. d) Kongensin A [163].
A new method for RIPK3 inhibition has also been revealed. Chaperone heat shock protein 90 (HSP90) was found to play a critical role in allowing signaling by the necrosome on multiple levels (summarized in detail in [161]). This includes regulation of stability of RIPK1, binding of HSP90/CDC37 to RIPK3 that is critical for RIPK3-mediated necroptosis downstream from RIPK1, and modulation of stability and ability to induce necroptosis by MLKL [162]. A natural product Kongensin A was recently shown to prevent RIPK3 activation upon TNFα stimulation [163] (Fig. 5d). This reflected an unusual mode of HSP90 inhibition, which involved formation of the covalent adduct with previously uncharacterized Cys420 residue and dissociation of the HSP90/CDC37 complex. Because Kongensin A was also shown to induce apoptosis in multiple cancer cell lines, likely through other targets of HSP90 besides RIPK3, this molecule may emerge as an interesting anti-cancer agent capable of attenuating RIPK/necroptosis-dependent inflammation while promoting putatively less pro-inflammatory apoptosis.
An important question stemming from the use of necroptosis inhibitors is whether these molecules may promote a switch to apoptosis, opposite to the activation of necroptosis in the presence of caspase inhibitors. In one example that we mentioned earlier, inhibition of Shikonin-induced necroptosis was shown to increase apoptosis [164]. Another interesting example comes from the recent report showing that Nec-1 promoted caspase-dependent neutrophil apoptosis in the presence of neutrophil survival signals such as GM-CSF and LPS [165]. This switch was associated with down-regulation of anti-apoptotic Mcl-1 protein and up-regulation Bax, promoting apoptosis. Induction of neutrophil apoptosis was proposed to contribute to the ability of Nec-1 to rapidly resolve established neutrophil-dependent inflammation in LPS-induced acute lung injury, although, the ability of Nec-1 to markedly reduce LPS-induced inflammatory gene expression [37] should also be considered. Overall, to the best of our knowledge, the switch from necroptosis-to-apoptosis has not been observed broadly in vitro or in vivo. However, the possibility of compensatory activation of apoptosis by necroptosis inhibitors cannot be completely disregarded.
Concluding Remarks
By virtue of their roles in negotiating a lytic cell death, RIPK1 and RIPK3 have garnered interest as targets to alleviate inflammation associated with pathologic cell damage and tissue injury. DAMPs released by permeabilized cells undergoing necroptosis are widely viewed as a major trigger for systemic inflammation associated with RIPK1 and RIPK3-dependent diseases [166, 167]. Accordingly, it is the new discoveries of the roles for RIPK1 and RIPK3 as drivers of cell-death independent inflammation that are particularly provocative as they prompt consideration of these proteins as potential drug targets for auto-inflammatory pathologies that do not have well-defined cell-death associated etiologies, such multiple sclerosis, neutrophilic dermatosis, ALS, cancer and others, as discussed in the review. In other cases, the sum of the diverse functions attributable to RIPK1 and RIPK3, including cell-death dependent and independent processes, may cooperatively contribute to inflammatory, degenerative and autoimmune pathologies when homeostasis is perturbed. We have described some of the striking recently published examples of such cooperation between cell death-dependent and cell death-independent signaling by RIPK1 and RIPK3, e.g. during progressive demyelination in the CNS or antigen cross-presentation [34, 35, 152].
Complex wiring of RIPK-dependent responses comes to the forefront when considering potential therapeutic targets. A focus on inhibition of RIPK1 kinase activity seems generally well justified, given tolerability of Nec-1s, lack of notable phenotype in kinase-dead animals, and efficient protection observed in multiple animal models of human pathology upon inhibition of RIPK1 kinase activity. In the long term perspective, targeting a particular downstream effector mechanism that plays a central role in the etiology of a particular disease may, hypothetically, offer benefits in efficacy and selectivity. For example, inhibition of RIPK3 may be beneficial under the circumstances when necroptosis is a primary driver of pathology because activation of RIPK3 may by-pass requirement for RIPK1 under some conditions and such inhibitors may not affect non-necroptotic functions of RIPK1. However, current inhibitors of RIPK3 as well as a kinase-dead RIPK3D161N/D161N mutant unexpectedly promote caspase-8-dependent apoptosis [51, 69]. The lack of this phenotype with RIPK3K51A/K51A suggests that non-toxic RIPK3 inhibitors may be an achievable goal [51], but it is not yet clear which mechanistic class of inhibitors is needed to realize this target.
