

Abstract
Crystallographic data of the dimeric and octameric forms of fragaceatoxin C (FraC) suggested the key role of a small hydrophobic protein–protein interaction surface for actinoporins oligomerization and pore formation in membranes. However, site‐directed mutagenesis studies supporting this hypothesis for others actinoporins are still lacking. Here, we demonstrate that disrupting the key hydrophobic interaction between V60 and F163 (FraC numbering scheme) in the oligomerization interface of FraC, equinatoxin II (EqtII), and sticholysin II (StII) impairs the pore formation activity of these proteins. Our results allow for the extension of the importance of FraC protein–protein interactions in the stabilization of the oligomeric intermediates of StII and EqtII pointing out that all of these proteins follow a similar pathway of membrane disruption. These findings support the hybrid pore proposal as the universal model of actinoporins pore formation. Moreover, we reinforce the relevance of dimer formation, which appears to be a functional intermediate in the assembly pathway of some different pore‐forming proteins.
Keywords: pore‐forming toxins, dimeric intermediate, oligomerization in membranes, molecular dynamic simulations, protein–lipid pore
Abbreviations
- AA
acrylamide
- CAS
computational alanine scanning
- CD
circular dichroism
- CF
carboxyfluorescein
- EM
energy minimization
- EqtII
equinatoxin II
- FraC
fragaceatoxin C
- ΔGbind
binding free energy
- ΔGpolar
polar desolvation energy
- ΔGnonpolar
nonpolar desolvation energy
- ΔGsolvat
solvation free energy
- HA
hemolytic activity
- Ksv
SternVolmer constant
- m
slope of regression fit/line
- MD
molecular dynamic
- MLV
multilamellar vesicles
- PFP
pore‐forming protein
- PFT
pore‐forming toxins
- POPC
1‐palmitoyl‐2‐oleylphosphatidylcholine
- SM
sphingomyelin
- StII
sticholysin II
- SUV
small unilamellar vesicles
- ΔVele
electrostatic energy
- ΔVvdw
van der Waals Energy; π, surface pressure
- π0
initial surface pressure
- Δπ
increment in surface pressure.
Introduction
The organization into supramolecular complexes or oligomers that pierce the membrane is a common feature of pore forming toxins (PFTs). These proteins undergo essential conformational changes from a water‐soluble state to a transmembrane oligomeric state and form cytolytic pores presumably involving the evolution of oligomeric intermediates.1, 2, 3, 4 However, the molecular details in the pathway of self‐assembly and the role of relevant intermediates remain unclear for most PFTs. Actinoporins are among the most studied eukaryotic PFTs.4, 5, 6 To date, the most studied actinoporins have been equinatoxin II (EqtII) from Actinia equina, sticholysins I and II (StI and II) from Stichodactyla helianthus, and fragaceatoxin C (FraC) from Actinia fragacea.4, 7 The 3D structure of the soluble forms of these actinoporins contain a hydrophobic β‐sandwich core flanked by two α‐helices, including one located in the N‐terminus, which has been identified as the pore‐forming domain [Fig. 1(A)].
Figure 1.
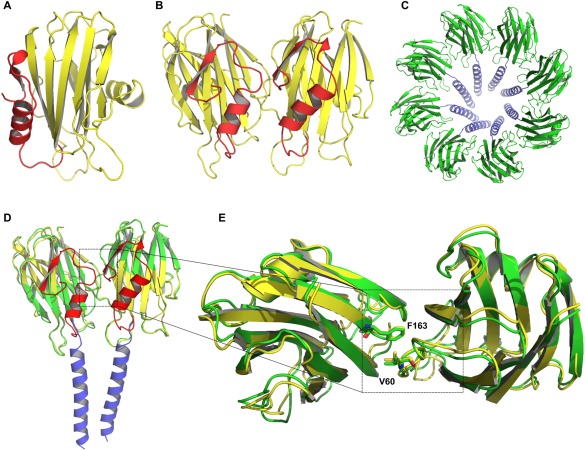
Representation of FraC in different states during the mechanism of pore formation. (A) Monomeric structure (PDB: 3VWI), (B) dimeric structure (PDB: 4TSL); hydrophobic β‐sandwich core (yellow), red α‐helix shows the pore forming domain. (C) Octameric pore structure; protein core (green), extended N‐terminal region (blue), (PDB: 4TSY). (D) Superposition of the dimeric (red and yellow) and octameric (blue and green) forms of FraC. The superposition was performed considering the carbon alpha atoms of both proteins. (E) Top view of the superposition of the dimeric and octameric structures. Zoom: superposition of the residues F163 and V60, which are highly conserved among actinoporins family.6 N‐terminal helices are not shown for the sake of clarity.
Understanding of the assembly mechanism of actinoporins has dramatically increased after determination of the crystal structure of FraC at different stages of the lytic process,8 complementing a body of biochemical and biophysical data previously obtained by different groups.9, 10, 11, 12, 13, 14, 15, 16 It is now generally accepted that actinoporins bind to membranes mainly as monomers10, 17, 18, 19, 20 and then undergo a conformational change that involves only the N‐terminal segment.4, 8, 14 Then, monomeric units oligomerize to form pores in which the N‐terminal α‐helix lines the walls in conjunction with lipids.8 The average radius of actinoporin pores has been determined by different approaches and have a fixed size (∼ 2 nm of diameter).8, 10, 21, 22 However, oligomeric structures seem to quickly switch among multiple functional stages4 and are transient and electrically heterogeneous.4, 23
There are mainly two hypothetical models under debate which describe the architecture of the pore formed by these toxins: the classical toroidal pore11, 12, 13, 24 and the hybrid protein–lipid pore.8, 25 These models differ in the stoichiometry of the pore and in the relevance of the protein–protein interactions for pore stability. The toroidal pore model was initially proposed and is mainly based on functional studies with sticholysins and EqtII.11, 13 Kinetic and electrophysiological studies and more recently single molecule imaging have revealed that predominantly 3‐4 monomers of these proteins form functional pores.10, 16, 21, 24, 26 Moreover, crystalografic data of a prepore like structure of StII in lipid monolayers shows that the units that form the assembly do not establish relevant protein–protein interactions.24 In contrast, the crystal structure of FraC shows an octameric pore where each protein molecule is associated with three molecules of lipids in a hybrid protein–lipid architecture.8 In this architecture, both protein–lipid and protein–protein interactions contribute to the pore stability.8
The relevance of dimers as an assembly intermediate has been highlighted by different studies.8, 16, 27 In general, data supporting either the toroidal or the hybrid protein–pore model are in agreement with the relevance of dimers in the mechanism of actinoporins cell death induction.4 It is important to note that the structure of dimeric FraC exhibits a similar protein–protein interface to that of the octameric pore, which reinforces its mechanistic relevance [Fig. 1(B–E)].8 It is also significant that both the dimeric and the octameric interfaces proposed from structural data are highly conserved in other members of the actinoporin family.8 However, there is no functional data validating the conserved role of the dimeric interface and protein–protein interactions in the mechanism of assembly of actinoporins.
We previously published that the structure and activity of FraC and EqtII are highly similar, indeed their N‐terminus segment, which is responsible for pore formation is identical.28 Thus, we would not expect that these proteins build different pore architectures as reported.8, 13, 24 Because this is a controversial topic in the literature, we decided to compare the structural and functional properties of three of the most studied actinoporins (FraC, EqtII, and StII) in more detail. Here, we combined mutagenesis studies with molecular dynamic simulations to test the hypothesis that the dimeric interface of actinoporins is conserved and plays a key role in pore formation. First, a double mutant that disrupts the putative dimerization interface of actinoporins was designed by combining sequence and structure analysis with free energy calculations. We cloned, expressed, and purified the double mutants FraCV60D/F163D, EqtIIV60D/F163D, and StIII58D/I161D, and characterized their structure by spectroscopic techniques and their function by biophysical tools. Although the mutants maintained their main structural features and the capacity to bind lipidic membranes of the wildtype, their pore‐forming ability was abolished. Our work reinforces the relevance of dimer formation as a necessary intermediate in the mechanism of action of actinoporins. We proposed that actinoporins follow a common pathway of assembly where protein–protein interactions stabilize the oligomeric structure in the membrane.
Results
The impairment of the interaction between V60 and F163 reduces the FraC dimer stability
The dimeric and the octameric structures of FraC exhibit similar protein–protein interfaces.8 This indicates that the dimeric structure is a functionaly relevant intermediate in the actinoporins self‐assembly mechanism8 [Fig. 1(D,E)]. Therefore, the dimer is the simplest relevant oligomeric structure for free energy calculations to identify warm/hot‐spots at the protein–protein oligomerization interface. Computational Alanine Scanning (CAS) calculations indicated that the residues D58 (ΔΔG efe = 7.55 ± 0.15 kcal/mol), W149 (ΔΔG efe = 10.30 ± 0.05 kcal/mol) and F163 (ΔΔG efe = 6.66 ± 0.03 kcal/mol) were the major contributors to the binding free energy of the dimer intermediate (Fig. 2; Supporting Information Table SI).
Figure 2.
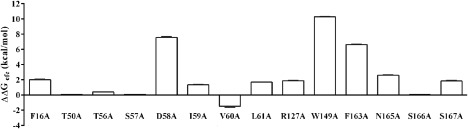
Identification of the warm/hot‐spots at the interface of the dimeric FraC using computational Ala scanning. Energy contributions of the residues belonging to the protein–protein interface are shown. Positive ΔΔG values mean substitutions that decrease the stability of the dimeric form of FraC, while negative ΔΔG values mean substitutions that increase the stability of the dimer.
