

Abstract
Most knowledge of protein structure and function is derived from experiments performed with purified protein resuspended in dilute, buffered solutions. However, proteins function in the crowded, complex cellular environment. Although the first four levels of protein structure provide important information, a complete understanding requires consideration of quinary structure. Quinary structure comprises the transient interactions between macromolecules that provides organization and compartmentalization inside cells. We review the history of quinary structure in the context of several metabolic pathways, and the technological advances that have yielded recent insight into protein behavior in living cells. The evidence demonstrates that protein behavior in isolated solutions deviates from behavior in the physiological environment.
Keywords: metabolon, protein stability, protein structure, quinary structure
Introduction
The first four levels of protein structure were proposed by Linderstrøm–Lang in 1952.1 The amino acid sequence defines the primary structure.2, 3 Secondary structure comprises individual α‐helices, β‐strands and turns that are stabilized by intramolecular hydrogen bonds.4 The three‐dimensional arrangement of atoms5 forms the tertiary structure, which is also stabilized by hydrophobic interactions.6, 7, 8 Quaternary structure describes the assembly of individual polypeptide chains in multimeric proteins.9
These four levels formed the basis for discovering the relationships between protein folding and function.10 This hierarchy, however, is only sufficient to describe proteins in isolation. Evidence for cellular compartmentalization of metabolic pathways was established in the 1960s.11 Quinary structure, the fifth level of protein structure, comprises the interactions between macromolecules that organize the cellular interior (Figure 1). The term was coined three times. In 1973, Vaïnshtein described quinary structure as the level of organization of the “combination of molecules of proteins, nucleic acids and nucleoproteins into aggregates.”12 In 1980, Edelstein extended the four levels of protein structure to include quinary structure to describe the “marked plasticity and inequivalence in the juxtaposition of constituent molecules.”13 In 1982, McConkey reasoned that the conservation of isoelectric points among homologous proteins was “a consequence of complexities of intracellular organization and the numerous macromolecular interactions in which most polypeptides participate,” which he called “quinary structure.”14
Figure 1.
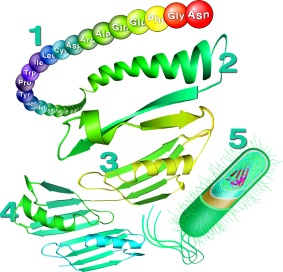
Five levels of protein structure by Chris A. Pielak. The tertiary and quanternary structures are based the B1 domain of protein G (PDB entries 1GB115 and 2RMM16).
The cellular interior is a crowded, dynamic environment; quinary structure is crucial for controlling and maintaining the diverse biochemical processes and signaling cascades. Quinary structure facilitates the assembly of multi‐enzyme complexes such the Krebs cycle enzymes,17, 18 the purinosome,19 and the respirasome.20, 21 Here, we illustrate quinary structure in the context of these systems. We then discuss in‐cell NMR and other recent contributions to understanding quinary structure. Finally, we address outstanding questions in the field.
Quinary Structure and Metabolic Pathways
Citric acid cycle
Evidence for quinary structure was initially reported in 1948 when Green isolated an aggregated supramolecular complex of the Krebs cycle enzymes, which he called the cyclophorase system.22 It was shown later that Green had actually isolated mitochondria,23 but the evidence for intracellular organization of the enzymes continued to accumulate.
The formation of multienzyme complexes facilitates substrate channeling, the direct transfer of metabolites between active centers.24, 25, 26, 27, 28 The bifunctional tryptophan synthase, which catalyzes the last two reactions in L‐tryptophan synthesis, is the simplest example of a complex that channels a metabolite between reaction centers.29, 30, 31 A tunnel within the protein transfers the metabolite from the first reaction center to the second. In 1973, Srere demonstrated the kinetic advantage of a microenvironment and reported higher rates of citrate formation by a group of three immobilized Krebs cycle enzymes than by their soluble counterparts.32 In 1977, he reported specific interactions between a couple of these enzymes in the presence of polyethylene glycol.33 Numerous groups have since described specific interactions among these enzymes in solution34, 35, 36, 37 and in vivo.38
In 1985, Srere coined the term “metabolon” to describe a “supramolecular complex of sequential metabolic enzymes and cellular structural elements” that was “formed by quinary interactions of complementary surfaces.”39 These dynamic complexes40 facilitate substrate channeling by protecting intermediates41, 42, 43 and result in enhanced rates of catalysis and metabolic flux.44 Brownian dynamics simulations attribute the transfer efficiency within a fusion protein of two tricarboxylic acid (TCA) cycle enzymes to electrostatic channeling of negatively charged substrates between positively charged active sites.45, 46 The simulation was experimentally validated by demonstrating that high salt concentrations eliminate substrate channeling between the two proteins.47
Recently, all eight enzymes in the TCA cycle metabolon were identified by in vivo crosslinking and mass spectrometry.48 Residues important for quinary interactions were identified. In addition, surface electrostatic potentials of the enzymes showed formation of a positively charged channel upon association, in agreement with the studies on the fusion protein.45, 46, 47 Site‐directed mutagenesis was used to identify a single arginine residue in citrate synthase crucial for interactions with malate dehydrogenase.49 While changes to this residue have a negligible effect on enzyme activity, they significantly decrease the overall channeling probability and metabolic flux. These studies provide important information about quinary structure and its role in governing metabolic pathways.
Purinosome
The purinosome comprises the enzymes responsible for the ten‐step biosynthesis of purine.50 The first evidence for quinary interactions between these enzymes was reported in 1978 when the Benkovic group copurified formyl‐methenyl‐methylenetetrahydrofolate synthetase and glycineamide ribonucleotide transformylase, raising “the possibility that the two proteins may specifically aggregate or be covalently linked in the native state.”51 Two years later, they reported the copurification of glycineamide ribonucleotide transformylase with two other enzymes.52 In both instances, each enzyme exhibited decreased activity upon isolation. The instability of the first intermediate,53 suggestive of a direct transfer between enzymes, and the identification of multifunctional polypeptide chains54 provided further evidence for a multi‐enzymatic complex.
In 2008, fluorescence microscopy revealed compartmentalization of all six enzymes in the cytoplasm of human cells.19 An et al. found that formation of the six‐enzyme complex was dynamic, reversible and depended on purine levels. The Benkovic group coined the term “purinosome” to describe this complex. Its formation is cell‐cycle dependent55 and regulated by G‐protein‐coupled receptors,56 kinases,57 and specific signaling pathways.58 Live cell imaging59, 60, 61 was employed to characterize the spatial organization of the purinosome and revealed that its formation was embedded within the microtubule network.62 Spatial colocalization between the purinosome and mitochondria has since been confirmed.63
Application of the Tango assay64 to characterize protein–protein interactions within the purinosome identified specific interactions between the six enzymes, and suggested that the first three form the “core.”65 The first quantitative measurements of purinosome formation dynamics66 were made using fluorescence recovery after photobleaching.67, 68 Under purine‐rich conditions, each enzyme exhibited its own diffusion coefficient, showing that the enzymes diffuse independently. Under purine‐depleted conditions, the diffusion coefficient had two components. These observations were interpreted as dynamic partitioning of the enzymes: the fast component reflected free diffusion, while the second component implied purinosome formation.66
Respirasome
The respiratory chain is another example of a pathway governed by quinary structure. The chain comprises four complexes (I through IV) in the inner mitochondrial membrane that drive oxidative phosphorylation. The notion that respiratory enzymes form higher order complexes was proposed in 1947 by Keilin.69 The organization of the respiratory chain was described initially by the solid state model, which suggested permanent interactions between complexes.69, 70 This model was supported by the isolation of assemblies of multiple complexes.71, 72, 73 Further analyses suggested the complexes assembled stoichiometrically.74, 75, 76
However, the isolation of independent, functional complexes77, 78, 79 supported the random collision model, in which the individual complexes assemble dynamically.80 This model was supported by reports of independent diffusion of the complexes,80, 81, 82, 83 and reports of higher order assemblies containing various distributions of complexes.84, 85 Ultimately, the plasticity model86 prevailed, which synthesized the two previous models and “envisions the mitochondrial respiratory chain as a combination of free complexes and which are able to adapt to changing conditions.”87, 88 Thus, quinary interactions play a pivotal role in orchestrating the organization and functionality of the respiratory chain.
