

Abstract
MPT63, a major secreted protein from Mycobacterium tuberculosis, has been shown to have immunogenic properties and has been implicated in virulence. MPT63 is a β‐sandwich protein containing 11 β strands and a very short stretch of 310 helix. The detailed experimental and computational study reported here investigates the equilibrium unfolding transition of MPT63. It is shown that in spite of being a complete β‐sheet protein, MPT63 has a strong propensity toward helix structures in its early intermediates. Far UV‐CD and FTIR spectra clearly suggest that the low‐pH intermediate of MTP63 has enhanced helical content, while fluorescence correlation spectroscopy suggests a significant contraction. Molecular dynamics simulation complements the experimental results indicating that the unfolded state of MPT63 traverses through intermediate forms with increased helical characteristics. It is found that this early intermediate contains exposed hydrophobic surface, and is aggregation prone. Although MPT63 is a complete β‐sheet protein in its native form, the present findings suggest that the secondary structure preferences of the local interactions in early folding pathway may not always follow the native conformation. Furthermore, the Gly25Ala mutant supports the proposed hypothesis by increasing the non‐native helical propensity of the protein structure.
Keywords: protein folding, protein aggregation, fluorescence correlation spectroscopy, single molecule fluorescence, molecular dynamics simulation
Introduction
A globular protein can fold spontaneously to its native structure, and this process may involve both local and nonlocal interactions. The local interactions occuring early in the folding pathway have been shown to result in non‐native conformations.1, 2, 3 Kuwajima et al. earlier found the formation of a helical intermediate state in the predominantly β‐sheet protein β‐lactoglobulin.4 Although this 162 residue globular protein primarily contains β‐strands, by using stopped flow circular dichroism (CD) kinetics, Hamada et al suggested that there is a prominent accumulation of helical structure in its early folding.5 The presence of a helical intermediate has been observed in the folding landscape of intestinal fatty acid binding protein.6, 7 In this study, we have shown an early enhancement of helical characteristics in the unfolding pathway of a Mycobacterium tuberculosis protein, MPT63. MPT63 is a small, almost entirely β‐sheet protein, which is being used in our laboratory as a convenient model protein for folding studies.8
The X‐ray crystal structure of MPT63 has been solved at 1.5Å resolution.9 MPT63, the secreted antigen of M. tuberculosis, has been reported to have high level of humoral responses in tuberculosis infected patients.10, 11 It has significant sequence and structural similarity with other members of the immunoglobulin superfamily. MPT63 is a 130 residue β sandwich protein consisting of two antiparallel β sheets that are made up of 11 β strands and a very short stretch (3 amino acids) of 310 helix at the start of sixth β strand. The longer stranded β‐sheet is made up of four antiparallel β‐strands, whereas the shorter stranded β‐sheet consists of five β‐strands. It is noteworthy that the amino‐terminal β‐strand of this protein forms a parallel β‐sheet with the carboxy‐terminal β‐strand. The only helical secondary structure is a 310 helix comprising three residues (Val65–Gln67). We have recently used phosphorescence spectroscopy to show that this protein is unfolded by guanidinium hydrochloride in a stepwise manner.12
Acid resistance of different enteric pathogens such as Helicobacter pylori, Escherichia coli, and Vibrio cholerae has been extensively studied.13, 14, 15, 16 These pathogens survive in the extremely low pH environment of the stomach (pH values between 1 and 3). In contrast, tuberculosis (TB) is mainly a pulmonary infection and therefore M. tuberculosis predominantly resides inside macrophages and endures a low pH of around 4.17 However, there have been reports of extrapulmonary TB incidents, among which abdominal TB is the most common type.18 In this case, tuberculosis infections occur in multiple components of the gastro intestinal (GI) system, including the GI track, liver, spleen, and pancreas.19 In the case of GI TB, M. tuberculosis is expected to encounter the acidic pH of stomach (as low as pH 1–3). Interestingly, the response of an intracellular pathogen such as M. tuberculosis in such low pH has never been studied. As MPT63 is immunogenic in TB, its conformational study at low pH may provide initial insights into the mechanism of GI TB pathogenesis.
Detailed far‐UV CD, steady‐state fluorescence spectroscopy, FTIR, and fluorescence correlation spectroscopy (FCS) studies have been carried out to monitor the equilibrium unfolding transitions of MPT63. Far‐UV CD and steady‐state fluorescence results suggest that the addition of 2M guanidinium hydrochloride (Gdn.HCl) results in significant unfolding. FCS data at acidic pH of 1.25 shows a decrease in the hydrodynamic radius of the protein. These data, in combination with far UV CD and FTIR, clearly suggest the formation of an intermediate at pH 1.25, which contains significant helical structure. The helical structure can be stabilized further by adding 2,2,2‐trifluoroethanol (TFE). Computational studies using molecular dynamics (MD) simulations support the experimental results. The early time scales in the refolding simulations (∼100 ns) suggest a greater extent of helical structure when compared to the native MPT63. MD simulations predict the presence of three helices near the proximity of residues 20, 80, and 130 that are absent from the native structure. A glycine to alanine substitution in the helix‐prone N‐terminal region of MPT63 enhances the helical propensity of this segment. To validate the computational results, the alanine mutant of MPT63 (Gly25AlaMPT63) has been prepared. Interestingly, spectroscopic measurements of this mutant support the MD simulation results.
Results and Discussion
pH‐induced conformational changes of MPT63 analyzed by far‐UV CD, steady‐state fluorescence and FT‐IR
The far‐UV CD spectrum of wild type (WT) MPT63 in its native buffer (20 mM sodium phosphate buffer at pH 7.5) is shown in Figure 1(A) (black curve). The observed spectrum is characterized by a prominent negative peak at 216 nm confirming its overall β‐sheet secondary structure.9 Interestingly, the far‐UV CD spectrum of MPT63 contains another small negative peak at 231 nm. Although the exact reason for the presence of this peak is not known, we believe that its appearance is a result of tertiary interactions of the aromatic residues inside the hydrophobic core of the protein.20 It has been shown recently that Trp26, one of the four tryptophan residues present in MPT63, contributes significantly to the 231 nm peak. A mutation at Trp26 (Trp26Phe) removes the 231 nm peak.8 It may be noted that similar peaks around 230 nm have been observed in the case of other proteins with immunoglobulin‐like folds.9, 21
Figure 1.
