

Abstract
Background
Undervirilized 46,XY males with bifid scrotum often pose a diagnostic challenge, and the majority of cases typically do not receive a genetic diagnosis. NR5A1 mutations can be seen in 10–20% of cases and are a relatively common cause of undervirilization.
Methods
Whole-exome sequencing was utilized to study ten undervirilized 46,XY subjects with bifid scrotum.
Results
Exome sequencing identified novel NR5A1 variants, both impacting exon 7, in two of the ten subjects with bifid scrotum. Subject One had a heterozygous frameshift variant, c.1150delC, p.Leu384fsTer1 within the ligand-binding domain inherited from his unaffected father. Subject Two had a novel splice-site variant c.1139-2T>C, affecting the canonical splice acceptor site for exon 7 and also disrupting the ligand-binding domain. Both subjects had serum testosterone within the normal range as infants.
Conclusions
We describe two novel NR5A1 variants, demonstrating mutations in this gene as a common cause of milder cases of 46,XY undervirilization. Whole-exome sequencing results yielded the diagnosis in 2 out of 10 cases without a previous diagnosis, supporting the value of this approach. Significant genotype-phenotype variability was also noted with subject one’s paternal inheritance from his unaffected father.
Key terms: NR5A1, DSD, bifid scrotum, undervirilization, exome sequencing
Introduction
Undervirilized 46,XY males are a challenging group of patients to evaluate, particularly when searching for an underlying genetic diagnosis. The broad phenotype can range from female-appearing external genitalia with or without mild clitoromegaly, to ambiguous genitalia, to typical male-appearing genitalia with hypospadias. While the more severe cases are readily recognized as disorders of sex development (DSD)[1], milder cases of hypospadias and bifid scrotum may not lead to a DSD label. Studies of exome sequencing applied to 46,XY DSD cases with frankly ambiguous genitalia has demonstrated the success of this approach in establishing a diagnosis in nearly one-third of cases, potentially avoiding expensive and extensive diagnostic odysseys[2]. It remains unclear if this diagnostic yield extends to milder cases.
This study applied whole exome sequencing to 46,XY males with mild/moderate undervirilization, a group not previously assessed using exome sequencing. We selected 46,XY subjects with bifid scrotum as a narrowly defined phenotype who undergo varying degrees of evaluation and often do not receive genetic diagnoses. The majority of subjects with bifid scrotum were also noted to have hypospadias. For many, the phenotype is sufficiently mild that they do not receive a formal DSD workup; indeed, 40% are seen solely by surgeons in the setting of hypospadias repair and undergo few if any diagnostic tests (manuscript in preparation, Boston Children’s Hospital). The initial clinical workup for these patients can vary, but may involve ruling out congenital adrenal hyperplasia (such as 21-hydroxylase deficiency or 3 beta-hydroxysteroid dehydrogenase deficiency), performing ultrasound imaging, obtaining a karyotype, and ordering targeted genetic testing[3]. Partial androgen insensitivity with an androgen receptor (AR) mutation is one of the most frequently identified genetic causes of undervirilization[4]. Mutations in NR5A1, the gene encoding steroidogenic factor-1 (SF-1), is another cause of undervirilization, seen in as many as 10–20% of cases of undervirilization[5,6]. The phenotypic spectrum of disease with NR5A1 is quite variable. Early reports of biallelic NR5A1 mutations were found in patients with complete gonadal dysgenesis and adrenal failure[7–9]. In recent years, there have been a number of publications focused on the wide phenotypic spectrum associated with monoallelic (heterozygous) NR5A1 mutations, including cases of undervirilization without adrenal insufficiency[10–12]. The relatively common 46,XY undervirilized phenotype associated with testicular dysgenesis in the setting of heterozygous NR5A1 mutations appears to be much more common than the rare homozygous cases with associated adrenal insufficiency[5].
This study addresses the diagnostic yield of whole-exome sequencing in a cohort of 10 subjects with bifid scrotum. To our knowledge, this is the first study to apply systematic exome-wide evaluation of mild to moderate undervirilization to better understand the underlying molecular genetics. The findings also have the potential to yield insights into whether these cases share a genetic etiology with more classical DSD presentations.
