

Abstract
The innate immune response constitutes the first line of defense against infections by pathogens. Successful pathogens such as human papillomaviruses (HPVs) have evolved mechanisms that target several points in these pathways including sensing of viral genomes, blocking the synthesis of interferons and inhibiting the action of JAK/STAT transcription factors. Disruption of these inhibitory mechanisms contributes to the ability of HPVs to establish persistent infections, which is the major etiological factor in the development of anogenital cancers. Interestingly, HPVs also positively activate several members of these pathways such as STAT-5 that are important for their differentiation-dependent life cycle. STAT-5 activation induces the ATM and ATR DNA damage response pathways that play critical roles in HPV genome amplification. Targeting of these pathways by pharmaceuticals can provide novel opportunities to inhibit infections by these important human pathogens.
Keywords: interferons, JAK-STAT pathway, inflammasome, PAMPs
1. Introduction
The host defense against pathogens is mediated by two basic systems: the innate immune pathway and the adaptive immune response. The innate immune response is the first line of defense against pathogens such as bacteria, fungi, or viruses. Following entry, pathogens are recognized by pattern recognition receptors (PRRs) that can identify small non-self molecular motifs referred to as pathogen-associated molecular patterns or PAMPs (Abbas et al., 2015). PAMPs include lipopolysaccharides and endotoxins on the surface of bacteria and nucleic acid motifs present on viral genomes. For viruses, recognition by PRRs leads to the activation of a cascade of downstream signaling pathways, including the interferon regulator factor (IRF) signaling pathway and the signal transducer and activator of transcription (STAT) pathway, as well as the production of many cytokines such as interferons (IFNs) and interleukin-1 (IL-1) (Abbas et al., 2015). IFNs are named for their ability to protect cells from viral infection by acting as messengers between cells to trigger expression of antiviral genes. Following infection of keratinocytes by human papillomaviruses (HPVs) an innate immune response is triggered and viral proteins act to modulate this response to establish persistent infections (Hebner and Laimins, 2006). Prior to describing what is known about how HPV modulates the innate immune response, it is first important to provide a general description of the cellular pathways involved. We will describe how viruses are detected by the innate immune response and how this leads to synthesis of IFNs and other cytokines. This is followed by a discussion of IRF signaling as well as the effects of IFNs on the JAK/STAT pathway and the roles of these factors in regulating the productive life cycle of HPVs. In addition, the effects of HPV proteins on cytokine expression will be addressed.
2. Summary of the innate immune response
2.1 Sensing of viral infection
The first step in mounting an innate immune response against viral infections is the detection of viral genomes following entry into cells, which as discussed is mediated by PRRs. PRRs are classified as cell surface PRRs or cytosolic according to their cellular locations (Abbas et al., 2015). The PRRs that detect RNA genomes are better understood than those that recognize DNA genomes and some of these act on both types of genomes. Following entry, viral RNAs can be recognized by membrane-bound Toll-like receptors (TLRs), such as TLR3, 7, and 8, as well as RIG-I-like receptors (RLRs), such as retinoic acid-inducible gene-I protein (RIG-I), melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (Lgp2) (Seth et al., 2006). TLR3 regulates the activation of the transcription factor IRF3, which induces expression of IFNs (Doan et al., 2008). Other TLRs act through the myeloid differentiation primary response gene 88 (MyD88) pathway that results in recruitment of downstream signaling molecules such as kinases that phosphorylate the inhibitor of nuclear factor kappa-B kinase subunit β (IKKβ) to induce nuclear factor kappa-light chain-enhancer of activated B cells (NF-κB) signaling (Seth et al., 2006). Another arm of RNA sensing is RIG-I/MDA5 pathway which can induce NF-κB activation as well as IFN production (Loo and Gale, 2011).
The DNA viral sensors include DNA-dependent activator of IFN-regulatory factors (DAI), Absent in melanoma 2 (AIM2), NACHT, LRR and PYD domains-containing protein 3 (NLRP3), γ-interferon-inducible protein 16 (IFI16), cyclic GMP-AMP synthase (cGAS) and other factors (Chan and Gack, 2016; Chen and Ichinohe, 2015). DAI responds to poly(dA:dT) or viral double-stranded DNAs (dsDNAs) to induce IFN expression (Takaoka et al., 2007). In a similar manner AIM2 and IFI16 are cytosolic DNA sensors that contribute to the activation of IFN signaling. cGAS recognizes and binds to dsDNA to produce cyclic GMP-AMP, which activates the interferon synthesis pathway through the stimulator of interferon genes (STING) proteins (Gao et al., 2013; Sun et al., 2013). Though these DNA sensors have been shown to be associated with many viruses, only a few studies have characterized their effects on HPV infection and this will be discussed below.
