

Abstract
The tumor‐suppressor protein BRCA1 works with BARD1 to catalyze the transfer of ubiquitin onto protein substrates. The N‐terminal regions of BRCA1 and BARD1 that contain their RING domains are responsible for dimerization and ubiquitin ligase activity. This activity is a common feature among hundreds of human RING domain‐containing proteins. RING domains bind and activate E2 ubiquitin‐conjugating enzymes to promote ubiquitin transfer to substrates. We show that the identity of residues at specific positions in the RING domain can tune activity levels up or down. We report substitutions that create a structurally intact BRCA1/BARD1 heterodimer that is inactive in vitro with all E2 enzymes. Other substitutions in BRCA1 or BARD1 RING domains result in hyperactivity, revealing that both proteins have evolved attenuated activity. Loss of attenuation results in decreased product specificity, providing a rationale for why nature has tuned BRCA1 activity. The ability to tune BRCA1 provides powerful tools for understanding its biological functions and provides a basis to assess mechanisms for rescuing the activity of cancer‐associated variations. Beyond the applicability to BRCA1, we show the identity of residues at tuning positions that can be used to predict and modulate the activity of an unrelated RING E3 ligase. These findings provide valuable insights into understanding the mechanism and function of RING E3 ligases like BRCA1.
Keywords: BRCA1, BARD1, RING domain, ubiquitin, ubiquitin E3 ligase
Introduction
Evidence for the existence of a gene involved in breast cancer susceptibility was first reported in 1990.1 Four years later, the gene dubbed BRCA1 (Breast Cancer‐1) was identified by positional cloning,2 and cloned by several groups.3 Unfortunately, the sequence of BRCA1 provided little insight into its function, as there was only a single previously recognized domain within its 1863‐residue sequence. The domain, called “RING” for Really Interesting New Gene, had been defined in 1991 as a domain that was likely to bind zinc ions based on its eight conserved Cys and/or His residues,4 but a biochemical function had yet to be defined. Nevertheless, the existence of missense mutations in the BRCA1 RING domain identified in families with high breast cancer risk motivated us to determine the three‐dimensional structure of the N‐terminal (RING‐containing) region of BRCA1.
An essential protein partner for BRCA1, BARD1 (BRCA1‐Associated RING Domain protein‐1), was identified through a yeast two‐hybrid screen as a stoichiometric binding partner for BRCA1.5 Like BRCA1, BARD1 has a RING domain at its N‐terminal end and this region is responsible for its tight association with BRCA1. Although each RING domain contains ∼60 residues, the structural units defined by limited proteolysis encompass the first ∼100 residues of each protein.6 Therefore, we determined the structure of the heterodimeric complex formed between the first ∼110 residues of BRCA1 and BARD1 by NMR [Fig. 1(A)].7 The structure revealed that the two proteins do not interact directly through their RING domains, as had been expected, but rather through a four‐helix bundle formed by BRCA1 and BARD1 helices that flank each RING domain [Fig. 1(A)]. Subsequent structures of other heterodimeric RINGs, as well as homodimeric complexes, are highly similar in architecture.8, 9 As we completed structural analysis of the BRCA1/BARD1 heterodimer, it was discovered that RING domains, including the BRCA1 RING domain, function as ubiquitin E3 ligases.10 Formation of a BRCA1/BARD1 heterodimer exhibits substantially greater E3 ligase activity in vitro than BRCA1 alone.11 Surprisingly, though both BRCA1 and BARD1 have RING domains, NMR binding studies showed that only BRCA1 interacts with the E2.12 These studies provided a biochemical function for BRCA1's RING. Importantly, all of the known cancer‐associated missense mutations in the N‐terminal region of BRCA1 yield ligase‐dead forms of BRCA1.12 With these structural and biochemical insights in hand, we sought to further characterize BRCA1 interactions with ubiquitin machinery.
Figure 1.
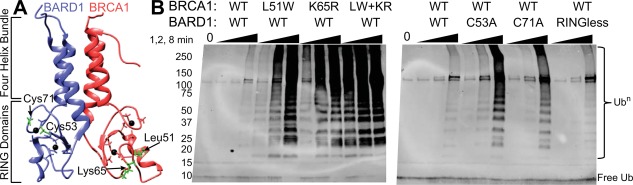
Hyperactive mutants of BRCA1 and BARD1. (A) Location of the RING domain hyperactive mutations are shown as green sticks on the BRCA1/BARD1 lowest energy solution structure (pdb ID 1JM7). Zinc coordinating residues are shown as sticks. (B) Western blot for HA tag on ubiquitin reveals the ubiquitin chain‐building activity of wild‐type (WT), and BRCA1 mutants: L51W, K65R, L51W/K65R (labeled LW + KR) (left panel) and BARD1 mutants: C53A, C71A, RINGless BARD1 (right panel). Time points were taken at 1, 2, and 8 min after ATP addition. Assay component concentrations were as follows: 2.5 μM E1, 5 μM Ube2d3, 5 μM E3, 20 μM HA‐Ub, 5 mM ATP/Mg.
