

Abstract
Purpose of review
The human body produces and removes 1011 platelets daily to maintain a normal steady-state platelet count. Platelet production must be tightly regulated to avoid spontaneous bleeding or arterial occlusion and organ damage. Multifaceted and complex mechanisms control platelet removal and production in physiological and pathological conditions. This review will focus on different mechanisms of platelet clearance, with focus on the biological significance of platelet glycans.
Recent findings
The Ashwell-Morrell receptor (AMR) recognizes senescent, desialylated platelets under steady state conditions. Desialylated platelets and the AMR are the physiological ligand-receptor pair regulating hepatic thrombopoietin (TPO) mRNA production, resolving the longstanding mystery of steady state TPO regulation. The AMR-mediated removal of desialylated platelets regulates TPO synthesis in the liver by recruiting JAK2 and STAT3 to increase thrombopoiesis.
Summary
Inhibition of TPO production downstream of the hepatic AMR-JAK2 signaling cascade could additionally contribute to the thrombocytopenia associated with JAK1/2 treatment, which is clinically used in myeloproliferative neoplasms.
Keywords: Ashwell-Morrell receptor, desialylated platelets, JAK2, STAT3, thrombopoietin
Introduction
While the primary function of platelets is hemostasis, platelets also participate in antimicrobial host defense, secrete cytokines that can induce inflammation and growth factors that aid tissue repair. Chronic inflammation is often associated with reactive high platelet counts, and responses to acute infections may be accompanied by sudden reduction or increase of platelets (thrombocytopenia or thrombocytosis, respectively), placing platelets as reporters of disease progression or healing. A steady platelet supply is ensured by a continuous platelet clearance and production of ~1011 platelets daily, and the rate of production rises sharply under conditions of platelet destruction to maintain levels of 150,000–400,000 platelets per microliter of blood. Platelet clearance and production must be therefore regulated to avoid spontaneous bleeding or arterial occlusion and organ damage, however both processes remain poorly understood. This review will focus on the current knowledge of platelet clearance and briefly address mechanisms of production.
Thrombopoiesis
Platelet production is a complex process that requires differentiation of hematopoietic stem cells (HSCs) into specialized progenitors and their organized interplay with the bone marrow microenvironment and hematopoietic cytokines. Data support the existence of two anatomical and functional marrow microenvironmental “niches”: the osteoblastic niche and the vascular niche [1, 2]. Megakaryocyte (MK) maturation and platelet formation are dependent on cellular migration from the osteoblastic to the vascular niche, where once adequately mature, MKs extend proplatelet processes through or between cells of the sinusoidal endothelial layer and shed platelets into the bloodstream [3]. Marrow stromal cells are an integral part of these local microenvironments through expression of soluble and surface-bound cytokines, counter-receptors for integrins and other adhesion molecules on the surface of hematopoietic cells, and the secretion of extracellular macromolecules [4]. In reference to MK development, marrow stromal cells have been shown to secrete thrombopoietin (TPO), the primary regulator of thrombopoiesis [5], and CXCL12, a primary chemokine that attracts MKs and other hematopoietic cells to the marrow microenvironment [6, 7]. Additionally, CXCL12 acts to stimulate MKs to express cell surface SCF [8], which synergistically promotes MK growth with TPO [9], and to express VCAM-1 and fibronectin, which promote cell growth through their binding to the MK integrin α4β1 [10, 11]. The interaction of microenvironmental Von Willebrand Factor (VWF) and its MK receptor glycoprotein (GP) Ib-IX appears important for platelet formation and release, whereas in contrast, type I collagen, which localizes to the osteoblastic niche, prevents platelet formation [12]. Recent studies point to the fact that glycans, specifically glycan synthesis by the β1,4-galactosyltransferase 1, are key elements in hematopoiesis regulating HSC function and MK migration (S.G., K.M.H. et al, unpublished data).
A major milestone in understanding the molecular mechanisms of thrombopoiesis was the discovery of TPO in 1994. TPO is the primary regulator of platelet production, supporting the survival, proliferation and differentiation of the platelet precursors, bone marrow MKs [13–15]. Since the discovery of TPO many molecular mechanisms of thrombopoiesis have been identified, including the development of polyploidy and proplatelet formation, the final fragmentation of the MK cytoplasm to yield blood platelets, and the regulation of this process [14, 16–19]. Although much progress has been made towards the understanding of thrombopoiesis, multiple unanswered questions remain. One unanswered question is the regulation of TPO production under steady state and under pathologic conditions.