2.2 Interferon and the JAK/STAT pathway
Recognition of viral infection by PRRs results in activation of the IRF family of transcription factors, which consists of nine members (IRF1-9). Following activation, these factors bind to the promoters of interferon-stimulated genes (ISGs) through specific sequences called interferon stimulatory response elements (ISRE) to activate transcription (Abbas et al., 2015). The IRFs are initially localized inactive in the cytoplasm and following activation migrate to the nucleus to activate interferon transcription as well as expression of other cellular genes such as p53. The IFN family consists of three major types. For human, Type I IFNs include IFN-α, IFN-β, IFN-ε, IFN-κ, and IFN-ω. Type II is IFN-γ whereas type III IFNs include IFN-λ1, IFN-λ2, and IFN-λ3 (Abbas et al., 2015). Type I IFNs are synthesized following recognition of viral genomes by PRRs and subsequent activation of IRF transcription factors. Type II IFNs are synthesized primarily by natural killer (NK) cells and natural killer T cells along with CD4 and CD8 T cells (Doan et al., 2008). Type III IFNs work similar to type I but act through different cytokine receptor complexes. IRF1, 3, 5 and 7 are the primary activators of Type I IFNs while IRF2 is a negative regulator of IRF1 activity (Doan et al., 2008).
After synthesis and secretion of IFNs from the infected cells, extracellular IFNs bind to receptors on adjacent cells leading to the activation of the JAK-STAT pathway to stimulate expression of hundreds of genes that act to block viral propagation. In the canonical type I IFN pathway, IFN-α and IFN-β bind to a heterodimeric transmembrane receptor termed IFNAR, which consists of IFNAR1 and IFNAR2 subunits (Abbas et al., 2015). Engagement of IFNAR activates the receptor-associated Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), which phosphorylate inactive STAT proteins located in the cytoplasm. Similarly, IFN-γ binds to IFNGR to activate JAK1 and JAK2, which then phosphorylate STAT proteins.
The STAT protein family has seven mammalian members: STAT-1, STAT-2, STAT-3, STAT-4, STAT-5α, STAT-5β, and STAT-6. STAT-1 and 2 are primarily responsible for mediating the response to type I and II IFNs while STAT-5 responds to cytokines and activates a different set of downstream genes (Doan et al., 2008). The other STATs play roles in development of cancers as well as inflammation and respond to different signaling molecules. In this review we will focus the discussion on the activities of STAT-1, 3 and 5 as they have been shown to impact HPV pathogenesis.
In uninfected cells, unphosphorylated STAT proteins are localized in the cytoplasm. Following receptor binding of IFNs, the STAT proteins become phosphorylated by receptor associated kinases, such as JAK1, 2 and TYK2 (Abbas et al., 2015). Upon phosphorylation, STAT-1and STAT-2 form heterodimers that also recruit IRF-9, to form a complex referred to as interferon stimulated gene factor (ISGF3). The ISGF3 complex migrates to the nucleus to bind to the interferon stimulated elements (ISRE) located in promoter regions of over 100 anti-viral genes (referred to as ISGs) to induce their expression. These ISGs include myxovirus resistance 1 (MX1), IFN-inducible double-stranded RNA-dependent protein kinase (PKR), 2′-5′-oligoadenylate synthease (OAS), IFN-induced protein with tetratricopeptide repeats (IFIT), IFN-induced transmembrane proteins (IFITMs) (Diamond and Farzan, 2013) and the tripartite motif-containing (TRIM) family of molecules (Rajsbaum et al., 2014). Two important factors are PKR that blocks translation of viral proteins and RNase L that mediates degradation of viral RNAs. Following binding of IFNγ to its receptors, bound JAK kinases are activated to phosphorylate STAT-1 proteins that then form homodimers, migrate to the nucleus and bind to interferonγ-activated sequence (GAS) elements in the promoters to induce the expression of ISGs. An overlapping set of genes becomes activated by ISGF3 complexes and STAT-1 homodimers. Similarly STAT-5 is localized in an unphosphorylated form in the cytoplasm and become activated by phosphorylation following binding of cytokines to the cytokine receptors resulting in the formation of either homo or heterodimers between its two isoforms STAT-5α and STAT-5β (Heltemes-Harris and Farrar, 2012). These STAT-5 complexes activate a different set of downstream genes than those targeted by STAT-1 and STAT-2. Interestingly, recent studies have also shown that unphosphorylated STAT-1 can associate with unphosphorylated STAT-2 as well as IRF9 to induce expression of a small subset of genes that have antiviral or immune-regulatory functions (Cheon and Stark, 2009).