The covalent attachment of ubiquitin (Ub) to a protein requires the activity of three enzymes: an E1 Ub‐activating enzyme, an E2 Ub‐conjugating enzyme and an E3 Ub ligase.13 E3 ligases that contain a RING‐type domain comprise the largest family of E3s, with over 600 members in the human proteome.14 Though the original biochemical assays using BRCA1 as an E3 included the human E2 Ube2d, this E2 tends to be nondiscriminating in terms of the E3s with which it works in vitro. Therefore, we tested whether any of the other ∼36 human E2s could work with BRCA1/BARD1. Under the presumption that an E2 must bind to an E3 to activate Ub transfer, we devised a yeast two‐hybrid strategy to identify E2s that bind to the BRCA1/BARD1 RING heterodimer.15 To our surprise, not only did the three isozymes of Ube2d bind, but we found a total of ten E2s that bind to BRCA1/BARD1. Of these ten E2s, nine are active with BRCA1/BARD1 in in vitro auto‐ubiquitylation assays. Furthermore, the type of products generated (mono‐ubiquitylated, or specific types of poly‐Ub chains) depended on the E2 with which BRCA1/BARD1 was paired. This revelation opened the possibility that different E2/BRCA1 pairs might work together to modify different substrates or target the same substrates with different types of modifications, leading to different cellular outcomes.
Through structural and biochemical studies from numerous labs, a general mechanism for how RING‐type E3 ligases activate E2∼Ub conjugates to transfer Ub to lysine residues has been defined.16 A surface on a RING domain serves as the binding site for an E2.12, 17, 18, 19, 20, 21, 22 If the E2 carries activated Ub, binding to the RING promotes a “closed” conformation of the E2∼Ub where the Ub makes additional noncovalent contacts with the E2.23, 24, 25, 26, 27 The closed conformation is more reactive towards nucleophilic attack by amino groups, thereby activating the E2 to transfer its Ub to a lysine sidechain. Most RING‐type E3s use a conserved residue, referred to as the “linchpin,” to activate E2∼Ub. The linchpin residue is usually an Arg or Lys whose side chain interacts with hydrogen bond acceptor groups on both the E2 and the C‐terminal tail of Ub, promoting the closed state.23, 24, 25, 26, 27 Though variations are emerging as more RING‐type E3s are studied in detail, this main theme appears to be quite general.
We have sought to use these insights to discover ways to modulate BRCA1/BARD1 activity and to test our understanding of its mechanism. Although only the RING of BRCA1 interacts directly with an E2, we report here that mutations in either the BRCA1 RING or the BARD1 RING are capable of increasing ligase activity. We use these hyperactive BRCA1 mutants to demonstrate the importance of native BRCA1/BARD1's finely tuned activity. In addition, these studies identify possible avenues for rescuing the activity of cancer‐associated BRCA1 variants and modulating the activity of other RING E3 ligases.
Results
BRCA1 and BARD1 RING domains have evolved attenuated activity
To better understand how the BRCA1/BARD1 heterodimer activates transfer of Ub, we sought to define what is required to destroy activity and what is required to enhance it. In the former case, well‐known cancer‐associated mutations in the BRCA1 RING domain are profoundly dead as ligases in vitro.12 However, each of these mutations substitutes a Cys residue that is responsible for binding Zn2+ and, therefore, critical for structural integrity of the RING domain.28 On the basis of our original characterization of the binding of the human E2, Ube2d3, to BRCA1/BARD1, we designed a mutant BRCA1 (BRCA1‐I26A) that selectively disrupts this interaction, but maintains the structure of the BRCA1/BARD1 heterodimer.12 This mutation has since become the standard “ligase‐dead” mutant of BRCA1 for studies both in vitro and in vivo.29, 30, 31, 32 However, our subsequent discovery that eight other human E2s can also function with BRCA1/BARD1 reopened the question as to how to make a BRCA1/BARD1 heterodimer that is truly ligase‐dead with all E2s. We found that three mutations are required to completely eliminate BRCA1 activity without disrupting its structure: I26A, L63A, and the allosteric linchpin residue K65A. Unlike the I26A mutation alone, this combination of three mutations eliminates detectable activity with all of the E2s shown to work with BRCA1 (Supporting Information Fig. S1). Future studies using this structurally‐intact but truly‐dead triple‐mutant BRCA1 may clear up conflicting reports about the importance of ligase activity in vivo.32, 33
Building on what we learned about how BRCA1/BARD1 binds to and activates E2s, coupled with deep mutational screening of BRCA1 activity, we have been able to generate new gain‐of‐function BRCA1 mutants with enhanced E3 ligase activity.12, 27, 34, 35 The mutational screen identified several positions of BRCA1 that increase E3 ligase activity upon mutation.34 The two most activating mutations were L51W and K65R. We purified recombinant RING domains containing these mutations and confirmed that indeed they have significantly enhanced ability to form ubiquitin chains in vitro when compared to wild‐type BRCA1/BARD1 [Fig. 1(B)]. Previous NMR studies demonstrated that L51 is located in the E2‐binding interface,12 while K65 is the allosteric linchpin residue that allows for activation of the E2∼Ub conjugate27 [positions shown in Fig. 1(A)]. Therefore, one would predict that these two mutations enhance BRCA1 activity through different mechanisms: altering E2∼Ub conjugate binding in one case and allosteric activation in the other. Consistent with this notion, ubiquitylation assays show the mutations have an additive effect [Fig. 1(B)]. The double mutant L51W/K65R BRCA1 is much more active than either of the individual activating mutants.