The pwEFED protocol was employed to gain insight into the energetic contribution of those hot spots in terms of pair‐wise interactions with neighboring residues. Interactions of V60–F163, V60–W149, D58–N165, and D58–S167 between each protomer significantly contribute to the formation of the dimeric structure [Fig. 3(A,B)]. Among these residues, the pair of interacting residues V60 and F163 showed the largest and most stable energy contribution to dimer formation (ΔG efeV60,F163 = −3.14 ± 0.40 kcal/mol). Remarkably, V60 was able to establish interaction with F163 that was three times stronger, in terms of the binding free energies, than W149 (ΔG efeV60,F163 = −1.03 ± 0.24 kcal/mol) [Fig. 3(A); Supporting Information Table SII]. van der Waals interactions and nonpolar solvation energies were identified as the major contributors to the formation of this pair (V60–F163) [Fig. 3(A), Supporting Information Table SII]. Hydrophobic residues such as V60 and F163 are highly conserved among the actinoporins family.8 We designed the double mutant FraCV60D/F163D, in order to evaluate the detrimental impact of disrupting the interaction between V60 and F163 in the dimer assembly and pore forming ability of FraC.
Figure 3.
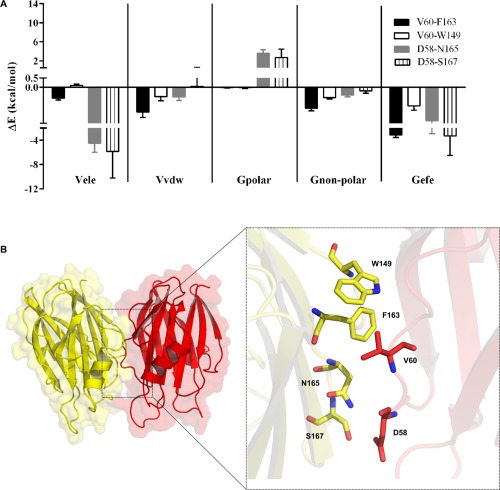
Per‐residue energy contributions to the formation of the dimeric structure of FraC. (A) Energetic contribution of previous hot‐spots residues in terms of their pair‐wise interactions with neighboring residues. (B) FraC dimeric structure illustrated by two protomers (yellow and red) is shown in surface and New Cartoon representations. Zoom of the spatial disposition of the residues that contribute more to the dimer formation.
We calculated the RMSD values for the subset of heavy atoms belonging to the interface residues of FraC and the double mutant FraCV60D/F163D with respect to the initial structure over 100 ns of simulation [Fig. 4(A)]. The double mutant FraCV60E/W149A, which is known to have 16 percent of the activity compared to the wildtype protein,8 was also simulated for comparison [Fig. 4(A)]. To further investigate the stability of the interaction of this pair of residues, the distance between them was calculated as a function of simulation time [Fig. 4(B)]. A stable RMSD time profile (<0.2 nm) for the wildtype protein showed good complementarity of the dimeric interface during the simulation [Fig. 4(A–C)]. Conversely, the time profiles of RMSD values of the double mutant FraCV60D/F163D (>0.5 nm) indicated conformational changes at the dimeric interface during the MD simulation [Fig. 4(A,B)]. After 70 ns simulation, the distance between the side chains of D60 and D163 notably increased due to electrostatic repulsion, which affects the stability of the FraCV60D/F163D interface [Fig. 4(A,B,D)]. The conformational changes of FraCV60E/W149A (RMSD < 0.4 nm), at the dimeric interface was lower than FraCV60D/F163D [Fig. 4(A,B,D)]. Interestingly, we detected two water molecules mediating hydrogen bonds between residues E60 of one protomer and E173 of another protomer of FraCV60E/W149A [Fig. 4(F)]. This fact might explain the reduced effect of this mutation compared to FraCV60D/F163D.
Figure 4.
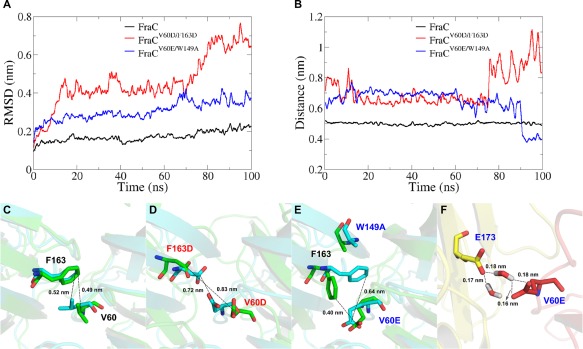
Impact of V60D/F163D mutations on the FraC dimer stability. (A) Root mean square deviation (RMSD) of heavy atoms of the dimer interface residues in the wildtype FraC and the mutants. (B) Distance between the residues in positions 60 and 163 of both protomers as a function of the simulation time; V60/F163 in the wildtype protein (black line), D60/D163 in the FraCV60D/F163 mutant (red line), and E60/F163 in the FraCV60E/W149A mutant (blue line). (C–E) Distance changes of the wildtype and mutated residues during the MD simulation (initial distance—cyan) and (final distance—green). The distance and RMSD values are cumulative averages considering sliding windows of 50 frames. (F) Two water molecules between E173 (yellow) and V60 (red) indicate hydrogen bond formation between those residues in FraCV60E/W149A. Colors (red and yellow) represent different protomers of the dimeric form.
Free energy calculations with the MM‐GBSA method using the GBn2 implicit solvation model (igb = 8) were performed to understand the differences in the stability among the dimeric wildtype FraC and the double mutants (FraCV60D/F163D and FraCV60E/W149A). Polar‐desolvation and electrostatic energies as well as the van der Waals and nonpolar desolvation energies associated with the formation of dimeric structures in solution are shown in Table 1. Taking the wildtype protein as a reference, the energy of dimer formation in the double mutant FraCV60D/F163D (ΔΔG efe = 6.37 kcal/mol) was 4.8‐fold lower than in the double mutant FraCV60E/W149A (ΔΔG efe = 1.33 kcal/mol) (Table 1). The van der Waals interaction energies (ΔΔΔE vdw = 4.78 kcal/mol) were the major energetic contributors to the stability differences between the dimeric form of both mutants (ΔΔΔG efe = 5.04 kcal/mol) (Table 1). The less drastic conformational changes at the dimer interface observed with the FraCV60E/W149A, in which only one negatively charged amino acid residue was introduced [Fig. 4(A,B,E)] might be a consequence of the lower electrostatic repulsion, and may also be due to an additional stabilization mediated by hydrogen bonds created through two water molecules between E60 and the E173 [Fig. 4(F); Table 1]. This finding explains why this mutant, although less active, still retains 16% of the wildtype pore‐forming ability.8 Taking into account all of these in silico results, we designed double‐residues mutants in equivalent position of the three most studied actinoporins, FraC, EqtII, and StII, to verify its impact on the dimerization step within the mechanism of pore formation.
Table 1.
MM‐GBSA Energy Components Associated with the Dimer Formation in Solution
| System |
ΔE
ele
a
(kcal/mol) |
ΔE
vdw
b
(kcal/mol) |
ΔG
polar
c
(kcal/mol) |
ΔG
nonpolar
d
(kcal/mol) |
ΔG
efe
e
(kcal/mol) |
|---|---|---|---|---|---|
| FraC | 513.15 ± 0.86 | −85.69 ± 0.23 | −442.12 ± 0.86 | −10.54 ± 0.02 | −25.21 ± 0.25 |
| FraCV60D/F163D | 178.55 ± 0.52 | −49.27 ± 0.11 | −141.68 ± 0.48 | −6.45 ± 0.01 | −18.84 ± 0.05 |
| ΔΔE f (kcal/mol) | −334.6 | 36.42 | 300.44 | 4.09 | 6.37 |
| FraCV60E/W149A | 418.30 ± 0.40 | −54.05 ± 0.10 | −381.34 ± 0.36 | −6.80 ± 0.01 | −23.88 ± 0.06 |
| ΔΔE g (kcal/mol) | −94.85 | 31.64 | 60.78 | 3.74 | 1.33 |
| ΔΔΔE h (kcal/mol) | −239.75 | 4.78 | 239.66 | 0.35 | 5.04 |
Electrostatic interaction energy.
van der Waals interaction energy.
Polar desolvation free energy.
Nonpolar desolvation free energy.
Effective binding free energy.
ΔΔE f difference between the energy component values of the same column of FraCV60D/F163D and FraC.
ΔΔE g difference between the energy component values of the same column of FraCV60E/W149A and FraC. ΔΔΔE h = ΔΔE f − ΔΔE g difference between the energy component values of the same column.