Methods and Subjects
Subjects and Clinical History
This study was approved by the Boston Children’s Hospital Institutional Review Board. All subjects provided written informed consent and assent (when appropriate). All probands were known to have a 46,XY karyotype and no identified genetic etiology. The majority of cases had physical findings of bifid scrotum with proximal hypospadias, consistent with mild to moderate undervirilization. Presence and location of testes and penile measurements were extracted from medical records. These individuals were all assigned a male gender at birth and had evaluations of hormonal and gonadal function to varying extents depending on clinical providers.
Subject One was born at 39 1/7 weeks gestation by Cesarean section and shortly after delivery was noted to have bifid scrotum, penoscrotal hypospadias, and chordee. The parents were expecting a female infant based on prenatal imaging. Both testes were palpable in prominent, rugated labioscrotal folds. His stretched phallic length was 2.4 cm and his midshaft diameter was 1.1 cm. Family history was unremarkable and parents were not consanguineous. An ultrasound confirmed the presence of inguinal testes bilaterally, and revealed absence of Müllerian structures. Laboratory assessment for adrenal steroid biosynthetic defects due to 3β-hydroxysteroid dehydrogenase deficiency, 17 α-hydroxylase deficiency and 17,20 desmolase deficiency ruled out these conditions. He was found to have a 46,XY karyotype, supporting a male gender assignment. His first testosterone measurement on day of life (DOL)3 was 21 ng/dL. Levels were remeasured in the setting of an hCG stimulation test at approximately 1 month of age. His baseline and stimulated testosterone levels were 195 (reference 233 +/− 88) and 349 ng/dL respectively, and his dihydrotestosterone levels were 193 (reference 50–600) and 415 pg/mL. He also had a cortisol level of 18.6 mcg/dL and ACTH level of 30 pg/mL (reference <45) included in post-hCG results. These results were interpreted as demonstrating robust testicular function and normal conversion of testosterone to dihydrotestosterone. This supported the presumed male gender assignment. Subject one also had androgen receptor (AR) sequencing completed in the course of his clinical care and this testing did not show any pathogenic variants. He was followed until age 2 without any additional testing. He underwent urologic surgery to repair his hypospadias during this time. At age 2, his penis remained small, and his clinical providers treated him with testosterone enanthate 50 mg IM monthly × 3 doses. This resulted in an increased stretched penile length from 3.2 cm to 4.6 cm, and his midshaft diameter increased from 1.6 cm to 2.1 cm, demonstrating a robust response to testosterone. He was otherwise developmentally appropriate for age.
Subject Two was born at 40 6/7 weeks gestation by vaginal delivery and was also noted to have bifid scrotum, penoscrotal hypospadias, and bilateral 1 mL scrotal gonads. Prenatal ultrasounds had suggested the baby appeared female. His scrotum was rugated and pigmented. His stretched phallic length was 2 cm and his midshaft diameter was 1 cm. He had incomplete scrotal fusion and an anogenital ratio of 0.75. Family history without any fertility issues or genitourinary abnormalities. Mother of proband still has regular menses as of age 33. Maternal grandmother with hot flashes, headaches, and sleep issues starting at age 39. Menses stopped by age 44. An ultrasound showed normal-appearing scrotal testes with a blind-ending vaginal pouch and no Müllerian structures. He had an evaluation of adrenal hormones and precursors that ruled out 17β -hydroxysteroid dehydrogenase deficiency, 17,20-lyase deficiency and 3β -HSD deficiency. His karyotype returned as 46,XY. He had a serum testosterone of 48 ng/dL on DOL1 and a stimulated cortisol level of 26.8 mcg/dL at 2 weeks, indicating normal adrenal function. At age 3 weeks, LH was 8.93 IU/L, FSH was 7.97 IU/L, testosterone was 177 ng/dL and dihydrotestosterone was 301 pg/mL. Following 3 days of hCG stimulation, his testosterone rose to 231 ng/dL and dihydrotestosterone to 500 pg/mL, consistent with appropriate testicular function. By one month of age, his penis size increased without treatment to a stretched length of 2.5 cm with a midshaft diameter of 1 cm. This improvement was thought to be related to his endogenous testosterone and, given the appropriate dimensions for age, he did not receive exogenous IM testosterone nor was it recommended that he have androgen receptor sequencing.