Although BRCA1 must dimerize with BARD1 via helices adjacent to their RING domains to become an efficient E3 ligase, little is known about the role of the BARD1 subunit in Ub transfer.11 NMR studies have shown that while BARD1 contains a RING domain that is structurally similar to BRCA1, it does not bind directly to an E2.7, 12 To ascertain the role that the BARD1 RING domain plays in E3 ligase function we created constructs in which a zinc‐coordinating cysteine in the RING domain was mutated to Ala (C53A or C71A). Analogous variants in the BRCA1 RING domain retain their ability to bind zinc, but have significant structural perturbations and loss of E3 ligase function.7, 28 To our surprise, we found that BRCA1/BARD1 with C53A‐ or C71A‐BARD1 mutations have increased E3 ligase activity [Fig. 1(B)]. This suggests that the BARD1 RING domain may serve to attenuate BRCA1 ligase activity. To further investigate this possibility, we designed a “RINGless” BARD1 construct where the RING domain of BARD1 is replaced with a five‐residue linker to directly connect the helices that normally flank the RING domain. Remarkably, even without the RING domain, the BARD1 helices retain the ability to bind and promote BRCA1 E3 ligase activity to levels similar to the wild‐type enzyme [Fig. 1(B)]. These findings along with those from a recent report indicating an important role for a BARD1 RING residue in ligase activity36 highlight the need for further investigation into the role of BARD1, the under‐appreciated half of the BRCA1/BARD1 heterodimeric E3.
BRCA1 activity is tuned for target specificity
BRCA1 has been shown to function as an E3 ligase for nine different human E2s.15 Because BRCA1 activates an E2∼Ub conjugate to transfer Ub directly to the substrate, the product largely depends on the E2. For example, the E2 Ube2w mono‐ubiquitylates the N‐terminus of target substrates, while the E2 Ubc13/Ube2v2 adds Ub to Lys 63 of another Ub molecule to build K63‐linked chains.37, 38 Auto‐ubiquitylation of BRCA1 provides a convenient assay for monitoring differences in activity and product formation. Figure 2(A) shows Ube2e1 and Ube2w both add one Ub to BRCA1, as evidenced by the band that appears at ∼50kDa in the gel. In contrast, Ubc13 and Ube2k can only modify BRCA1 if used in conjunction with Ube2w. Ube2w adds the first Ub to prime the system, and the Ubc13 or Ube2k can then build chains on that Ub [Fig. 2(A)]. When the hyperactive BRCA1 is used in the same assays, there is a pronounced change in the observed products. The mono‐ubiquitylating E2 Ube2e1 now produces high molecular weight products, signifying attachment of multiple Ub moieties. Ube2w primarily mono‐ubiquitylates hyperactive BRCA1, but higher molecular weight products are also observed. Unlike with wild‐type BRCA1, both Ubc13 and Ube2k now modify hyperactive BRCA1 with high molecular weight products in the absence of a priming E2 [red box in Fig. 2(A)]. These data demonstrate that the hyperactive BRCA1 mutations lead to a loss of E2 product specificity typically catalyzed by wild‐type BRCA1.
Figure 2.
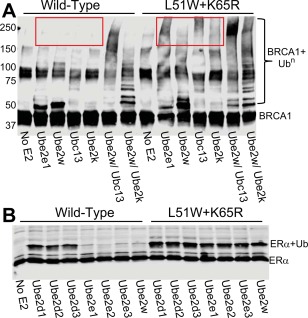
Hyperactive BRCA1 mutants mask specificity of the E2. (A) Western blot for the FLAG tag present on BRCA1 shows auto‐ubiquitylation activity after 1 h with multiple E2 enzymes (listed below lanes). Where Ubc13 is listed, Ube2v2 is also present. Red boxes highlight key differences in E2 specificity with the hyperactive BRCA1 E3 ligase (L51W + K65R) compared to wild‐type. Mono‐Ub‐BRCA1 appears as a band that runs with an apparent MW of ∼50 kDa on the gel (just above the band for unmodified BRCA1). (B) Western blot for the V5 tag present on the ERα LBD in an ubiquitylation assay after 1 h using the BRCA1 construct listed above the lanes and the E2 enzymes listed below the lanes. Hyperactive BRCA1 ubiquitylates ERα independent of the E2 enzyme used.
Hyperactivity causes a similar loss of E2 specificity toward the BRCA1 substrate estrogen receptor (ERα). Ubiquitylation of the ERα ligand‐binding domain (LBD) can turn off estrogen signaling, but does not lead to degradation of ERα in cancer cells.39, 40 While the E2 required for this activity in vivo is unknown, it is clear that specificity is required to avoid modifying ERα with Lys 48‐linked Ub chains that would lead to its destruction. In vitro ubiquitylation assays of ERα show that of all the E2 enzymes that can function with wild‐type BRCA1, only the Ube2d family adds Ub to ERα LBD efficiently [Fig. 2(B)]. However, the hyperactive L51W/K65R BRCA1 mutant is able to activate the Ube2e family and Ube2w to transfer Ub to ERα, again demonstrating loss of specificity. Loss of E2 product‐specificity could decrease the fidelity of signaling by allowing multiple products (and therefore multiple signals) to be generated, providing a rationale for the fine‐tuning of BRCA1 activity.