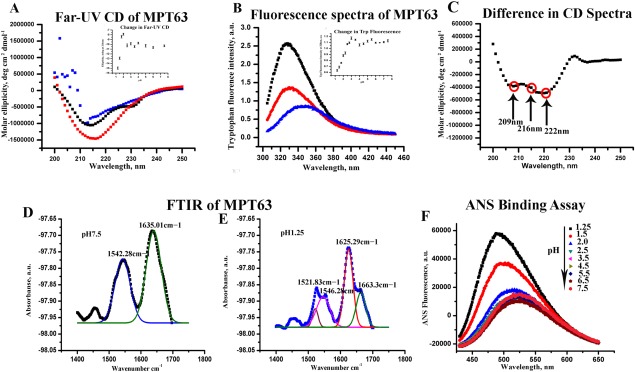
Changes in the [A] far‐UV CD and [B] steady‐state tryptophan fluorescence spectra at different solution conditions; pH 7.5 buffer (black), pH 1.25 (red), and 2M GdnHCl (blue). The variations in [A] the ellipticity at 216 nm and [B] the changes in fluorescence emission intensity at 328 nm with pH are shown in the inset. [C] The difference far‐UV CD (calculated by subtracting the CD spectrum at pH 1.25 from that measured at pH 7.5) spectrum show prominent double minimum at 209 and 222 nm and a dip at 216 nm. FTIR spectra of MPT63 at [D] pH 7.5 (two peaks fit are shown in blue and green) and at [E] pH 1.25 (four peaks fit shown in grey, pink, red, and green). [F] Fluorescence spectra of ANS in the presence of MPT63 obtained at different pH values ranging from 7.5 to 1.25. The ANS fluorescence assay shows a large increase at low pH indicating the presence of exposed hydrophobic surface at these solution conditions.
In the presence of 2M Gdn.HCl at pH 7.5, the ellipticity decreases significantly and the far‐UV CD spectrum becomes featureless [Fig. 1(A), blue curve]. This characteristic is typically observed in the case of unfolding induced by a denaturant, like Gdn.HCl. In contrast, at low pH [Fig. 1(A)], the far‐UV CD spectrum of MPT63 has been found to be quite different. At low pH (pH 1.25), the ellipticity of the protein increases significantly [Fig. 1(A), red curve], a feature not commonly observed for an acid‐induced unfolded protein.22 Typically, far‐UV CD shows a decrease in the ellipticity (not an increase), when a protein is subjected to low pH as evident for the intestinal fatty acid binding proteins.23
Steady‐state Trp fluorescence is a convenient method to monitor the local conformation of a protein around its tryptophan residues. Figure 1(B) shows the steady‐state fluorescence emission spectra of MPT63 in the presence of different solution conditions. We use an excitation wavelength of 295 nm to exclude the contributions of the four tyrosine residues, which are present in MPT63. In the native solution condition, the maximum of the steady‐state fluorescence spectrum has been found at 328 nm, which is typical for a well‐folded globular protein. This emission maximum typically indicates that the tryptophan residues are situated inside a hydrophobic environment. In the presence of 2M Gdn.HCl, the steady‐state emission maximum shifts to 350 nm as a result of chemical unfolding of the protein. This shift in the emission maximum in the presence of 2M Gdn.HCl is accompanied by a large quenching of the fluorescence intensity. In contrast, at low pH of 1.25, the fluorescence emission maximum does not change much, although a significant decrease in the emission intensity is observed [Fig. 1(B)].
The inset of Figure 1(A) plots the changes in the ellipticity at 216 nm, which is obtained by subjecting WT MPT63 to different pH conditions. The data show no or insignificant change in the ellipticity between pH 7.5 and pH 2.5. However, the values of ellipticity at 216 nm show a small but prominent decrease (become less negative) between pH 2.5 and 2. This change is followed by a large increase in the ellipticity, which occurs between pH 2 and 1.25. In contrast, the variation in fluorescence intensity with pH [inset in Fig. 1(B)] shows a large decrease between pH 3 and pH 1.25. As far‐UV CD is a probe for the secondary structure, it typically shows a prominent decrease as a result of protein unfolding. The decrease in the far‐UV CD that we observe in the presence of 2M Gdn.HCl is hence expected. As low pH generally leads to a partial or complete unfolding of proteins, the increase in the ellipticity observed between pH 2 and pH 1.25 is surprising.
To obtain more insights into the nature of this low pH form, the difference in ellipticity of the CD spectrum between pH 1.25 and 7.5 has been measured [Fig. 1(C)]. The difference far‐UV CD spectrum at low pH clearly shows the appearance of two negative minima at 222 nm and 209 nm. This is accompanied by a decrease in the ellipticity at 216 nm [Fig. 1(C)]. As alpha helical proteins are characterized by double minima at 222 nm and 209 nm and proteins containing beta sheet show a negative minimum at 216 nm, the above results clearly suggest a low‐pH‐induced increase in helical structure, which is accompanied by a simultaneous decrease in beta sheet. The discussed results suggest that the low‐pH intermediate of MTP63 has enhanced helical content.
FT‐IR spectroscopy has been used to complement the far‐UV CD data. IR spectroscopy probes universally available amide (peptide) bonds, which display distinct IR signals for differently folded peptides and proteins. Figure 1(D,E) shows the IR spectra of MPT63 at pH 7.5 and 1.25, respectively. A strong absorbance at the amide I region is seen at 1635 cm−1, which can be correlated with the presence of β‐sheets. Interestingly, at pH 1.25, there are two distinct absorbance peaks at 1625 and at 1663 cm−1. The later one (1663 cm−1) indicates the presence of α‐helix, whereas the peak at 1625 cm−1 represents β‐turns.24 At pH 7.5, another peak is found at 1542 cm−1, which corresponds to helical structure from amide II region (1500–1600 cm−1).24 On the other hand, at pH 1.25, MPT63 shows two distinct peaks at1546 cm−1 and 1521 cm−1 representing alpha helix from amide II region.
The far‐UV CD and FT‐IR experiments described above suggest the formation of an intermediate at low pH with increased alpha helical content. This is interesting as the WT protein in its native state is almost entirely a beta sheet protein (with the exception of a short 310 helix). It can be speculated that the protein may have an inherent tendency to form local helical structure, which is destabilized by hydrogen bonding and other long range contacts which play important roles in maintaining the native state at pH 7.5. At low pH, which disfavors both the long distance interactions and hydrogen bonding, the contributions of the local interactions may become predominant. These local contacts, which are expected to be hydrophobic, are probably responsible for the entirely different alpha helical structure observed at low pH. It may be noted that the predominant hydrophobic interactions at low pH lead to partial folding and formation of “molten globule” intermediates for a large number of proteins.25 As suggested by Lim et al., even proteins with native β‐sheet structures may possess strong potential to form helices.26 Lim's hypothesis suggests that helical structure accumulation could be possible early in the folding of a complete β‐sheet protein, which may subsequently get destabilized as the folding progresses. Goto et al. observed the accumulation of early helices in the case of a predominantly β‐sheet protein β‐lactoglobulin.5 Earlier it has been shown that the formation of hydrophobic contacts and the generation of local secondary structure depend on several factors including the nature of solvent27 and mutation at the area of hydrophobic clustering.23
To get more insights into the conformation changes at different pH, we have studied the binding of 1‐anilino‐8‐naphthalene‐sulfonate (ANS) to MPT63. ANS binding has been extensively used as a reporter to the exposed hydrophobic surface of a protein. The binding of ANS to the protein is insignificant at pH 7.5 [Fig. 1(F)]. In contrast, an increase in the fluorescent intensity and a blue shift of the emission maxima have been observed at pH value lower than 2. This large increase in the extent of binding is seen at pH 1.5 and 1.25, suggesting the presence of exposed hydrophobic surface at low pH.