2.3 The NF–κB pathway
The NF–κB (nuclear factor kappa-light chain-enhancer of activated B cells) complex of proteins plays a central role in regulating the immune response to viral and bacterial infections. It regulates transcription of cytokines and other factors in response to stress, external cytokines, UV as well as viral and bacterial antigens (Abbas et al., 2015). NF–κB is present in the cytoplasm in an inactive state in complex with the inhibitor IκBα. Following activation of the kinase IKK by external stimuli sensed by membrane receptors such as the TLRs, IκBα becomes phosphorylated resulting in ubiquitination and degradation. The unbound NF–κB then migrates to the nucleus to bind to specific response elements in the promoter regions of a set of responsive genes including those involved in innate immune regulation (Doan et al., 2008). The NF–κB family of proteins consists of NF–κB1 and 2 that are made up of p50 and p52 subunits as well as three REL (proto-oncogene c-Rel) proteins that can form either homo or heterodimers (Abbas et al., 2015). Importantly the NF–κB pathway is often suppressed by viruses during infection.
3. Human papillomaviruses
3.1 HPV viral proteins and its life cycle
HPVs infect stratified epithelia and are the primary causative agents of cervical, anal as well as many oropharyngeal cancers. Over 200 types of HPVs have been identified and approximately one third of these infect the genital tract (zur Hausen, 2002). Approximately 9 types of HPV are considered as high risk types, including HPV16, 18, 31, and 45 that are the causative agents of most anogenital cancers. HPVs’ genomes are about 8 kb in length and contain at least eight open reading frames that encode for E1, E2, E4, E5, E6, E7 early proteins, as well as two capsid proteins L1 and L2 (Munger and Howley, 2002). E1 and E2 regulate HPV genome replication, while E4 and E5 modulate late viral functions. The E6 and E7 proteins regulate cell cycle regulatory activities in both undifferentiated and differentiated cells as well as play critical roles in maintenance of viral episomes. In the high-risk types, E6 and E7 act as oncogenes that are necessary for the development of anogenital cancers. One of the important targets of the high-risk E6 protein is the transcription factor p53. E6 binds to the cellular E3 ubiquitin ligase E6-associated protein (E6AP) to degrade p53 (Huibregtse et al., 1991; Scheffner et al., 1993; 1990) as well as to inhibit p53 function by blocking acetylation (Hebner et al., 2007). The E7 protein regulates cell cycle events by binding to retinoblastoma protein (pRb) to facilitate the activities of E2F family members that regulate expression of many S-phase genes (Dyson et al., 1989; Longworth et al., 2005; Munger et al., 1989). A more detailed discussion of the action of the HPV viral proteins is described in other chapters in this volume.
To understand how HPVs modulate the innate immune response, it is important to first briefly describe the viral life cycle. The HPV life cycle is dependent on epithelial differentiation (Bodily and Laimins, 2011). Since these viruses encode only a small number of proteins they must utilize the host regulatory and replication enzymes to facilitate viral replication (Hebner and Laimins, 2006). HPVs infect cells in the basal layers of stratified squamous epithelia that become exposed upon trauma. Following entry, HPV genomes are established as low copy episomes (around 100 copies per cell) in the nuclei of basal cells. In infected basal cells, HPV episomes are replicated together with cellular chromosomes and distributed equally to the new basal cell and the daughter cell that will undergo differentiation. As this infected daughter cell undergoes differentiation, the viral genomes are replicated to thousands of copies per cell in a process referred to as amplification. While little is known how HPV modulates the innate immune response during initial viral entry, information is available about how viral proteins target these pathways once early genes are expressed.
3.2 Sensing HPV by the host DNA sensors
HPV utilizes several distinct ways to escape innate immune surveillance to establish persistent infections. First, HPV targets the DNA sensors that trigger downstream events to impair viral infection. Second, HPV interferes with the activities of IRF proteins that regulate interferon expression. Third, HPV proteins block activation of the JAK-STAT pathway to suppress expression of downstream effector genes. We will discuss each of these mechanisms in detail below.
Several studies reported that nuclear and cytoplasmic DNA sensors recognize HPV genomes. A role for the nuclear DNA sensor, IFI16, in detecting HPV genomes has been suggested by studies in which its overexpression impairs HPV18 transient replication while silencing increases replication (Lo Cigno et al., 2015). Interestingly, examination of the biopsy material from patients demonstrates increased levels of IFI16 in HPV-related cancers (Mazibrada et al., 2014; Reinholz et al., 2013), suggesting this factor may also play a role in cancer progression perhaps by interfering with viral replication. The HPV18 E7 protein has been reported to bind to cytoplasmic DNA sensor STING and inhibit its activity, while silencing of E7 restored cGAS-STING activation. No studies, however, have reported the effects of cGAS-STING on viral replication or stable maintenance of episomes so it is unclear how this binding affects these activities. In addition, RIG-I/MDA5 expression is down-regulated in HPV-positive cells (Reiser et al., 2011), however, neither E6 nor E7 associates with members of the RIG-I pathway.