In 2000, Schägger and Pfeiffer isolated complexes I, III, and IV from mammalian mitochondria and coined the term “respirasome” to describe this 1.7 MDa supercomplex.84 The respirasome (Figure 2) comprises 81 polypeptide chains divided among the three multisubunit respiratory chain complexes and establishes the proton gradient across the inner mitochondrial membrane that is essential for ATP synthesis and energy conversion. Multiple configurations of this supercomplex have been reported.89, 90 The supercomplex I1III2IV1 attracted the most attention.91, 92, 93, 94 Recently, two groups used single particle cryo‐electron microscopy to produce high‐resolution structures of the mammalian respirasome that reveal important information about the spatial arrangements of the individual complexes and the key motifs necessary for higher order assembly.20, 21 Gu et al.20 reported a 5.4 Å resolution structure purified from porcine heart. Letts et al.21 reported structures of both a “tight” and “loose” conformation (5.8 Å and 6.7 Å, respectively) of the ovine respirasome. In the loose conformation, CIV forms well‐defined contacts with CI, whereas in the tight conformation, there are well‐defined contacts between all three complexes. Interconversion between the tight and loose conformations may depend on cardiolipin, which stabilizes CIII and CIV interactions.95 The authors note the absence of scaffold proteins, and demonstrate that the active sites of the complexes are well positioned for substrate channeling.
Figure 2.
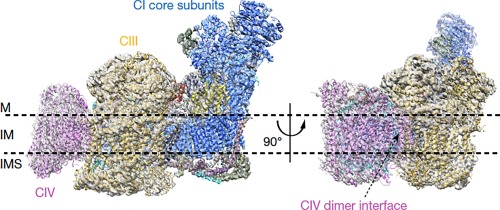
Mammalian respirasome. Core subunits are colored in blue (CI), golden (CIII) and magenta (CIV). M, matrix; IM, inner membrane; IMS, intermembrane space. Reprinted by permission from Macmillan Publishers Ltd.20
Characterizing Quinary Structure
Most protein biochemistry research is carried out at protein concentrations <10 g L−1, yet Escherichia coli cells contain over 300 g L−1 of macromolecules.96 Eukaryotic cells contain a greater variety of macromolecules that participate in more complex cellular processes, and compartmentalization within organelles emphasizes the important of quinary structure.97, 98, 99 In 1984, Clegg asserted “it is no longer reasonable to assume that molecules in cells experience an aqueous cytoplasmic microenvironment comparable to the test tube.”100 Despite early observations that transient chemical interactions modulate protein behavior in cells, interest in quinary structure remained dormant, partially because technology to study proteins in cells did not yet exist.
The dynamic and reversible assembly of the purinosome, TCA cycle metabolon and the respirasome illustrate the intricacy and essential role of quinary structure. But what do we know about the fundamental nature of these interactions? How do they change protein behavior in cells compared to buffer? Next, we answer some of these questions, and discuss some of the questions that remain.
In‐cell NMR spectroscopy
The transient nature of quinary interactions and the challenges associated with studying proteins in living cells has limited fundamental information about the effects of quinary interactions on protein properties. In 2001, Dötsch used heteronuclear nuclear magnetic resonance (NMR) spectroscopy to observe 15N‐enriched‐enriched proteins overexpressed in living Escherichia coli cells,101 asserting “in‐cell NMR spectroscopy will open new avenues of research into protein conformations in their natural environment, as affected by protein‐protein interactions, reversible small molecule binding, and post translation modifications.”
Investigators, however, quickly realized that in‐cell NMR (Figure 3) is limited by the fact that many globular proteins cannot be seen in cells.103 Background resonances from E. coli macromolecules are not to blame; the absence or presence of resonances depends on the degree of protein overexpression and the rotational correlation time of the molecule.104 Disordered proteins, which exhibit fast internal motions, almost always yield high quality in‐cell HSQC spectra.103 Our group constructed a fusion protein comprising a disordered protein, α‐synuclein, and a globular protein, ubiquitin, and compared the in‐cell HSQC spectra.105 The ubiquitin portion failed to produce an in‐cell HSQC spectrum and the only observable crosspeaks belonged to α‐synuclein. However, both sets of crosspeaks were visible in the cell lysate, which is diluted compared to the in‐cell sample. This result demonstrates that the success or failure of in‐cell NMR depends on a molecule's rotational correlation time, which for globular proteins is increased in the cellular environment, resulting in faster transverse relaxation and line broadening. The increased rotational correlation time of folded, globular proteins in cells is attributed to quinary interactions, which increase the effective molecular weight.
Figure 3.
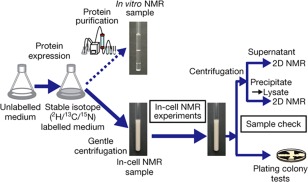
Scheme for in‐cell NMR. Reprinted by permission from Macmillan Publishers Ltd.102
Cytochrome c is an important model protein,106, 107 but does not yield an in‐cell 15N‐1H HSQC spectrum, nor is the spectrum detectable in cell lysates.108 Crowley et al. attributed these observations to interactions between highly basic (pI >10) cytochrome c and the excess of primarily negatively charged macromolecules in the cellular environment. They demonstrated this “stickiness” by subjecting whole cell lysates to size exclusion chromatography and found that cytochrome c eluted at a volume corresponding to over ten times its true molecular weight. Interestingly, a charge reversal on the protein's surface altered its elution profile. Replacing three basic residues with glutamates weakened quinary interactions such that the triple variant was detectable in cell lysates. Similar results were obtained with a GB1 fusion protein.109 This observation suggests that electrostatic interactions are an important component of quinary structure, an idea we discuss later in this review.
Similarly, Gierasch et al. investigated the effects of quinary structure on in‐cell NMR spectra by characterizing the rotational diffusion of three small globular proteins, GB1, NmerA and ubiquitin, in E. coli.110 These proteins are roughly the same size (6.2, 6.9 and 8.5 kDa, respectively), but have different surface properties. GB1 and NmerA could be observed in cells, but as discussed above, crosspeaks from ubiquitin were absent. Despite their similar size, GB1 yielded a significantly higher quality in‐cell 15N‐1H HSQC spectrum than did NmerA. The authors constructed a GB1‐L15‐NmerA fusion protein and, despite equal levels of protein expression, NmerA suffered severe line broadening. Moreover, the in‐cell spectrum of a fusion protein linking two GB1 domains (12.5 kDa) was still of higher quality than that of NmerA. These experiments ruled out protein size as the main contributing factor to the increased rotational correlation time. GB1 is negatively charged (pI 4.2), while both NmerA (pI 7.2) and ubiquitin (pI 7.3) have a net charge of about zero near physiological pH. However, mutational studies revealed that the differences in in‐cell NMR spectral quality are due to the differences in distribution of hydrophobic residues on the surfaces of NmerA and ubiquitin. These results demonstrate that protein surfaces determine the ability to acquire high quality in‐cell NMR spectra and that hydrophobic interactions also play a role in quinary structure.