Multiple organs display RNA transcripts, with hepatocytes having the highest levels and being the primary cells responsible for the production and secretion of TPO into the bloodstream. In one model hepatic TPO production has been identified as constitutive, where TPO serum levels are maintained solely by its uptake and metabolism by platelets and MKs [20–24]. Thus, the removal and destruction of TPO released into the bloodstream is mediated by expression of its receptor, Mpl. In patients with thrombocytopenia little of the hepatocyte produced TPO is presumed to be removed by platelets and TPO blood levels rise; in contrast, thrombocytosis should be accompanied by low steady state levels of blood TPO, because platelet mediated TPO destruction surpasses TPO production [20–24]. Additional support for this model comes from Thpo+/− mice [25], where loss of one Thpo allele leads to a 40% reduction of platelet counts, and from a rabbit model of busulfan-induced thrombocytopenia [20].
A growing body of evidence lends credence to the assertion that platelet TPO metabolism is not the sole determinant of plasma TPO levels, implying that TPO levels are regulated in a more complex fashion. A number of inflammatory states are associated with thrombocytosis and increased blood TPO levels (e.g., ulcerative colitis, rheumatoid arthritis, ovarian cancer) [5, 26–34]. The inflammation-induced increase in TPO expression is mediated by IL-6, which stimulates hepatocyte TPO production both in vitro and in vivo [31, 32, 35, 36]. Selective liver irradiation also stimulates hepatocyte TPO production in humans [37]. In contrast to the “autoreguation” model of blood TPO levels, serum TPO levels are lower than expected in patients with Immune Thrombocytopenia (ITP) [38, 39], and high in patients with Essential Thrombocythemia (ET) [34, 40]. The notion that TPO production is regulated, rather than autonomous, is further supported by data showing that marrow stromal cell produce TPO in response to thrombocytopenia both in mice and in humans [5, 41].
In addition to its effects on MK progenitors and mature cells, TPO affects HSCs in vitro, especially when used in combination with IL-3 or SCF [42, 43]. HSCs express Mpl on their surface, indicating the stem cell effects of TPO are direct [44, 45]. Recently, mice lacking Mpl expression on MKs and platelets but expressing Mpl normally on stem/progenitor cells (Mplfl/fl Pf4-Cre mice) were generated. Despite lacking Mpl on MKs and platelets, Mplfl/fl Pf4-Cre mice displayed profound megakaryocytosis and thrombocytosis with a remarkable expansion of MK-committed and multipotential progenitor cells, the latter displaying biological responses and a gene expression signature indicative of chronic TPO overstimulation as the underlying causative mechanism. Even more surprising was the normal circulating TPO levels in these mice. Thus, the authors concluded that TPO signaling in MKs is dispensable for platelet production. The key role of TPO signaling is in controlling platelet numbers via generation and stimulation of the bipotential MK precursors. On the other hand, Mpl expression in MKs and platelets is essential to prevent megakaryocytosis and myeloproliferation by restricting the amount of TPO available to stimulate the production of MKs from the progenitor cell pool [46]. This is an intriguing finding as Mplfl/fl Pf4-Cre mice were obviously able to “bypass” the lack of Mpl on the MK lineage, presenting more evidence that circulating TPO levels are regulated in a complicated manner.
A new model (detailed below) furthers our understanding of the regulation of blood TPO levels and thrombopoiesis: desialylated, senescent platelet clearance via the hepatic Ashwell-Morrell Receptor (AMR) enhances hepatic TPO production. In support of this notion, injection of desialylated platelets in rabbits stimulates platelet production [47], presumably by stimulating liver TPO secretion.
Platelet clearance
Several mechanisms mediate platelet clearance. One mechanism appears to function via aging (senescence) induced signals, via glycan degradation and apoptotic mechanisms. Platelets are also removed by immune (antibody) responses. Whether these mechanisms converge at a certain points during platelet-life time is unclear.