The low‐pH intermediate is aggregation prone
Subsequently, we wanted to find out if MPT63 or its low‐pH intermediate has any propensity to form amyloid‐like aggregates. To investigate this, thioflavin‐T (ThT), a dye which binds to cross beta structures, was used. ThT exhibits enhanced fluorescence upon binding with amyloid fibrils. No significant increase in the fluorescence intensity was observed when the protein is incubated at pH 7.5 buffer. In contrast, a large increase in the fluorescence intensity was seen at pH 1.25. We have measured ThT binding of MPT63 at different pH and plotted this together with the CD data at 222 nm (Fig. 2). The data suggest a direct relationship between amyloid formation and helical structure accumulation at low pH. At pH 1.25, the ThT fluorescence intensity is found to increase with time, with a plateau after 4 h (inset of the figure). No large change in intensity between 4 h and 24 h was observed. There is no change in fluorescence intensity at pH 7.5. The ThT binding data suggest that the low‐pH intermediate is aggregation prone.
Figure 2.
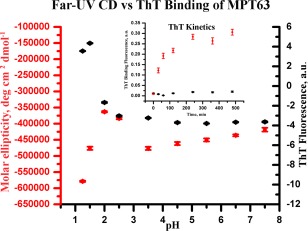
Far‐UV CD at 222 nm (left Y‐axis) plotted together with the changes in ThT binding of MPT63 at different pH (right Y‐axis). The inset shows the ThT binding kinetics of MPT63 at pH 7.5 (black) and pH 1.25 (red).
Fluorescence correlation spectroscopy (FCS) suggests a compaction of the protein at low pH
FCS experiments have been carried out to study the translational diffusion of the folded and low‐pH intermediate states of MPT63. FCS measures fluorescence intensity fluctuations as the proteins diffuse in and out of the confocal volume. Optimum analyses of the correlation function can provide sensitive information on the hydrodynamic radius (r H) of a protein at single molecule sensitivity.28 Figure 3 shows the autocorrelation functions obtained with the Alexa488Maleimide‐labeled Gly20Cys mutant of MPT63 (Alexa488MPT63) at the native folded condition (pH 7.5), at low pH (pH 1.25), and at denatured condition (in the presence of a chemical denaturant, Gdn.HCl). We previously observed that MPT63Gly20Cys shows similar structural and conformational features as the native WT protein when monitored by far‐UV CD and steady‐state fluorescence.29 Correlation functions optimally fit a single component diffusion model. The goodness of the fit is determined using the randomness of the residual distributions.
Figure 3.
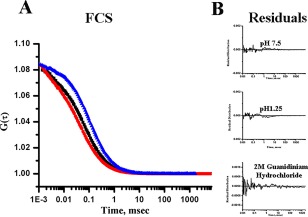
[A] Normalized autocorrelation functions obtained from the FCS experiments with Alexa488Maleimide‐labeled MPT63 G20C at pH 7.5 (black), pH 1.25 (red), and in 2M Gdn.HCl (blue). [B] The residual distributions of the fits at different conditions are shown.
Table 1 shows the values of r H and τ D obtained from the FCS experiments. The theoretical hydrodynamic radius of MPT63 has been estimated to be 19.48 Å using the HYDROPRO program.30 This value matches well with the experimental r H value 20.2 Å (Table 1) calculated from the τ D value obtained by the FCS experiments. The Stokes–Einstein equation Eq. (5) has been used to calculate the experimental r H value from τ D. Thus, a model with a single diffusion component adequately fits the correlation function data observed with Alexa488MPT63. The value of r H of a folded globular protein can be calculated from the equation suggested by Wilkins et al.31
| (1) |
where, Na is the number of amino acid residues of a protein. The r H value calculated from this equation is 19.5 Å correlates with the r H value calculated from the FCS data (20.2 Å). In the presence of 2M Gdn.HCl, the r H value increases significantly (30 Å) (Table 1). The r H value of a chemically denatured (using urea or Gdn.HCl) protein has been shown to follow the following relationship.
| (2) |
Table 1.
Different rH and τD values found by fitting the autocorrelation function to Eq. (1)
| MPT63 protein | τ D (usec) | r H (Å) |
|---|---|---|
| pH 7.5 | 87 | 20.2 |
| pH 1.25 | 63 | 14.6 |
| 2M Gdn.HCl | 130 | 30.1 |
In the case of MPT63, the calculated r H for the chemically unfolded protein has been found to be 35.4 Å. This value correlates well with the r H value calculated from the FCS data of Gdn.HCl treated MPT63 (30 Å).
Interestingly, at pH 1.25, MPT63 has been found to diffuse faster and the estimated r H value (14.6 Å) (Table 1) is less than that of the native protein (20.2 Å). Unfolding at low pH generally increases the values of τ D increasing r H. This is expected because low pH is supposed to increase charge repulsion between positively charged residues leading to the formation of an extended state. In contrast, the decrease in diffusion time observed here suggests the formation of a more compact structure at pH 1.25. As helices are predicted to diffuse faster, this compact structure of MPT63 can be a result of helical structure accumulation at pH 1.25. Lim's hypothesis predicted that the helical bundles formed at early folding pathways would diffuse faster.26
TFE induced conformational changes in MPT63
TFE is known to stabilize helical structures of proteins and peptides. TFE, as a hydrogen bond donor, participates in forming direct hydrogen bonds with carbonyl backbone of proteins, which has been shown to enhance helix structure in model protein systems.5, 32 Figure 4(A) shows the far‐UV CD spectra of MPT63 at pH 1.25 in the presence of different concentrations of TFE. A stable helical structure of appreciable extent is found to form at low pH in the presence of TFE. The change in ellipticity at 222 nm at pH 1.25 has been plotted in Figure 4(B).
Figure 4.
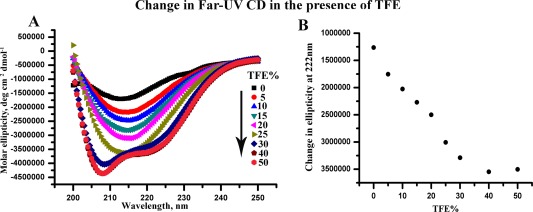
[A] Changes in far‐UV CD spectra at pH 1.25 with increasing percentage of TFE. [B] Changes in ellipticity at 222 nm with TFE concentrations at pH 1.25.
The experimental results discussed above suggest that at low pH, MPT63 assumes an intermediate structure with significant helical content. The propensity of the helix formation is further enhanced at low pH in the presence of TFE. Incidentally, no helix formation has been observed in the native or completely unfolded (in the presence of chemical denaturants) states of MPT63. These observations indicate that the partially folded (and not the completely unfolded or folded) states of MPT63 may have significant propensity to form helical intermediates, which can be further stabilized by using appropriate solution conditions, like the presence of TFE. In the following paragraphs, we discuss our computational results to support experimental observation that MPT63 may have existing bias toward helical structures, particularly at the partially unfolded states.