V60D and F163D mutations do not affect the folding of the wildtype proteins in solution
Next, we designed homologous mutants to FraCV60D/F163D in the related proteins EqtII and StII. The solution structure of the mutants (FraCV60D/F163D, EqtIIV60D/F163D, and StIII58D/I161D) were compared with those of the wildtype variants (FraC, EqtII, and StII). UV‐CD spectra of the toxins and the mutants were recorded. Typical far UV‐CD spectra of wildtype FraC and FraCV60D/F163D are shown in Figure 5. The spectra of the wildtype protein and the mutant was highly similar, exhibiting a positive band centered at 195 nm and a minimum around 217 nm, typical of proteins containing mainly β‐sheet structure.29 Deconvolution of these spectra according to the CONTIN30, 31 and SELCON332, 33 algorithms located at the DICHROWEB Internet server34, 35, 36 provided the contents of secondary structure given in Table 2. The structure of the wild‐type proteins and their mutants consist predominantly of β‐sheet structures, indicating that mutation of the selected residues by Asp did not significantly modify their secondary structure in solution. Moreover, we did not observe differences in folding among the three actinoporins (Table 2). The results are in agreement with data derived from far‐UV‐CD studies of actinoporins27, 37, 38 and also with their three‐dimensional structures.8, 22, 24, 39, 40
Figure 5.
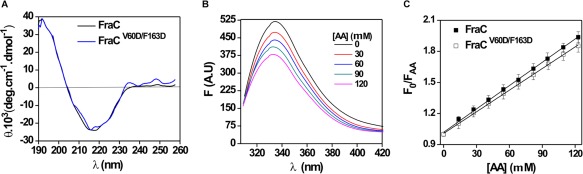
Structural properties of the wildtype proteins and mutants. (A) Far UV‐CD spectra of FraC and FraCV60D/F163D. Far UV‐CD spectra were recorded in water, at RT. λ: 190–260, cuvettes: 15 mm path length. Protein concentration: 10 µM. The base line was corrected by using similarly prepared solutions without the protein. The reported spectra are the average of six scans. (B) Tryptophan fluorescence spectra of FraC in solution and in the presence of AA. Spectra were recorded in 10 mM Tris–HCl buffer, pH 7.4, λ: 300–450 nm, excitation: 295 nm. Slit widths of nominal band pass of 5 nm were used both in the excitation and emission beams. Protein concentration: 2 µM. Background intensities measured in samples without protein were subtracted. (C) Acrylamide quenching of the intrinsic fluorescence of FraC and FraCV60D/F163D. Fluorescence intensities measured in absence and in the presence of AA are plotted according to the Stern–Volmer equation (1). Lines represent the best linear fit of the F 0/F AA as a function of AA concentration. Experiments were done in triplicate (R 2 > 0.99).
Table 2.
Conformational and Functional Properties of the Mutants
| FraC | FraCV60D/F163D | EqtII | EqtIIV60D/F163D | StII | StIII58D/I161D | ||
|---|---|---|---|---|---|---|---|
| CDa | α‐helix | 10 ± 2 | 9 ± 1 | 10 ± 0 | 9 ± 2 | 10 ± 0 | 10 ± 0 |
| β‐sheet | 59 ± 4 | 58 ± 3 | 57 ± 0 | 58 ± 4 | 58 ± 0 | 58 ± 0 | |
| Random coil | 31 ± 3 | 33 ± 3 | 33 ± 0 | 33 ± 3 | 32 ± 0 | 32 ± 0 | |
| λ max (nm) in solutionb | 334.5 ± 0.4 | 334± 0.25 | 335 ± 0.3 | 334.7 ± 0.2 | 334.1± 0.5 | 333.4 ± 0.4 | |
| F max in solutionb | 305.2± 3.1 | 303.6 ± 1.9 | 304.27 ± 0.36 | 303.8 ± 0.5 | 305.3 ± 0.41 | 304.1 ± 0.52 | |
| Ksv (M−1) in solutionc | 7.44 ± 0.41 | 7.05 ± 0.48 | 7.3 ± 0.5 | 6.51 ± 0.6 | 4.3 ± 0.39 | 4.8 ± 0.4 | |
| Ksv (M−1) in the presence of SUVc | 2.57 ± 0.2 | 2.7 ± 0.0.2 | 2.53 ± 0.1 | 2.42 ± 0.18 | 1.96 ± 0.2 | 1.77 ± 0.2 | |
| πc (mN m−1)d | 35.33 ± 0.49 | 29.95 ± 0.68 | 36.77 ± 0.48 | 32.1 ± 0.37 | 33.5 ± 0.98 | 29.59 ± 1.21 | |
| m [Link] | 0.56 ± 0.02 | 0.57 ± 0.01 | 0.55 ± 0.03 | 0.57 ± 0.02 | 0.57 ± 0.03 | 0.54 ± 0.02 | |
| F/F 0max[Link] | 1.33 ± 0.02 | 1.32 ± 0.01 | 1.28 ± 0.01 | 1.28 ± 0.02 | 1.33 ± 0.02 | 1.34 ± 0.02 | |
| Lip50 [Link] | 3.29 ± 0.4 | 2.98 ± 0.4 | 3.62 ± 0.4 | 3.35 ± 0.3 | 3.54 ± 0.2 | 3.29 ± 0.3 | |
| C50 (nM)[Link] | 81.21 ± 0.03 | ∞ | 82.37 ± 0.04 | ∞ | 48,91 ± 0.04 | ∞ | |
| HC50 (nM)i | 0.89 ± 0.02 | ∞ | 0.91 ± 0.01 | ∞ | 0.22 ± 0.01 | ∞ | |
The secondary structure content was estimated by deconvolution of the CD spectra using the algorithms CONTIN and SELCON. The media ± standard deviation of both estimations are shown.
Parameters from the fluorescence spectra in solution.
Stern–Volmer constants (K SV) calculated from the Stern–Volmer equation [Eq. (1)].82
Critical pressure (πc) and eslope (m) obtained from the linear fitting of the increase in pressure (Δπ) as a function of π0. πc corresponds to the pressure that must be applied to avoid incorporation of the toxin to the monolayer and is directly correlated with its affinity for the lipids.30
Highest fluorescence intensity ratio (F/F 0).
Amount of lipid necessary to bind half of the total protein (Lip50) estimated from the Boltzman function. F 0 and F are the fluorescence intensities in the absence and presence of vesicles, respectively.
C50 is the toxin concentration necessary to achieve 50% of vesicles permeabilization estimated according to Eq. (2).
HC50 is the toxin concentration necessary to achieve the lysis of 50% of the red blood cells estimated according to Eq. (3). ∞ refers no activity of the proteins. All determinations were done in triplicate. Calculation were made using Origin 8.0 (Microcal, USA). SUV and monolayers were composed by PC:SM (50:50).
Tryptophan emission spectra of wildtype proteins and mutants were also determined. The mutations did not modify the average degree of Trp exposure to solvent since the fluorescence maximum wavelength were very similar to those of the wildtype proteins (Table 2). The average location of the Trp residues was also estimated from fluorescence quenching experiments employing AA as a soluble quencher. Figure 5(B) shows the fluorescence spectra of FraC in the presence of increasing amounts of AA. Stern–Volmer plots were linear, indicating that the emitting residues can be treated as an approximately homogeneous population [Fig. 5(C)]. As shown in Figure 5(C) for FraC, the mutations did not perturb the exposure of Trp residues to the quencher suggesting that the folding was not affected. Moreover, the Stern–Volmer curves and values obtained with the wildtype protein and the mutant were similar [Fig. 5(C) for FraC] (Table 2) and significantly different from the protein completely denaturalized by heat (data not shown).
Double mutants retain binding ability to monolayers and liposomes
To obtain insight into the interaction of wildtype proteins and their mutants with membranes, we studied their binding to lipid monolayers and bilayers of PC:SM (50:50). This has been the most used lipid composition to study the interaction of actinoporins with membrane‐mimetic systems.10, 17, 37, 48
The increase in surface pressure (Δπ) elicited by the association of a toxin to a previously formed lipid monolayer can be employed to characterize its ability to interact with organized lipids.41 The Δπ was evaluated at several initial pressures (π0) of the lipid monolayer keeping the monolayer area constant. The injection of the toxin into the subphase of the lipidic monolayer triggered a Δπ which stabilized after ∼400 s [shown in Fig. 6(A) for FraC]. The Δπ at equilibrium caused by FraC and FraCV60D/F163D as a function of the π0 are shown in Figure 6(B). As expected, Δπ decreased with increasing π0 for all proteins because tighter lipid packing prevented insertion.
Figure 6.
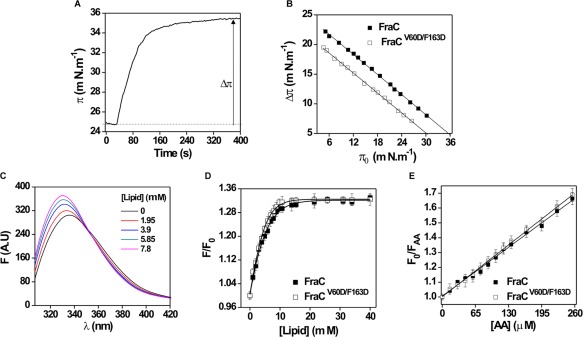
Interaction of wildtype protein with lipid membranes. (A) Temporal course of the surface pressure increase in the water/air surface following the addition of FraC in a PC:SM (50:50) monolayer. (B) Critical pressure induced by FraC and FraCV60D/F163Don the lipidic monolayer. Lines represent the best linear fit of the Δπ as a function of the πo, calculated using Origin 8.0, Microcal (USA). Experiments were carried out under constant stirring. Protein concentration: 0.8 μM. (C) Tryptophan fluorescence spectra of FraC in solution and in the presence of SUV composed by PC:SM (50:50). Spectra were recorded in 10 mM Tris–HCl buffer, pH 7.4. λ: 300–450 nm, excitation: 295 nm. Slit widths of nominal band pass of 5 nm were used both in the excitation and emission beams. Protein concentration: 2 µM. Background intensities measured in samples without protein were subtracted. (D) FraC and FraCV60D/F163D intrinsic fluorescence intensity as a function of lipid concentration. Curves were fitted to a Boltzman function using Origin 8.0 (Microcal, USA). Increasing concentrations of PC:SM (50:50) SUV were added to 2 µM toxin solutions. (E) FraC and FraCV60D/F163D fluorescence quenching by AA in the presence of SUV. Lipid concentration: 400 µM. Fo and FAA correspond to the fluorescence in the absence and the presence of AA, respectively. The spectra were corrected for light dispersion by the vesicles. Lines represent the best linear fit of F 0/F AA as a function of AA concentration, calculated using Origin 8.0 (Microcal, USA) (R 2 > 0.99). All the experiments were done in triplicate.