A series of studies have reported HPV-induced changes in the levels of Toll-like receptors though the effects differ with the HPV types. Some studies indicate TLR9 levels are increased in HPV16-positive cells, while others report a decrease (Cannella et al., 2015; Hasan et al., 2013). In addition, no changes are seen in TLR9 levels in HPV18-positive cells while TLR3 levels are reduced (Reiser et al., 2011). Moreover, HPV38 down-regulates TLR9 levels through the action of the transcriptional regular ΔNp73 (Pacini et al., 2015). It is therefore difficult to make general conclusions about TLRs and HPV infections as effects may be type specific.
3.3 Suppression of IFN signaling by HPV
In addition to targeting DNA sensors, HPV deregulates the synthesis of interferons by regulating IRF transcription factors (Figure 1B). HPV16 E7 has been shown to block IRF-1 expression via an HDAC-dependent mechanism (Park et al., 2000). Similarly, overexpression of HPV38 E6E7 leads to downregulation of IRF-1 as well as MHC I heavy chain expression (Cordano et al., 2008). HPV16 E6 also targets IRFs and has been shown to bind to IRF-3 to inhibit its transcriptional activity (Ronco et al., 1998). As a consequence of these and other as yet uncharacterized interactions, IRFs are deregulated by HPV proteins to reduce expression of IFN-α (Chang and Laimins, 2000), IFN-β (Park et al., 2000; Ronco et al., 1998), as well as IFN-κ (Rincon-Orozco et al., 2009). IFN-κ is a unique IFN that is specifically expressed in skin tissue and may be a major type affecting HPV infections. HPV E6 has also been shown to regulate methylation of the IFN-κ promoter so as to down-regulate its expression (Rincon-Orozco et al., 2009), which also leads to changes of the expression pattern of PRRs (Reiser et al., 2011). Interestingly, IRFs can also function as part of a feedback loop to directly regulate HPV transcription as IRF-2 can activate the promoter of E6E7 in keratinocytes (Lace et al., 2010), while IRF-3 suppresses HPV8 expression (Oldak et al., 2011).
Figure 1. HPV modulates STAT pathways.
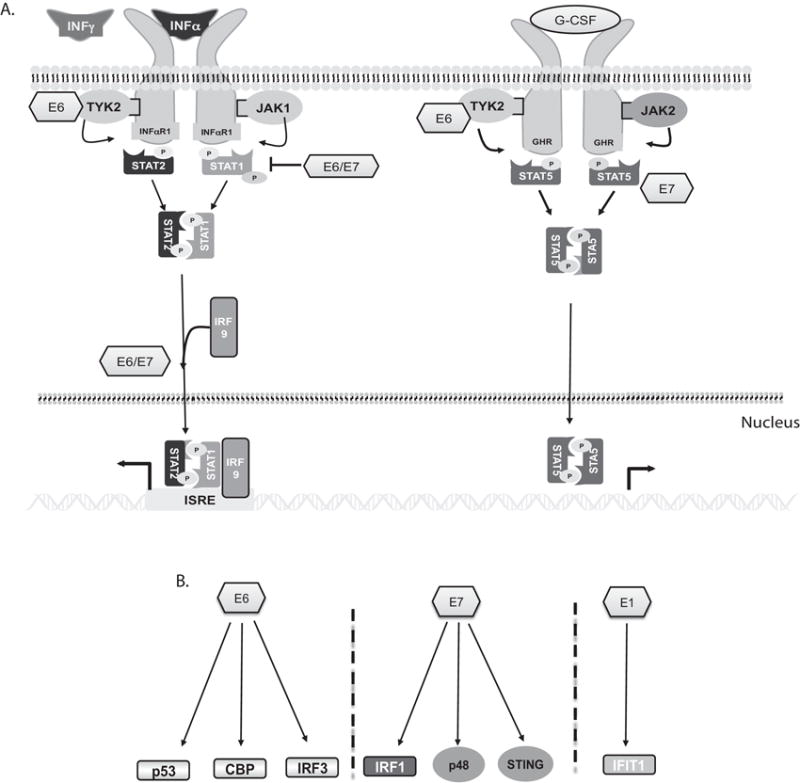
A. HPV suppresses STAT-1 expression while it activates STAT-5. STAT-1 phosphorylation is induced following binding of type I or II IFNs to receptors and activation of associated kinases. The TYK2 kinase can be regulated by E6 protein. The phosphorylated STAT-1 forms heterodimers with STAT-2 or homodimers that migrate to the nucleus to activate expression of over 100 genes. HPV E6 or E7 suppress STAT-1 transcription. In contrast, HPV promotes STAT-5 phosphorylation in the absence of exogenous activating signals.