Nevertheless, in‐cell NMR has provided the first atomic level detail of protein structure in cells. In‐cell NMR was used to determine the three dimensional structure of several proteins in their physiological state.102, 111 Quinary structure is unlikely to affect the tertiary structure of globular proteins because Anfinsen showed the structure of a globular protein represents the lowest free energy state,10 and Richards showed that the inside of a globular protein is almost as efficiently packed as perfectly packed spheres.112
On the other hand, FlgM is a protein that is disordered in solution, but gains structure in cells.113 There has also been much debate about the oligomeric state of α‐synuclein,114 but careful in‐cell NMR and EPR data reveal its physiological state is a disordered monomer.115, 116
Although prokaryotic cells are established systems that enable rapid overexpression of proteins, they lack the breadth of biological activities and post‐translational modifications of eukaryotic cells. In 2006, Selenko et al. injected the B1 domain of protein G (GB1), a model protein, into Xenopus laevis oocytes.117, 118 The utility of in‐cell NMR has also been extended to proteins overexpressed in human cells.119, 120 Majumder et al. used in‐cell NMR to identify key residues involved in quinary interactions for four different proteins in both prokaryotic and eukaryotic cells.121 The key residues comprise hydrophobic and hydrophilic residues on the protein surface, and were highly conserved across species. RNA was also identified as a component of quinary structure. Similarly, Barbieri et al. showed that replacing positively charged residues on the surface of the human protein profilin 1 with neutral or negatively charged residues improved the quality of in‐cell HSQC spectra in both bacterial and human cells.122 They were able to differentiate between residues involved in specific interactions and nonspecific interactions, contributing to the mounting evidence that electrostatic interactions play an important role in forming quinary structure.
19F NMR spectroscopy is another approach for studying protein structure and stability,123, 124 and fluorine was one the first nuclei exploited for in cell NMR.38, 125, 126, 127, 128 Fluorine is rarely found in biology,129, 130 and the 19F nucleus is NMR‐active, extremely sensitive and does not perturb protein structure. Fluorine‐containing amino acids can be incorporated into proteins at a specific site,127 or by feeding E. coli fluorine‐containing precursors, such a fluoroindole.131 Because these strategies incorporate fluorine at one or more specific residues, depending on the protein, spectral analysis is simplified and 19F chemical shifts and line widths provide information on site‐specific structure and dynamics. One dimensional 19F NMR experiments are less time‐consuming than traditional two‐dimensional methods; spectra can be acquired in minutes with protein concentrations as low as 100 μM.128 Although 19F NMR does not offer the breadth of information afforded by heteronuclear multidimensional in‐cell NMR experiments, it is a simple approach for obtaining general information about proteins in cells, including larger proteins that may not afford two‐dimensional in‐cell spectra. Furthermore, 19F NMR can be used to study protein–ligand interactions132, 133 with biomedical applications.134, 135, 136, 137
Quantitative assessments of quinary structure
In 2000, the Fitzgerald and Oas laboratories developed a mass spectrometry based method to measure the stability of unpurified proteins from rates of hydrogen‐deuterium exchange (SUPREX).138 Briefly, they resuspended unpurified protein extracts in deuterium‐containing buffer and used mass spectrometry to measure protein mass as a function of time. The data were fit to an exponential function to obtain the rate of exchange and to estimate protein stability. A year later, Ghaemmaghami and Oas adapted SUPREX to provide the first quantitative comparison of protein stability in cells and in buffered solution.139 They used the SUPREX method to show that the stability of monomeric λ repressor in E. coli is comparable to the stability in vitro, which was a radical idea at that time, because crowding was thought only to stabilize proteins.
In 2014, our lab presented the “quenched lysate method” to quantify protein stability in living E. coli cells (Figure 4).140 Adapted from SUPREX,139 the quenched lysate method pairs amide proton exchange and NMR spectroscopy141, 142 to yield residue level information about protein stability in cells. Poor signal‐to‐noise ratio prevented quantification of exchange directly from in‐cell NMR. Instead, the protein of interest is overexpressed in 15N‐containing media, and the cells are transferred to D2O‐containing buffer after chloramphenicol is added to halt protein expression. Aliquots are removed at discrete time points and hydrogen exchange is quenched while cells are lysed simultaneously. The lysate is transferred to an NMR tube and a 15N‐1H HSQC spectrum is acquired. Samples are collected until the signal decays two complete half‐lives.
Figure 4.
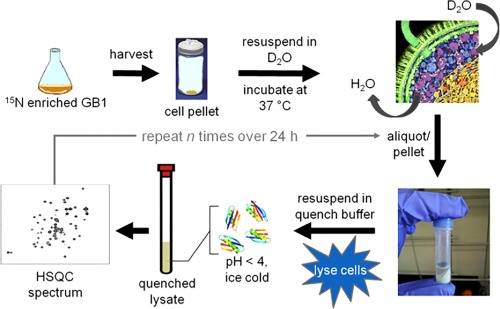
Scheme for quantifying protein stability in cells using the quenched lysate method.140
The quenched lysate method enabled quantification of the stability of several GB1 variants in cells. In buffer, mutations to residue D40, a solvent exposed residue not involved in secondary structure formation, had a negligible effect on GB1 stability. However, as the mutations became increasingly positively charged, GB1 stability decreased (Figure 5).143 These results support observations that surface charge plays an important role in quinary structure.108, 110
Figure 5.
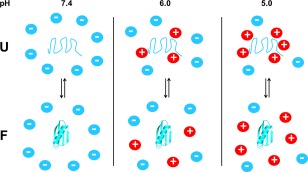
Cartoon depicting the pH‐dependence of quinary interactions. Reprinted with permission from the Journal of the American Chemical Society.145
To explore the role of charge‐charge interactions in quinary structure, we quantified GB1 stability in E. coli cells at different pH values. Using the K10H variant of GB1 to report on intracellular pH and a buffer to control the intracellular pH, we showed that the quality of the in‐cell HSQC spectrum degrades at lower pH values.144 We used the quenched lysate method to quantify GB1 stability at different pH values to show that lowering the pH destabilizes the protein to a greater extent in cells than in buffer.145 At pH 5.0, the protein is destabilized by over 1 kcal/mol in cells compared to buffer at 37°C, providing further evidence of an electrostatic contribution to quinary structure.
The fact that GB1 is not natural to E. coli is also important. The lack of homologs in E. coli means there are no specific interactions to interfere with the proposed studies of how the intracellular environment affects the protein. Simply put, the proposed work defines a path to knowledge about the potential of quinary interactions. The situation is analogous to the early days of site‐directed mutagenesis with its focus on model proteins such as T4 lysozyme,146 GB1,147 eglin c148 and the barnase/barstar complex.149 This classic work delivered vital information about key protein properties, including the propensity of amino acids to form α‐helices and β‐sheets, the importance of packing, and the energetics of protein complex formation. Studies of heterologous systems like GB1 have the same potential to advance both fundamental and practical knowledge about how proteins behave in cells.