Computational studies in support of the experimental observation
To explore the presence of any additional helices, we have unfolded the protein at elevated temperature, which is followed by folding at low temperature. Our objective is to identify any helical intermediate that the protein might populate while traversing from the unfolded to its native configuration. To attain the unfolded state computationally, molecular dynamics simulations have been performed at 550 K for 20 ns, where the backbone RMSD attains a value of ∼18 Å at the end of the simulation [Fig. 5(A)]. Our reported R g (14 Å) of the native protein calculated from simulation data is in agreement with R g obtained from the crystal structure of MPT63 (15.1 Å) as computed by HYDROPRO.30 The R g of 15.1 Å approximately corresponds to a hydrodynamic radius of 19.48 Å, as calculated using the published relationship.31 The computational results also reveal a significant decrease in the secondary structure content (particularly the beta sheet) as a result of the heat‐induced unfolding [Fig. 5(B) and Table 2]. The extended end point structure of this simulation has been used as the starting structure for conducting the refolding runs at 300 K.
Figure 5.
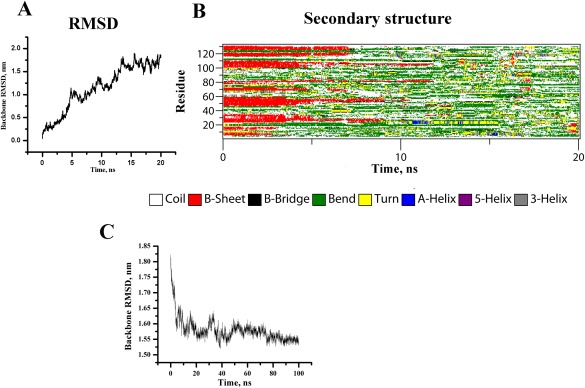
Time evolution of [A] backbone RMSD and [B] the secondary structural elements of MPT63 at 550 K (DSSP classification). [C] Backbone RMSD of MPT63 during refolding at 300 K.
Table 2.
Average secondary structure contents from DSSP analysis in WT and computationally unfolded MPT63 obtained by the MD simulations. Refold and RefoldGly25Ala correspond to the average values calculated from two refolding runs for the WT and the Gly25Ala mutant, respectively. Secondary structure content obtained from the crystal structure is also shown
| Coil | β‐Sheet | β‐Bridge | Bend | Turn | α‐helix | 310 helix | |
|---|---|---|---|---|---|---|---|
| Crystal | 39 [29.77%] | 63 [48.09%] | 4 [3.05%] | 12 [9.16%] | 10 [7.63%] | 0 | 3 [2.29%] |
| MPT63 | 35.40 ± 2.7 [27.02%] | 62.21 ± 2.5 [47.48%] | 2.94 ± 1.0 [2.24%] | 20.91 ± 2.6 [15.96%] | 7.31 ± 2.5 [5.58%] | 0 | 2.22 ± 1.3 [1.69%] |
| Unfolded | 55.29 ± 9.1 [42.20%] | 16.15 ± 19.2 [12.32%] | 3.38 ± 2.3 [2.58%] | 42.96 ± 9.7 [32.79%] | 10.63 ± 5.3 [8.11%] | 0.71 ± 1.6 [0.54%] | 0.6 ± 2.4 [0.45%] |
| Refold | 47.23 ± 2.8 [36.05%] | 5.16 ± 1.2 [3.93%] | 1.46 ± 1.0 [1.11%] | 56.43 ± 2.9 [43.07%] | 15.69 ± 2.6 [11.97] | 1.23 ± 1.1 [0.94%] | 6.64 ± 1.7 [5.06%] |
| RefoldG25A | 50.95 ± 3.2 [38.89%] | 2.04 ± 2.0 [1.55%] | 2.88 ± 1.3 [2.19%] | 53.21 ± 4.0 [40.61%] | 13.49 ± 3.7 [10.3%] | 2.09 ± 2.1 [1.60%] | 7.34 ± 2.4 [5.60%] |
Percentage values are enclosed by square brackets.
The refolding runs [Fig. 5(C)] obtained by the molecular dynamic simulations show that the helical elements of protein secondary structure have evolved within 100 ns. It may be noted that biophysical measurements and computer simulation studies have already demonstrated that many of the local elements of protein structures can be generated very rapidly. It has been reported that the formation of helix can take place as early as within 100 ns.33, 34 Therefore, our results of local helix formation within the time scale of 100 ns are well correlated with previous experimental and computational findings. A total of eight simulations with different time lengths have been performed. Five refolding simulations continued for 100 ns, while three other simulations were carried out for 200, 500, and 800 ns, respectively. In all cases, an overall increase in the total helical content was observed as the protein was allowed to refold. The percentage of helices was found to increase to ∼6%, compared with ∼1% present in the computationally unfolded starting structure for refolding simulations. The average secondary structure content for the last 20 ns of the trajectories is shown in Table 2. Table 2 also reports average secondary structure content of native crystal structure. To provide more extensive characterization of the secondary structure content of MPT63 produced in these simulations, we calculated the propensity of helices as a function of amino acid residue within the MPT63 sequence [Fig. 6(A)]. From the figure, it is clearly evident that MPT63 generates three additional helices near the vicinity of residues 20, 80, and 130 during refolding. These are found to alternate between 310 helix and alpha‐helices. Several independent simulations were carried out using different random velocities to validate the results. We realize that the heat‐induced denatured starting structure which has been used in our simulation for the refolding simulation may have some residual structures that can bias the result of occurrence of helices. To investigate the effect of these residual structures, assistance from ab‐initio peptide folding study has also been adopted where peptide structure evolves from linear chain of amino acids. The PEP‐Fold3 server,35, 36 an online resource for de novo peptide structure prediction was used for this purpose. These calculations also predict the existence of three probable helices near residues 20, 80, and 130 [Fig. 6(A)]. Although the prediction from the PEP‐Fold3 (without any residual structure) is very similar to a simulation beginning from the heat denatured state, there can be subtle differences between the helical structures. It is difficult for us to understand at this point whether the difference occurs due to the presence of any residual structure, or due the difference in the prediction algorithm. Finally, to find the probable stretches having helix forming potential, we attempt bioinformatics sequence analysis of MPT63. Sequence analysis reveals the existence of eight stretches with chameleon signature within MPT63 such as 19VGQVVL24, 27KVSDLK32, 28VSDLKS33, 34SDLKSST40, 56AIRGSV61, 52ATVNAI57, 51TATVNA56, and 69NARTAD74 as determined from ChSeq database.37 These chameleon sequences show the potential to attain both helical and strand conformations but they are comparatively short to be stabilized by local interaction alone. These sequences principally exist as β‐sheet or coil in the original crystal structure, but may acquire non‐native helical conformation in an intermediate state of the same protein or in a different protein. Interestingly, MPT63 shows the presence of two short overlapping stretches of chameleon sequences KVSDLK (residues 27–32) and VSDLKS (residues 28–33) near the N‐terminus with propensity to form helices as visualized both from refolding simulation and the ab initio study discussed above.
Figure 6.