Hyperactive substitutions can rescue activity of cancer‐associated variants
Cancer‐associated variations in the BRCA1 RING domain lead to a loss of E3 ligase activity.12 BRCA1 ligase activity is important for homologous recombination and genome stability which have clear implications for tumorigenesis.36, 41 Here we show that hyperactive mutations are capable of rescuing the ligase activity of two cancer‐associated variants, C61G and C64G (Fig. 3). BRCA1 that contains both a cancer variation and the hyperactive substitutions activates the E2 Ube2d to build poly‐Ub chains as well as wild‐type BRCA1. Both activating substitutions (L51W and K65R) are necessary to restore activity of the cancer variants to the level seen in wild‐type (Fig. 3, Supporting Information S2). This rescue of activity is surprising given that each of the cancer‐associated mutations, C61G and C64G, result in a loss of a zinc‐coordinating ligand and, therefore, a destabilization that is in close proximity to the hyperactive substitution, K65R. Future studies to confirm the rescue phenotype in vivo are needed, but the finding provides a conceptual basis for searching for a small molecule capable of rescuing the activity of even profoundly ligase‐dead cancer‐associated variants of BRCA1.
Figure 3.
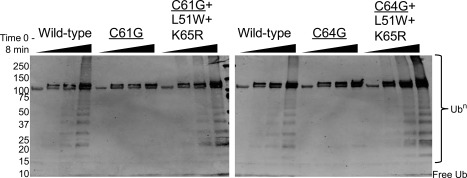
Hyperactive mutations (L51W and K65R) can rescue the activity of cancer variants. Ubiquitin chain‐building (Ubn) visualized by blotting for the HA tag on ubiquitin (Ub). BRCA1 variants are listed above the lanes. Time points were taken at 0, 1, 2, and 8 min after ATP addition. Assay component concentrations were as follows: 2.5 μM E1, 5 μM Ube2d3, 20 μM HA‐Ub, 10 μM E3, 5 mM ATP/Mg.
BRCA1 tuning residues are applicable to other RING E3 ligases
There are hundreds of predicted RING E3 ligases in the human genome and the enzymatic activity of most of them has yet to be characterized. Fortunately, many aspects of the BRCA1 RING E3 mechanism can be used to characterize other RINGs. For example, the positively charged allosteric linchpin residue in other RINGs, K65 in BRCA1, is a conserved and essential part of the mechanism for many different RINGs.27 Notably, many RING E3s have a positively charged Arg in this position instead of Lys and/or a native Trp at the position equivalent to L51 in BRCA1. In support of the general importance of these positions to the functioning of RING‐type E3s, mutation of the native R1143 of E4BU (K65 equivalent in BRCA1) or W408 of cCbl (L51 equivalent in BRCA1) is reported to lead to significant loss of ligase activity.27, 42
To determine if the activity of uncharacterized RING E3s can be predicted and modulated using these insights, we chose the monomeric RING E3 Listerin1 (Ltn1). Ltn1 contains a native Trp and Arg at the positions equivalent to L51 and K65 in BRCA1 [Fig. 4(A)]. We will refer to these residues using the BRCA1 numbering scheme for clarity. Under identical conditions and concentrations, wild‐type Ltn1 is more effective at activating Ube2d2 when compared to BRCA1/BARD1, as would be predicted by the presence of the activity‐enhancing residues, W51 and R65 [Fig. 4(B)]. As the residues are in a different context in Ltn1, we asked if they can still modulate ligase activity. Indeed, substitutions at either of the tuning positions substantially decrease Ltn1 activity [Fig. 4(B)]. The cellular function of Ltn1 is to ubiquitylate and degrade nascent proteins that are stalled during translation.43 Therefore, Ltn1's function requires it to ubiquitylate any stalled peptide without specificity, providing a possible rationale for why it has evolved enhanced rather than attenuated activity. Hence, the identity of the residue at these positions can be used as starting points for modulating the activity of other RING E3s, providing a useful strategy to characterize their cellular functions.
Figure 4.
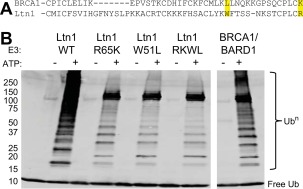
Listerin (Ltn1) natively contains “hyperactive” residues at equivalent positions and its activity mimics the hyperactive BRCA1 as measured by Ub chain‐building. (A) Position of activity‐determining residues (51 and 65 in BRCA1) highlighted in yellow. (B) Western blot for HA tag on ubiquitin reveals the ubiquitin chain‐building activity of wild‐type and mutant Ltn1 compared to BRCA1/BARD1 after 1 h. Mutation of Ltn1 residues to the residues found in BRCA1 at highlighted positions decreases activity. Assay component concentrations were as follows: 1 μM E1, 20 μM HA‐Ub, 2 μM Ube2d3, 2 μM E3, 10 mM ATP/MgCl2. Western blotted for HA tag on Ub.