Additional methods have been developed to quantify protein thermodynamics in cells. In 2015, Danielsson et al. used in‐cell NMR to quantify the stability of a marginally stable β‐barrel protein (I135A SOD1) in mammalian cells.150 Because crosspeaks are observable for both the folded and unfolded state of this protein, the ratio of crosspeak volumes was used to quantify stability. Their work shows that the protein is destabilized in cells relative to buffer. The temperature‐dependence of the stability shows that the destabilization is enthalpic.
Smith et al.151 used a similar approach to quantify the stability of the N‐terminal SH3 domain of Drosophila signal transduction protein drk (SH3). SH3 has a ΔG°'U of ∼0 kcal mol−1 at 37°C, meaning that the folded and the unfolded states are equally populated at equilibrium. By incorporating 5‐fluoroindole at a single residue, they used 19F NMR to show that the stability of SH3 in cells is comparable to, or less than, its stability in buffer. These results show that attractive quinary interactions offset, and perhaps dominate, the stabilizing effect of hard‐core repulsions.152
Studies of protein folding in living cells using Förster resonance energy transfer experiments have also yielded important results. Gruebele's group showed that phosphoglycerate kinase (PGK) has a more compact structure and experiences accelerated folding rates in the nucleus of U2OS bone tissue cancer cells, compared to buffered solutions.153 In contrast, the cytoplasm slows PGK folding relative to buffer. These results emphasize the differential effects resulting from cellular compartmentalization. These authors also showed that PGK (pI 7.8) is stabilized in cells, while the temperature‐dependence of the folding kinetics remains similar to those in buffer.154 In contrast, a truncation of the Variable major protein‐Like sequence Expressed (VlsE) is destabilized and its folding kinetics are slowed in U2OS cells compared to buffer.155 While PGK and VlsE are similar in size and isoelectric point, the authors suggest “hydrophobic contacts of unfolded VlsE… could stabilize the unfolded state when it sticks to cellular crowders” to explain the different effects of the cellular environment on the stability of the two proteins.
Conclusions and Perspectives
Evidence of quinary structure emerged over half a century ago, but efforts to characterize and understand the fundamental physical chemistry of these transient interactions were undertaken only recently.156 In‐cell NMR spectroscopy is a valuable tool for obtaining both molecular‐level and residue‐level information about proteins in cells. In recent years, we have learned that quinary interactions can impede the acquisition of in‐cell NMR spectra of globular proteins, largely due to electrostatic and hydrophobic interactions between the test protein and the intracellular environment. In‐cell NMR has also been used to identify key surface residues that form quinary interactions. The cellular environment also changes the folding landscape of proteins, and hydrophobic interactions in the unfolded state must also be considered when trying to understand quinary structure. Most importantly, we have learned that the protein surfaces, both in the folded or unfolded state, mediate interactions with the intracellular environment, and that the effects of quinary structure depend on the inherent properties of the protein of interest.
These same ideas concerning the importance of protein surfaces are also applicable to the formulation of biologics. The ability to successfully formulate these life‐saving, protein‐based drugs depends on the lack of what the pharmaceutical industry calls higher order structure157 in highly concentrated solutions. Such structure can result in highly viscous solutions that are impossible to inject. It is known that the surface properties of biologics contribute to viscosity.158
Although textbooks discuss the four levels of protein structure, they now must include a fifth. Quinary structure is responsible for the elegant organization of the dynamic, complex cellular environment and governs the billions of signaling pathways and cascades vital for cell survival. Experiments using purified proteins have formed the basis for our knowledge about protein structure, function and dynamics; however, this information must be put in the context of the physiological environment to fully understand the implications of these characteristics. We believe the key to understanding protein behavior in nature lies at the surface, in the residues that mediate interactions with the rest of the cellular environment to form quinary structure.
Acknowledgments
GJP thanks the Protein Society for the 2016 Carl Brändén Award, his students, postdocs, and colleagues whose efforts made it possible, and the NIH and NSF whose support kept the wolf from the door. The authors thank past and present members of the Pielak group and our other colleagues at UNC, especially Linda Spremulli and Matthew Redinbo for helpful feedback and years of moral support. They also thank Elizabeth Pielak and Peter B. Crowley for comments on the manuscript and Chris A. Pielak for making Figure 1.
References
- 1. Linderstrøm‐Lang KU (1952) Proteins and enzymes: Lane medical lectures. Stanford, CA: Stanford University Publications. [Google Scholar]
- 2. Fischer E (1902) Über die hydrolyse der proteinstoffe. Chem Zeitung 26:939–940. [Google Scholar]
- 3. Hofmeister F (1902) Ueber den bau des Eiweissmoleküla. Naturwissenschaftliche Rundschau 17:529–545. [Google Scholar]
- 4. Pauling L, Corey RB, Branson HR (1951) The structure of proteins: two hydrogen‐bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA 37:235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wrinch D (1939) The structure of the globular proteins. Nature 143:482–483. [Google Scholar]
- 6. Kauzmann W (1956) Structural factors in protein denaturation. J Cell Physiol Suppl 47:113–131. [DOI] [PubMed] [Google Scholar]
- 7. Kauzmann W (1959) Some factors in the interpretation of protein denaturation. Adv Protein Chem 14:1–63. [DOI] [PubMed] [Google Scholar]
- 8. Kauzmann W (1964) The three dimensional structures of proteins. Biophys J 4:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bernal JD (1958) General introduction structure arrangements of macromolecules. Discuss Faraday Soc 25:7–18. [Google Scholar]
- 10. Anfinsen CB (1973) Principles that govern the folding of protein chains. Science 181:223–230. [DOI] [PubMed] [Google Scholar]
- 11. McBrien DCH, Moses V (1968) Compartmentation of the metabolism of lactose, galactose and glucose in Escherichia coli . J Gen Microbiol 51:159–172. [DOI] [PubMed] [Google Scholar]
- 12. Vaïnshteïn BK (1973) Three‐dimensional electron microscopy of biological macromolecules. Physics‐Uspekhi 16:185–206. [PubMed] [Google Scholar]
- 13. Edelstein SJ (1980) Patterns in the quinary structures of proteins. Biophys J 32:347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McConkey EH (1982) Molecular evolution, intracellular organization, and the quinary structure of proteins. Proc Natl Acad Sci USA 79:3236–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gronenborn AM, Filpula DR, Essig NZ, Achari A, Whitlow M, Wingfield PT, Clore GM (1991) A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science 253:657–660. [DOI] [PubMed] [Google Scholar]