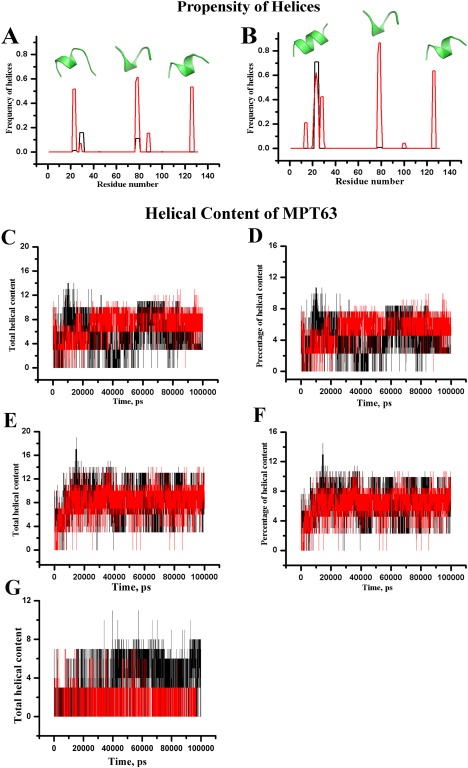
α‐helix (black) and 310 helix (red) as a function of amino acid residues within [A] WT MPT63 and [B] Gly25Ala mutant of MPT63 for refolding run at 300 K staring from an extended conformation of native and mutant MPT63 proteins respectively. Corresponding PEP‐Fold3 server generated helical conformations obtained from linear sequences are shown by green colored cartoon representation. [C] Total helical content and [D] percentage of total helix content of WT MPT63 are plotted as a function of time for two representative refolding simulations as shown by red and black. [E] Total helical content and [F] the percentage of total helix content of Gly25Ala mutant of MPT63. [G] Total helical content of Val23Gly (red) and Leu24Gly (black) MPT63 are plotted as a function of time.
The effect of the Gly25Ala mutation on conformational behavior of MPT63
The simulation studies reported here also indicate that the N‐terminal helix typically contains residues like Val22, Val23, Leu24, and Gly25. It has been noted that the sequence of MPT63 has residues Trp26, Lys27, Val28, Ser29, Asp30, Leu31, and Lys32 next to the Gly25. It is known that Trp, Asp, and Leu have higher propensities toward helix formation,38 and there are several of these neighboring Gly25. As the propensity of Gly to form alpha helix is very low, we mutated this residue to Ala (which has the highest propensity toward helix formation) [Fig. 6(B)]. It has been hypothesized that this mutation would form a continuous alpha helix containing Ala25 and the surrounding residues. As Gly has the lowest helical propensity, we have alternatively designed two Gly substitution mutants (Leu24Gly and Val23Gly) with the expectation that these would result in less helical content.
The Gly25Ala mutant was then subjected to several refolding simulations to determine if this mutation could affect the overall helical content of MPT63. The total and percentage of helical content for both WT MPT63 and Gly25AlaMPT63 have been calculated [Fig. 6(C–F), respectively]. Figure 6 suggests that the helical content of Gly25Ala is enhanced upon Ala substitution. Additionally, we found that the propensity of the adjoining residues to form helices was also increased. Therefore, the substitution of Gly by Ala appears to promote more helical content in the overall conformation of MPT63. The Pep‐Fold3 server also portrays well‐defined helical conformation upon Ala substitution [Fig. 6(B)]. Interestingly, the folding pathways of other two mutants (Leu24Gly and Val23Gly), as studied by time evolution MD simulation, suggest significant decrease in the overall helical content [Fig. 6(G)].
The distributions of the backbone (ϕ, ψ) angles for each residue corresponding to three probable helical regions of WT MPT63 have been analyzed. Data for these residues are pooled together. The distributions of the Ramachandran (ϕ, ψ) angles collected from the simulations indicate that these residues mostly populate helical regions. These data reconfirm the appearance of three intermediate helices while the protein undergoes folding from an unfolded state [Fig. 7(A–C)]. The distributions of ϕ, ψ angles in a Ramachandran plot for Gly25AlaMPT63 [Fig. 7(D–F)] also illustrate the existence of three prominent helices that the mutant may visit in the folding energy landscape. The Gly to Ala substitution (in the Gly25Ala mutant) has been found to be within the allowed region of helical structure of Ramachandran plot. Our results show that the N‐terminal helix is maintained throughout the simulation up to 100 ns for all the refolding runs performed for the Gly25Ala mutant.
Figure 7.
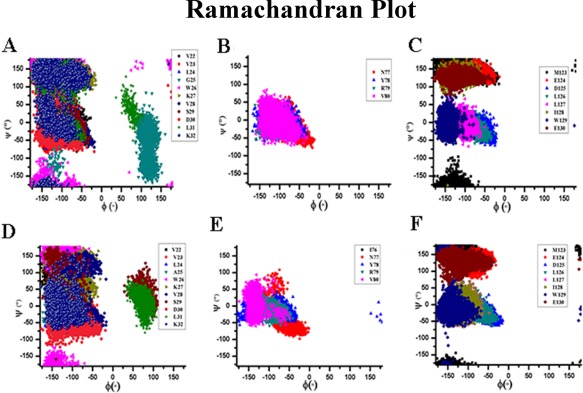
Distribution of Ramachandran plot for residues of three probable helical regions within MPT63 for refolding run at 300 K staring from an extended conformation of [A–C] native MPT63 and [D–F] Gly25Ala mutant MPT63.
Subsequently, we have also used site‐directed mutagenesis to prepare the Gly25Ala mutant to experimentally validate the computational findings. Far‐UV CD at pH 7.5 shows that both WT and the Gly25Ala mutant contain similar secondary structures [Fig. 8(A–C)]. However, at low pH in both the absence and presence of TFE, there is a large increase in the helical content of the Gly25Ala mutant, when compared with the data obtained with the WT protein. The far‐UV CD data has been complemented by FT‐IR measurements [Fig. 8(D,E)].
Figure 8.
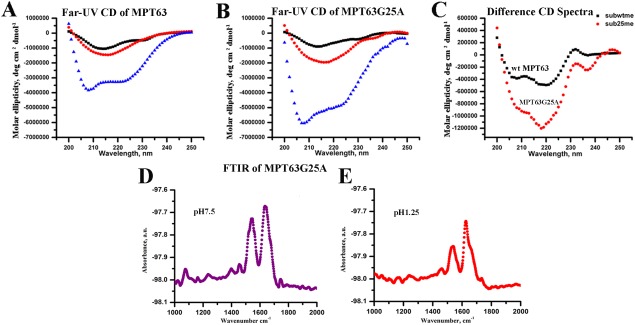
Far‐UV CD data of both [A] WT and [B] Gly25Ala mutant of MPT63 at pH7.5 (black), 1.25 (red), and in the presence of 50% TFE (blue); [C] Difference CD spectrum (obtained by subtracting pH 7.5 data from pH 1.25 data) of WT (black) and Gly25Ala mutant (red). FTIR analysis of MPT63Gly25Ala at [D] pH 7.5 and [E] pH 1.25.