Discussion
With over 600 members, human RING E3s are the largest class of Ub E3 ligases, and even eclipse the number of kinases in the human proteome. Many advances have been made to decipher how RING E3s work. Although there are some examples of RING E3s that are inactive and require an external factor to activate an E2∼Ub,16, 44, 45 most RING E3s are constitutively active. A defining feature of RINGs is that they lack a single active site catalytic residue. Instead, RINGs act as allosteric activators of E2∼Ub conjugates, facilitating Ub transfer from the E2 directly onto a substrate. Results presented here show that the degree to which a given RING E3 activates an E2∼Ub conjugate can be dialed up or down by the identity of residues at particular positions in the RING domain. Our results with BRCA1 show it has not evolved to obtain maximal E3 ligase activity. Instead, substitution of key residues (i.e., L51W and K65R) results in dramatic increases in activity. Analysis of over 1,600 RING sequences revealed that nearly half (46%) have Arg as their linchpin compared with 14% that have Lys at this position. This suggests that other E3s may also have attenuated activity. While the biological driving force for lower activity may vary among RING E3s, our in vitro data suggest that the ability to maintain product specificity through differential activity with multiple E2s is important. Hyperactive BRCA1 produces high molecular weight modifications with E2s that are normally limited to mono‐ubiquitylation with wild‐type BRCA1. BRCA1 has been fine‐tuned through evolution to prevent the generation of off‐pathway products, particularly auto‐ubiquitylation that could result in the unintended degradation of BRCA1. BRCA1 is expressed in cells at low levels, making such a suicidal tendency especially catastrophic, and providing a hint as to why maximal E3 ligase activity is not always optimal.
The heterodimeric RING partner, BARD1, is essential to BRCA1's ligase activity, so it was surprising to find that the BARD1 RING itself appears to play an attenuating role on ligase activity. The helices adjacent to the RING domains of BRCA1 and BARD1 are important for stabilizing the BRCA1/BARD1 heterodimer, but the role of the BARD1 RING has remained enigmatic. We find that disruption of the BARD1 RING domain through mutation of zinc coordinating ligands can unexpectedly increase BRCA1 ligase activity. Indeed, elimination of the BARD1 RING altogether still supports BRCA1 ligase activity as long as the BRCA1‐binding helices of BARD1 are present. A modulatory role for the non‐E2‐binding RING partner of heterodimeric RING E3s has been reported for the polycomb group ring finger (PCGF) family of six homologs that bind to RING1A/B in the gene silencing polycomb repressive complex 1. Analogous to what we observe for BARD1, the level of ligase activity of RING1A/B depends on the identity of the non‐E2‐binding partner. Strikingly, RING1A/B ligase activity is attenuated when paired with the canonical partner, PCGF4 (also called BMI1), compared with the other five homologs.46 In the case of BRCA1, which has only a single obligate heterodimeric RING partner, the complex has evolved attenuated activity. This raises the intriguing possibility that BRCA1 activity could be further modulated by some post‐translational modification of the BARD1 RING.
The lack of an essential active site residue that can be mutated to ablate activity in RING E3s poses a substantial challenge in designing molecular tools to decipher cellular function. For instance, use of the I26A BRCA1 mutant in a mouse knock‐in experiment led to the provocative conclusion that BRCA1's E3 ligase function is not required for its tumor suppressor function.32 While this conclusion may indeed be correct, we show here that the I26A‐BRCA1 is not ligase‐dead with all E2s, reopening the question. Only by mutating the linchpin residue and two residues within the E2 binding site can we generate a BRCA1 mutant that is structurally intact and not active with any E2 enzymes. This mutant affords the opportunity to distinguish between phenotypes associated with BRCA1 E3 ligase activity from its other roles as a protein scaffold. Our findings with BRCA1 are relevant to other E3 ligases. For instance, mutation of equivalent residues in other RINGs provides a good starting place to identify their cellular functions. Along these lines, we show with an unrelated RING E3, Ltn1, we can predict and modulate RING activity by targeting specific RING tuning residues.
A second question that continues to cloud the field is the issue of RING E3 specificity. Here we use a hyperactive BRCA1 mutant to show that not only does the type of ubiquitin modification made on a product depend on the E2 that is paired with a RING E3, but it also depends on (1) the degree to which an E3 can activate the given E2 and (2) the substrate itself. Substrate‐dictated specificity is observed in other RING‐type E3s such as RING1/PCGF complexes, which robustly auto‐ubiquitylate themselves with poly‐ubiquitin chains but mostly mono‐ubiquitylate nucleosomes.46 Insight into the different specificity comes from the recent structure of the E2/E3/nucleosome complex showing that the E2 active site is positioned in close proximity to the target lysine.47 This specific mode of binding indicates that after ubiquitin is added to the nucleosome it may sterically hinder binding of a second E2∼Ub conjugate. Even if a second E2∼Ub can bind the ubiquitylated nucleosome, this would place the E2 active site too close to the conjugated ubiquitin's C‐terminus and more distant from the lysines in the conjugated ubiquitin. This is in contrast to auto‐ubiquitylation which likely does not involve specific substrate binding or addition to a specific lysine. More structural and biochemical data on E3/E2 complexes and their substrates are needed to better understand the source E3 ligase specificity.