- 16. Jee J, Byeon IJ, Louis JM, Gronenborn AM (2008) The point mutation a34f causes dimerization of gb1. Proteins 71:1420–1431. [DOI] [PubMed] [Google Scholar]
- 17. Krebs HA, Johnson WA (1937) Acetopyruvic acid (αγ‐diketovaleric acid) as an intermediate metabolite in animal tissues. Biochem J 31:772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krebs HA, Johnson WA (1937) Metabolism of ketonic acid in animal tissues. Biochem J 31:645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. An S, Kumar R, Sheets ED, Benkovic SJ (2008) Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320:103–106. [DOI] [PubMed] [Google Scholar]
- 20. Gu J, Wu M, Guo R, Yan K, Lei J, Gao N, Yang M (2016) The architecture of the mammalian respirasome. Nature 537:639–643. [DOI] [PubMed] [Google Scholar]
- 21. Letts JA, Fiedorczuk K, Sazanov LA (2016) The architecture of respiratory supercomplexes. Nature 537:644–648. [DOI] [PubMed] [Google Scholar]
- 22. Green DE, Loomis WF, Auerbach VH (1948) Studies of the cyclophorase system: the complete oxidation of pyruvic acid to carbon dioxide and water. J Biol Chem 172:389–404. [PubMed] [Google Scholar]
- 23. Green DE (1951) The cyclophorase complex of enzymes. Biol Rev 26:410–455. [Google Scholar]
- 24. Floridi A (1978) Regulation processes in biological systems. II. Role of compartmentation and of organized multienzyme systems. Acta Vitaminol Enzymol 32:67–109. [PubMed] [Google Scholar]
- 25. veer Reddy GP, Pardee AB (1980) Multienzyme complex for metabolic channeling in mammalian DNA replication. Proc Natl Acad Sci USA 77:3312–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ovádi J (1991) Physiological significance of metabolic channelling. J Theor Biol 152:1–22. [PubMed] [Google Scholar]
- 27. Cascante M, Sorribas A, Canela EI (1994) Enzyme‐enzyme interactions and metabolite channelling: alternative mechanisms and their evolutionary significance. Biochem J 298:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Winkel BSJ (2004) Metabolic channeling in plants. Annu Rev Plant Biol 55:85–107. [DOI] [PubMed] [Google Scholar]
- 29. Hyde C, Ahmed S, Padlan EA, Miles EW, Davies DR (1988) Three‐dimensional structure of the tryptophan synthase α2β2 multienzyme complex from Salmonella typhimurium . J Biol Chem 263:17857–17871. [PubMed] [Google Scholar]
- 30. Mozzarelli A, Peracchi A, Rossi GL, Ahmed SA, Miles EW (1989) Microspectrophotometric studies on single crystals of the tryptophan synthase α2β2 complex demonstrate formation of enzyme‐substrate intermediates. J Biol Chem 264:15774–15780. [PubMed] [Google Scholar]
- 31. Hyde CC, Miles EW (1990) The tryptophan synthase multienzyme complex: exploring structure–function relationships with X‐ray crystallography and mutagenesis. Nat Biotechnol 8:27–32. [DOI] [PubMed] [Google Scholar]
- 32. Srere PA, Mattiasson B, Mosbach K (1973) An immobilized three‐enzyme system: a model for microenvironmental compartmentation in mitochondria. Proc Natl Acad Sci USA 70:2534–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Halper LA, Srere PA (1977) Interaction between citrate synthase and mitochondrial malate dehydrogenase in the presence of polyethylene glycol. Arch Biochem Biophys 184:529–534. [DOI] [PubMed] [Google Scholar]
- 34. Beeckmans S, Kanarek L (1981) Demonstration of physical interactions between consecutive enzymes of the citric acid cycle and of the aspartate‐malate shuttle: a study involving fumarase, malate dehydrogenase, citrate synthase and aspartate aminotransferase. Eur J Biochem 117:527–535. [DOI] [PubMed] [Google Scholar]
- 35. Greksak M, Lopes‐Cardozo M, Van den Bergh SG (1982) Citrate synthesis in intact rat‐liver mitochondria is irreversible. Eur J Biochem 122:423–427. [DOI] [PubMed] [Google Scholar]
- 36. Srere PA (1982) The structure of the mitochondrial inner membrane‐matrix. Trends Biochem Sci 7:375–378. [Google Scholar]
- 37. Barnes SJ, Weitzman PDJ (1986) Organization of citric acid cycle enzymes into a multienzyme cluster. FEBS Lett 201:267–270. [DOI] [PubMed] [Google Scholar]
- 38. Haggie PM, Brindle KM (1999) Mitochondrial citrate synthase is immobilized in vivo . J Biol Chem 274:3941–3945. [DOI] [PubMed] [Google Scholar]
- 39. Srere PA (1985) The metabolon. Trends Biochem Sci 10:109–110. [Google Scholar]
- 40. Laursen T, Borch J, Knudsen C, Bavishi K, Torta F, Martens HJ, Silvestro D, Hatzakis NS, Wenk MR, Dafforn TR, Olsen CE, Motawia MS, Hamberger B, Møller BL, Bassard J‐E (2016) Characterization of a dynamic metabolon producing the defense compound dhurrin in sorghum. Science 354:890–893. [DOI] [PubMed] [Google Scholar]
- 41. Vélot C, Mixon MB, Teige M, Srere PA (1997) Model of a quinary structure between Krebs TCA cycle enzymes: a model for the metabolon. Biochemistry 36:14271–14276. [DOI] [PubMed] [Google Scholar]
- 42. Morgunov I, Srere PA (1998) Interaction between citrate synthase and malate dehydrogenase: substrate channeling of oxaloacetate. J Biol Chem 273:29540–29544. [DOI] [PubMed] [Google Scholar]
- 43. Miles EW, Rhee S, Davies DR (1999) The molecular basis of substrate channeling. J Biol Chem 274:12193–12196. [DOI] [PubMed] [Google Scholar]
- 44. Robinson JB, Inman L, Sumegi B, Srere PA (1987) Further characterization of the Krebs tricarboxylic acid cycle metabolon. J Biol Chem 262:1786–1790. [PubMed] [Google Scholar]
- 45. Elcock AH, McCammon JA (1996) Evidence for electrostatic channeling in a fusion protein of malate dehydrogenase and citrate synthase. Biochemistry 35:12652–12658. [DOI] [PubMed] [Google Scholar]
- 46. Elcock AH, Huber GA, McCammon JA (1997) Electrostatic channeling of substrates between enzyme active sites: comparison of simulation and experiment. Biochemistry 36:16049–16058. [DOI] [PubMed] [Google Scholar]
- 47. Shatalin K, Lebreton S, Rault‐Leonardon M, Vélot C, Srere PA (1999) Electrostatic channeling of oxaloacetate in a fusion protein of porcine citrate synthase and porcine mitochondrial malate dehydrogenase. Biochemistry 38:881–889. [DOI] [PubMed] [Google Scholar]
- 48. Wu F, Minteer S (2015) Krebs cycle metabolon: structural evidence of substrate channeling revealed by cross‐linking and mass spectrometry. Angew Chem Int Ed 54:1851–1854. [DOI] [PubMed] [Google Scholar]
- 49. Bulutoglu B, Garcia KE, Wu F, Minteer SD, Banta S (2016) Direct evidence for metabolon formation and substrate channeling in recombinant TCA cycle enzymes. ACS Chem Biol 11:2847–2853. [DOI] [PubMed] [Google Scholar]
- 50. Buchanan JM (1951) Biosynthesis of the purines. J Cell Physiol Suppl 38:143–171. [DOI] [PubMed] [Google Scholar]
- 51. Caperelli CA, Chettur G, Lin LY, Benkovic SJ (1978) Purification of glycineamide ribonucleotide transformylase. Biochem Biophys Res Commun 82:403–410. [DOI] [PubMed] [Google Scholar]
- 52. Caperelli CA, Benkovic PA, Chettur G, Benkovic SJ (1980) Purification of a complex catalyzing folate cofactor synthesis and transformulation in de novo purine biosynthesis. J Biol Chem 255:1885–1890. [PubMed] [Google Scholar]