Two suitable parameters for the characterization of the protein–lipid interaction are the critical pressure (πc) and the slope of the linear fitting (m) obtained from plotting Δπ versus π0 (Table 2). In particular, πc is obtained by extrapolating to Δπ = 0 [Fig. 5(B)] and is directly related to the protein affinity for the lipids in the monolayer.42 Each toxin and its respective mutant showed curves with similar values of slope (m). Similar values of m indicate that the effect of the mutation on binding is independent of the actual surface pressure value and consequently of the lipid concentration.
The πc for the wildtype proteins were similar, in the range ∼33–37 mN m−1. Those values were slightly lower for the mutants (∼30–32 mN m−1) but are still in the range where they can theoretically30, 42, 43, 44 insert into a biological membrane (Table 2). Since m was similar between wildtypes and mutants, the higher changes in surface pressure obtained with the wildtype do not seem to arise from differences in lipid affinity, but rather from differences in protein–lipid packing (i.e., oligomerization) at the interface. Our results are in agreement with previous observations with the mutants FraC45 and StI27, 46 in which variations in the oligomeric state affected the πc values, but the slope of the curve remained almost identical. At this point, it is tempting to speculate that the monolayer system is able to detect variations in the oligomeric state of the proteins as a consequence of the variation of the overall packing of the lipid and protein molecules within the liquid–air interface.
Lipid vesicles are good models to mimic the behavior of natural membranes. These aggregates allow for the evaluation of protein association to lipids and the protein function by measuring the release of entrapped solutes.47 To obtain further insight into the insertion mechanism of the proteins into lipid membranes, binding to SUV was assessed by measuring the effect of lipid concentration on Trp intrinsic fluorescence [Fig. 6(C)]. Addition of liposomes progressively increased the fluorescence intensity and induced a blue‐shift of the spectra until a plateau was reached, indicating quantitative association of the proteins [Fig. 6(D)]. The curves in Figure 6(D) show a similar behavior for the wildtype FraC and its respective mutant. The magnitude of the change in the fluorescence intensity due to the protein binding to lipids (F/F 0) can be extrapolated to high lipid concentrations to determine F/F 0 max. Moreover, the lipid concentration is related to the strength of the protein/lipid interaction and this can be characterized by the Lip50 value (Table 2). These values were similar when compared to the wildtype FraC, EqtII, and StII, as well as the mutants.
Stern–Volmer constant (Ksv) values were obtained by employing AA as fluorescence quencher (Table 2). This parameter was derived from the slopes of Stern–Volmer type plots, like those shown in Fig. 6(E). Fluorescence attenuation with AA were carried out in the presence of saturating lipid concentrations in order to ensure that all the protein's molecules were bound to liposomes. For all the proteins, Ksv values in the presence of liposomes were lower than those obtained in solution. This decrease indicated protein association to the lipids since AA accessibility to the Trp is partially blocked when the protein was bound to the membrane.37 Moreover, Ksv values from wild‐type proteins were similar to the mutant variants suggesting that the mutations did not affect the step of binding to the membrane (Table 2).
Double mutants do not show pore‐forming ability in liposomes and cells
The pore‐forming capacity of the toxins was assessed by evaluating their ability to promote the release of the fluorescent dye CF encapsulated in liposomes (Fig. 7). It is well known that actinoporins promote the release of fluorescent solutes entrapped in liposomes.5, 12, 30 The fraction of vesicles bearing at least one pore (f) versus time was calculated assuming that the release of CF is simultaneous with pore formation [Fig. 7(A)]. The dose‐dependent permeabilization of liposomes elicited by the toxins exhibited a sigmoidal shape [Fig. 7(B)]. This feature indicates that vesicles were not efficiently permeabilized until a critical number of monomers per vesicle was reached.10 The C50 parameter was estimated by fitting the data to a Hill sigmoid (Table 2). C50 characterizes the protein concentration necessary to promote the release of 50% of the dye entrapped into the vesicles. As we previously reported, the three actinoporins differs in activity, with StII as the most active one, followed by EqtII and FraC, which did not differ.30 In contrast to the wildtype proteins, the mutants did not show permeabilizing activity in the concentration range assessed [Fig. 7(B); Table 2].
Figure 7.
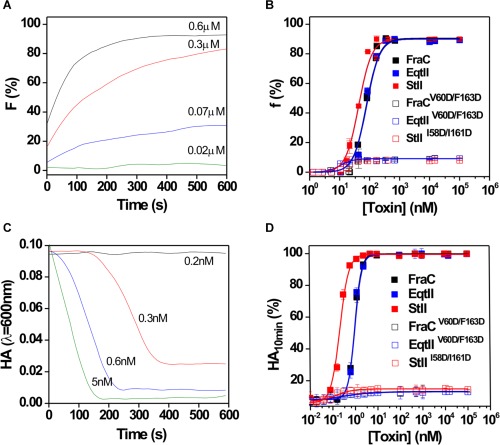
Permeabilizing and hemolytic activity of wildtype proteins and their mutants. (A) Time course of vesiclepermeabilization induced by FraC. (B) Fraction of vesicles in which at least one pore was formed (f) as a function of initial toxin concentration; f was calculated using eq. (2) at 10 min after toxin addition. Buffer: TBS, lipid composition: PC:SM (50:50), lipid concentration: 10 μM. (C) Time course of the lysis of the red blood cells promoted by FraC. (D) Dose‐dependence curve of the lysys of red blood cell after 10 min of assay (AH10 min). The time course of hemolysis was followed by the decrease in turbidity of a cell suspension initially adjusted to an apparent absorbance of 0.1 at 600 nm. Buffer: TBS, cell concentration: 105 cell mL−1. Curves in B and D were fitted to a Hill function using Origin 8.0, Microcal (USA). Experiments were done in triplicate (R 2 > 0.99).
The hemolytic activity of the proteins toward human red blood cells was also determined to quantitatively measure the pore‐forming activity of the three actinoporins in a cell model.21, 37 Figure 7(C) shows the time course of FraC‐elicited hemolysis at different concentrations. The hemolytic activity of the wildtype toxins increased with a clear sigmoidal relationship to toxin concentration [Fig. 7(D)]. The HC50 was determined by fitting the experimental data to a Hill sigmoid model (Table 2). This parameter represents the protein concentration needed to promote lysis of 50% of the cells in the assay and is inversely correlated to toxin activity. Similar to what was obtained with vesicles, wildtype proteins differ in activity and the mutants were not active against cells in the concentration range evaluated.
Discussion
The exact sequence of events that take place during the assembly of actinoporins and functionally relevant intermediates in the membrane remains under debate. Some studies suggest the relevance of protein–protein interactions8, 22, 25 while others assume that there is no need for such protein interfaces to stabilize oligomeric intermediates for the final pore assembly.24, 48 However, evidence supports the structural and functional importance of dimeric structures in the transition of the monomer structure from solution to the pore state.16, 21, 27, 49
The key role of dimers in actinoporins pore‐formation has been investigated from indirect or direct observations over the last decades.16, 21, 27, 49 Recently, a high resolution crystal structure of a dimeric form of FraC was obtained in a lipidic environment.8 In this dimeric structure the putative transmembrane α‐helices were found to be still attached to the protein core, but the oligomerization interface resembled the one of the octameric pore [Fig. 1(D,E)]. This finding suggested dimer formation as an important structural step in the initial stages of the assembly of actinoporins channel.8
In this study, we have revealed the driving forces for the spontaneous formation of FraC dimeric structure using free energy calculations with the MM‐GBSA approach.50 In particular, V60–F163 is predicted as the most relevant pair of interacting residues in the dimeric structure [Fig. 3(A); Supporting Information Table SII]. van der Waals interactions and nonpolar desolvation are the most relevant energetic components of this interacting pair, which suggests a key role of the geometric complementarity between these residues in the stability of the dimer [Fig. 3(A); Supporting Information Table SII]. FraC oligomers have a small protein–protein oligomerization surface and are held together by shape complementarity and hydrophobic interactions,8 two features that are easily compromised and result in stability reduction.45 These features are in marked contrast with other PFTs that form pores exclusively built by proteins segments (i.e., ClyA) stabilized by electrostatic interactions.51, 52, 53, 54
We predicted that the introduction of anionic amino acid residues (D60 and D163) in the dimeric interface of FraC allows its partial disruption as a consequence of electrostatic repulsion [Fig. 4(A,B,D); Table 1]. The reorganization observed generated an incompatible orientation of one protomer to detach its N‐terminal segment once the dimer binds to the membrane. As V60 and F163 are residues of FraC conserved among the actinoporins,8 we evaluated the effect of introducing homologous mutations in EqtII and StII in order to verify our hypothesis in others proteins of the family. These proteins differ in pore‐forming activity both in cells and liposomes [Fig. 7(B,D; Table 2].30 However, these differences have been correlated with variations in their N‐terminal segment, which accounts for mechanistic differences in the membrane insertion step and does not seem to be involved in oligomerization or binding steps.30 Introducing two anionic charged amino acid residues at positions 60 and 163 dramatically abolished the activity of these three actinoporins [Fig. 7(B,D; Table 2]. The complete activity loss of the double mutants is not due to the impairment of protein folding in solution (Fig. 5) and seems not to be related with differences in the affinity of the mutants for the lipid bilayer [Figs. 5(A,B) and 6(C–E)].