In summary, we have built on the current understanding of RING structure and mechanism to design and characterize hyperactive and ligase‐dead mutants of BRCA1/BARD1 that shed light on their E3 ligase function. These results provide tools that can be used to study E2 enzyme specificity, predict and modulate the activity of other RING E3s, and potentially develop activity‐enhancing small molecules for RING E3s in the future.
Materials and Methods
BRCA1 constructs and purification
The growth, co‐expression, and co‐purification of BRCA1/BARD1 constructs were carried out as described previously.48 Briefly, RING domain constructs consisted of residues 1–112 of BRCA1 and 26–140 of BARD1, while long constructs used in auto‐ubiquitylation assays (Fig. 2, Supporting Information S1) consisted of residues 1–304 of BRCA1 and 26–327 of BARD1. The RINGless BARD1 construct was made using the Agilent Quikchange protocol with a primer designed to delete BARD1 residues 50–98 and loop in a five residue linker in place of the RING domain (SerGlyGlySerGly). All BRCA1 and BARD1 constructs are co‐expressed in BL21 Escherichia coli cells and purified with nickel affinity resin and size exclusion chromatography. ERα LBD, Ub, HA‐tagged Ub, E1 and various E2s used for assays were expressed and purified as described previously.15, 39, 49
The RING domain of Ltn1 (residues 1716–1766) was cloned from a construct generously provided by C. Joazeiro downstream of an N‐terminal histidine tagged SUMO with a H3C protease cleavable attachment site. Expression of SUMO‐fused Ltn1 was induced in BL21 Escherichia coli cells using 0.2 mM IPTG at 16°C overnight. SUMO‐Ltn1 fusion was purified using a nickel affinity column per manufacturer protocol (Invitrogen). Imidazole was removed via dialysis at 4°C into the recommended nickel affinity resin binding buffer while SUMO was cleaved from Ltn1 using a GST tagged H3C protease. The H3C protease and cleaved SUMO were removed with GST and nickel affinity resins, respectively, while Ltn1 remained unbound. Size exclusion chromatography with SDX75 resin in 25 mM sodium phosphate pH 7.0, 150 mM sodium chloride was used as a final purification step. Ltn1 was stored at 4°C and assayed within a week of purification. All BRCA1, BARD1 and Ltn1 mutations were made using Agilent Quikchange protocol and purified using the same methods as the wild‐type enzymes.
Ubiquitylation assays
All ubiquitylation assays were carried out in 25 mM sodium phosphate pH 7.0, 150 mM sodium chloride at 37°C with shaking. All western blots were imaged using an Odyssey infrared imaging system (Licor). The ability of E3 RING domain constructs to enhance the rate of ubiquitin chain formation was measured by combining E1, E2 (Ube2d3), E3, HA‐tagged Ub, ATP, and MgCl2 in the concentrations listed in the figure captions. The reaction is stopped by addition of denaturing and reducing SDS‐PAGE load dye at the time after addition of ATP listed in the figure captions. Samples were run on SDS‐PAGE and visualized by western‐blotting for the HA tag on Ub using an antibody from Bethyl Laboratories (A190‐108A).
BRCA1 auto‐ubiquitylation with various E2 enzymes was measured by combining 1 μM E1, 2 μM E2, 3 μM BRCA1/BARD1 long constructs, 40 μM Ub, 5 mM ATP, and 5 mM MgCl2 for 1 h and then stopping the reaction by addition of denaturing and reducing SDS‐PAGE load dye. A FLAG tag was appended to the N‐terminus of BRCA1. Samples were run on SDS‐PAGE and western blots performed using a FLAG antibody from Sigma–Aldrich (F3165).
Ubiquitylation of the ligand binding domain (LBD) of ERα containing a V5 tag with various E2s was measured by combining 2 μM ERα LBD, 1 μM E1, 2 μM E2, 3 μM BRCA1/BARD1 long constructs, 20 μM Ub, 5 mM ATP, and 5 mM MgCl2 for 1 h and then stopping the reaction by addition of denaturing and reducing SDS‐PAGE load dye. Samples were run on SDS‐PAGE and western blots performed using V5 antibody from Invitrogen (46‐0705).
Supporting information
Supporting Information
Acknowledgments
The authors thank P. Littlefield and J. Keefe for the cloning and initial characterization of RINGless BARD1.
References
- 1. Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC (1990) Linkage of early‐onset familial breast cancer to chromosome 17q21. Science 250:1684–1689. [DOI] [PubMed] [Google Scholar]
- 2. Miki Y, Swensen J, Shattuck‐Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, Bell R, Rosenthal J, Hussey C, Tran T, McClure M, Frye C, Hattier T, Phelps R, Haugen‐Strano A, Katcher H, Yakumo K, Gholami Z, Shaffer D, Stone S, Bayer S, Wray C, Bogden R, Dayananth P, Ward J, Tonin P, Narod S, Bristow PK, Norris FH, Helvering L, Morrison P, Rosteck P, Lai M, Barrett JC, Lewis C, Neuhausen S, Cannon‐Albright L, Goldgar D, Wiseman R, Kamb A, Skolnick MH (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66–71. [DOI] [PubMed] [Google Scholar]
- 3. Arney K (2012) Tracking down the BRCA genes. Cancer Research UK. http://scienceblog.cancerresearchuk.org/2012/02/28/high-impact-science-tracking-down-the-brca-genes-part-1/.