The aging (senescence) induced platelet clearance
Studies have shown that platelet surface glycans mediate platelet clearance [48, 49]. Recently, loss of sialic acid has been identified as a determinant of senescent platelet removal [50]. Platelets lose sialic acid during circulation and are cleared via the hepatic AMR, a transmembrane heteroligomeric glycoprotein complex composed of ASGPR1 (CLEC4H1, HL-1) and ASGPR2 (CLEC4H2, HL-2) subunits. This highly conserved receptor has been largely regarded as an endocytic receptor [51], and since its discovery four decades ago the regulatory role of the hepatic AMR has remained largely unclear. Specifically, mice lacking either ASGPR1 or ASGPR2 subunit do not accumulate plasma proteins or lipids lacking sialic acid, which has been the predicted outcome of eliminating one of the AMR subunits [51]. It has therefore been a surprising discovery that platelets with reduced α2,3-linked sialic acid during sepsis, after cold storage (in vitro aging), or in mice lacking the sialyltransferase ST3GalIV are cleared by the hepatic endocytic AMR [52–55].
These findings led subsequently to the discovery that removal of senescent, sialic acid deprived platelets drives hepatic TPO mRNA expression in vivo and in vitro via JAK2 and STAT3 to increase MK numbers and de novo platelet production. The notion that loss of sialic acid determines platelet life span is not entirely novel [52, 53, 55–58], however, the recent study elucidates that aged, desialylated platelets regulate hepatic TPO mRNA production in vivo via the AMR. This feedback mechanism presents the AMR-desialylated platelet pair as the critical control point for TPO homeostasis and shows that TPO expression in hepatocytes is regulated and not constitutive (Figure 1). Importantly, disruption of AMR-desialylated platelet signaling by the JAK1/2 inhibitors AZD1480, TG101348 and BMS911543 adversely affect hepatic TPO mRNA expression and secretion in hepatocytes in vitro and in vivo [50]. Thrombocytopenia is a common adverse event of JAK1/2 inhibitor treatment, which is clinically used in myeloproliferative neoplasms (MPNs) [59, 60]. JAK1/2 inhibitors target hematopoietic stem and precursor cell mutant JAK2-V617F as well as wild-type JAK2, activation of which is essential for red blood cell and platelet production [61, 62]. This new study indicates that inhibition of TPO production downstream of the hepatic AMR-JAK2 signaling cascade could additionally contribute to the thrombocytopenia associated with JAK1/2 treatment. Clinical studies are necessary to investigate this notion.
Figure 1. Scheme of hepatic TPO production via JAK2-STAT3 signaling after desialylated platelet uptake by the AMR.
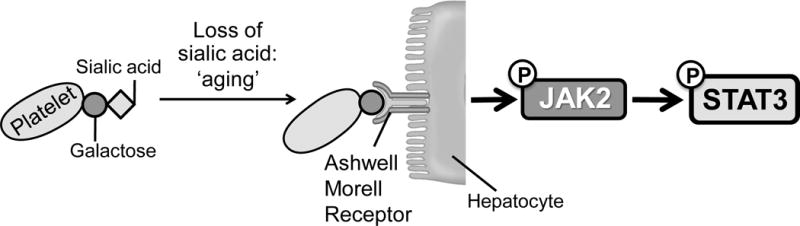
Desialylated, senescent platelets are recognized by the hepatic AMR to regulate hepatic TPO production, bone marrow homeostasis and thrombopoiesis. Bone marrow MKs produce and release young sialic acid (purple ring) containing platelets into the blood stream. Young platelets maximally internalize TPO through Mpl receptors. Circulating platelets become desialylated by active blood-borne sialidases as they age (dashed purple ring), thereby becoming ligands for the AMR and being ingested by hepatocytes. Desialylated platelet ingestion positively stimulates hepatic TPO mRNA expression via JAK2-STAT3 signaling and TPO release in the circulation, thereby regulating bone marrow homeostasis and thrombopoiesis.
The AMR-IL-6R connection
Interestingly, the AMR signaling cascade, which involves JAK2 phosphorylation and STAT3 phosphorylation and translocation to the nucleus shares similarities with that of IL-6 [63]. IL-6 stimulates TPO mRNA expression in hepatocytes in vivo and in HepG2 and Hep3B cells in vitro [31, 32, 35, 64]. These data suggest that both desialylated platelets and IL-6 lead to STAT3-mediated hepatic TPO mRNA expression downstream of the AMR-JAK2 and IL6 receptor (IL-6R)-JAK1 signaling cascades, respectively.