Similarly, given a critical role of caspase-8 in many injuries and potential benefits of necroptosis activation in cancer, the development of new selective inhibitors of pro-apoptotic caspase-8 homodimers and anti-necrotic caspase-8/FLIPL heterodimers will be worthwhile. These inhibitors can be expected to have significant value as research tools. Therapeutically, however, inhibition of caspase-8 may carry risks. Genetic loss of caspase-8 was found to result in activation of necroptosis and either animal death or extensive local tissue injury [168, 169]. On the other hand, pan-caspase inhibitor Emricasan (IDN-6556) was well tolerated, suggesting that caspase inhibitors might not be too far away from being recognized as clinically useful drug targets to modulate RIPKs [141]. Importantly, these dichotomous observations may reflect tissue-specific regulation, whereby cells and tissues exposed to physiologic necroptosis-initiating signals (e.g. coming from commensal microbiota) may have limited exposure to Emricasan, due to the specific biodistribution profile of this drug. Accordingly, the pharmacologic profile of future caspase inhibitors may be critical to their tolerability and efficacy in patients. It may also be worthwhile to consider that the toxicity of caspase-8-/- may reflect additional scaffolding roles of this caspase, in which case small molecule inhibitors may be tolerated.
Along these lines, even though the loss of both RIPK3 and caspase-8 alleviates many acute injuries, it leads to autoimmune disease, which is further exacerbated in Mlkl-/-;caspase-8-/- animals [112, 113]. Overall, we interpret these and other published data to indicate that effector mechanisms activated by RIPK1 kinase are carefully balanced and their modulation should be considered with caution as it may fundamentally change the wiring of the regulation, leading to unexpected detrimental responses. Extending on our general model, normally innocuous pro-Complex IIb signals, e.g. from intestinal microbiota [41, 77, 78], could promote some degree of RIPK1 kinase activation in vivo. However, formation of stable pro-death complexes could be restricted by the presence of active caspase-8 in heterodimers with FLIP(L) [25] and RIPK3 in the active DFG-in/Glu-in conformation [170]. Inhibition of caspase-8 or conformational change to RIPK3, induced by an inhibitor or D161N mutation, may alter the balance and promote robust initiation of either apoptosis or necroptosis. Thus, while attractive, targeting of downstream effectors of Complex IIb for therapeutic gain remains a risky area (See Outstanding Questions).
In this new complexity, the roles for RIPK1 and RIPK3 as drivers of cell death might seem most straight-forward based on the available data. However, this is not entirely the case as the roles of these proteins also differ in a context-dependent manner. For example, in some cases, activation of RIPK1 kinase-dependent apoptosis, has been primarily linked to the development of the injury, while in other cases a combination of apoptosis and necroptosis was observed. Curiously, this is sometimes observed when activation of RIPK1 signaling is mediated by the same event but in different tissues, as has been reported upon deletion of NEMO in intestinal epithelial cells versus hepatocytes [41, 105]. In some instances, preferential activation of a particular response was traced to the deficiency in caspase-8 activation or low expression of MLKL or RIPK3, but it seems likely that additional signals that affect the direction of Complex IIb responses may exist. Similarly, the molecular mechanisms leading to the formation of pro-apoptotic (ripoptosome) and pro-necroptotic (necrosome) versions of Complex IIb also remain to be fully understood. For example, addition of TNFα alone does not result in cell death in most cell types, as this requires additional factors promoting formation of Complex IIb. Curiously, while induction of RIPK1 kinase-dependent apoptosis by TNFα requires inhibition of IAPs, this is not absolutely required for necrosome formation, which can occur upon caspase-8 inhibition in the absence of IAP inhibitors [125]. This may reflect protection of CYLD by caspase-8 inhibition [27], resulting in a more robust de-ubiquitinase activity associated with necrosomes, making inhibition of ubiquitination in Complex I less essential. Nevertheless, these observations may also indicate that distinct molecular events may be ultimately required for the formation of productive ripoptosomes and necrosomes, representing a departure from the current linear model where necrosome complexes are often considered as an extension of ripoptosomes by the addition of RIPK3.