- 53. Schendel FJ, Cheng YS, Otvos JD, Wehrli S, Stubbe J (1988) Characterization and chemical properties of phosphoribosylamine, an unstable intermediate in the de novo purine biosynthetic pathway. Biochemistry 27:2614–2623. [DOI] [PubMed] [Google Scholar]
- 54. Henikoff S, Keene MA, Sloan JS, Bleskan J, Hards R, Patterson D (1986) Multiple purine pathway enzymes activites are encoded at a single genetic locus in Drosophila . Proc Natl Acad Sci USA 83:720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chan CY, Zhao H, Pugh RJ, Pedley AM, French J, Jones SA, Zhuang X, Jinnah H, Huang TJ, Benkovic SJ (2015) Purinosome formation as a function of the cell cycle. Proc Natl Acad Sci USA 112:1368–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Verrier F, An S, Ferrie AM, Sun H, Kyoung M, Deng H, Fang Y, Benkovic SJ (2011) GPCRs regulate the assembly of a multienzyme complex for purine biosynthesis. Nat Chem Biol 7:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. An S, Kyoung M, Allen JJ, Shokat KM, Benkovic SJ (2010) Dynamic regulation of a metabolic multi‐enzyme complex by protein kinase CK2. J Biol Chem 285:11093–11099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sundaram A, An S (2015) Investigating the role of 3‐phosphoinositide‐dependent protein kinase 1 in the spatiotemporal regulation of the purinosome. FASEB J 29:573.26. [Google Scholar]
- 59. Huang B, Jones SA, Brandenburg B, Zhuang X (2008) Whole‐cell 3D STORM reveals interactions between cellular structures with nanometer‐scale resolution. Nat Methods 5:1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang B, Wang W, Bates M, Zhuang X (2008) Three‐dimensional super‐resolution imaging by stochastic optical reconstruction microscopy. Science 319:810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rust MJ, Bates M, Zhuang X (2006) Sub‐diffraction‐limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3:793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. An S, Deng Y, Tomsho JW, Minjoung K, Benkovic SJ (2010) Microtubule‐assisted mechanism for functional metabolic macromolecular complex formation. Proc Natl Acad Sci USA 107:12872–12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. French JB, Jones SA, Deng H, Pedley AM, Kim D, Chan CY, Hu H, Pugh RJ, Zhao H, Zhang Y, Huang TJ, Fang Y, Zhuang X, Benkovic SJ (2016) Spatial colocalization and functional link of purinosomes and mitochondria. Science 351:733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Alex R, Lee KJ (2008) The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci USA 105:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Deng Y, Gam J, French JB, Zhao H, An S, Benkovic SJ (2012) Mapping protein–protein proximity in the purinosome. J Biol Chem 287:36201–36207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kyoung M, Russell SJ, Kohnhorst CL, Esemoto NN, An S (2015) Dynamic architecture of the purinosome involved in human de novo purine biosynthesis. Biochemistry 54:870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stavreva DA, McNally JG (2004) Fluorescence recovery after photobleaching (FRAP) methods for visualizing protein dynamics in living mammalian cell nuclei. Methods Enzymol 375:443–455. [DOI] [PubMed] [Google Scholar]
- 68. Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW (1976) Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J 16:1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Keilin D, Hartree EF (1947) Activity of the cytochrome system in heart muscle preparations. Biochem J 41:500–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chance B, Estabrook RW, Lee C (1963) Electron transport in the oxysome. Science 140:379–380. [DOI] [PubMed] [Google Scholar]
- 71. Hatefi Y, Rieske JS (1967) The preparation and properties of DPNH‐cytochrome c reductase (complex I‐III of the respiratory chain). Methods Enzymol 10:225–231. [Google Scholar]
- 72. Tisdale HD (1967) Preparation and properties of succinic—cytochrome c reductase (complex II–III). Methods Enzymol 10:213–215. [Google Scholar]
- 73. Blair PV (1967) Preparation and properties of repeating units of mitochondrial electron transfer. Methods Enzymol 10:208–212. [Google Scholar]
- 74. Fowler L, Hatefi Y (1961) Reconstitution of the electron transport system III. Reconstitution of DPNH oxidase, succinic oxidase, and DPNH, succinic oxidase. Biochem Biophys Res Commun 5:203–208. [DOI] [PubMed] [Google Scholar]
- 75. Hatefi Y, Haavik AG, Fowler LR, Griffiths DE (1962) Studies on the electron transfer system: XIII. Reconstitution of the electron transfer systems. J Biol Chem 237:2661–2669. [PubMed] [Google Scholar]
- 76. Ragan CI, Heron C (1978) The interaction between mitochondrial NADH‐ubiquinone oxidoreductase and ubiquinol‐cytochrome c oxidoreductase. Evidence for stoichiometric association. Biochem J 174:783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hatefi Y, Haavik A, Griffiths D (1961) Reconstitution of the electron transport system: I. Preparation and properties of the interacting enzymes complexes. Biochem Biophys Res Commun 4:441–446. [DOI] [PubMed] [Google Scholar]
- 78. Hatefi Y, Haavik AG, Griffiths DE (1962) Studies on the electron transfer system: XI. Preparation and properties of mitochondrial DPNH‐coenzyme Q reductase. J Biol Chem 237:1676–1680. [PubMed] [Google Scholar]
- 79. Hatefi Y, Rieske JS (1967) Preparation and properties of DPNH‐coenzyme Q reductase (complex I of the respiratory chain). Methods Enzymol 10:235–239. [Google Scholar]
- 80. Hackenbrock CR, Chazotte B, Gupte SS (1986) The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J Bioenerg Biomembr 18:331–368. [DOI] [PubMed] [Google Scholar]
- 81. Gupte S, Wu E‐S, Hoechli L, Hoechli M, Jacobson K, Sowers AE, Hackenbrock CR (1984) Relationship between lateral diffusion, collision frequency, and electron transfer of mitochondrial inner membrane oxidation‐reduction components. Proc Natl Acad Sci USA 81:2606–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gupte S, Hackenbrock CR (1988) Multidimensional diffusion modes and collision frequencies of cytochrome c and its redox partners. J Biol Chem 263:5241–5247. [PubMed] [Google Scholar]
- 83. Hochman J, Ferguson‐Miller S, Schindler M (1985) Mobility in the mitochondrial electron transport chain. Biochemistry 24:2509–2516. [DOI] [PubMed] [Google Scholar]
- 84. Schägger H, Pfeiffer K (2000) Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 19:1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schägger H, Pfeiffer K (2001) The ratio of oxidative phosphorylation complexes I–V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J Biol Chem 276:37861–37867. [DOI] [PubMed] [Google Scholar]
- 86. Acín‐Pérez R, Fernández‐Silva P, Peleato ML, Pérez‐Martos A, Enriquez JA (2008) Respiratory active mitochondrial supercomplexes. Mol Cell 32:529–539. [DOI] [PubMed] [Google Scholar]
- 87. Porras CA, Bai Y (2015) Respiratory supercomplexes: plasticity and implications. Front Biosci 20:621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Acín‐Pérez R, Enriquez JA (2014) The function of the respiratory supercomplexes: the plasticity model. Biochim Biophys Acta 1837:444–450. [DOI] [PubMed] [Google Scholar]