Genetic Analyses
Whole-exome sequencing of blood or saliva-derived genomic DNA was performed at the Broad Institute (Cambridge, Massachusetts, USA) on ten probands as well as parents for six of the probands. For hybrid selection, we used the custom Illumina Content Exome capture kit (Illumina, San Diego, California, USA). Sequencing reads were aligned to the hg19 reference genome[13]. We applied the Genome Analysis Toolkit (GATK) for base quality score recalibration and indel (insertion-deletion) realignment[14]. Variant quality score recalibration was simultaneously performed for SNP and indel discovery according to GATK Best Practices recommendations [15,16]. We used SnpEff (http://snpeff.sourceforge.net/) for functional annotation. We filtered for variants that were novel or found in less than 1% of the reference population based on allele frequencies from the Exome Aggregation Consortium (http://exac.broadinstitute.org/)[17]. Based on the quality standards at the Broad Institute, at least 80% of the exome had 20X coverage. When parental sequencing was performed, we additionally filtered for de novo variants that were absent in ExAC. Results were reviewed for variants in known DSD related genes previously reported[2] as well as genes near hypospadias GWAS loci[18]. Sanger sequencing was performed to confirm the variants of interest along with SRY confirmation when indicated.
Hormonal Assays
Luteinizing hormone (LH) and follicle stimulating hormone (FSH) were analyzed on the Roche e170 analyzer using a chemiluminescent immunoassay (Roche Diagnostics, Indianapolis, IN). Testosterone and dihydrotestosterone (DHT) were measured by quantitative liquid chromatography/tandem mass spectrometry at Boston Children’s Hospital (Boston, MA) and ARUP Laboratories (Salt Lake City, Utah), respectively.
Results
The clinical characteristics and laboratory measurements for the ten subjects with bifid scrotum who had exome sequencing performed are detailed in Table 1. Most subjects had severe hypospadias in addition to bifid scrotum. Laboratory measurements during the first few months of life, when the gonadotropin axis is active, were available for half of the subjects.
Table 1.
| ID | GA | Clinic Visit |
Phallic Stretched Length** |
Phallic Midshaft Diameter** |
Hypospadias Degree |
Gonadal Location |
Testosterone*** (ng/dL) |
DHT*** (pg/mL) |
LH*** (IU/L) |
FSH*** (IU/L) |
Clinical AR Sequencing |
Non-GU Abnormalities |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| 1* | 39.1 | Both | 2.1 | 0.9 | Penoscrotal | Scrotal | 195 | 193 | 4.34 | NR | Y, no diagnosis | None |
|
| ||||||||||||
| 2* | 40.9 | Both | 2 | 1 | Perineal | Scrotal | 177 | 301 | 8.93 | 7.97 | N | None |
| 3 | 40.0 | Urology | NR | NR | No hypospadias | Scrotal | NR | NR | NR | NR | N | Tethered cord, anteriorly placed anus, renal abnormalities |
|
| ||||||||||||
| 4 | 33.0 | Both | 2.5 | 1.1 | Penoscrotal | Scrotal | 132 | 462 | 3 | 3.34 | N | Intrauterine growth restriction |
|
| ||||||||||||
| 5 | 41.0 | Both | 3.2 | 1.1 | Perineal | Inguinal | NR | NR | NR | NR | N | None |
| 6 | 34.7 | Both | NR | NR | Scrotal | Scrotal | 207 | 71 | NR | NR | Y, no diagnosis | None |
|
| ||||||||||||
| 7 | 37.0 | Both | NR | NR | Scrotal | Scrotal | NR | NR | NR | NR | N | Global developmental delay |
| 8 | 35.3 | Urology | NR | NR | Penoscrotal | Scrotal | NR | NR | NR | NR | N | None |
|
| ||||||||||||
| 9 | 39.1 | Urology | NR | NR | Glanular | Scrotal | NR | NR | NR | NR | N | Ventricular Septal Defect |
|
| ||||||||||||
| 10 | 39.0 | Both | 2 | 1.1 | Perineal | Scrotal | 85.9 | 29 | 5.23 | 4.62 | N | None |
GA, Gestational age in weeks
NR, not reported
Clinic visit: Endocrinology, Urology or both (at any point in the EMR)
Testosterone: ng/dL * 0.03467 = nmol/L
Dihydrotestosterone: pg/mL * 0.003443 = nmol/L