It should also be noted that the tools, such as the RIPK1 inhibitor Nec-1s and Ripk3-/- mice, that have been used to interrogate necroptosis, are not exclusive for this mechanism. In lieu, the specific role of necroptosis may be specifically addressed with the availability of Mlkl-/- mice. Although the data generated using these mice are still limited, it appears that they show some overlap with protection by Ripk3-/- as well as Nec-1s and RIPK1D138N/D138N; however, there are also differences, likely reflecting a role for necroptosis as just one of the alternative Complex IIb-directed mechanisms [83].
In sum, the data summarized in this review highlights the tremendous therapeutic promise of targeting RIPK1 and RIPK3 regulation. These reports illustrate the complexity of this regulation which also needs to be considered in interpreting experimental results and strategizing for new therapeutic avenues. Importantly, additional tools and biomarkers, targeting activation of specific effector mechanisms will be helpful and, this, in turn, will necessitate further mechanistic interrogation of necrosome functions.
Outstanding questions box.
How is Complex IIb regulated in disease? Evidence indicates that Complex IIb can control multiple disease-associated processes, including caspase-8-dependent apoptosis, RIPK3-MLKL-dependent necroptosis, and pro-inflammatory gene expression. In some cases, a particular response plays a predominant role in the etiology of the disease. In other cases, combinations of responses, acting cooperatively, modulate disease progression. Moving forward, it remains to be determined which molecular events define the direction of Complex IIb-dependent responses in pathology.
Is there a single Complex IIb? The molecular composition of Complex IIb is not definitively known. It remains to be determined whether it is a single complex, promoting multiple context-dependent responses, or alternatively, there are several complexes, mediating distinct responses.
What are the roles of Complex IIb in pathologic injury and disease? Initial view postulated that RIPK1 and RIPK3 kinases specifically and exclusively mediate necroptosis. However, new evidence suggests that we need to examine contributions of Complex IIb in contexts that are not limited to overt cell death phenotypes, such as auto-inflammatory diseases and cancer. This will require better tools and specific markers to understand contributions of different modes of Complex IIb signaling in disease.
Can we target components of this pathway other than RIPK1 therapeutically? Targeting specific downstream effectors of RIPK1, such as RIPK3 kinase or caspase-8, may improve efficacy under some conditions. However, serious concerns remain whether such strategies can be efficacious or well tolerated.
How do RIPK1 kinase inhibitors compare with anti-TNF agents? The TNF-dependent auto-amplification loop is a critical feature of RIPK1-associated pathologies. Theoretically, inhibitors of RIPK1 can terminate pathologic signaling by TNFα, but have yet to be directly compared to anti-TNF drugs.
Trends box.
RIPK1 and RIPK3 kinases first emerged as mediators of a process of regulated necrosis, termed necroptosis.
These factors also promote caspase-8-dependent apoptosis and proinflammatory gene expression.
These responses are controlled in a context-dependent manner by Complex IIb, formation of which requires kinase activity of RIPK1.
Signaling by Complex IIb is controlled on multiple levels, including phosphorylation and ubiquitination of complex components.
Complex IIb-associated cell death and inflammation both contribute to disease and represent a target for therapeutic interventions. In particular, RIPK1 kinase activity has emerged as a driver of multiple degenerative, inflammatory and auto-immune pathologies.
Multiple inhibitors of RIPK1 and RIPK3 have been developed, and represent useful tools to study their biology as well as potential leads for drug development.