Is there a cross-talk between the AMR and IL-6R? Binding of IL-6 to its hepatic receptor engages the signal transducing subunit gp130, leading to STAT3 tyrosine phosphorylation and activation by gp130-associated JAK1. Whether JAK2 and STAT3 directly associate with the AMR or require gp130 remains to be determined. A tyrosine kinase of 127 kDa (JAK2?) constitutively associates with the AMR ASGPR1 subunit in HepG2 cells [65]. It is therefore possible that both IL-6 and desialylated platelets lead to STAT3-mediated hepatic TPO mRNA expression downstream of JAK1 and JAK2, respectively. Hepatic STAT3 controls the transcription of mRNA for acute phase plasma proteins [66]. Since both the AMR and IL-6R share signaling through STAT3, it is therefore tempting to speculate that acute phase proteins are produced in response to AMR ligation, which would establish clearance of desialylated platelets as a component of the acute phase response. Consistent with this hypothesis, the AMR-mediated removal of desialylated platelets improves the probability of host survival during sepsis [54, 55]. Separate studies have shown that liver regeneration following injury is promoted by platelets [67] and requires AMR and hepatic STAT3 function [68, 69]. Thus, the platelet-AMR-STAT3 signaling cascade may connect desialylated platelets to inflammatory responses.
Apoptosis as a platelet clearance mechanism
Platelet survival also depends on the interplay between prosurvival and proapoptotic members of the Bcl-2 family, which are critical regulators of the intrinsic apoptotic pathway. Platelet survival is extended in mice lacking the proapoptotic proteins Bak and Bax, whereas platelet clearance is accelerated in mice lacking the prosurvival proteins Bcl-2, Bcl-xL and Mcl-1, and in mice treated with the BH3 mimetic ABT-737 (inhibitor of prosurvival Bcl-2, Bcl-xL and Bcl-w) [70–72]. Whether members of the Bcl-2 family alter platelet surface sialic acid content is unclear. Interestingly, the primary platelet clearance site following administration of ABT-737 is the liver in dogs presumably via scavenger receptors [73], whereas the spleen does not appear to regulate the platelet life span in mice [72]. More data are needed to establish if glycan degradation in vivo, i.e. sialic acid loss, triggers the intrinsic apoptotic machinery in platelets, linking glycan degradation and intrinsic apoptotic machinery in the clearance mechanisms regulating platelet survival. Interestingly, data show that newborn and adult mice have similar platelet production rates, but neonatal platelets survived 1 day longer in circulation [74]. A study of pro-apoptotic and anti-apoptotic Bcl-2 family proteins shows that neonatal platelets have higher levels of the anti-apoptotic protein Bcl-2 and are more resistant to apoptosis induced by the Bcl-2/Bcl-xL inhibitor ABT-737 than adult platelets. However, genetic ablation or pharmacologic inhibition of Bcl-2 alone does not shorten neonatal platelet survival or reduce platelet counts in newborn mice, indicating the existence of redundant or alternative mechanisms mediating the prolonged lifespan of neonatal platelets [74]. Whether glycans (increase in platelet surface sialic acid) play a role in the prolonged survival of neonatal platelets remains to be established.
Antibody mediated platelet clearance
ITP is a common bleeding disorder caused primarily by autoantibodies directed against platelet integrin αIIbβ3 (GPIIb-IIIa) and/or the GPIb-IX complex. The prevailing model posits that antibody-mediated platelet destruction occurs in the spleen [75], where the interaction between the Fc portion of platelet-associated autoantibodies and Fcγ receptors (FcγRs) on macrophages initiates phagocytosis. However, data show that, in contrast to anti-αIIbβ3-mediated ITP, anti-GPIbα-mediated ITP is often refractory to therapies targeting FcγR pathways or splenectomy. Recent findings show that certain anti-GPIbα-antibodies trigger platelet desialylation, a process that deviates platelet clearance from splenic macrophage Fc-receptors to the liver, likely via the AMR [76], showing that FcγR-independent mechanisms of ITP exist [77]. In this regard, an adult chronic ITP patient with an anti-GPIb-IX autoantibody, who was resistant to corticosteroids, IVIG, recombinant human TPO, rituximab, danazol and vindesine (Eldisine®), has been successfully treated with oseltamivir phosphate, a sialidase inhibitor used to treat influenza [78]. The mechanism how anti-GPIbα antibody binding leads to desialylation remains to be established. It is likely that platelets secrete active sialidases (i.e. Neu1 and Neu3) upon platelet antibody binding and/or activation [79]. The notion that the AMR plays a significant role in the clearance of anti-GPIbα-opsonized and desialylated platelets provides a potential explanation for refractoriness to splenectomy, as well as to steroid and IVIG therapies. Recent data also show that platelet destruction in ITP patients is mediated by CD8+ cytotoxic T lymphocytes (CTLs) [80].