Subjects found to have NR5A1 presumed pathogenic variants
In cm, initial measurement reported between birth and 3 months of age)
Baseline measurement between 2 weeks and 3 months
Exome sequencing identified two subjects with presumed pathogenic mutations in known DSD-associated genes – both in NR5A1 (Figure 1)[6,19], which had not been sequenced as part of the clinical evaluation in any of the 10 subjects. Subject One was found to have a heterozygous frameshift variant, c.1150delC, p.Leu384fsTer1 within the ligand binding domain. This frameshift variant is predicted to result in premature cessation of translation, with nearly 80 fewer amino acids than the wild-type protein product. This variant was not present in any of the 60,706 exomes in ExAC. A nearly identical frameshift variant, c.1151del, p.Leu384Argfs*7, was recently reported in a 46,XY female with clitoromegaly and was shown to have significantly decreased activity of the protein product SF-1 on functional assays[20]. Sequencing of Subject One’s parents revealed the same variant in the subject’s father. The presence of SRY was confirmed in Subject One’s father to ensure that parental samples were not switched. Evaluation of the father’s exome sequencing results did not show any evidence of somatic mosaicism – the frameshift variant was found in 8 of 11 sequence reads. The father denied any history of genital abnormalities, known hormonal abnormalities, or surgeries.
Figure 1.
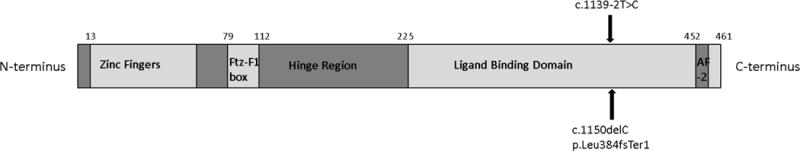
NR5A1 gene structure, with location of heterozygous mutations in Subjects One and Two. Both mutations involved exon 7 in the ligand binding domain. (adapted from Wong, J Mol Endocrinol, 1996 and Camats, JCEM, 2012) AF-2: activation function domain 2
*Figure not according to scale.
Subject Two was found to have a heterozygous novel splice-site variant, c.1139-2T>C. The variant disrupts a canonical splice acceptor site and is predicted to perturb the normal splicing of exon 7, which encodes a portion of the ligand binding domain of SF-1. Subject Two’s mother was found to have the same NR5A1 variant. As shown in Table 1, there were no obvious clinical differences between the subjects found to have NR5A1 variants as compared to the rest of the cohort.
No other pathogenic or likely pathogenic variants in known DSD genes[2], including AR, were identified in the cohort of 10 subjects.
Discussion
Establishing genetic diagnoses in individuals with undervirilization has the potential to improve the care of these patients. As next generation sequencing costs continue to decrease, more comprehensive sequencing approaches have already started to supplant targeted single-gene testing. Prior exome-based studies have focused on classic DSD cases with ambiguous genitalia[2]. To our knowledge, this study is the first to apply whole-exome sequencing to a cohort of cases with bifid scrotum and hypospadias but not frank genital ambiguity.
In our study of bifid scrotum cases, 2 of the 10 patients were found to have NR5A1 variants that are likely to be pathogenic. This finding is consistent with previous reports that found pathogenic NR5A1 variants in 10–20% of DSD cases with varying degrees of undervirilization without adrenal insufficiency[5]. In a related chart review, we found that androgen receptor mutations were identified in 2 of 10 patients with bifid scrotum during standard clinical evaluation (Swartz et al., manuscript in preparation). Thus, either whole-exome sequencing or targeted sequencing of AR and NR5A1 may provide a diagnosis in a significant proportion of 46,XY cases with bifid scrotum. This information can guide understanding of recurrence risk in a family and help inform expected progression of symptoms, appropriate dose of hormonal supplementation (if needed), tumor risk, and fertility.