- 4. Freemont PS, Hanson IM, Trowsdale J (1991) A novel cysteine‐rich sequence motif. Cell 64:483–484. [DOI] [PubMed] [Google Scholar]
- 5. Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang M‐CW, Hwang L‐Y, Bowcock AM, Baer R (1996) Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet 14:430–440. [DOI] [PubMed] [Google Scholar]
- 6. Meza JE, Brzovic PS, King MC, Klevit RE (1999) Mapping the functional domains of BRCA1. Interaction of the ring finger domains of BRCA1 and BARD1. J Biol Chem 274:5659–5665. [DOI] [PubMed] [Google Scholar]
- 7. Brzovic PS, Rajagopal P, Hoyt DW, King M‐C, Klevit RE (2001) Structure of a BRCA1‐BARD1 heterodimeric RING‐RING complex. Nat Struct Mol Biol 8:833–837. [DOI] [PubMed] [Google Scholar]
- 8. Buchwald G, van der Stoop P, Weichenrieder O, Perrakis A, van Lohuizen M, Sixma TK (2006) Structure and E3‐ligase activity of the Ring–Ring complex of Polycomb proteins Bmi1 and Ring1b. embo J 25:2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang A, Hibbert RG, de Jong RN, Das D, Sixma TK, Boelens R (2011) Symmetry and asymmetry of the RING‐RING dimer of Rad18. J Mol Biol 410:424–435. [DOI] [PubMed] [Google Scholar]
- 10. Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM (1999) RING fingers mediate ubiquitin‐conjugating enzyme (E2)‐dependent ubiquitination. Proc Natl Acad Sci USA 96:11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T (2001) The RING heterodimer BRCA1‐BARD1 is a ubiquitin ligase inactivated by a breast cancer‐derived mutation. J Biol Chem 276:14537–14540. [DOI] [PubMed] [Google Scholar]
- 12. Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D, Fukuda M, Ohta T, Klevit R (2003) Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin‐ligase complex. Proc Natl Acad Sci USA 100:5646–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hershko A, Heller H, Elias S, Ciechanover A (1983) Components of ubiquitin‐protein ligase system. Resolution, affinity purification, and role in protein breakdown. J Biol Chem 258:8206–8214. [PubMed] [Google Scholar]
- 14. Deshaies RJ, Joazeiro CAP (2009) RING domain E3 ubiquitin ligases. Annu Rev Biochem 78:399–434. [DOI] [PubMed] [Google Scholar]
- 15. Christensen DE, Brzovic PS, Klevit RE (2007) E2‐BRCA1 RING interactions dictate synthesis of mono‐ or specific polyubiquitin chain linkages. Nat Struct Mol Biol 14:941–948. [DOI] [PubMed] [Google Scholar]
- 16. Metzger MB, Pruneda JN, Klevit RE, Weissman AM (2014) RING‐type E3 ligases: master manipulators of E2 ubiquitin‐conjugating enzymes and ubiquitination. Biochim Biophys Acta 1843:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng N, Wang P, Jeffrey PD, Pavletich NP (2000) Structure of a c‐Cbl‐UbcH7 complex: RING domain function in ubiquitin‐protein ligases. Cell 102:533–539. [DOI] [PubMed] [Google Scholar]
- 18. Mace PD, Linke K, Feltham R, Schumacher FR, Smith CA, Vaux DL, Silke J, Day CL (2008) Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin‐conjugating enzyme (E2) recruitment. J Biol Chem 283:31633–31640. [DOI] [PubMed] [Google Scholar]
- 19. Xu Z, Kohli E, Devlin KI, Bold M, Nix JC, Misra S (2008) Interactions between the quality control ubiquitin ligase CHIP and ubiquitin conjugating enzymes. BMC Struct Biol 8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benirschke RC, Thompson JR, Nomine Y, Wasielewski E, Juranic N, Macura S, Hatakeyama S, Nakayama KI, Botuyan MV, Mer G (2010) Molecular basis for the association of human E4B U box ubiquitin ligase with E2‐conjugating enzymes UbcH5c and Ubc4. Structure 18:955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bentley ML, Corn JE, Dong KC, Phung Q, Cheung TK, Cochran AG (2011) Recognition of UbcH5c and the nucleosome by the Bmi1/Ring1b ubiquitin ligase complex. EMBO J 30:3285–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang L, Fairall L, Goult BT, Calkin AC, Hong C, Millard CJ, Tontonoz P, Schwabe JW (2011) The IDOL‐UBE2D complex mediates sterol‐dependent degradation of the LDL receptor. Genes Dev 25:1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT (2012) BIRC7–E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol 19:876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Plechanovova A, Jaffray EG, Tatham MH, Naismith JH, Hay RT (2012) Structure of a RING E3 ligase and ubiquitin‐loaded E2 primed for catalysis. Nature 489:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Branigan E, Plechanovova A, Jaffray EG, Naismith JH, Hay RT (2015) Structural basis for the RING‐catalyzed synthesis of K63‐linked ubiquitin chains. Nat Struct Mol Biol 22:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS (2016) E2 enzymes: more than just middle men. Cell Res 26:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. JN Pruneda, PJ Littlefield, SE Soss, KA Nordquist, WJ Chazin, PS Brzovic, RE Klevit (2012) Structure of an E3:E2∼Ub complex reveals an allosteric mechanism shared among RING/U‐box ligases. Mol Cell 47:933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brzovic PS, Meza J, King M‐C, Klevit RE (1998) The cancer‐predisposing mutation C61G disrupts homodimer formation in the NH2‐terminal BRCA1 RING finger domain. J Biol Chem 273:7795–7799. [DOI] [PubMed] [Google Scholar]