Conclusion
Platelet counts are controlled in a multifaceted, complex manner. Recent evidence shows that the AMR recognizes senescent, desialylated platelets under steady state conditions. Desialylated platelets and the AMR are the physiological ligand-receptor pair regulating hepatic TPO mRNA production, resolving the longstanding mystery of steady state TPO regulation. The AMR-mediated removal of desialylated platelets regulates TPO synthesis in the liver by recruiting JAK2 and STAT3 to increase thrombopoiesis. Senescent platelets are also removed from the circulation by apoptotic signals. Platelets are cleared by antibody binding to platelets via macrophage FcγRs during pathologic conditions. Recent findings suggest that antibodies can induce platelet desialylation, thereby converging signals for platelet removal with immune mediated platelet removal. Many questions remain concerning the mechanisms governing platelet numbers. How do the above processes work together to maintain platelet numbers? Do clearance systems communicate with the bone marrow environment to ensure adequate thrombopoiesis? Research as well as clinical trials will continue to elucidate the mechanisms that regulate billions of circulating platelets.
Key points.
The AMR recognizes senescent, desialylated platelets under steady state conditions.
Desialylated platelets and the AMR are the physiological ligand-receptor pair regulating hepatic TPO mRNA production.
The AMR-mediated removal of desialylated platelets regulates TPO synthesis in the liver by recruiting JAK2 and STAT3 to increase thrombopoiesis.
Acknowledgments
None
Financial Support and Sponsorship
This work was supported by National Institutes of Health grants HL089224 and HL107146 (to K.M.H.) and the Brigham Research Institute Fund to Sustain Research Excellence (to H.F.).
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Calvi L, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Natute. 2003;425(6960):841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 2.Kiel MJ, Morrison SJ. Maintaining hematopoietic stem cells in the vascular niche. Immunity. 2006;25(6):862–4. doi: 10.1016/j.immuni.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Junt T, et al. Dynamic visualization of thrombopoiesis within bone marrow. Science. 2007;317(5845):1767–70. doi: 10.1126/science.1146304. [DOI] [PubMed] [Google Scholar]
- 4.Malara A, et al. Megakaryocytes contribute to the bone marrow-matrix environment by expressing fibronectin, type IV collagen, and laminin. Stem Cells. 2014;32(4):926–37. doi: 10.1002/stem.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarty J, et al. Murine thrombopoietin mRNA levels are modulated by platelet count. Blood. 1995;86(10):3668–3875. [PubMed] [Google Scholar]
- 6.Hodohara K, et al. Stromal cell-derived factor-1 (SDF-1) acts together with thrombopoietin to enhance the development of megakaryocytic progenitor cells (CFU-MK) Blood. 2000;95(3):769–75. [PubMed] [Google Scholar]
- 7.Sugiyama T, et al. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–88. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 8.Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90(4):1345–64. [PubMed] [Google Scholar]
- 9.Broudy VC, Lin NL, Kaushansky K. Thrombopoietin (c-mpl ligand) acts synergistically with erythropoietin, stem cell factor, and interleukin-11 to enhance murine megakaryocyte colony growth and increases megakaryocyte ploidy in vitro. Blood. 1995;85(7):1719–26. [PubMed] [Google Scholar]
- 10.Avraham H, et al. Characterization of adhesive interactions between human endothelial cells and megakaryocytes. J Clin Invest. 1993;91(6):2378–84. doi: 10.1172/JCI116470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox NE, Kaushansky K. Engagement of integrin alpha4beta1 enhances thrombopoietin-induced megakaryopoiesis. Exp Hematol. 2005;33(1):94–9. doi: 10.1016/j.exphem.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Balduini A, et al. Adhesive receptors, extracellular proteins and myosin IIA orchestrate proplatelet formation by human megakaryocytes. J Thromb Haemost. 2008;6(11):1900–7. doi: 10.1111/j.1538-7836.2008.03132.x. [DOI] [PubMed] [Google Scholar]