A number of NR5A1 mutations have been previously described in the literature. Heterozygous or homozygous NR5A1 mutations can cause DSD, and its gene product SF-1 plays a role in both gonadal and adrenal development as well as steroidogenesis. SF-1 is an orphan nuclear receptor with a central role as a transcription factor for regulation of adrenal and gonadal developmental genes including SOX9, NR0B1, and AMH as well as many genes encoding enzymes essential for steroid hormone synthesis[21]. Previous reports have noted that the degree of undervirilization appears greater than the degree of testicular dysgenesis in many of the described cases[21].
The observation of paternal Inheritance of the NR5A1 mutation in Subject One is an unusual finding from this study. To date, the vast majority of familial NR5A1 mutations have been inherited maternally[22,23], as in the case of Subject Two. There is one report of a missense NR5A1 variant inherited from an unaffected father with normal external genitalia and maintained fertility, but in that case the father was believed to have low level somatic mosaicism[22]. There are also reports of paternal inheritance of frameshift variants from affected fathers with hypospadias but intact fertility[24], as well as reports of an extended family with an NR5A1 missense variant transmitted via an affected male with hypospadias[25]. Our finding of a frameshift variant inherited from Subject One’s apparently unaffected father is very unusual in the NR5A1 literature, especially given the lack of evidence of somatic mosaicism on sequencing results. This may be an example of the incomplete penetrance in NR5A1 cases. Alternatively, digenic inheritance is a possibility with an additional yet to be identified gene influencing the phenotypes of different family members. Monoallelic expression is also a potential factor, though enough cases have been reported in the literature that this pattern would potentially have been identified already. It is also possible Subject One’s father has a mild phenotype with some degree of endocrine dysfunction and/or subclinical oligospermia. The splice site variant is Subject Two is also relatively uncommon in the NR5A1 literature, with only a few reports of splice site variants in NR5A1 published to date [12,26].
This study highlights that normal testosterone levels during infancy, either at baseline or after hCG stimulation, do not rule out NR5A1 mutations. Most prior reports found low levels of testosterone in undervirilized NR5A1 cases, though there have been some reports of normal androgen biosynthesis in such patients [6,11,12,24,27]. The evidence of undervirilization at birth indicates decreased androgen signaling during early fetal development, but this signaling abnormality may not be evident on later testing. This raises a yet-unanswered question about the role of SF-1 in steroidogenesis during early fetal life compared to postnatal life. This may be an indication that intra-individual variability on the impact of SF-1 variants on steroidogenesis at different developmental times (e.g., in utero vs postnatal life) may be as relevant as interindividual variability seen within families with the same NR5A1 variants[23].
This study also demonstrates the utility of genetic testing in patients with bifid scrotum and hypospadias. Exome sequencing was shown to be high-yield in more severe DSD cases, with a likely genetic diagnosis being identified in about one-third of patients with 46,XY DSD [2]. Our study suggests that exome sequencing is similarly high yield in milder cases of undervirilization. There is evidence that a significant fraction of bifid scrotum cases may have an identifiable genetic mutation that will improve disease understanding as well as be important for families interested in having additional children. These findings have significant implications for families and their management in the clinical setting. The recommendations for testing have been evolving in recent years with the decreasing costs of whole-exome sequencing. Although targeted single gene testing is reasonable when there is a high pre-test probability of a pathogenic result, the cost of multiple single gene Sanger sequencing quickly outpaces the cost of next generation sequencing approaches (either disease specific panel or whole-exome)[28]. This will be a more and more significant consideration as the costs of next generation sequencing continue to decline. Even with these factors to consider, challenges remain significant given the difficulty in obtaining insurance coverage for clinical whole-exome sequencing. Based on our findings, we feel it is reasonable to perform a genetic evaluation for all children with bifid scrotum and hypospadias, even those with a normal hormonal profile, as the genetic evaluation is reasonably likely to establish a diagnosis, which in turn can guide counseling about risk for future siblings, treatment, and prognosis.
Established Facts
Whole exome sequencing has been used effectively in severe DSD cases to yield a diagnosis.
Heterozygous mutations in NR5A1 have been identified as a cause of undervirilization in 46,XY males without adrenal insufficiency.
The vast majority of inherited NR5A1 cases reported in the literature in 46,XY males are from carrier mothers.
Novel Insights
Whole-exome sequencing performed on ten 46,XY subjects with bifid scrotum led to the identification of potentially pathogenic heterozygous NR5A1 mutations in two cases.