Structural data supporting the toroidal pore model assumes that there are no relevant protein–protein interactions and that proteins and lipid alternate in the final pore structure.24, 48 In contrast, the recently proposed hybrid pore model argues against this property and assumes that the pore assemble is held together through relevant protein–protein and protein–lipid interactions.8 Here, we extend the importance of FraC protein–protein interactions in the stabilization of the oligomeric intermediates to StII and EqtII pointing out that all of these proteins follow a similar pathway of membrane disruption. We found that the interface leading to the octameric assembly seen in the FraC crystal structure8 is critical for actinoporins pore formation. However, from our data we cannot rule out the existence of pore‐forming states with different stoichoimetries.26 Future work aimed to correlate oligomer stoichiometry with pore function should shed light about the relevance of intermediate stages, including dimeric structures, for actinoporins pore formation. Moreover, our results reinforce a model of pore assembly based on the addition of dimeric units recently proposed for the actinoporin EqtII.16 In fact, dimer formation has been assumed as a more efficient mechanism of oligomer assembly compared with simple sequential monomer addition.55
The dimer interface obtained by Tanaka et al.8 is been described as the basis for the octameric pore observed in the FraC structure and, more recently, for an octameric prepore structure of this toxin.25 However, from our results and previous published data,16 we cannot rule out the possibility that this dimeric interface can nucleate up to 8 units by the sequential addition of single or dimeric units to complete a final ring‐like assembly. EqtII was described to be organized in the cell surface as a mixture of oligomeric species mostly including those formed by even number of units.16 However, the mutant EqtII–L26C is able to oligomerize by an alternative pathway based on the addition of monomeric units even though this alteration decreased its toxic activity. This finding, obtained in conditions closer to those existing in physiological systems, highlights the relevance of dimers for EqtII assembly and toxicity. However, the fact that the EqtII mutant was able to oligomerize through monomers addition indicates that, although more efficient, dimers nucleation could be dispensable for complex formation.16 Moreover, it would be possible that different intermediate oligomeric states could form arc‐like toroidal pores with one edge filled by protein complexes and the other with bend lipids,55 similar to those observed in the MACPF/CDC protein superfamily56, 57 or for the Bcl2 protein Bax.58 Further studies should confirm this hypothesis since current high quality data obtained by electron microscopy or atomic force microscopy at high protein concentration do not support the formation of arc‐like structures in the mechanism of action of actinoporins.8, 22, 25
One of the main differences between the toroidal and hybrid models is the stoichiometry of the final oligomeric state of the pore. While the toroidal model assumes that the channel is formed by 3–4 monomers of proteins, the hybrid pore claims for an octameric molecularity. In this regard, Gilbert59 kindly advocated a second inspection of StII tetramers pointing out that such structures could be masking an octamer, similar to the one obtained for FraC.8 Then, each protein unit identified in the StII toroidal pore model would correspond to dimeric structures rather than monomers. From this perspective, a consensus point between the toroidal and the hybrid pore models might be the contribution of a priming dimeric structure to trigger pore opening,83 which is in agreement with our findings.
Based on recent evidence and our functional data, we can generalize the actinoporin model of pore formation by proposing that (i) dimeric units are the simplest structural intermediate with functional relevance which drive the assembly of bigger oligomeric species, (ii) the oligomerization interface is conserved across the family, and stabilized by shape complementarity and hydrophobic interactions, and consequently, (iii) the introduction of two anionic residues on this interface (V60D/F163D) dramatically abolishes the pore‐forming activity. This last result clearly indicates the importance of the key hydrophobic pair V60–F163 in stabilizing the structure of the actinoporins pore.11, 12, 13
Conclusions
In this work, we identified two mutations in FraC (Val60D and F163D) which completely hinder dimer formation and dramatically abolish the pore‐forming activity. Of mechanistic significance, we extend the importance of the FraC oligomerization interface to other members of the actinoporins family and reveal that they follow a common pathway of assembly and membrane disruption. It is highly likely that the complete activity loss of the mutants arose from their inability to form the lowest functionally relevant oligomer, the dimer, being the monomer the only molecular entity found in the membrane. Future work must be addressed to unravel this question.
Materials and Methods
Chemicals and reagents
Egg SM and 1‐palmitoyl‐2‐oleylphosphatidylcholine (POPC) were purchased from Avanti Polar Lipids (Alabaster, AL, USA) with 99% of purity and used without further purification. Solvents and chemicals were of the highest commercial purity available.
Methods
Molecular dynamics simulations
All Energy Minimizations (EM) and Molecular Dynamics (MD) simulations were carried out using the GROMACS software package (version 4.6.5)60 with the AMBER99sb force field,61 and the TIP3P water model.62 The simulation systems consist of the dimer (PDB: 4TSL) solvated in a truncated dodecahedrical box with 13640 water molecules. The protonation states of ionizable residues were assigned at pH 7.4 with the program PDB2PQR (http://nbcr-222.ucsd.edu/pdb2pqr_1.8/), which uses PROPKA for the prediction of pK avalues. Cl− ions were added to achieve electroneutrality of the system. At each step, the electrostatic interactions were calculated with the particle‐mesh Ewald method.63 Van der Waals interactions were described by a Lennard–Jones potential with a cutoff of 1.1 nm that was switched to zero between 1.0 and 1.1 nm. Dispersion corrections for energy and pressure were applied. The SETTLE algorithm64 was used to constrain bonds and angles of water molecules and LINCS65 was used for all other bonds, allowing a time step of 2 fs. The equilibrium simulations were sampled for 100 ns, using the stochastic dynamic integrator66 at 310 K and a constant pressure of 1 atm, using the weak coupling to the velocity rescaling thermostat67 and Parrinello–Rahman barostat,68, 69 respectively. The friction coefficient (ξ) was set to 0.5 ps−1 in all systems as recommended.70 Snapshots were saved at 10 ps intervals.
MM‐GBSA free energy calculations
T s (i.e., the molecular energy in the gas phase (ΔE gas), the solvation free energy (ΔG solv), and the entropy contribution (−T·ΔS)). When the third term was neglected, the computed value is an effective free energy (ΔG eff), which usually suffices for comparing the relative affinities of a series of similar ligands for a given receptor because in that case the entropy term is approximately constant.71 The term ΔE gas, which includes the internal (ΔE int), the van der Waals (ΔE vw), and the electrostatic (ΔE ele) energies, was derived from the force‐field equations. The term ΔG solv was further decomposed into two components (i.e., the polar‐solvation free energy (ΔG polar) and the nonpolar solvation free energy (ΔG nonpolar).71 The former was calculated using the Generalized Born‐Neck2 implicit‐solvation model (GBn2, igb = 8),72 whereas the latter was obtained by:
| (1) |
in which ΔG nonpolar (X) represents the solvent‐accessible surface area changeof the solute molecule X upon complex formation, while γ and β are empirical constants whose values for GB models are almost always 0.0072 kcal Å−2 mol−1 and 0, respectively.71, 73 T·ΔS is frequently computed by normal‐mode analysis and is, therefore, the most computationally‐demanding step of the MM‐GBSA method.71, 73
The MMPBSA.py program of the Amber12 package was used for MM‐GBSA free energy calculations71, 73 after converting the GROMACS trajectories into the Amber format using VMD v1.9.1.74 In all cases, we followed the single trajectory (ST) approach,71, 73 in which the trajectories for both protomers were extracted from that of the dimeric form of FraC. In the ST approach only a single trajectory is required to generate all three ensembles. Furthermore, the internal potential terms (e.g., bonds, angles, and dihedrals) cancel exactly, because the conformations in the bound and unbound ensembles are the same, leading to lower fluctuations, and easier convergence of binding free calculations with the MM‐GBSA approach.71, 73 The GBn2 implicit‐solvation model (igb = 8) was employed to calculate the ΔG GB value of each complex.71, 72 Topologies were obtained with the Amber program tleap using mbondi3 radii for GBn2 models.71, 72 Salt concentrations of 0.05 mol/L were set for each system and default solvent and solute dielectric constants (ε w = 78.3 and ε in = 1, respectively) and rgbmax cutoff (rgbmax = 25 Å) values were used in all GB calculations.71 In all cases, the ΔSA values were determined using the linear combination of pairwise overlaps algorithm.75 ΔG nonpolar was then estimated through Eq. (1).50 Finally, ΔE gas was estimated from AMBER99SB parameters.61 Mean values of the energy terms were obtained by averaging over the snapshots extracted every 10 ps from each production MD simulation. Snap shots were considered as statistically independent from each other, since previous work has determined a typical correlation time for MM‐GB(PB)SA energy terms of ∼5 ps or less.76