B. Regulation of host innate immune factors by HPV viral proteins. HPV E6 binds to p300/CBP to inhibit its acetylation of p53. E6 can also regulate CBP as well as IRF-3. E7 inhibits ISGF3 complex formation to suppress STAT-1-dependent induction of downstream genes. E7 also regulates p48 and STING proteins. E1 can bind to IFIT1 and this interaction results in a dampened eukaryotic initiation factor 3 (eIF3) activity.
In addition to repressing interferon expression, HPV proteins also interfere with the induction of the JAK-STAT pathway (Figure 1A). HPV E6 interferes with activation of STAT proteins by directly interacting with TYK2 to impair phosphorylation of STAT-1 and STAT-2 (Li et al., 1999). Similarly, E7 has been reported to bind p48, which is a component of ISGF3 complex, to block the complex translocation to the nucleus and inhibiting ISG gene expression (Barnard and McMillan, 1999). Microarray analysis of HPV31 and 16 cervical keratinocytes has shown that STAT-1 but not STAT-2 transcription is repressed by E6 and E7 proteins (Nees et al., 2001). This results in suppression of STAT-1 expression in HPV31-positive keratinocytes and correspondingly reduced transcription of downstream genes (Chang and Laimins, 2000). High-risk HPV proteins suppress the expression of a broad set of ISGs including MX1, IFIT1, OAS, and defensin-β1 (Chang and Laimins, 2000). Upon exposure to exogenously added IFNs, the expression of these genes can be restored to that seen in normal cells. The IFN-inducible IFIT1 has been shown to bind to E1 replication protein and sequester it to the cytoplasm, resulting in a decrease in HPV replication (Terenzi et al., 2008). Unlike IFIT1, other members of the IFIT family, such as IFITM proteins, do not have any inhibitory effects on HPV infection (Warren et al., 2014). The dsRNA-activated protein kinase (PKR) is commonly targeted by viral proteins and PKR expression is also suppressed by HPV proteins leading to impaired regulation of the eukaryotic translation initiation factor 2α(eIF2α) (Hebner et al., 2006; Kazemi et al., 2004). In addition, HPV18 E6 can relocalize PKR to cytoplasmic P-bodies to further inhibit PKR activities (Hebner et al., 2006).
Inhibition of STAT-1 expression by HPV proteins is critical for the stable maintenance of viral episomes as well as amplification in differentiating cells (Hong et al., 2011). When cells that maintain HPV episomes were transfected with STAT-1 expression vectors so as to restore levels to those seen in normal keratinocytes, the amount of viral genomes was rapidly decreased, differentiation-dependent amplification was inhibited and cells with integrated genomes became the predominant species. Whether the critical regulator that needs to be suppressed is STAT-1 itself or a downstream target is, however, unclear. In addition, HPV acts on the co-activators associated with STAT-1 proteins such as p300/CBP and MCM5. HPV E6 binds to p300/CBP to inhibit its ability to acetylate p53, resulting in blocking cell growth arrest induced by IFNs (Hebner et al., 2007). While the levels of MCM5 are increased in all grades of HPV-positive squamous and glandular dysplasia (Murphy et al., 2005), the specific role of MCM5 plays in the HPV life cycle is not yet clear. Overall, these studies demonstrate the importance of suppressing STAT-1 in HPV infections.
Other members of the STAT family, such as STAT-3 and STAT-5 are regulated differently than STAT-1 in HPV-positive cells. Both STAT-3 and STAT-5 are involved in controlling cellular proliferation, differentiation, as well as the innate immune response. Studies have shown the enhanced expression of STAT-3 in HPV-positive cells is the result of E6 and E7 directed decreases in the levels of miRNA-125a that regulates STAT-3 synthesis (Fan et al., 2015). Furthermore, inhibition of STAT-3 activities by siRNAs or pharmacological inhibitors lead to an accumulation of p53 as well as pRb and reduced HPV gene expression (Shukla et al., 2013). Silencing of HPV E6 by E6-specific siRNAs results in abrogation of STAT-3 signaling (Tyagi et al., 2016), whereas HPV E7 controls CDC91L1 (Guo et al., 2004) which in turn increases STAT-3 phosphorylation.
Expression of high-risk HPV proteins has also been shown to induce the constitutive activation of STAT-5 while only minimally affecting total levels. This activation of STAT-5 in HPV positive cells results in induction of the Ataxia Telangiectasia Mutated (ATM) DNA damage repair pathway (Hong and Laimins, 2013). Activation of the ATM pathway is required for HPV genome amplification but has only a minimal effect on the stable maintenance of episomes (Moody and Laimins, 2009). HPV proteins act through PPARγ, GSK3β, as well as Tip60 to induce ATM activation (Hong et al., 2015; Hong and Laimins, 2013). Furthermore, high-risk HPV proteins also activate the Ataxia Telangiectasia and Rad3-related (ATR) pathway, which mediates repair of single strand DNA breaks, to further regulate HPV viral replication. STAT-5 regulates ATR activation, in part, by controlling the transcription of the topoisomerase II -binding protein 1 (TopBP1), a binding partner of ATR (Hong et al. 2015).