- 89. Wittig I, Carrozzo R, Santorelli FM, Schägger H (2006) Supercomplexes and subcomplexes of mitochondrial oxidative phosphorylation. Biochim Biophys Acta 1757:1066–1072. [DOI] [PubMed] [Google Scholar]
- 90. Bultema JB, Braun H‐P, Boekema EJ, Kouřil R (2009) Megacomplex organization of the oxidative phosphorylation system by structural analysis of respiratory supercomplexes from potato. Biochim Biophys Acta 1787:60–67. [DOI] [PubMed] [Google Scholar]
- 91. Althoff T, Mills DJ, Popot J‐L, Kühlbrandt W (2011) Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1 . EMBO J 30:4652–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schäfer E, Dencher NA, Vonck J, Parcej DN (2007) Three‐dimensional structure of the respiratory chain supercomplex I1III2IV1 from bovine heart mitochondria. Biochemistry 46:12579–12585. [DOI] [PubMed] [Google Scholar]
- 93. Dudkina NV, Kudryashev M, Stahlberg H, Boekema EJ (2011) Interaction of complexes I, III, and IV within the bovine respirasome by single particle cryoelectron tomography. Proc Natl Acad Sci USA 108:15196–15200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Schafer E, Seelert H, Reifschneider NH, Krause F, Dencher NA, Vonck J (2006) Architecture of active mammalian respiratory chain supercomplexes. J Biol Chem 281:15370–15375. [DOI] [PubMed] [Google Scholar]
- 95. Mileykovskaya E, Dowhan W (2014) Cardiolipin‐dependent formation of mitochondrial respiratory supercomplexes. Chem Phys Lipids 179:42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zimmerman SB, Trach SO (1991) Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli . J Mol Biol 222:599–620. [DOI] [PubMed] [Google Scholar]
- 97. Conlon I, Raff M (2003) Differences in the way a mammalian cell and yeast cells coordinate cell growth and cell‐cycle progression. J Biol 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zeskind BJ, Jordan CD, Timp W, Trapani L, Waller G, Horodincu V, Ehrlich DJ, Matsudaira P (2007) Nucleic acid and protein mass mapping by live‐cell deep‐ultraviolet microscopy. Nat Methods 4:567–569. [DOI] [PubMed] [Google Scholar]
- 99. Cheung MC, LaCroix R, McKenna BK, Liu L, Winkelman J, Ehrlich DJ (2013) Intracellular protein and nucleic acid measured in eight cell types using deep‐ultraviolet mass mapping. Cytometry A 83:540–551. [DOI] [PubMed] [Google Scholar]
- 100. Clegg JS (1984) Properties and metabolism of the aqueous cytoplasm and its boundaries. Am J Physiol 246:133–151. [DOI] [PubMed] [Google Scholar]
- 101. Serber Z, Keatinge‐Clay A, Ledwidge R, Kelly A, Miller S, Dötsch V (2001) High‐resolution macromolecular NMR spectroscopy in living cells. J Am Chem Soc 123:2446–2447. [DOI] [PubMed] [Google Scholar]
- 102. Sakakibara D, Sasaki A, Ikeya T, Hamatsu J, Hanashima T, Mishima M, Yoshimasu M, Hayashi N, Mikawa T, Walchli M, Smith BO, Shirakawa M, Guntert P, Ito Y (2009) Protein structure determination in living cells by in‐cell NMR spectroscopy. Nature 458:102–105. [DOI] [PubMed] [Google Scholar]
- 103. Li C, Charlton LM, Lakkavaram A, Seagle C, Wang G, Young GB, Macdonald JM, Pielak GJ (2008) Differential dynamical effects of macromolecular crowding on an intrinsically disordered protein and a globular protein: implications for in‐cell NMR spectroscopy. J Am Chem Soc 130:6310–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Serber Z, Ledwidge R, Miller SM, Dötsch V (2001) Evaluation of parameters critical to observing proteins inside living Escherichia coli by in‐cell NMR spectroscopy. J Am Chem Soc 123:8895–8901. [DOI] [PubMed] [Google Scholar]
- 105. Barnes CO, Monteith WB, Pielak GJ (2011) Internal and global protein motion assessed with a fusion construct and in‐cell NMR spectroscopy. ChemBioChem 12:390–391. [DOI] [PubMed] [Google Scholar]
- 106. Bertini I, Cavallaro G, Rosato A (2006) Cytochrome c: occurrence and functions. Chem Rev 106:90–115. [DOI] [PubMed] [Google Scholar]
- 107. Pielak G, Auld D, Betz S, Hilgen‐Willis S, Garcia L. Nuclear magnetic resonance studies of class I cytochrome c In: Scott R, Mauk A, Eds. (1996) Cytochrome c: a multidisciplinary approach. Sausalito: University Science Books, pp 203–284. [Google Scholar]
- 108. Crowley PB, Chow E, Papkovskaia T (2011) Protein interactions in the Escherichia coli cytosol: an impediment to in‐cell NMR spectroscopy. ChemBioChem 12:1043–1048. [DOI] [PubMed] [Google Scholar]
- 109. Kyne C, Ruhle B, Gautier VW, Crowley PB (2015) Specific ion effects on macromolecular interactions in Escherichia coli extracts. Protein Sci 24:310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang Q, Zhuravleva A, Gierasch LM (2011) Exploring weak, transient protein–protein interactions in crowded in vivo environments by in‐cell nuclear magnetic resonance spectroscopy. Biochemistry 50:9225–9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Reardon PN, Spicer LD (2005) Multidimensional NMR spectroscopy for protein characterization and assignment inside cells. J Am Chem Soc 127:10848–10849. [DOI] [PubMed] [Google Scholar]
- 112. Richards FM (1977) Areas, volumes, packing and protein structure. Annu Rev Biophys Bioeng 6:151–176. [DOI] [PubMed] [Google Scholar]
- 113. Dedmon MM, Patel CN, Young GB, Pielak GJ (2002) FlgM gains structure in living cells. Proc Natl Acad Sci USA 99:12681–12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Alderson TR, Bax A (2016) Parkinson's disease: disorder in the court. Nature 530:38–39. [DOI] [PubMed] [Google Scholar]
- 115. Binolfi A, Theillet FX, Selenko P (2012) Bacterial in‐cell NMR of human alpha‐synuclein: a disordered monomer by nature? Biochem Soc Trans 40:950–954. [DOI] [PubMed] [Google Scholar]
- 116. Theillet F, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P (2016) Structural disorder of monomeric α‐synuclein persists in mammalian cells. Nature 530:45–50. [DOI] [PubMed] [Google Scholar]
- 117. Serber Z, Selenko P, Hansel R, Reckel S, Lohr F, Ferrell JE Jr., Wagner G, Dötsch V (2006) Investigating macromolecules inside cultured and injected cells by in‐cell NMR spectroscopy. Nat Protocols 1:2701–2709. [DOI] [PubMed] [Google Scholar]
- 118. Selenko P, Serber Z, Gadea B, Ruderman J, Wagner G (2006) Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proc Natl Acad Sci USA 103:11904–11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Barbieri L, Luchinat E, Banci L (2016) Characterization of proteins by in‐cell NMR spectroscopy in cultured mammalian cells. Nat Protocols 11:1101–1111. [DOI] [PubMed] [Google Scholar]
- 120. Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, Takeuchi T, Futaki S, Ito Y, Hiroaki H, Shirakawa M (2009) High‐resolution multi‐dimensional NMR spectroscopy of proteins in human cells. Nature 458:106–109. [DOI] [PubMed] [Google Scholar]
- 121. Majumder S, Xue J, DeMott CM, Reverdatto S, Burz DS, Shekhtman A (2015) Probing protein quinary interactions by in‐cell nuclear magnetic resonance spectroscopy. Biochemistry 54:2727–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Barbieri L, Luchinat E, Banci L (2015) Protein interaction patterns in different cellular environments are revealed by in‐cell NMR. Sci Rep 5:14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gerig J (1994) Fluorine NMR of proteins. Prog Nucl Magn Reson Spectrosc 26:293–370. [Google Scholar]