Conclusion
The native structure of the protein is determined by both local interactions between nearby regions and nonlocal interactions mediated by residues distant from each other. Although there exist many controversies regarding the early events of protein folding, it is generally believed that different processes may compete, resulting in significant heterogeneity. It has recently been shown that the hydrophobic clustering and the generation of secondary structure may occur in subsequent steps or in concert depending on solution conditions.27 We have also found that hydrophobic mutation at the hydrophobic core can speed up early contact formation, resulting in misfolding; while similar increase in hydrophobicity elsewhere in the same protein did not seem to have any significant effect.23 At the early stage of folding, local hydrophobic contact formation is expected to predominate over the long distant interactions, which is presumably the reason for the observed helical structure in the intermediate state. As folding proceeds further, these non‐native helices would potentially encounter multiple possible pathways: they may fold correctly into the native structure or they may misfold and aggregate. Alternatively, they may fold into a predominately helical conformer provided sufficient helix inducing conditions (like adding TFE, or using a protein template in which nonlocal interactions support the early helix formation) are present.
In the case of the first possible pathway (folding into the correct β‐sheet structure), the protein needs to break the misfolded helical contacts to form the correct structure. Depending on the position of the rate‐limiting steps of a folding landscape, this process may slow down the overall folding kinetics. The position and resulting effect of rate‐limiting step on the folding kinetics are extensively studied and widely debated. Examples of the second possible pathway may come from the aggregation of alpha synuclein, a natively unfolded protein which aggregates through the formation of a helix intermediate in the presence of lipid membrane.39 Although proteins, particularly those based on beta‐sheet structure, can take a long time to fold,34 we can trace the intermediate conformations that a protein may visit in the folding energy landscape in early folding events. Sequence analysis of MPT63 shows the presence of eight stretches with chameleon signature. These chameleon sequences can fold locally to form either alpha or beta‐sheet secondary structure. The results described here suggest that MPT63 primarily contains three regions with high propensity for helices. One of them is situated at the N‐terminal region and Ala (which is more helically preferable residue) substitution at this region leads to increased helical propensity. This is observed both computationally and experimentally. Formation of such non‐native intermediate structure was also found in another predominantly β‐sheet protein β‐lactoglobulin by the Kuwajima et al. and Goto et al.4, 5 They investigated the folding kinetics of β‐lactoglobulin and revealed a burst‐phase intermediate with non‐native helices with a radius of gyration close to the native protein. From the present studies, it can be suggested that a complete β‐sheet protein MPT63 has a unique helical intermediate during the early stages of its folding which is assumed to be further converted to mature β‐sheet structure.
Materials and Methods
Expression and purification of MPT63 and its mutants
The plasmid pQE30 containing the WT MPT63 gene was kindly provided by Dr. David Eisenberg (University of California, Los Angeles, U.S.). Plasmids were transformed in E. coli XL1‐Blue cells. Site‐directed mutagenesis (Gly20Cys and Gly25Ala) was carried out using a Quick Change Lighting site‐directed mutagenesis kit (Agilent Technologies). The mutations were verified by DNA sequencing.pQE30 was transformed into XL1‐Blue cells and these transformed strains were used to express and purify WT as well as mutants of MPT63. Single colony of transformed XL1‐Blue cells were grown aerobically at 37°C in an LB medium containing 100 μg/mL ampicillin and induced with 1 mM IPTG for 4 h when the absorbance of the medium reaches 0.5 at 600 nm. Cells were harvested by centrifugation at 8000 rpm for 10 min at 4°C and resuspended in 40 mL of sonication buffer (50 mM potassium phosphate, 300 mM KCl, 10 mM imidazole, pH 7.8). Cells were subjected to one cycle of freezing–thawing and 1 mM phenylmethylsulfonyl fluoride (PMSF) was added to the sonication buffer prior to sonication. Cell lysates were then centrifuged at 12,500 rpm for 45 min at 4°C and supernatants were mixed with 2 mL of Ni‐NTA agarose resin (Qiagen), previously equilibrated in sonication buffer, and stirred at 4°C for 1 h. The protein and resin slurry were then loaded onto a column and washed with 10 volumes of sonication buffer followed by 10 volumes of wash buffer (50 mM potassium phosphate K3PO4, 300 mM KCl, 20 mM imidazole, pH 7.8). Protein was eluted in the same buffer with a gradient of 20–500 mM imidazole. Fractions were then analyzed by SDS‐PAGE and those containing the protein were pooled and dialyzed overnight against ResourceQ start buffer (30 mM Tris–HCl, pH 8.7). This dialyzed sample was then applied to a ResourceQ FPLC column pre‐equilibrated in ResourceQ start buffer (30 mM Tris–HCl, 100 mM KCl, pH 8.7). The column was washed with 20 volumes of the same buffer, and the protein was eluted with a 0–500 mM KCl gradient. Fractions containing purified protein as assessed by SDS‐PAGE (purity >90%) were pooled and dialyzed against 20 mM sodium phosphate buffer pH 7.5 and aliquots were stored at −20°C until required.
Circular dichroism spectroscopy
Circular dichroism (CD) spectra were recorded using a Jasco J720 spectropolarimeter (Japan Spectroscopic Ltd.). Far‐UV CD measurements (between 200 and 250 nm) were performed with 10 μM protein using a cuvette with the path length of 1 mm. Eight spectra were collected in continuous mode and averaged. Protein solutions were made in sodium phosphate buffer pH 7.5.
Steady‐state fluorescence spectroscopy
The steady‐state fluorescence emission measurements of MPT63 in different pH conditions (7.5–1.25) were carried out with a PTI (Photon Technology International) Felix32 fluorometer in a cuvette of 1 cm path length and using 1 µM protein concentration at 25°C. For tryptophan fluorescence measurements, the samples were excited at 295 nm to avoid any contribution of tyrosine residues and emission spectra were recorded between 300 and 450 nm.
FTIR spectroscopy
FTIR spectra of MPT63 were measured using a Bruker Tensor 27 FTIR spectrometer. The FTIR spectral readouts were taken at pH 7.5 and 1.25 immediately after dispensing the proteins in respective buffer solutions. Buffer baseline was subtracted before taking each spectrum. The deconvolution of raw spectra in the amide I region (1600–1700 cm−1) and amide II region (1500–1400 cm−1) was done by least‐squares iterative curve fitting to Gaussian line shapes. The assignment of peaks was done using previously described spectral components associated with different secondary structures.40
Fluorescence correlation spectroscopy (FCS) experiments and data analysis
FCS experiments were carried out using an LSM 510 Meta (Carl Zeiss, Germany) with 40× water immersion objective. The Gly20Cys mutant of MPT63 was labeled with Alexa488Maleimide using a previously published procedure.41 The labeled protein samples were excited using an argon laser at 488 nm.