Acknowledgments
This work was supported in part by grants from NIH to A.D. (RO1GM080356 and RO1GM084205). A.D. is a consultant for Denali Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thompson C. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267(5203):1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 2.Laster SM, et al. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141(8):2629–34. [PubMed] [Google Scholar]
- 3.Vercammen D, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187(9):1477–85. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vercammen D, et al. Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine. 1997;9(11):801–8. doi: 10.1006/cyto.1997.0252. [DOI] [PubMed] [Google Scholar]
- 5.Holler N, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura H, et al. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151(6):1247–56. doi: 10.1083/jcb.151.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanden Berghe T, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17(6):922–30. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 8.Chan FK, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278(51):51613–21. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 9.Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 10.Degterev A, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaczmarek A, et al. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Thapa RJ, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110(33):E3109–18. doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He S, et al. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108(50):20054–9. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Upton JW, et al. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7(4):302–13. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanden Berghe T, et al. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 16.Vanlangenakker N, et al. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012;19(1):75–86. doi: 10.1038/cdd.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Micheau O, Tschopp J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell. 2003;114(2):181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 18.Wong WW, et al. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ. 2010;17(3):482–7. doi: 10.1038/cdd.2009.178. [DOI] [PubMed] [Google Scholar]
- 19.Varfolomeev E, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283(36):24295–9. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerlach B, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471(7340):591–6. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 21.Dondelinger Y, et al. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol Life Sci. 2016;73(11-12):2165–76. doi: 10.1007/s00018-016-2191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peltzer N, et al. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016;26(6):445–61. doi: 10.1016/j.tcb.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Justus SJ, Ting AT. Cloaked in ubiquitin, a killer hides in plain sight: the molecular regulation of RIPK1. Immunol Rev. 2015;266(1):145–60. doi: 10.1111/imr.12304. [DOI] [PubMed] [Google Scholar]
- 24.Dickens LS, et al. The ‘complexities’ of life and death: death receptor signalling platforms. Exp Cell Res. 2012;318(11):1269–77. doi: 10.1016/j.yexcr.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Dillon CP, et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell reports. 2012;1(5):401–7. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moquin DM, et al. CYLD Deubiquitinates RIP1 in the TNFalpha-Induced Necrosome to Facilitate Kinase Activation and Programmed Necrosis. PLoS One. 2013;8(10):e76841. doi: 10.1371/journal.pone.0076841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Donnell MA, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nature cell biology. 2011;13(12):1437–42. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legarda D, et al. CYLD Proteolysis Protects Macrophages from TNF-Mediated Auto-necroptosis Induced by LPS and Licensed by Type I IFN. Cell Rep. 2016;15(11):2449–61. doi: 10.1016/j.celrep.2016.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onizawa M, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol. 2015;16(6):618–27. doi: 10.1038/ni.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–9. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 31.Bellail AC, et al. A20 ubiquitin ligase-mediated polyubiquitination of RIP1 inhibits caspase-8 cleavage and TRAIL-induced apoptosis in glioblastoma. Cancer Discov. 2012;2(2):140–55. doi: 10.1158/2159-8290.CD-11-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meng L, et al. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci U S A. 2015;112(35):11007–12. doi: 10.1073/pnas.1514730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353(6299):603–8. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ofengeim D, et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10(11):1836–49. doi: 10.1016/j.celrep.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berger SB, et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192(12):5476–80. doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Najjar M, et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity. 2016 doi: 10.1016/j.immuni.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McQuade T, et al. Positive and Negative Phosphorylation Regulates RIP1 and RIP3-Induced Programmed Necrosis. Biochem J. 2013 doi: 10.1042/BJ20130860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dondelinger Y, et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell. 2015;60(1):63–76. doi: 10.1016/j.molcel.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 40.O'Donnell MA, et al. NEMO inhibits programmed necrosis in an NFkappaB-independent manner by restraining RIP1. PLoS One. 2012;7(7):e41238. doi: 10.1371/journal.pone.0041238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vlantis K, et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-kappaB-Dependent and -Independent Functions. Immunity. 2016;44(3):553–67. doi: 10.1016/j.immuni.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dondelinger Y, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20(10):1381–92. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanlangenakker N, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18(4):656–65. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blonska M, et al. TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappaB activation. J Biol Chem. 2005;280(52):43056–63. doi: 10.1074/jbc.M507807200. [DOI] [PubMed] [Google Scholar]