- 29. Choudhury AD, Xu H, Baer R (2004) Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J Biol Chem 279:33909–33918. [DOI] [PubMed] [Google Scholar]
- 30. Yu X, Fu S, Lai M, Baer R, Chen J (2006) BRCA1 ubiquitinates its phosphorylation‐dependent binding partner CtIP. Genes Dev 20:1721–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reid LJ, Shakya R, Modi AP, Lokshin M, Cheng JT, Jasin M, Baer R, Ludwig T (2008) E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology‐directed repair of double‐strand DNA breaks. Proc Natl Acad Sci USA 105:20876–20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin C‐S, deRooij DG, Hirsch S, Ravi K, Hicks JB, Szabolcs M, Jasin M, Baer R, Ludwig T (2011) BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334:525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, Klijn C, van der Heijden I, van der Gulden H, Wientjens E, Pieterse M, Catteau A, Green P, Solomon E, Morris JR, Jonkers J (2011) BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 20:797–809. [DOI] [PubMed] [Google Scholar]
- 34. Starita LM, Pruneda JN, Lo RS, Fowler DM, Kim HJ, Hiatt JB, Shendure J, Brzovic PS, Fields S, Klevit RE (2013) Activity‐enhancing mutations in an E3 ubiquitin ligase identified by high‐throughput mutagenesis. Proc Natl Acad Sci USA 110:E1263–E1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, Fields S (2015) Massively parallel functional analysis of BRCA1 RING domain variants. Genetics 2015;200:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza‐Martin M, Fletcher A, Blair‐Reid S, Beesley J, Johal B, Pearl LH, Neely R, Keep NH, Watts FZ, Morris JR (2016) Human BRCA1‐BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol 23:647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hofmann RM, Pickart CM (1999) Noncanonical MMS2‐encoded ubiquitin‐conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell 96:645–653. [DOI] [PubMed] [Google Scholar]
- 38. Scaglione KM, Basrur V, Ashraf NS, Konen JR, Elenitoba‐Johnson KSJ, Todi SV, Paulson HL (2013) The ubiquitin conjugating enzyme (E2) Ube2w ubiquitinates the N‐terminus of substrates. J Biol Chem. 2013;288:18784–18788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eakin CM, MacCoss MJ, Finney GL, Klevit RE (2007) Estrogen receptor α is a putative substrate for the BRCA1 ubiquitin ligase. Proc Natl Acad Sci USA 104:5794–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG, Tomita YA, Rosen EM (2010) BRCA1 regulates acetylation and ubiquitination of estrogen receptor‐α. Mol Endocrinol 24:76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM (2011) BRCA1 tumour suppression occurs via heterochromatin‐mediated silencing. Nature 477:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC (1999) The tyrosine kinase negative regulator c‐Cbl as a RING‐type, E2‐dependent ubiquitin‐protein ligase. Science 286:309–312. [DOI] [PubMed] [Google Scholar]
- 43. Bengtson MH, Joazeiro CAP (2010) Role of a ribosome‐associated E3 ubiquitin ligase in protein quality control. Nature 467:470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DaRosa PA, Wang Z, Jiang X, Pruneda JN, Cong F, Klevit RE, Xu W (2015) Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP‐ribosyl)ation signal. Nature 517:223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harrison JS, Cornett EM, Goldfarb D, DaRosa PA, Li ZM, Yan F, Dickson BM, Guo AH, Cantu DV, Kaustov L, Brown PJ, Arrowsmith CH, Erie DA, Major MB, Klevit RE, Krajewski K, Kuhlman B, Strahl BD, Rothbart SB (2016) Hemi‐methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by UHRF1. eLife 5:e17101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taherbhoy AM, Huang OW, Cochran AG (2015) BMI1‐RING1B is an autoinhibited RING E3 ubiquitin ligase. Nat Commun 6:7621. [DOI] [PubMed] [Google Scholar]
- 47. McGinty RK, Henrici RC, Tan S (2014) Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514:591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, Klevit RE (2006) A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1‐directed ubiquitination. Mol Cell 21:873–880. [DOI] [PubMed] [Google Scholar]
- 49. Pickart CM, Raasi S (2005) Controlled synthesis of poly‐ubiquitin chains: Methods Enzymol. 2005;399:21–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information