For a single‐component system, the diffusion time (τ D) of a fluorophore and the number of particles (N) can be calculated by fitting the correlation function [G(τ)] to Eq. (3):
| (3) |
where, N is the number of particles in the observation volume and S is the structure parameter, which is the depth‐to‐diameter ratio of the Gaussian observation volume. The diffusion coefficient (D) of the molecule can be calculated from τ D using Eq. (4):
| (4) |
where ω is the beam radius of the observation volume, which can be obtained by measuring the τ D of a fluorophore with a known D. The hydrodynamic radius (r H) of a labeled molecule can be calculated from D using the Stokes–Einstein equation [Eq. (5)]:
| (5) |
where k is the Boltzmann constant, T is the temperature, and η corresponds to the viscosity of the solution.
ANS fluorescence measurements
1‐Aniline‐8‐naphthalene sulfonic acid (ANS) stock solution was prepared in methanol. Final ANS and protein concentration used in the experiments were 100 and 1 µM, respectively. Prior to the experiments, ANS was added to the samples and the solutions were incubated for 10 min before the measurements of ANS fluorescence. Fluorescence spectra of ANS‐containing samples were recorded using an excitation wavelength of 370 nm and emission spectra were recorded between 450 and 650 nm at 25°C.
Thioflavin T assay
Thioflavin T (ThT) stock solution was prepared in 20 mM sodium phosphate buffer, pH 7.5. Protein solution containing 20 µM of MPT63 was incubated in different buffer solutions. Aliquots were taken at different time incubation and diluted to a final protein concentration of 1 µM. Then ThT solution was added (final ThT concentration 5 µM) and mixed well before fluorescence measurements. Fluorescence measurements were performed at 25°C by exciting the sample at 450 nm and monitoring the fluorescence emission from 460 to 550 nm.
Molecular dynamics simulation
The 1.5 Å resolution crystal structure of Mycobacterium tuberculosis‐MPT63 containing 131 residues (PDB ID# 1LMI)9 was chosen as the starting configuration for explicit all atom simulations. Crystallographic water molecules were removed from the system. Simulations were performed at room temperature (300 K) and pH 7.5. To equilibrate the protein to final pH conditions, protonation states of the acidic and basic residues were adjusted based on site‐specific pKa values estimated using the PROPKA framework in the PDB2PQ web server.42 The protein was immersed in a triclinic box of SPC/E43 type explicit water molecules. All protein atoms were at a distance equal to 10 Å from the box edges. The system at physiological pH was neutralized by adding 4 Na+ ions. Energy minimization was carried out using the steepest descent method followed by equilibration. All systems were equilibrated in two stages, applying position restraints to all heavy atoms. The first phase employed a canonical (NVT) ensemble for 100 ps. The second phase of equilibration employed an isothermal–isobaric ensemble (NPT) ensemble for 100 ps. Temperature and pressure were maintained at 300 K and 1 Bar, respectively. The equilibrated system was then subjected to production MD simulation.
All molecular dynamics simulations were carried out using GROMACS 4.6.144 with OPLS force‐field45, 46 in an isothermal–isobaric (NPT) ensemble using periodic boundary conditions. Constant temperature and pressure was maintained using v‐rescale47 and Parrinello–Rahman48 algorithms, respectively, with coupling constants of 0.1 and 2 ps, respectively. Long‐range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method49, 50 and a 10 Å cut‐off was set for van der Waals and short‐range electrostatic interactions. Equations of motion were integrated every 2 fs using the leap‐frog algorithm.
The nonbonded pair list was updated every 10 steps and conformations were stored every 20 ps. Secondary structure analysis was performed using the program DSSP.51 Other analyses were performed using scripts included with the GROMACS (D. van der Spoel, E. Lindahl, B. Hess, and the GROMACS development team, GROMACS User Manual version 4.6.5, www.gromacs.org, 2013) distribution. The visual analysis of protein structures was carried out using Rasmol52 and Pymol (W.L. DeLano, The PyMOL Molecular Graphics System, 2002, Internet http://www.pymol.org).
One high‐temperature unfolding simulation of MPT63 was performed at 550 K for 20 ns. The end point of that simulation was used as starting structure for conducting several refolding runs at 300 K. Eight independent trajectories were produced by generating different random velocities at the outset of NVT. The Gly25 residue of the denatured end point simulation structure of MPT63 derived from 550 K simulation was mutated to alanine using the mutagenesis utility of Pymol. In a similar manner, two other mutants (Val23Gly and Leu24Gly) were constructed. The mutant proteins were again subjected to several refolding simulations at 300 K.
Acknowledgments
This study has been funded by CSIR network project grant HOPE. KC thanks the director, CSIR‐IICB, for his support. AK thanks the UGC for a fellowship. SK thanks the Start‐up Research Grant for Young Scientists. Access to the supercomputing facility of CSIR‐4Pi is gratefully acknowledged.
References
- 1. Wright PE, Dyson HJ, Lerner RA (1988) Conformation of peptide fragments of proteins in aqueous solution: implications for initiation of protein folding. Biochemistry 27:7167–7175. [DOI] [PubMed] [Google Scholar]
- 2. Dyson HJ, Sayre JR, Merutka G, Shin H‐C, Lerner RA, Wright PE (1992) Folding of peptide fragments comprising the complete sequence of proteins: models for initiation of protein folding II. Plastocyanin. J Mol Biol 226:819–835. [DOI] [PubMed] [Google Scholar]
- 3. Dyson HJ, Merutka G, Waltho JP, Lerner RA, Wright PE (1992) Folding of peptide fragments comprising the complete sequence of proteins: models for initiation of protein folding I. Myohemerythrin. J Mol Biol 226:795–817. [DOI] [PubMed] [Google Scholar]
- 4. Kuwajima K, Yamaya H, Sugai S (1996) The burst‐phase intermediate in the refolding of β‐lactoglobulin studied by stopped‐flow circular dichroism and absorption spectroscopy. J Mol Biol 264:806–822. [DOI] [PubMed] [Google Scholar]
- 5. Hamada D, Segawa S‐i, Goto Y (1996) Non‐native α‐helical intermediate in the refolding of β‐lactoglobulin, a predominantly β‐sheet protein. Nat Struct Mol Biol 3:868–873. [DOI] [PubMed] [Google Scholar]
- 6. Chen E, Everett ML, Holzknecht ZE, Holzknecht RA, Lin SS, Bowles DE, Parker W (2010) Short‐lived α‐helical intermediates in the folding of β‐sheet proteins. Biochemistry 49:5609–5619. [DOI] [PubMed] [Google Scholar]
- 7. Sarkar‐Banerjee S, Chowdhury S, Paul SS, Dutta D, Ghosh A, Chattopadhyay K (2016) Non‐native helical intermediate state may accumulate at low pH in the folding/aggregation landscape of the intestinal fatty acid binding protein. Biochemistry 55:4457–4468. [DOI] [PubMed] [Google Scholar]
- 8. Ghosh R, Mukherjee M, Chattopadhyay K, Ghosh S (2012) Unusual optical resolution of all four tryptophan residues in MPT63 protein by phosphorescence spectroscopy: assignment and significance. J Phys Chem B 116:12489–12500. [DOI] [PubMed] [Google Scholar]