Energetically‐relevant residues (i.e., warm‐ and hot‐spots, at the interfaces of the studied complexes) were predicted by using the computational alanine scanning (CAS) protocol.50, 71 Briefly, Ala single‐point mutations were generated at specific positions using the Modeller 9.14 software77 and the topologies of the mutated complexes were obtained using tleap of Amber12. Subsequently, relative free energy values (ΔΔG) between the native and mutated complexes were determined using MMPBSA.py.50, 71 These calculations were performed under the ST approach in which the trajectory of the mutated complex is generated from that of the native complex by simply truncating the side‐chain of the residue of interest and replacing the Cγ atom by a hydrogen.78, 79 Finally, the pairwise effective free energy decomposition (pwEFED) protocol of MMPBSA.py was employed to calculate interaction energies between pairs of residues (ΔG r1,r2).50, 71, 78
Cloning, expression, and isolation of the mutants
fraC, eqtII, and stII coding genes were cloned in the expression vector pET21a+ (Entelchon, Bad Abbach, Germany) without any tag to avoid affecting protein activity. The double mutants FraCV60D/F163D, EqtIIV60D/F163D, and StIII58D/I161D were obtained by replacing the corresponding residues by the anionic charged aspartic acid aminoacid. Recombinant proteins and mutants were producedin E. coli BL21 (DE3) competent cells(F − ompThsdSB(rB − mB−) gal dcm met (DE3)). For purification we followed the protocol described by Pazos et al.,80 with a slight modification. Briefly, we used cation‐exchange chromatography in CM–cellulose column (Sigma‐Aldrich, Munich, Germany) employing a NaCl continuous linear gradient (from 0 to 1M) in 0.05M ammonium acetate buffer, pH 5. The identity of the recombinant proteins and mutants were verified by mass spectrometry in a MALDI TOF/TOF™5800 mass (Darmstadt, Germany). Protein concentration was determined employing absorption coefficients of 42,530 cm– 1 M−1 (FraC),38 36,100 cm−1 M−1 (EqtII),81 and 37,400 cm−1 M−1 (StII).82
Circular dichroism studies
Circular dichroism (CD) spectra were acquired from 190 to 260 nm in 1 mm path length cuvettes, at room temperature using a JobinYvon CD6 spectropolarimeter (JobinYvon, Longjumeau, France). The instrument was routinely calibrated with an aqueous solution of recrystallized D‐10‐camphorsulfonic acid. The reported spectra are averages of 6 scans. Samples were prepared at 10 µM in PBS buffer and spectra were baseline corrected by using control samples of solutions not containing protein. The percentage of secondary structure was calculated using the Dichroweb Internet server.34, 35, 36
Surface pressure measurements on lipid monolayers
Surface pressure (π) measurements were carried out with a μThrough‐S system (Kibron, Helsinki, Finland) at room temperature under constant stirring. The aqueous phase consisted of 300 µL of Tris‐buffered saline (TBS: 145 mM NaCl, 10 mM Tris–HCl, pH 7.4). The lipid mixture of POPC:SM (50:50) was predissolved in chloroform:methanol (2:1, v:v) and was gently spread over the surface. Then, the desired initial surface pressure (π0) was attained by changing the amount of lipid applied to the air–water interface. The proteins were injected into the subphase to achieve 0.8 μM protein final concentration at which no effect on surface tension of the air–water interface was observed.83 The increment in surface pressure (Δπ) was recorded as a function of the elapsed time until a stable signal was obtained.
Preparation of vesicles
The lipid mixture of POPC:SM (50:50) was dissolved in chloroform:methanol (2:1, v:v), mixed and evaporated thoroughly at 50°C, and submitted to vacuum for not less than 2 h. For permeabilizing assays, multilamellar vesicles (MLV) were obtained by subsequent hydration in the presence of 80 mM carboxyfluorescein (CF), pH 7.4 in water and subjected to six cycles of freezing and thawing. For binding assays, MLV were hydrated in TBS. Small unilamellar vesicles (SUV) were prepared through sonication of MLV suspensions using an ultrasonicator (Branson 450, Danbury, USA) equipped with a titanium tip and subjected to 15 cycles of 1 min sonication between resting intervals of 1 min. Titanium particles released from the probe were removed by further centrifugation at 10 000g for 10 min. To remove untrapped CF in the permeabilization assay, the vesicles were filtered through mini‐columns (Pierce, Rockford, USA) loaded with Sephadex G‐50 (medium) pre‐equilibrated with TBS. Phospholipid concentration was measured by inorganic phosphate quantification.84
Intrinsic fluorescence measurements
Fluorescence spectra were recorded in a Hitachi F‐4500 spectrofluorimeter, using 1 cm path length quartz cuvettes at room temperature. Slit widths of 5 nm nominal band pass were used both in the excitation and emission beams. Intrinsic fluorescence emission spectra of the proteins (2 µM) were recorded from 300 to 450 nm in solution. Excitation was performed at 295 nm to obtain fluorescence spectra derived only from Trp residues. Changes in the intrinsic fluorescence of the proteins upon addition of increasing quantities of SUV composed of POPC: SM (50:50) were also measured. Background intensities measured in samples without proteins were subtracted.
Quenching of proteins was achieved by adding the water‐soluble quencher acrylamide (AA) in the presence of buffer or saturating concentration of SUV (40 µM). The maximum fluorescence intensities without and with the quencher (F0 and FAA, respectively) were determined and F 0/F AA values were plotted as a function of acrylamide concentration ([AA]). The experimental data were analyzed according to the Stern–Volmer equation:
| (2) |
The slopes of the best‐fit linear plots were used to determine the Stern–Volmer quenching constants (K SV).85
Leakage studies from CF‐containing SUV
SUV permeabilization was assessed at room temperature, using a FLUOstar OPTIMA microplate reader (BMG Labtech, Offenburg, Germany) by measuring the fluorescence (exc at 490 nm and em at 520 nm) of CF released. The toxins were two‐fold serially diluted in the well microplates in a final volume of 100 μL of TBS and the reaction was started by adding the same volume of SUV (10 μM of final lipid concentration). After mixing vesicles and toxins, the release of CF produced an increase in fluorescence (f) due to the dequenching of the dye into the external medium, which was resolved in time. Spontaneous (toxin‐free) leakage of dye was negligible under these conditions. Maximum release was always obtained by adding 1 mM Triton X‐100 (final concentration) and provided the fluorescence value f max. The fraction of fluorophore release (f) was calculated as follows:
| (3) |
where f 0 and f t represent the value of fluorescence before and at time t after toxin addition, respectively. C50was calculated by fitting dose‐dependence curves of permeabilization induced by the toxins to a Hill sigmoid using Origin 8.0, Microcal (USA).
Hemolytic activity
Hemolytic activity (HA) was evaluated turbidimetrically at 600 nm at room temperature in a microplate reader (Labsystems, Helsinki, Finland). Erythrocyte suspension was prepared using pooled fresh human red blood cells collected intravenously from at least four healthy volunteers. Cells were washed by repeated centrifugation (600g, 15 min), the cell pellet resuspended in TBS and finally diluted to an apparent absorbance of 0.1 at 600 nm. Toxins were twofold serially diluted in a flat‐bottom 96‐well microplate in a final volume of 100 µL of TBS. The hemolysis was started by adding the same volume of red blood cell suspension. The decrease in optical density (O.D) was recorded as a function of time with intermittent shaking. The hemolytic activity (HA) was calculated as follows:
| (4) |
where ODt, OD0, and ODmin represent the O.D. at time t, at time zero, and in the presence of an excess of toxin, respectively.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
DPT is an Alberta Innovates Health Solutions Scientist and Alberta Innovates Technology Futures Strategic Chair in (Bio)Molecular Simulation. H.M.G and K.H.D.M acknowledge ELAP (Canada) scholarship for financial support to visit the Centre for Molecular Simulation, University of Calgary, Canada. H.M.G, K.H.D.M, U.R and P.A.V acknowledge Dr. Michael Paolillo for the language edition of the manuscript.
Contributor Information
Uris Ros, Email: uris.ros@ifib.uni-tuebingen.de.