- 13.Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115(12):3339–33947. doi: 10.1172/JCI26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaushansky K. Determinants of platelet number and regulation of thrombopoiesis. Hematology Am Soc Hematol Educ Program. 2009:147–52. doi: 10.1182/asheducation-2009.1.147. [DOI] [PubMed] [Google Scholar]
- 15.Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med. 2009;60:193–206. doi: 10.1146/annurev.med.60.042307.181154. [DOI] [PubMed] [Google Scholar]
- 16.Machlus KR, Thon JN, Italiano JE., Jr Interpreting the developmental dance of the megakaryocyte: a review of the cellular and molecular processes mediating platelet formation. Br J Haematol. 2014;165(2):227–36. doi: 10.1111/bjh.12758. [DOI] [PubMed] [Google Scholar]
- 17.Bender M, et al. Dynamin 2-dependent endocytosis is required for normal megakaryocyte development. Blood. 2014 doi: 10.1182/blood-2014-07-587857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, et al. A novel role of sphingosine 1-phosphate receptor S1pr1 in mouse thrombopoiesis. J Exp Med. 2012;209(12):2165–81. doi: 10.1084/jem.20121090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaushansky K. Thrombopoiesis. Semin Hematol. 2015;52(1):4–11. doi: 10.1053/j.seminhematol.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Kuter DJ, Rosenberg RD. The reciprocal relationship of thrombopoietin (c-Mpl ligand) to changes in the platelet mass during busulfan-induced thrombocytopenia in the rabbit. Blood. 1995;85(10):2720–30. [PubMed] [Google Scholar]
- 21.Cohen-Solal K, et al. Constitutive expression of Mpl ligand transcripts during thrombocytopenia or thrombocytosis. Blood. 1996;88(7):2578–28. [PubMed] [Google Scholar]
- 22.Fielder P, et al. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood. 1996;87(6):2154–61. [PubMed] [Google Scholar]
- 23.Shinjo K, et al. Serum thrombopoietin levels in patients correlate inversely with platelet counts during chemotherapy-induced thrombocytopenia. Leukemia. 1998;12(3):295–300. doi: 10.1038/sj.leu.2400946. [DOI] [PubMed] [Google Scholar]
- 24.Engel C, et al. Endogenous thrombopoietin serum levels during multicycle chemotherapy. Br J Haematol. 1999;105(3):832–838. doi: 10.1046/j.1365-2141.1999.01459.x. [DOI] [PubMed] [Google Scholar]
- 25.de Sauvage F, et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med. 1996;183(2):651–666. doi: 10.1084/jem.183.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sungaran R, Markovic B, Chong B. Localization and regulation of thrombopoietin mRNa expression in human kidney, liver, bone marrow, and spleen using in situ hybridization. Blood. 1997;89(1):101–107. [PubMed] [Google Scholar]
- 27.Qian S, et al. Primary role of the liver in thrombopoietin production shown by tissue-specific knockout. Blood. 1998;92(6):2189–2191. [PubMed] [Google Scholar]
- 28.Wolber EM, et al. Hepatic thrombopoietin mRNA levels in acute and chronic liver failure of childhood. Hepatology. 1999;29(6):1739–42. doi: 10.1002/hep.510290627. [DOI] [PubMed] [Google Scholar]
- 29.McIntosh B, Kaushansky K. Transcriptional regulation of bone marrow thrombopoietin by platelet proteins. Exp Hematol. 2008;36(7):799–806. doi: 10.1016/j.exphem.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolber EM, et al. Hepatic thrombopoietin mRNA is increased in acute inflammation. Thromb Haemost. 2001;86(6):1421–4. [PubMed] [Google Scholar]
- 31.Kaser A, et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood. 2001;98(9):2720–5. doi: 10.1182/blood.v98.9.2720. [DOI] [PubMed] [Google Scholar]
- 32.Stone RL, et al. Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med. 2012;366(7):610–8. doi: 10.1056/NEJMoa1110352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerutti A, et al. Thrombopoietin levels in patients with primary and reactive thrombocytosis. Br J Haematol. 1997;99(2):281–4. doi: 10.1046/j.1365-2141.1997.3823196.x. [DOI] [PubMed] [Google Scholar]
- 34.Wang JC, et al. Blood thrombopoietin levels in clonal thrombocytosis and reactive thrombocytosis. Am J Med. 1998;104(5):451–5. doi: 10.1016/s0002-9343(98)00090-4. [DOI] [PubMed] [Google Scholar]