- 45.Kanayama A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15(4):535–48. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 46.Zhang SQ, et al. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12(3):301–11. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 47.Wu CJ, et al. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8(4):398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 48.de Almagro MC, et al. Coordinated ubiquitination and phosphorylation of RIP1 regulates necroptotic cell death. Cell Death Differ. 2016 doi: 10.1038/cdd.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150(2):339–50. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen W, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem. 2013;288(23):16247–61. doi: 10.1074/jbc.M112.435545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mandal P, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56(4):481–95. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy JM, et al. The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism. Immunity. 2013;39(3):443–53. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 53.Cook WD, et al. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014;21(10):1600–12. doi: 10.1038/cdd.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orozco S, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21(10):1511–21. doi: 10.1038/cdd.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu XN, et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014;21(11):1709–20. doi: 10.1038/cdd.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su L, et al. A plug release mechanism for membrane permeation by MLKL. Structure. 2014;22(10):1489–500. doi: 10.1016/j.str.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun L, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 58.Wang H, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–46. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 59.Wu J, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell research. 2013;23(8):994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cai Z, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105–21. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dondelinger Y, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971–81. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 63.Quarato G, et al. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol Cell. 2016;61(4):589–601. doi: 10.1016/j.molcel.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hildebrand JM, et al. Flicking the molecular switch underlying MLKL-mediated necroptosis. Mol Cell Oncol. 2015;2(3):e985550. doi: 10.4161/23723556.2014.985550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Degterev A, Linkermann A. Generation of small molecules to interfere with regulated necrosis. Cell Mol Life Sci. 2016;73(11-12):2251–67. doi: 10.1007/s00018-016-2198-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Polykratis A, et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193(4):1539–43. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelliher MA, et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8(3):297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 68.Li H, et al. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006;281(19):13636–43. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 69.Newton K, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343(6177):1357–60. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 70.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell. 2011;145(1):92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 71.Christofferson DE, et al. A novel role for RIP1 kinase in mediating TNFalpha production. Cell Death Dis. 2012;3:e320. doi: 10.1038/cddis.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hitomi J, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135(7):1311–23. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McNamara CR, et al. Akt Regulates TNFalpha synthesis downstream of RIP1 kinase activation during necroptosis. PLoS One. 2013;8(3):e56576. doi: 10.1371/journal.pone.0056576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong WW, et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood. 2014;123(16):2562–72. doi: 10.1182/blood-2013-06-510743. [DOI] [PubMed] [Google Scholar]
- 75.Dillon CP, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189–202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Upton JW, et al. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11(3):290–7. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaiser WJ, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A. 2014;111(21):7753–8. doi: 10.1073/pnas.1401857111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takahashi N, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513(7516):95–9. doi: 10.1038/nature13706. [DOI] [PubMed] [Google Scholar]