Pedro A. Valiente, Email: valiente@fbio.uh.cu
REFERENCES
- 1. Kristan KC, Viero G, Dalla Serra M, Macek P, Anderluh G (2009) Molecular mechanism of pore formation by actinoporins. Toxicon 54:1125–1134. [DOI] [PubMed] [Google Scholar]
- 2. Frazao B, Vasconcelos V, Antunes A (2012) Sea anemone (Cnidaria, Anthozoa, Actiniaria) toxins: an overview. Mar Drugs 10:1812–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderluh G, Macek P (2002) Cytolytic peptide and protein toxins from sea anemones (Anthozoa: Actiniaria) . Toxicon 40:111–124. [DOI] [PubMed] [Google Scholar]
- 4. Rojko N, Dalla Serra M, Macek P, Anderluh G (2016) Pore formation by actinoporins, cytolysins from sea anemones. Biochim Biophys Acta 1858:446–456. [DOI] [PubMed] [Google Scholar]
- 5. Álvarez C, Mancheno JM, Martinez D, Tejuca M, Pazos F, Lanio ME (2009) Sticholysins, two pore‐forming toxins produced by the Caribbean Sea anemone Stichodactyla helianthus: their interaction with membranes. Toxicon 54:1135–1147. [DOI] [PubMed] [Google Scholar]
- 6. García‐Ortega L, Alegre‐Cebollada J, Garcia‐Linares S, Bruix M, Martinez‐Del‐Pozo A, Gavilanes JG (2011) The behavior of sea anemone actinoporins at the water‐membrane interface. Biochim Biophys Acta 1808:2275–2288. [DOI] [PubMed] [Google Scholar]
- 7. Ros U, Garcia‐Saez AJ (2015) More than a pore: The interplay of pore‐forming proteins and lipid membranes. J Membr Biol 248:545–561. [DOI] [PubMed] [Google Scholar]
- 8. Tanaka K, Caaveiro JM, Morante K, Gonzalez‐Manas JM, Tsumoto K (2015) Structural basis for self‐assembly of a cytolytic pore lined by protein and lipid. Nat Commun 6:6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hong Q, Gutiérrez‐Aguirre I, Barlic A, Malovrh P, Kristan K, Podlesek Z, Macek P, Turk DA, González‐Mañas JM, Lakey JH (2002) Two‐step membrane binding by Equinatoxin II, a pore‐forming toxin from the sea anemone, involves an exposed aromatic cluster and a flexible helix. J Biol Chem 277:41916–41924. [DOI] [PubMed] [Google Scholar]
- 10. Tejuca M, Serra MD, Ferreras M, Lanio ME, Menestrina G (1996) Mechanism of membrane permeabilization by sticholysin I, a cytolysin isolated from the venom of the sea anemone Stichodactyla helianthus. Biochemistry 35:14947–14957. [DOI] [PubMed] [Google Scholar]
- 11. Álvarez C, Dalla Serra M, Potrich C, Bernhart I, Tejuca M, Martinez D, Pazos F, Lanio ME, Menestrina G (2001) Effects of lipid composition on membrane permeabilization by sticholysin I and II, two cytolysins of the sea anemone Stichodactyla helianthus. Biophys J 80:2761–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alvarez C, Casallanovo F, Shida CS, Nogueira LV, Martinez D, Tejuca M, Pazos IF, Lanio ME, Menestrina G, Lissi E, Schreier S (2003) Binding of sea anemone pore‐forming toxins sticholysins I and II to interfaces—modulation of conformation and activity, and lipid–protein interaction. Chem Phys Lipids 122:97–105. [DOI] [PubMed] [Google Scholar]
- 13. Anderluh G, Dalla Serra M, Viero G, Guella G, Macek P, Menestrina G (2003) Pore formation by equinatoxin II, a eukaryotic protein toxin, occurs by induction of nonlamellar lipid structures. J Biol Chem 278:45216–45223. [DOI] [PubMed] [Google Scholar]
- 14. Rojko N, Kristan KC, Viero G, Zerovnik E, Macek P, Dalla Serra M, Anderluh G (2013) Membrane damage by an alpha‐helical pore‐forming protein, Equinatoxin II, proceeds through a succession of ordered steps. J Biol Chem 288:23704–23715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Antonini V, Perez‐Barzaga V, Bampi S, Penton D, Martinez D, Dalla Serra M, Tejuca M (2014) Functional characterization of sticholysin I and W111C mutant reveals the sequence of the actinoporin's pore assembly. PLoS One 9:e110824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subburaj Y, Ros U, Hermann E, Tong R, Garcia‐Saez AJ (2015) Toxicity of an alpha‐pore‐forming toxin depends on the assembly mechanism on the target membrane as revealed by single molecule imaging. J Biol Chem 290:4856–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barlic A, Gutierrez‐Aguirre I, Caaveiro JM, Cruz A, Ruiz‐Arguello MB, Perez‐Gil J, Gonzalez‐Manas JM (2004) Lipid phase coexistence favors membrane insertion of equinatoxin‐II, a pore‐forming toxin from Actinia equina. J Biol Chem 279:34209–34216. [DOI] [PubMed] [Google Scholar]
- 18. Bakrac B, Gutierrez‐Aguirre I, Podlesek Z, Sonnen AF, Gilbert RJ, Macek P, Lakey JH, Anderluh G (2008) Molecular determinants of sphingomyelin specificity of a eukaryotic pore‐forming toxin. J Biol Chem 283:18665–18677. [DOI] [PubMed] [Google Scholar]
- 19. Pedrera L, Fanani ML, Ros U, Lanio ME, Maggio B, Alvarez C (2014) Sticholysin I‐membrane interaction: an interplay between the presence of sphingomyelin and membrane fluidity. Biochim Biophys Acta 1838:1752–1759. [DOI] [PubMed] [Google Scholar]
- 20. Rojko N, Cronin B, Danial JS, Baker MA, Anderluh G, Wallace MI (2014) Imaging the lipid‐phase‐dependent pore formation of equinatoxin II in droplet interface bilayers. Biophys J 106:1630–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Belmonte G, Pederzolli C, Macek P, Menestrina G (1993) Pore formation by the sea anemone cytolysin equinatoxin II in red blood cells and model lipid membranes. J Membr Biol 131:11–22. [DOI] [PubMed] [Google Scholar]
- 22. Mechaly AE, Bellomio A, Gil‐Carton D, Morante K, Valle M, Gonzalez‐Manas JM, Guerin DM (2011) Structural insights into the oligomerization and architecture of eukaryotic membrane pore‐forming toxins. Structure 19:181–191. [DOI] [PubMed] [Google Scholar]
- 23. Rojko N, Anderluh G (2015) How lipid membranes affect pore forming toxin activity. Acc Chem Res 48:3073–3079. [DOI] [PubMed] [Google Scholar]
- 24. Mancheno JM, Martin‐Benito J, Martinez‐Ripoll M, Gavilanes JG, Hermoso JA (2003) Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure 11:1319–1328. [DOI] [PubMed] [Google Scholar]
- 25. Morante K, Bellomio A, Gil‐Carton D, Redondo‐Morata L, Sot J, Scheuring S, Valle M, Gonzalez‐Manas JM, Tsumoto K, Caaveiro JM (2016) Identification of a membrane‐bound prepore species clarifies the lytic mechanism of actinoporins. J Biol Chem 291:19210–19219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baker MA, Rojko N, Cronin B, Anderluh G, Wallace MI (2014) Photobleaching reveals heterogeneous stoichiometry for equinatoxin II oligomers. Chembiochem 15:2139–2145. [DOI] [PubMed] [Google Scholar]
- 27. Valle A, Lopez‐Castilla A, Pedrera L, Martinez D, Tejuca M, Campos J, Fando R, Lissi E, Alvarez C, Lanio ME, Pazos F, Schreier S (2011) Cys mutants in functional regions of Sticholysin I clarify the participation of these residues in pore formation. Toxicon 58:8–17. [DOI] [PubMed] [Google Scholar]
- 28. Ros U, Rodriguez‐Vera W, Pedrera L, Valiente PA, Cabezas S, Lanio ME, Garcia‐Saez AJ, Alvarez C (2015) Differences in activity of actinoporins are related with the hydrophobicity of their N‐terminus. Biochimie 116:70–78. [DOI] [PubMed] [Google Scholar]
- 29. Venyaminov SY, Yang JT (1996) Determination of protein secondary structure. Circular dichroism and the conformational analysis of biomolecules. New York: Springer, pp 69–107. [Google Scholar]
- 30. Provencher SW, Glockner J (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20:33–37. [DOI] [PubMed] [Google Scholar]
- 31. van Stokkum IH, Spoelder HJ, Bloemendal M, van Grondelle R, Groen FC (1990) Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal Biochem 191:110–118. [DOI] [PubMed] [Google Scholar]
- 32. Sreerama N, Woody RW (1993) A self‐consistent method for the analysis of protein secondary structure from circular dichroism. Anal Biochem 209:32–44. [DOI] [PubMed] [Google Scholar]
- 33. Sreerama N, Venyaminov SY, Woody RW (1999) Estimation of the number of [alpha]‐helical and [beta]‐strand segments in proteins using circular dichroism spectroscopy. Protein Sci 8:):370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lobley A, Whitmore L, Wallace BA (2002) DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 18:211–212. [DOI] [PubMed] [Google Scholar]
- 35. Whitmore L, Wallace BA (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res 32:W668–6W673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whitmore L, Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89:392–400. [DOI] [PubMed] [Google Scholar]
- 37. Martínez D, Campos AM, Pazos F, Alvarez C, Lanio ME, Casallanovo F, Schreier S, Salinas RK, Vergara C, Lissi E (2001) Properties of St I and St II, two isotoxins isolated from Stichodactyla helianthus: a comparison. Toxicon 39:1547–1560. [DOI] [PubMed] [Google Scholar]
- 38. Bellomio A, Morante K, Barlic A, Gutiérrez‐Aguirre I, Viguera AR, González‐Mañas JM (2009) Purification, cloning and characterization of fragaceotoxina C, a novel actinoporin from the sea anemone Actinia fragacea . Toxicon 54:869–880. [DOI] [PubMed] [Google Scholar]
- 39. Hinds MG, Zhang W, Anderluh G, Hansen PE, Norton RS (2002) Solution structure of the eukaryotic pore‐forming cytolysin equinatoxin II: implications for pore formation. J Mol Biol 315:1219–1229. [DOI] [PubMed] [Google Scholar]