- 9. Goulding CW, Parseghian A, Sawaya MR, Cascio D, Apostol MI, Gennaro ML, Eisenberg D (2002) Crystal structure of a major secreted protein of Mycobacterium tuberculosis—MPT63 at 1.5‐Å resolution. Protein Sci 11:2887–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lyashchenko K, Colangeli R, Houde M, Al Jahdali H, Menzies D, Gennaro ML (1998) Heterogeneous antibody responses in tuberculosis. Infect Immun 66:3936–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Manca C, Lyashchenko K, Wiker HG, Usai D, Colangeli R, Gennaro ML (1997) Molecular cloning, purification, and serological characterization of MPT63, a novel antigen secreted by Mycobacterium tuberculosis . Infect Immun 65:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mukherjee M, Ghosh R, Chattopadhyay K, Ghosh S (2016) Stepwise unfolding of a multi‐tryptophan protein MPT63 with immunoglobulin like fold: detection of zone‐wise perturbation during guanidine hydrochloride induced unfolding using phosphorescence spectroscopy. RSC Adv 6:61077–61087. [Google Scholar]
- 13. Czajkowsky DM, Iwamoto H, Cover TL, Shao Z (1999) The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc Natl Acad Sci USA 96:2001–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bauerfeind P, Garner R, Dunn B, Mobley H (1997) Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut 40:25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Small P, Blankenhorn D, Welty D, Zinser E, Slonczewski JL (1994) Acid and base resistance in Escherichia coli and Shigella flexneri: role of rpoS and growth pH. J Bacteriol 176:1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duret G, Simonet V, Delcour AH (2007) Modulation of Vibrio cholerae porin function by acidic pH. Channels 1:70–79. [DOI] [PubMed] [Google Scholar]
- 17. Vandal OH, Nathan CF, Ehrt S (2009) Acid resistance in Mycobacterium tuberculosis . J Bacteriol 191:4714–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhansali S (1977) Abdominal tuberculosis. Experiences with 300 cases. Am J Gastroenterol 67:324–337. [PubMed] [Google Scholar]
- 19. Marshall JB (1993) Tuberculosis of the gastrointestinal tract and peritoneum. Am J Gastroenterol 88:989–999. [PubMed] [Google Scholar]
- 20. Rucker AL, Pager CT, Campbell MN, Qualls JE, Creamer TP (2003) Host‐guest scale of left‐handed polyproline II helix formation. Proteins 53:68–75. [DOI] [PubMed] [Google Scholar]
- 21. Vuilleumier S, Sancho J, Loewenthal R, Fersht AR (1993) Circular dichroism studies of barnase and its mutants: characterization of the contribution of aromatic side chains. Biochemistry 32:10303–10313. [DOI] [PubMed] [Google Scholar]
- 22. Fink AL, Calciano LJ, Goto Y, Kurotsu T, Palleros DR (1994) Classification of acid denaturation of proteins: intermediates and unfolded states. Biochemistry 33:12504–12511. [DOI] [PubMed] [Google Scholar]
- 23. Sarkar S, Chattopadhyay K (2014) Studies of early events of folding of a predominately β‐sheet protein using fluorescence correlation spectroscopy and other biophysical methods. Biochemistry 53:1393–1402. [DOI] [PubMed] [Google Scholar]
- 24. Barth A (2007) Infrared spectroscopy of proteins. Biochim Biophys Acta 1767:1073–1101. [DOI] [PubMed] [Google Scholar]
- 25. Kuwajima K (1989) The molten globule state as a clue for understanding the folding and cooperativity of globular‐protein structure. Proteins 6:87–103. [DOI] [PubMed] [Google Scholar]
- 26. Lim V (1974) Structural principles of the globular organization of protein chains. A stereochemical theory of globular protein secondary structure. J Mol Biol 88:857–862. [DOI] [PubMed] [Google Scholar]
- 27. Haldar S, Chattopadhyay K (2012) Interconnection of salt‐induced hydrophobic compaction and secondary structure formation depends on solution conditions. Revisiting early events of protein folding at single molecule resolution. J Biol Chem 287:11546–11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ghosh S, Ghosh C, Nandi S, Bhattacharyya K (2015) Unfolding and refolding of a protein by cholesterol and cyclodextrin: a single molecule study. Phys Chem Chem Phys 17:8017–8027. [DOI] [PubMed] [Google Scholar]
- 29. Paramanik B, Kundu A, Chattopadhyay K, Patra A (2014) Study of binding interactions between MPT63 protein and Au nanocluster. RSC Adv 4:35059–35066. [Google Scholar]
- 30. Ortega A, Amorós D, de La Torre JG (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic‐and residue‐level models. Biophys J 101:892–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ (1999) Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 38:16424–16431. [DOI] [PubMed] [Google Scholar]
- 32. Hamada D, Kuroda Y, Tanaka T, Goto Y (1995) High helical propensity of the peptide fragments derived from β‐lactoglobulin, a predominantly β‐sheet protein. J Mol Biol 254:737–746. [DOI] [PubMed] [Google Scholar]
- 33. Snow CD, Nguyen H, Pande VS, Gruebele M (2002) Absolute comparison of simulated and experimental protein‐folding dynamics. Nature 420:102–106. [DOI] [PubMed] [Google Scholar]
- 34. Dobson CM (2003) Protein folding and misfolding. Nature 426:884–890. [DOI] [PubMed] [Google Scholar]
- 35. Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, Tufféry P (2016) PEP‐FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res 44:W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maupetit J, Derreumaux P, Tuffery P (2009) PEP‐FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res 37:W498–W503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li W, Kinch LN, Karplus PA, Grishin NV (2015) ChSeq: a database of chameleon sequences. Protein Sci 24:1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pace CN, Scholtz JM (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys J 75:422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trexler AJ, Rhoades E (2009) αSynuclein binds large unilamellar vesicles as an extended helix. Biochemistry 48:2304–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kong J, Yu S (2007) Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim Biophys Sinica 39:549–559. [DOI] [PubMed] [Google Scholar]
- 41. Chattopadhyay K, Saffarian S, Elson EL, Frieden C (2002) Measurement of microsecond dynamic motion in the intestinal fatty acid binding protein by using fluorescence correlation spectroscopy. Proc Natl Acad Sci USA 99:14171–14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 35:W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Berendsen H, Grigera J, Straatsma T (1987) The missing term in effective pair potentials. J Phys Chem 91:6269–6271. [Google Scholar]
- 44. Hess B, Kutzner C, Van Der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load‐balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447. [DOI] [PubMed] [Google Scholar]
- 45. Jorgensen WL, Tirado‐Rives J (1988) The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc 110:1657–1666. [DOI] [PubMed] [Google Scholar]
- 46. Jorgensen WL, Maxwell DS, Tirado‐Rives J (1996) Development and testing of the OPLS all‐atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225–11236. [Google Scholar]
- 47. Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:014101. [DOI] [PubMed] [Google Scholar]
- 48. Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. [Google Scholar]
- 49. Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. [Google Scholar]
- 50. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593. [Google Scholar]
- 51. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 52. Sayle RA, Milner‐White EJ (1995) RASMOL: biomolecular graphics for all. Trends Biochem Sci 20:374–376. [DOI] [PubMed] [Google Scholar]