- 79.Rickard JA, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157(5):1175–88. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 80.Dannappel M, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513(7516):90–4. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kearney CJ, et al. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 2015;22(8):1313–27. doi: 10.1038/cdd.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gunther C, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest. 2016;126(11):4346–4360. doi: 10.1172/JCI87545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Newton K, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23(9):1565–76. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nogusa S, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe. 2016;20(1):13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moriwaki K, et al. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J Immunol. 2015;194(4):1938–44. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moriwaki K, et al. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41(4):567–78. doi: 10.1016/j.immuni.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moriwaki K, et al. Border Security: The Role of RIPK3 in Epithelium Homeostasis. Front Cell Dev Biol. 2016;4:70. doi: 10.3389/fcell.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lawlor KE, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vince JE, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36(2):215–27. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 90.Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 91.Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol. 2014;35:14–23. doi: 10.1016/j.semcdb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 92.Smith CC, et al. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21(4):227–33. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 93.Oerlemans MI, et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. 2012;107(4):270. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- 94.Koudstaal S, et al. Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs. Eur J Clin Invest. 2015;45(2):150–9. doi: 10.1111/eci.12391. [DOI] [PubMed] [Google Scholar]
- 95.Luedde M, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. 2014;103(2):206–16. doi: 10.1093/cvr/cvu146. [DOI] [PubMed] [Google Scholar]
- 96.Zhang T, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22(2):175–82. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 97.Wang Z, et al. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148(1-2):228–43. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 98.Moriwaki K, et al. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. J Immunol. 2016;196(1):407–15. doi: 10.4049/jimmunol.1501662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu W, et al. Mitochondrial Protein PGAM5 Regulates Mitophagic Protection against Cell Necroptosis. PLoS One. 2016;11(1):e0147792. doi: 10.1371/journal.pone.0147792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen G, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54(3):362–77. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 101.Duprez L, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35(6):908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 102.Grewal IS. Overview of TNF superfamily: a chest full of potential therapeutic targets. Adv Exp Med Biol. 2009;647:1–7. doi: 10.1007/978-0-387-89520-8_1. [DOI] [PubMed] [Google Scholar]
- 103.Pierdomenico M, et al. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol. 2014;109(2):279–87. doi: 10.1038/ajg.2013.403. [DOI] [PubMed] [Google Scholar]
- 104.Liu ZY, et al. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am J Cancer Res. 2015;5(10):3174–85. [PMC free article] [PubMed] [Google Scholar]
- 105.Kondylis V, et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell. 2015;28(5):582–98. doi: 10.1016/j.ccell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lukens JR, et al. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature. 2013;498(7453):224–7. doi: 10.1038/nature12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meng L, et al. RIP3-dependent necrosis induced inflammation exacerbates atherosclerosis. Biochem Biophys Res Commun. 2016;473(2):497–502. doi: 10.1016/j.bbrc.2016.03.059. [DOI] [PubMed] [Google Scholar]
- 108.Rickard JA, et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife. 2014;3 doi: 10.7554/eLife.03464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Boisson B, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med. 2015;212(6):939–51. doi: 10.1084/jem.20141130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boisson B, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13(12):1178–86. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Re DB, et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81(5):1001–8. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang X, et al. MLKL and FADD Are Critical for Suppressing Progressive Lymphoproliferative Disease and Activating the NLRP3 Inflammasome. Cell Rep. 2016;16(12):3247–59. doi: 10.1016/j.celrep.2016.06.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alvarez-Diaz S, et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity. 2016;45(3):513–26. doi: 10.1016/j.immuni.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou Q, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2016;48(1):67–73. doi: 10.1038/ng.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moriwaki K, Chan FK. Necroptosis-independent signaling by the RIP kinases in inflammation. Cell Mol Life Sci. 2016;73(11-12):2325–34. doi: 10.1007/s00018-016-2203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Han W, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6(5):1641–9. doi: 10.1158/1535-7163.MCT-06-0511. [DOI] [PubMed] [Google Scholar]
- 118.Lehne G. P-glycoprotein as a drug target in the treatment of multidrug resistant cancer. Curr Drug Targets. 2000;1(1):85–99. doi: 10.2174/1389450003349443. [DOI] [PubMed] [Google Scholar]