- 40. Athanasiadis A, Anderluh G, Macek P, Turk D (2001) Crystal structure of the soluble form of equinatoxin II, a pore‐forming toxin from the sea anemone Actinia equina. Structure 9:341–346. [DOI] [PubMed] [Google Scholar]
- 41. Martínez D, Otero A, Alvarez C, Pazos F, Tejuca M, Lanio ME, Gutierrez‐Aguirre I, Barlic A, Iloro I, Arrondo JL, Gonzalez‐Manas JM, Lissi E (2007) Effect of sphingomyelin and cholesterol on the interaction of St II with lipidic interfaces. Toxicon 49:68–81. [DOI] [PubMed] [Google Scholar]
- 42. Ros U, Souto AL, de Oliveira FJ, Crusca E, Jr. , Pazos F, Cilli EM, Lanio ME, Schreier S, Alvarez C (2013) Functional and topological studies with Trp‐containing analogs of the peptide StII(1‐30) derived from the N‐terminus of the pore forming toxin sticholysin II: contribution to understand its orientation in membrane. Biopolymers 100:337–346. [DOI] [PubMed] [Google Scholar]
- 43. Caaveiro JM, Echabe I, Gutiérrez‐Aguirre I, Nieva JL, Arrondo JL, González‐Mañas JM (2001) Differential interaction of equinatoxin II with model membranes in response to lipid composition. Biophys J 80:1343–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brockman H (1999) Lipid monolayers: why use half a membrane to characterize protein–membrane interactions? Curr Opin Struct Biol 9:438–443. [DOI] [PubMed] [Google Scholar]
- 45. Morante K, Caaveiro JM, Viguera AR, Tsumoto K, Gonzalez‐Manas JM (2015) Functional characterization of Val60, a key residue involved in the membrane‐oligomerization of fragaceatoxin C, an actinoporin from Actinia fragacea. FEBS Lett 589:1840–1846. [DOI] [PubMed] [Google Scholar]
- 46. Valle A, Alvarado‐Mesén J, Lanio M, Álvarez C, Barbosa J, Pazos I (2015) The multigene families of actinoporins (part I): isoforms and genetic structure. Toxicon 103:176–187. [DOI] [PubMed] [Google Scholar]
- 47. Patel H, Tscheka C, Heerklotz H (2009) Characterizing vesicle leakage by fluorescence lifetime measurements. Soft Matter 5:2849–2851. [Google Scholar]
- 48. Mancheno JM, Martin‐Benito J, Gavilanes JG, Vazquez L (2006) A complementary microscopy analysis of Sticholysin II crystals on lipid films: atomic force and transmission electron characterizations. Biophys Chem 119:219–223. [DOI] [PubMed] [Google Scholar]
- 49. Macek P, Belmonte G, Pederzolli C, Menestrina G (1994) Mechanism of action of equinatoxin II, a cytolysin from the sea anemone Actinia equina L. belonging to the family of actinoporins. Toxicology 87:205–227. [DOI] [PubMed] [Google Scholar]
- 50. Miller BR, McGee TD, Jr Swails JM, Homeyer N, Gohlke H, Roitberg AE (2012) MMPBSA. py: an efficient program for end‐state free energy calculations. J Chem Theory Comput 8:3314–3321. [DOI] [PubMed] [Google Scholar]
- 51. Yamashita K, Kawai Y, Tanaka Y, Hirano N, Kaneko J, Tomita N, Ohta M, Kamio Y, Yao M, Tanaka I (2011) Crystal structure of the octameric pore of staphylococcal gamma‐hemolysin reveals the beta‐barrel pore formation mechanism by two components. Proc Natl Acad Sci USA 108:17314–17319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N (2009) The structure of a cytolytic alpha‐helical toxin pore reveals its assembly mechanism. Nature 459:726–730. [DOI] [PubMed] [Google Scholar]
- 53. Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE (1996) Structure of staphylococcal alpha‐hemolysin, a heptameric transmembrane pore. Science 274:1859–1866. [DOI] [PubMed] [Google Scholar]
- 54. De S, Olson R (2011) Crystal structure of the Vibrio cholerae cytolysin heptamer reveals common features among disparate pore‐forming toxins. Proc Natl Acad Sci USA 108:7385–7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gilbert RJ, Dalla Serra M, Froelich CJ, Wallace MI, Anderluh G (2014) Membrane pore formation at protein–lipid interfaces. Trends Biochem Sci 39:510–516. [DOI] [PubMed] [Google Scholar]
- 56. Sonnen AF, Plitzko JM, Gilbert RJ (2014) Incomplete pneumolysin oligomers form membrane pores. Open Biol 4:140044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Metkar SS, Marchioretto M, Antonini V, Lunelli L, Wang B, Gilbert RJ, Anderluh G, Roth R, Pooga M, Pardo J, Heuser JE, Serra MD, Froelich CJ (2015) Perforin oligomers form arcs in cellular membranes: a locus for intracellular delivery of granzymes. Cell Death Differ 22:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salvador‐Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, Garcia‐Saez AJ (2016) Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J 35:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gilbert RJ (2016) Protein–lipid interactions and non‐lamellar lipidic structures in membrane pore formation and membrane fusion. Biochim Biophys Acta 1858:487–499. [DOI] [PubMed] [Google Scholar]
- 60. Pall S, Abraham MJ, Kutzner C, Hess B, Lindahl E (2014) Tackling exascale software challenges in molecular dynamics simulations with GROMACS. Solving software challenges for exascale. New York: Springer, pp 3–27. [Google Scholar]
- 61. Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Price DJ, Brooks CL III (2004) A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys 121:10096–10103. [DOI] [PubMed] [Google Scholar]
- 63. Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N‐log (N) method for Ewald sums in large systems. J Chem Phys 98:10089. [Google Scholar]
- 64. Miyamoto S, Kollman PA (1992) Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 13:952–962. [Google Scholar]
- 65. Hess B, Bekker H, Berendsen HJ, Fraaije JG (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472. [Google Scholar]
- 66. Van Gunsteren WF, Berendsen HJC (1988) A leap‐frog algorithm for stochastic dynamics. Mol Simul 1:173–185. [Google Scholar]
- 67. Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:014101. [DOI] [PubMed] [Google Scholar]
- 68. Nosé S, Klein M (1983) Constant pressure molecular dynamics for molecular systems. Mol Phys 50:1055–1076. [Google Scholar]
- 69. Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. [Google Scholar]
- 70. Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 71. Case D, Darden T, Cheatham III TE, Simmerling C, Wang J, Duke R, Luo R, Walker R, Zhang W, Merz K (2012) AMBER 12. San Francisco: University of California. [Google Scholar]
- 72. Nguyen H, Roe DR, Simmerling C (2013) Improved generalized born solvent model parameters for protein simulations. J Chem Theory Comput 9:2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jayaram B, Sprous D, Beveridge D (1998) Solvation free energy of biomacromolecules: Parameters for a modified generalized Born model consistent with the AMBER force field. J Phys Chem B 102:9571–9576. [Google Scholar]
- 74. Hernandez Alvarez L, Naranjo Feliciano D, Hernandez Gonzalez JE, Soares RO, Barreto Gomes DE, Pascutti PG (2015) Insights into the interactions of Fasciola hepatica cathepsin L3 with a substrate and potential novel inhibitors through in silico approaches. PLoS Negl Trop Dis 9:e0003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Weiser J, Shenkin PS, Still WC (1999) Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem 20:217–230. [Google Scholar]
- 76. Fulle S, Withers‐Martinez C, Blackman MJ, Morris GM, Finn PW (2013) Molecular determinants of binding to the Plasmodium subtilisin‐like protease 1. J Chem Inform Model 53:573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sali A, Blundell TL (1993) Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol 234:779–815. [DOI] [PubMed] [Google Scholar]
- 78. Gohlke H, Kiel C, Case DA (2003) Insights into protein–protein binding by binding free energy calculation and free energy decomposition for the Ras–Raf and Ras–RalGDS complexes. J Mol Biol 330:891–913. [DOI] [PubMed] [Google Scholar]
- 79. Zoete V, Michielin O (2007) Comparison between computational alanine scanning and per‐residue binding free energy decomposition for protein–protein association using MM–GBSA: application to the TCR‐p‐MHC complex. Proteins 67:1026–1047. [DOI] [PubMed] [Google Scholar]
- 80. Pazos F, Valle A, Martinez D, Ramirez A, Calderon L, Pupo A, Tejuca M, Morera V, Campos J, Fando R, Dyszy F, Schreier S, Horjales E, Alvarez C, Lanio ME, Lissi E (2006) Structural and functional characterization of a recombinant sticholysin I (rSt I) from the sea anemone Stichodactyla helianthus. Toxicon 48:1083–1094. [DOI] [PubMed] [Google Scholar]
- 81. Macek P, Lebez D (1988) Isolation and characterization of three lethal and hemolytic toxins from the sea anemone Actinia equina L. Toxicon 26:441–451. [DOI] [PubMed] [Google Scholar]
- 82. Lanio ME, Morera V, Alvarez C, Tejuca M, Gomez T, Pazos F, Besada V, Martinez D, Huerta V, Padron G, de los Angeles Chavez M (2001) Purification and characterization of two hemolysins from Stichodactyla helianthus. Toxicon 39:187–194. [DOI] [PubMed] [Google Scholar]
- 83. Doyle JW, Kem WR, Vilallonga FA (1989) Interfacial activity of an ion channel‐generating protein cytolysin from the sea anemone Stichodactyla helianthus. Toxicon 27:465–471. [DOI] [PubMed] [Google Scholar]
- 84. Rouser G, Fkeischer S, Yamamoto A (1970) Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5:494–496. [DOI] [PubMed] [Google Scholar]
- 85. Lakowicz JR (2006) Principles of fluorescence spectroscopy. New York: Springer. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information