- 124. Danielson MA, Falke JJ (1996) Use of 19F NMR to probe protein structure and conformational changes. Annu Rev Biophys Biomol Struct 25:163–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Williams SP, Fulton AM, Brindle KM (1993) Estimation of the intracellular free ADP concentration by 19F NMR studies of fluorine‐labeled yeast phosphoglycerate kinase in vivo . Biochemistry 32:4895–4902. [DOI] [PubMed] [Google Scholar]
- 126. Williams SP, Haggie PM, Brindle KM (1997) 19F NMR measurements of the rotational mobility of protein in vivo . Biophys J 72:490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jackson JC, Hammill JT, Mehl RA (2007) Site‐specific incorporation of a 19F‐amino acid into proteins as an NMR probe for characterizing protein structure and reactivity. J Am Chem Soc 129:1160–1166. [DOI] [PubMed] [Google Scholar]
- 128. Li C, Wang GF, Wang Y, Creager‐Allen R, Lutz EA, Scronce H, Slade KM, Ruf RA, Mehl RA, Pielak GJ (2010) Protein 19F NMR in Escherichia coli . J Am Chem Soc 132:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Harper DB, O'Hagan D (1994) The fluorinated natural products. Nat Prod Rep 11:123–133. [DOI] [PubMed] [Google Scholar]
- 130. O'Hagan DB, Harper D (1999) Fluorine‐containing natural products. J Fluorine Chem 100:127–133. [Google Scholar]
- 131. Crowley PB, Kyne C, Monteith WB (2012) Simple and inexpensive incorporation of 19F‐tryptophan for protein NMR spectroscopy. Chem Commun 48:10681–10683. [DOI] [PubMed] [Google Scholar]
- 132. Entress RMH, Dancer RJ, O'Brien DP, Try AC, Cooper MA, Williams DH (1998) 19F NMR in the measurement of binding affinities of chloroeremomycin to model bacterial cell‐wall surfaces that mimic VanA and VanB resistance. Chem Biol 5:329–337. [DOI] [PubMed] [Google Scholar]
- 133. Richards KL, Rowe ML, Hudson PB, Williamson RA, Howard MJ (2016) Combined ligand‐observe 19F and protein‐observe 15N,1H‐HSQC NMR suggests phenylalanine as the key δ‐somatostatin residue recognized by human protein disulfide isomerase. Sci Rep 6:19518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gee CT, Arntson KE, Urick AK, Mishra NK, Hawk LM, Wisniewski AJ, Pomerantz WC (2016) Protein‐observed 19F‐NMR for fragment screening, affinity quantification and druggability assessment. Nat Protocols 11:1414–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Jordan JB, Whittington DA, Bartberger MD, Sickmier EA, Chen K, Cheng Y, Judd T (2016) Fragment‐linking approach using 19F NMR spectroscopy to obtain highly potent and selective inhibitors of β‐secretase. J Med Chem 59:3732–3749. [DOI] [PubMed] [Google Scholar]
- 136. Norton RS, Leung EW, Chandrashekaran IR, MacRaild CA (2016) Applications of 19F NMR in fragment‐based drug discovery. Molecules 21:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gee CT, Koleski EJ, Pomerantz WCK (2015) Fragment screening and druggability assessment for the CBP/p300 KIX domain through protein‐observed 19F NMR spectroscopy. Angew Chem Int Ed 54:3735–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ghaemmaghami S, Fitzgerald MC, Oas TG (2000) A quantitiative, high‐throughput screen for protein stability. Proc Natl Acad Sci USA 97:8296–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ghaemmaghami S, Oas TG (2001) Quantitative protein stability measurement in vivo . Nat Struct Biol 8:879–882. [DOI] [PubMed] [Google Scholar]
- 140. Monteith WB, Pielak GJ (2014) Residue level quantification of protein stability in living cells. Proc Natl Acad Sci USA 111:11336–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Englander SW, Kallenbach NR (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys 16:521–655. [DOI] [PubMed] [Google Scholar]
- 142. Miklos AC, Li C, Pielak GJ (2009) Using NMR‐detected backbone amide 1H exchange to assess macromolecular crowding effects on globular‐protein stability. Methods Enzymol 466:1–16. [DOI] [PubMed] [Google Scholar]
- 143. Monteith WB, Cohen RD, Smith AE, Guzman‐Cisneros E, Pielak GJ (2015) Quinary structure modules protein stability in cells. Proc Natl Acad Sci USA 112:1739–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Cohen RD, Guseman AJ, Pielak GJ (2015) Intracellular pH modulates quinary structure. Protein Sci 24:1748–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cohen RD, Pielak GJ (2016) Electrostatic contributions to protein quinary structure. J Am Chem Soc 138:13139–13142. [DOI] [PubMed] [Google Scholar]
- 146. Matthews BW (1995) Studies on protein stability with T4 lysozyme. Adv Protein Chem 46:249–278. [DOI] [PubMed] [Google Scholar]
- 147. Smith CK, Regan L (1995) Guidelines for protein design: the energetics of β sheet side chain interactions. Science 270:980. [DOI] [PubMed] [Google Scholar]
- 148. Lahr SJ, Broadwater A, Carter CW Jr., Collier ML, Hensley L, Waldner JL, Pielak GJ, Edgell MH (1999) Patterned library analysis: a method for the quantitative assessment of hypotheses concerning the determinants of protein structure. Proc Natl Acad Sci USA 96:14860–14865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Schreiber G, Fersht AR (1995) Energetics of protein‐protein interactions: analysis of the Barnase–Barstar interface by single mutations and double mutant cycles. J Mol Biol 248:478–486. [DOI] [PubMed] [Google Scholar]
- 150. Danielsson J, Mu X, Lang L, Wang H, Binolfi A, Theillet FX, Bekei B, Logan DT, Selenko P, Wennerstrom H, Oliveberg M (2015) Thermodynamics of protein destabilization in live cells. Proc Natl Acad Sci USA 112:12402–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Smith AE, Zhou LZ, Gorensek AH, Senske M, Pielak GJ (2016) In‐cell thermodynamics and a new role for protein surfaces. Proc Natl Acad Sci USA 113:1725–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Sarkar M, Li C, Pielak GJ (2013) Soft interactions and crowding. Biophys Rev 5:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Dhar A, Girdhar K, Singh D, Gelman H, Ebbinghaus S, Gruebele M (2011) Protein stability and folding kinetics in the nucleus and endoplasmic reticulum of eucaryotic cells. Biophys J 101:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Guo M, Xu Y, Gruebele M (2012) Temperature dependence of protein folding kinetics in living cells. Proc Natl Acad Sci USA 109:17863–17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Guzman I, Gelman H, Tai J, Gruebele M (2014) The extracellular protein VlsE is destabilized inside cells. J Mol Biol 426:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kyne C, Crowley PB (2016) Grasping the nature of the cell interior: From physiological chemistry to chemical biology. The FEBS Journal 283:3016–3028. [DOI] [PubMed] [Google Scholar]
- 157. Chen K, Long DS, Lute SC, Levy MJ, Brorson KA, Keire DA (2016) Simple NMR methods for evaluating higher order structures of monoclonal antibody therapeutics with quinary structure. J Pharm Biomed Anal 128:398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Shire SJ (2009) Formulation and manufacturability of biologics. Curr Opin Biotechnol 20:708–714. [DOI] [PubMed] [Google Scholar]