- 119.Huang C, et al. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One. 2013;8(6):e66326. doi: 10.1371/journal.pone.0066326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shahsavari Z, et al. Shikonin Induced Necroptosis via Reactive Oxygen Species in the T-47D Breast Cancer Cell Line. Asian Pac J Cancer Prev. 2015;16(16):7261–6. doi: 10.7314/apjcp.2015.16.16.7261. [DOI] [PubMed] [Google Scholar]
- 121.Fu Z, et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013;13:580. doi: 10.1186/1471-2407-13-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wada N, et al. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int J Oncol. 2015;46(3):963–72. doi: 10.3892/ijo.2014.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tenev T, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43(3):432–48. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 124.Feoktistova M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–63. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Najjar M, et al. Structure Guided Design of Potent and Selective Ponatinib-Based Hybrid Inhibitors for RIPK1. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li L, et al. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305(5689):1471–4. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 127.Varfolomeev E, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131(4):669–81. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 128.Vince JE, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131(4):682–93. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 129.Bake V, et al. Synergistic interaction of Smac mimetic and IFNalpha to trigger apoptosis in acute myeloid leukemia cells. Cancer Lett. 2014;355(2):224–31. doi: 10.1016/j.canlet.2014.08.040. [DOI] [PubMed] [Google Scholar]
- 130.Cristofanon S, et al. Identification of RIP1 as a critical mediator of Smac mimetic-mediated sensitization of glioblastoma cells for Drozitumab-induced apoptosis. Cell Death Dis. 2015;6:e1724. doi: 10.1038/cddis.2014.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Czaplinski S, et al. Differential role of RIP1 in Smac mimetic-mediated chemosensitization of neuroblastoma cells. Oncotarget. 2015;6(39):41522–34. doi: 10.18632/oncotarget.6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fulda S, et al. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med. 2002;8(8):808–15. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 133.Hehlgans S, et al. The SMAC mimetic BV6 sensitizes colorectal cancer cells to ionizing radiation by interfering with DNA repair processes and enhancing apoptosis. Radiat Oncol. 2015;10:198. doi: 10.1186/s13014-015-0507-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liese J, et al. Smac mimetic and oleanolic acid synergize to induce cell death in human hepatocellular carcinoma cells. Cancer Lett. 2015;365(1):47–56. doi: 10.1016/j.canlet.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 135.Lueck SC, et al. Smac mimetic induces cell death in a large proportion of primary acute myeloid leukemia samples, which correlates with defined molecular markers. Oncotarget. 2016 doi: 10.18632/oncotarget.10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Opel D, et al. Targeting inhibitor of apoptosis proteins by Smac mimetic elicits cell death in poor prognostic subgroups of chronic lymphocytic leukemia. Int J Cancer. 2015;137(12):2959–70. doi: 10.1002/ijc.29650. [DOI] [PubMed] [Google Scholar]
- 137.Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene. 2015;34(47):5796–806. doi: 10.1038/onc.2015.35. [DOI] [PubMed] [Google Scholar]
- 138.Seyfrid M, et al. Reactive oxygen species contribute toward Smac mimetic/temozolomide-induced cell death in glioblastoma cells. Anticancer Drugs. 2016;27(10):953–959. doi: 10.1097/CAD.0000000000000412. [DOI] [PubMed] [Google Scholar]
- 139.Steinwascher S, et al. Identification of a novel synergistic induction of cell death by Smac mimetic and HDAC inhibitors in acute myeloid leukemia cells. Cancer Lett. 2015;366(1):32–43. doi: 10.1016/j.canlet.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 140.Wagner L, et al. Smac mimetic sensitizes glioblastoma cells to Temozolomide-induced apoptosis in a RIP1- and NF-kappaB-dependent manner. Oncogene. 2013;32(8):988–97. doi: 10.1038/onc.2012.108. [DOI] [PubMed] [Google Scholar]
- 141.Brumatti G, et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci Transl Med. 2016;8(339):339ra69. doi: 10.1126/scitranslmed.aad3099. [DOI] [PubMed] [Google Scholar]
- 142.Hannes S, et al. Smac mimetic triggers necroptosis in pancreatic carcinoma cells when caspase activation is blocked. Cancer Lett. 2016;380(1):31–8. doi: 10.1016/j.canlet.2016.05.036. [DOI] [PubMed] [Google Scholar]
- 143.McComb S, et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci Transl Med. 2016;8(339):339ra70. doi: 10.1126/scitranslmed.aad2986. [DOI] [PubMed] [Google Scholar]
- 144.Koo GB, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25(6):707–25. doi: 10.1038/cr.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gerges S, et al. Cotreatment with Smac mimetics and demethylating agents induces both apoptotic and necroptotic cell death pathways in acute lymphoblastic leukemia cells. Cancer Lett. 2016;375(1):127–32. doi: 10.1016/j.canlet.2016.02.040. [DOI] [PubMed] [Google Scholar]