One of the heterozygous NR5A1 variants was inherited from an unaffected father, expanding the phenotypic spectrum associated with NR5A1 variants reported in the literature.
Acknowledgments
We are grateful to all of the patients and families willing to participate in this study – for their time and contributions in helping to advance the field. We also thank Dr. Christina Astley and Ms. Kara McLaughlin for their advice and assistance in completing this study. We would finally like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Grants or fellowships supporting the writing of the paper: This work was supported by National Institutes of Health (NIH) Grant 5T32DK007699-32 (J.S.), the March of Dimes Foundation, as well as the Manton Center for Orphan Disease Research. This work was also conducted with support from Harvard Catalyst. The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR001102) and financial contributions from Harvard University and its affiliated academic healthcare centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic healthcare centers, or the National Institutes of Health.
Footnotes
PES Membership: Jonathan Swartz, Yee-Ming Chan, Joel Hirschhorn
Disclosure summary: The authors have no relevant conflicts of interest.
References
- 1.Hughes I, Houk C, Ahmed SF, Lee P. Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group: Consensus statement on management of intersex disorders. J Pediatr Urol. 2006 Jun;2:148–62. doi: 10.1016/j.jpurol.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Baxter RM, Arboleda V, Lee H, Barseghyan H, Adam MP, Fechner PY, et al. Exome Sequencing for the Diagnosis of 46,XY Disorders of Sex Development. J Clin Endocrinol Metab. 2015 Nov 10;100:E333–E344. doi: 10.1210/jc.2014-2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arboleda V, Lee H, Sánchez FJ, Délot EC, Sandberg DE, Grody WW, et al. Targeted massively parallel sequencing provides comprehensive genetic diagnosis for patients with disorders of sex development. Clin Genet. 2013 Jan;83:35–43. doi: 10.1111/j.1399-0004.2012.01879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed SF, Bashamboo A, Lucas-Herald A, McElreavey K. Understanding the genetic aetiology in patients with XY DSD. Br Med Bull. 2013 Jan;106:67–89. doi: 10.1093/bmb/ldt008. [DOI] [PubMed] [Google Scholar]
- 5.Suntharalingham JP, Buonocore F, Duncan AJ, Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab. 2015;29:607–619. doi: 10.1016/j.beem.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camats N, Pandey AV, Fernández-Cancio M, Andaluz P, Janner M, Torán N, et al. Ten novel mutations in the NR5A1 gene cause disordered sex development in 46,XY and ovarian insufficiency in 46,XX individuals. J Clin Endocrinol Metab. 2012 Jul;97:E1294–306. doi: 10.1210/jc.2011-3169. [DOI] [PubMed] [Google Scholar]
- 7.Achermann JC, Ito M, Hindmarsh PC, Jameson JL. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999;22:125–6. doi: 10.1038/9629. [DOI] [PubMed] [Google Scholar]
- 8.Ito M, Achermann JC, Jameson JL. A naturally occurring steroidogenic factor-1 mutation exhibits differential binding and activation of target genes. J Biol Chem. 2000;275:31708–14. doi: 10.1074/jbc.M002892200. [DOI] [PubMed] [Google Scholar]
- 9.Achermann JC, Ozisik G, Ito M, Orun UA, Harmanci K, Gurakan B, et al. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab. 2002 Apr;87:1829–33. doi: 10.1210/jcem.87.4.8376. [DOI] [PubMed] [Google Scholar]