3.4 HPV and NF-κB
In addition to controlling IFN signaling, HPV also regulates the NF-κB pathway that crosstalks with IFN signaling. HPV E7 associates with IκB kinase complex and reduces activities of IKK to attenuate NF-κB activation (Spitkovsky et al., 2002). E7 also reduces the nuclear translocation and acetylation of the p65 subunit of NF-κB (Richards et al., 2015). Both E6 and E7 have been reported to compete with NF-κB/p65 to bind to p300/CBP to decrease IL-8 expression (Huang and McCance, 2002). Interestingly, at the same time HPV16 E6 has been reported to enhance the expression of NF-κB-dependent gene products such as TRAF-interacting proteins (Nees et al., 2001). HPV E6 is also responsible for prolonged hypoxia-induced NF-κB activation in cancer development via degradation of the CYLD lysine 63 (K63) deubiquitinase, which is a negative mediator for NF-κB pathway (An et al., 2008). The effects of HPV proteins on NF-κB activation are complex but important for viral pathogenesis.
3.5 Cytokines and HPV
Many cytokines play important roles in cell apoptosis as well as antiviral response. IL-1β is a key regulator of programmed cell death and a critical product of inflammasome activation. The inflammasome complex is a critical component of the innate immune response as it is responsible for induction of cell pyroptosis, recruitment of immune cells to the sites of infection as well as mediating antiviral response. The complex consists of NOD-like receptors (NLRs), apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and pro-caspase-1. The assembly of this complex leads to the formation of mature caspase-1 that processes pro-IL-1β and pro-IL-18 to cleaved forms that are secreted as active pro-inflammatory cytokines. It has been shown that HPV16 E6 down-regulates IL-1β expression via E6-AP and p53 through post-translational modifications and may interfere with inflammasome recruitment (Niebler et al., 2013). HPV E7 can also suppress expression of the cytokines that can activate migration of activated NKT cells (Cicchini et al., 2016).
4. Conclusion
HPVs must escape innate immune surveillance to establish persistent infections and viral proteins manipulate this process by several mechanisms. First, HPV reduces the levels of the DNA sensors that can recognize HPV. Second, HPV suppresses IRF signaling as well as production of IFNs to block this critical part of the innate immune response. Third, viral proteins suppress STAT-1 expression while activating STAT-5 both of which are necessary for HPV life cycle. Targeting these multiple factors of the innate immune response is therefore essential for persistence of human papillomavirus infections.
Highlights.
HPV reduces the levels of the DNA sensors such as TLR9 and PKR.
HPV proteins suppress IRF-1 and IRF-3 to reduce expression of type I IFNs including IFN-α, IFN-β, and IFN-κ.
IFN-κ is specifically expressed in epithelial cells and HPV E2 or E6 proteins can reduce IFN-κ expression levels.
Suppression of STAT-1 activity and STAT-1-dependent ISGs is necessary for HPV life cycle.
Activation of STAT-5 is required for HPV genome amplification as well as activation of DNA damage response
Acknowledgments
This work was supported by grants from the NCI (RO1CA142861, RO1CA59655) and NIAID (R21AI20492) to L.A.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. eighth. ELSEVIER SAUNDERS; 2015. [Google Scholar]
- An J, Mo D, Liu H, Veena MS, Srivatsan ES, Massoumi R, Rettig MB. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell. 2008;14:394–407. doi: 10.1016/j.ccr.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard P, McMillan NA. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology. 1999;259:305–313. doi: 10.1006/viro.1999.9771. [DOI] [PubMed] [Google Scholar]
- Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol. 2011;19:33–39. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannella F, Pierangeli A, Scagnolari C, Cacciotti G, Tranquilli G, Stentella P, Recine N, Antonelli G. TLR9 is expressed in human papillomavirus-positive cervical cells and is overexpressed in persistent infections. Immunobiology. 2015;220:363–368. doi: 10.1016/j.imbio.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol. 2016;14:360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YE, Laimins LA. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J Virol. 2000;74:4174–4182. doi: 10.1128/jvi.74.9.4174-4182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IY, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015;23:55–63. doi: 10.1016/j.tim.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci USA. 2009;106:9373–9378. doi: 10.1073/pnas.0903487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchini L, Westrich JA, Xu T, Vermeer DW, Berger JN, Clambey ET, Lee D, Song JI, Lambert PF, Greer RO, Lee JH, Pyeon D. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. MBio. 2016;7:e00270–16. doi: 10.1128/mBio.00270-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Cigno I, De Andrea M, Borgogna C, Albertini S, Landini MM, Peretti A, Johnson KE, Chandran B, Landolfo S, Gariglio M. The Nuclear DNA Sensor IFI16 Acts as a Restriction Factor for Human Papillomavirus Replication through Epigenetic Modifications of the Viral Promoters. J Virol. 2015;89:7506–7520. doi: 10.1128/JVI.00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordano P, Gillan V, Bratlie S, Bouvard V, Banks L, Tommasino M, Campo MS. The E6E7 oncoproteins of cutaneous human papillomavirus type 38 interfere with the interferon pathway. Virology. 2008;377:408–418. doi: 10.1016/j.virol.2008.04.036. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol. 2013;13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan T, Melvold R, Viselli S, Waltenbaugh C. Lippincott’s Illustrated Reviews: Immunology Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2008. [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Fan Z, Cui H, Xu X, Lin Z, Zhang X, Kang L, Han B, Meng J, Yan Z, Yan X, Jiao S. MiR-125a suppresses tumor growth, invasion and metastasis in cervical cancer by targeting STAT3. Oncotarget. 2015;6:25266–25280. doi: 10.18632/oncotarget.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Linn JF, Wu G, Anzick SL, Eisenberger CF, Halachmi S, Cohen Y, Fomenkov A, Hoque MO, Okami K, Steiner G, Engles JM, Osada M, Moon C, Ratovitski E, Trent JM, Meltzer PS, Westra WH, Kiemeney LA, Schoenberg MP, Sidransky D, Trink B. CDC91L1 (PIG-U) is a newly discovered oncogene in human bladder cancer. Nat Med. 2004;10:374–381. doi: 10.1038/nm1010. [DOI] [PubMed] [Google Scholar]
- Hasan UA, Zannetti C, Parroche P, Goutagny N, Malfroy M, Roblot G, Carreira C, Hussain I, Müller M, Taylor-Papadimitriou J, Picard D, Sylla BS, Trinchieri G, Medzhitov R, Tommasino M. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J Exp Med. 2013;210:1369–1387. doi: 10.1084/jem.20122394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- Hebner C, Beglin M, Laimins LA. Human papillomavirus E6 proteins mediate resistance to interferon-induced growth arrest through inhibition of p53 acetylation. J Virol. 2007;81:12740–12747. doi: 10.1128/JVI.00987-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol. 2006;16:83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- Hebner CM, Wilson R, Rader J, Bidder M, Laimins LA. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J Gen Virol. 2006;87:3183–3193. doi: 10.1099/vir.0.82098-0. [DOI] [PubMed] [Google Scholar]
- Heltemes-Harris LM, Farrar MA. The role of STAT5 in lymphocyte development and transformation. Curr Opin Immunol. 2012;24:146–152. doi: 10.1016/j.coi.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dutta A, Laimins LA. The acetyltransferase Tip60 is a critical regulator of the differentiation-dependent amplification of human papillomaviruses. J Virol. 2015;89:4668–4675. doi: 10.1128/JVI.03455-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Laimins LA. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013;9:e1003295. doi: 10.1371/journal.ppat.1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Mehta KP, Laimins LA. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol. 2011;85:9486–9494. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, McCance DJ. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J Virol. 2002;76:8710–8721. doi: 10.1128/JVI.76.17.8710-8721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4129–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi S, Papadopoulou S, Li S, Su Q, Wang S, Yoshimura A, Matlashewski G, Dever TE, Koromilas AE. Control of alpha subunit of eukaryotic translation initiation factor 2 (eIF2 alpha) phosphorylation by the human papillomavirus type 18 E6 oncoprotein: implications for eIF2 alpha-dependent gene expression and cell death. Mol Cell Biol. 2004;24:3415–3429. doi: 10.1128/MCB.24.8.3415-3429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lace MJ, Anson JR, Haugen TH, Turek LP. Interferon regulatory factor (IRF)-2 activates the HPV-16 E6-E7 promoter in keratinocytes. Virology. 2010;399:270–279. doi: 10.1016/j.virol.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Li S, Labrecque S, Gauzzi MC, Cuddihy AR, Wong AH, Pellegrini S, Matlashewski GJ, Koromilas AE. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-alpha. Oncogene. 1999;18:5727–5737. doi: 10.1038/sj.onc.1202960. [DOI] [PubMed] [Google Scholar]
- Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005;24:1821–1830. doi: 10.1038/sj.emboj.7600651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Gale M. Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazibrada J, Longo L, Vatrano S, Cappia S, Giorcelli J, Pentenero M, Gandolfo S, Volante M, dell’Oste V, Lo Cigno I, Biolatti M, Landolfo S, Papotti M. Differential expression of HER2, STAT3, SOX2, IFI16 and cell cycle markers during HPV-related head and neck carcinogenesis. New Microbiol. 2014;37:129–143. [PubMed] [Google Scholar]
- Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009;5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89:213–228. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy N, Ring M, Heffron CCBB, King B, Killalea AG, Hughes C, Martin CM, McGuinness E, Sheils O, O’Leary JJ. p16INK4A, CDC6, and MCM5: predictive biomarkers in cervical preinvasive neoplasia and cervical cancer. J Clin Pathol. 2005;58:525–534. doi: 10.1136/jcp.2004.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol. 2001;75:4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebler M, Qian X, Höfler D, Kogosov V, Kaewprag J, Kaufmann AM, Ly R, Böhmer G, Zawatzky R, Rösl F, Rincon-Orozco B. Post-translational control of IL-1β via the human papillomavirus type 16 E6 oncoprotein: a novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog. 2013;9:e1003536. doi: 10.1371/journal.ppat.1003536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldak M, Tolzmann L, Wnorowski A, Podgórska MJ, Silling S, Lin R, Hiscott J, Müller CSL, Vogt T, Smola H, Smola S. Differential regulation of human papillomavirus type 8 by interferon regulatory factors 3 and 7. J Virol. 2011;85:178–188. doi: 10.1128/JVI.00998-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacini L, Savini C, Ghittoni R, Saidj D, Lamartine J, Hasan UA, Accardi R, Tommasino M. Downregulation of Toll-Like Receptor 9 Expression by Beta Human Papillomavirus 38 and Implications for Cell Cycle Control. J Virol. 2015;89:11396–11405. doi: 10.1128/JVI.02151-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Kim EJ, Kwon HJ, Hwang ES, Namkoong SE, Um SJ. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem. 2000;275:6764–6769. doi: 10.1074/jbc.275.10.6764. [DOI] [PubMed] [Google Scholar]
- Rajsbaum R, García-Sastre A, Versteeg GA. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. Journal of molecular biology. 2014;426:1265–1284. doi: 10.1016/j.jmb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinholz M, Kawakami Y, Salzer S, Kreuter A, Dombrowski Y, Koglin S, Kresse S, Ruzicka T, Schauber J. HPV16 activates the AIM2 inflammasome in keratinocytes. Arch Dermatol Res. 2013;305:723–732. doi: 10.1007/s00403-013-1375-0. [DOI] [PubMed] [Google Scholar]
- Reiser J, Hurst J, Voges M, Krauss P, Münch P, Iftner T, Stubenrauch F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J Virol. 2011;85:11372–11380. doi: 10.1128/JVI.05279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KH, Wasson CW, Watherston O, Doble R, Blair GE, Wittmann M, Macdonald A. The human papillomavirus (HPV) E7 protein antagonises an Imiquimod-induced inflammatory pathway in primary human keratinocytes. Sci Rep. 2015;5:12922. doi: 10.1038/srep12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon-Orozco B, Halec G, Rosenberger S, Muschik D, Nindl I, Bachmann A, Ritter TM, Dondog B, Ly R, Bosch FX, Zawatzky R, Rösl F. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res. 2009;69:8718–8725. doi: 10.1158/0008-5472.CAN-09-0550. [DOI] [PubMed] [Google Scholar]
- Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12:2061–2072. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Res. 2006;16:141–147. doi: 10.1038/sj.cr.7310019. [DOI] [PubMed] [Google Scholar]
- Shukla S, Mahata S, Shishodia G, Pandey A, Tyagi A, Vishnoi K, Basir SF, Das BC, Bharti AC. Functional regulatory role of STAT3 in HPV16-mediated cervical carcinogenesis. PLoS ONE. 2013;8:e67849. doi: 10.1371/journal.pone.0067849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitkovsky D, Hehner SP, Hofmann TG, Möller A, Schmitz ML. The human papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the Ikappa B kinase complex. J Biol Chem. 2002;277:25576–25582. doi: 10.1074/jbc.M201884200. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Terenzi F, Saikia P, Sen GC. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J. 2008;27:3311–3321. doi: 10.1038/emboj.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi A, Vishnoi K, Mahata S, Verma G, Srivastava Y, Masaldan S, Roy BG, Bharti AC, Das BC. Cervical Cancer Stem Cells Selectively Overexpress HPV Oncoprotein E6 that Controls Stemness and Self-Renewal through Upregulation of HES1. Clin Cancer Res. 2016;22:4170–4184. doi: 10.1158/1078-0432.CCR-15-2574. [DOI] [PubMed] [Google Scholar]
- Warren CJ, Griffin LM, Little AS, Huang IC, Farzan M, Pyeon D. The antiviral restriction factors IFITM1, 2 and 3 do not inhibit infection of human papillomavirus, cytomegalovirus and adenovirus. PLoS ONE. 2014;9:e96579. doi: 10.1371/journal.pone.0096579. [DOI] [PMC free article] [PubMed] [Google Scholar]