- 146.Steinhart L, et al. Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell death & disease. 2013;4:e802. doi: 10.1038/cddis.2013.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lindemann C, et al. Smac Mimetic-Induced Upregulation of CCL2/MCP-1 Triggers Migration and Invasion of Glioblastoma Cells and Influences the Tumor Microenvironment in a Paracrine Manner. Neoplasia. 2015;17(6):481–9. doi: 10.1016/j.neo.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rettinger E, et al. SMAC Mimetic BV6 Enables Sensitization of Resistant Tumor Cells but also Affects Cytokine-Induced Killer (CIK) Cells: A Potential Challenge for Combination Therapy. Front Pediatr. 2014;2:75. doi: 10.3389/fped.2014.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Seifert L, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532(7598):245–9. doi: 10.1038/nature17403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu X, et al. Key roles of necroptotic factors in promoting tumor growth. Oncotarget. 2016;7(16):22219–33. doi: 10.18632/oncotarget.7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tortola L, et al. The Tumor Suppressor Hace1 Is a Critical Regulator of TNFR1-Mediated Cell Fate. Cell Rep. 2016;15(7):1481–92. doi: 10.1016/j.celrep.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yatim N, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350(6258):328–34. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Aaes TL, et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016;15(2):274–87. doi: 10.1016/j.celrep.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 154.Schmidt SV, et al. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1alpha release, and efficient paracrine dendritic cell activation. Oncotarget. 2015;6(11):8635–47. doi: 10.18632/oncotarget.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zou J, et al. Poly IC triggers a cathepsin D- and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity. 2013;38(4):717–28. doi: 10.1016/j.immuni.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 156.Zou C, et al. Design, Synthesis, and Biological Evaluation of 1-Benzyl-1H-pyrazole Derivatives as Receptor Interacting Protein 1 Kinase Inhibitors. Chem Biol Drug Des. 2016;87(4):569–74. doi: 10.1111/cbdd.12689. [DOI] [PubMed] [Google Scholar]
- 157.Degterev A, et al. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013;20(2):366. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Berger SB, et al. Characterization of GSK'963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov. 2015;1:15009. doi: 10.1038/cddiscovery.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Harris PA, et al. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J Med Chem. 2016;59(5):2163–78. doi: 10.1021/acs.jmedchem.5b01898. [DOI] [PubMed] [Google Scholar]
- 160.Xie T, et al. Structural basis of RIP1 inhibition by necrostatins. Structure. 2013;21(3):493–9. doi: 10.1016/j.str.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 161.Yang CK, He SD. Heat shock protein 90 regulates necroptosis by modulating multiple signaling effectors. Cell Death Dis. 2016;7:e2126. doi: 10.1038/cddis.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jacobsen AV, et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016;7:e2051. doi: 10.1038/cddis.2015.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li D, et al. Natural Product Kongensin A is a Non-Canonical HSP90 Inhibitor that Blocks RIP3-dependent Necroptosis. Cell Chem Biol. 2016;23(2):257–66. doi: 10.1016/j.chembiol.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 164.Han W, et al. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis. 2009;14(5):674–86. doi: 10.1007/s10495-009-0334-x. [DOI] [PubMed] [Google Scholar]
- 165.Jie H, et al. Necrostatin-1 enhances the resolution of inflammation by specifically inducing neutrophil apoptosis. Oncotarget. 2016 doi: 10.18632/oncotarget.8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–20. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 167.Silke J, et al. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16(7):689–97. doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- 168.Dillon CP, et al. Developmental checkpoints guarded by regulated necrosis. Cell Mol Life Sci. 2016;73(11-12):2125–36. doi: 10.1007/s00018-016-2188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Weinlich R, et al. Protective Roles for Caspase-8 and cFLIP in Adult Homeostasis. Cell Rep. 2013 doi: 10.1016/j.celrep.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Xie T, et al. Structural Insights into RIP3-Mediated Necroptotic Signaling. Cell Rep. 2013;5:70–78. doi: 10.1016/j.celrep.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 171.Jagtap PG, et al. Structure-activity relationship study of tricyclic necroptosis inhibitors. J Med Chem. 2007;50(8):1886–95. doi: 10.1021/jm061016o. [DOI] [PubMed] [Google Scholar]
- 172.Teng X, et al. Structure-activity relationship study of [1,2,3]thiadiazole necroptosis inhibitors. Bioorg Med Chem Lett. 2007;17(24):6836–40. doi: 10.1016/j.bmcl.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Teng X, et al. Structure-activity relationship and liver microsome stability studies of pyrrole necroptosis inhibitors. Bioorg Med Chem Lett. 2008;18(11):3219–23. doi: 10.1016/j.bmcl.2008.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang K, et al. Structure-activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5. Bioorg Med Chem Lett. 2007;17(5):1455–65. doi: 10.1016/j.bmcl.2006.11.056. [DOI] [PubMed] [Google Scholar]
- 175.Teng X, et al. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg Med Chem Lett. 2005;15(22):5039–44. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 176.Harris PA, et al. Discovery of Small Molecule RIP1 Kinase Inhibitors for the Treatment of Pathologies Associated with Necroptosis. ACS Med Chem Lett. 2013;4(12):1238–43. doi: 10.1021/ml400382p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fauster A, et al. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 2015;6:e1767. doi: 10.1038/cddis.2015.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kaiser WJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rodriguez DA, et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016;23(1):76–88. doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Li JX, et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014;5:e1278. doi: 10.1038/cddis.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hildebrand JM, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111(42):15072–7. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]