- 10.Lin L, Philibert P, Ferraz-de-Souza B, Kelberman D, Homfray T, Albanese A, et al. Heterozygous missense mutations in steroidogenic factor 1 (SF1/Ad4BP, NR5A1) are associated with 46,XY disorders of sex development with normal adrenal function. J Clin Endocrinol Metab. 2007;92:991–999. doi: 10.1210/jc.2006-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Köhler B, Lin L, Ferraz-de-Souza B, Wieacker P, Heidemann P, Schröder V, et al. Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46,XY patients with severe underandrogenization but without adrenal insufficiency. Hum Mutat. 2008 Jan;29:59–64. doi: 10.1002/humu.20588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Köhler B, Lin L, Mazen I, Cetindag C, Biebermann H, Akkurt I, et al. The spectrum of phenotypes associated with mutations in steroidogenic factor 1 (SF-1, NR5A1, Ad4BP) includes severe penoscrotal hypospadias in 46,XY males without adrenal insufficiency. Eur J Endocrinol. 2009 Aug;161:237–42. doi: 10.1530/EJE-09-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans [Internet] bioRxiv. 2015 Oct 30; doi: 10.1038/nature19057. Available from: http://biorxiv.org/content/early/2015/10/30/030338.abstract. [DOI] [PMC free article] [PubMed]
- 18.Geller F, Feenstra B, Carstensen L, Pers TH, van Rooij IaLM, et al. Genome-wide association analyses identify variants in developmental genes associated with hypospadias. Nat Genet. 2014 Aug 10; doi: 10.1038/ng.3063. [DOI] [PubMed] [Google Scholar]
- 19.Wong M, Ramayya MS, Chrousos GP, Driggers PH, Parker KL. Cloning and sequence analysis of the human gene encoding steroidogenic factor 1. J Mol Endocrinol. 1996;17:139–147. doi: 10.1677/jme.0.0170139. [DOI] [PubMed] [Google Scholar]
- 20.Woo KH, Cheon B, Kim JH, Cho J, Kim GH, Yoo HW, et al. Novel Heterozygous Mutations of NR5A1 and Their Functional Characteristics in Patients with 46,XY Disorders of Sex Development without Adrenal Insufficiency. Horm Res Paediatr. 2015;84:116–123. doi: 10.1159/000431324. [DOI] [PubMed] [Google Scholar]
- 21.Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev. 2008;2:200–209. doi: 10.1159/000152036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Philibert P, Polak M, Colmenares A, Lortat-Jacob S, Audran F, Poulat F, et al. Predominant Sertoli cell deficiency in a 46,XY disorders of sex development patient with a new NR5A1/SF-1 mutation transmitted by his unaffected father. Fertil Steril. 2011;95:5–9. doi: 10.1016/j.fertnstert.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 23.Warman DM, Costanzo M, Marino R, Berensztein E, Galeano J, Ramirez PC, et al. Three new SF-1 (NR5A1) gene mutations in two unrelated families with multiple affected members: Within-family variability in 46,XY subjects and low ovarian reserve in fertile 46,XX subjects. Horm Res Paediatr. 2011;75:70–77. doi: 10.1159/000320029. [DOI] [PubMed] [Google Scholar]
- 24.Baetens D, Mladenov W, Delle Chiaie B, Menten B, Desloovere A, Iotova V, et al. Extensive clinical, hormonal and genetic screening in a large consecutive series of 46,XY neonates and infants with atypical sexual development. Orphanet J Rare Dis. 2014;9:209. doi: 10.1186/s13023-014-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciaccio M, Costanzo M, Guercio G, De Dona V, Marino R, Ramirez PC, et al. Preserved Fertility in a Patient with a 46,XY Disorder of Sex Development due to a New Heterozygous Mutation in the NR5A1/SF-1 Gene: Evidence of 46,XY and 46,XX Gonadal Dysgenesis Phenotype Variability in Multiple Members of an Affected Kindred. Horm Res Paediatr. 2012;78:119–126. doi: 10.1159/000338346. [DOI] [PubMed] [Google Scholar]
- 26.Takagi M, Nishina N, Yagi H, Hasegawa Y. Functional Characterization of c 870+3_6delGAGT Splice Site Mutation in NR5A1. Horm Res pædiatrics. 2016;85:65–8. doi: 10.1159/000440862. [DOI] [PubMed] [Google Scholar]
- 27.Wu JY, McGown IN, Lin L, Achermann JC, Harris M, Cowley DM, et al. A novel NR5A1 variant in an infant with elevated testosterone from an Australasian cohort of 46,XY patients with disorders of sex development. Clin Endocrinol (Oxf) 2013;78:545–550. doi: 10.1111/cen.12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Achermann JC, Domenice S, Bachega TASS, Nishi MY, Mendonca BB. Disorders of sex development: effect of molecular diagnostics. Nat Rev Endocrinol. 2015;11:478–88. doi: 10.1038/nrendo.2015.69. [DOI] [PubMed] [Google Scholar]