- 35.Wolber E, et al. Hepatic thrombopoietin mRNA is increased in acute inflammation. Thromb Haemost. 2001;86(6):1421–1424. [PubMed] [Google Scholar]
- 36.Burmester H, et al. Thrombopoietin production in wild-type and interleukin-6 knockout mice with acute inflammation. J Interferon Cytokine Res. 2005;25(7):407–13. doi: 10.1089/jir.2005.25.407. [DOI] [PubMed] [Google Scholar]
- 37.Mouthon M, et al. Preferential liver irradiation enhances hematopoiesis through a thrombopoietin-independent mechanism. Radiat Res. 1999;152(4):390–397. [PubMed] [Google Scholar]
- 38.Kosugi S, et al. Circulating thrombopoietin level in chronic immune thrombocytopenic purpura. Br J Haematol. 1996;93(3):704–6. doi: 10.1046/j.1365-2141.1996.d01-1702.x. [DOI] [PubMed] [Google Scholar]
- 39.Ichikawa N, et al. Regulation of serum thrombopoietin levels by platelets and megakaryocytes in patients with aplastic anaemia and idiopathic thrombocytopenic purpura. Thromb Haemost. 1996;76(2):156–60. [PubMed] [Google Scholar]
- 40.Griesshammer M, et al. High levels of thrombopoietin in sera of patients with essential thrombocythemia: cause or consequence of abnormal platelet production? Ann Hematol. 1998;77(5):211–5. doi: 10.1007/s002770050445. [DOI] [PubMed] [Google Scholar]
- 41.Sungaran R, et al. The role of platelet alpha-granular proteins in the regulation of thrombopoietin messenger RNA expression in human bone marrow stromal cells. Blood. 2000;95(10):3094–101. [PubMed] [Google Scholar]
- 42.Ku H, et al. Thrombopoietin, the ligand for the Mpl receptor, synergizes with steel factor and other early acting cytokines in supporting proliferation of primitive hematopoietic progenitors of mice. Blood. 1996;87(11):4544–51. [PubMed] [Google Scholar]
- 43.Sitnicka E, et al. The effect of thrombopoietin on the proliferation and differentiation of murine hematopoietic stem cells. Blood. 1996;87(12):4998–5005. [PubMed] [Google Scholar]
- 44.Qian H, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1(6):671–84. doi: 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 45.Arai F, et al. Niche regulation of hematopoietic stem cells in the endosteum. Ann N Y Acad Sci. 2009;1176:36–46. doi: 10.1111/j.1749-6632.2009.04561.x. [DOI] [PubMed] [Google Scholar]
- 46■.Ng AP, et al. Mpl expression on megakaryocytes and platelets is dispensable for thrombopoiesis but essential to prevent myeloproliferation. Proc Natl Acad Sci U S A. 2014;111(16):5884–9. doi: 10.1073/pnas.1404354111. This report shows that Mpl expression by the megakaryocytes, i.e. TPO signaling in MKs, is dispensable for platelet production. The key role of TPO signaling is in controlling platelet numbers via generation and stimulation of the bipotential MK precursors. This is an intriguing finding as Mplfl/fl Pf4-Cre mice were obviously able to “bypass” the lack of Mpl on the MK lineage, presenting more evidence that circulating TPO levels are regulated in a complicated manner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karpatkin S, Shulman S. Asialo platelets enhance thrombopoiesis. Trans Assoc Am Physicians. 1980;93:244–250. [PubMed] [Google Scholar]
- 48.Rumjantseva V, Hoffmeister KM. Novel and unexpected clearance mechanisms for cold platelets. Transfus Apher Sci. 2010;42(1):63–70. doi: 10.1016/j.transci.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmeister KM. The role of lectins and glycans in platelet clearance. J Thromb Haemost. 2011;9(Suppl 1):35–43. doi: 10.1111/j.1538-7836.2011.04276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50■■.Grozovsky R, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med. 2015;21(1):47–54. doi: 10.1038/nm.3770. The AMR recognizes senescent, desialylated platelets under steady state conditions. Desialylated platelets and the AMR are the physiological ligand-receptor pair regulating hepatic TPO mRNA production, resolving the longstanding mystery of steady state TPO regulation. The AMR-mediated removal of desialylated platelets regulates TPO synthesis in the liver by recruiting JAK2 and STAT3 to increase thrombopoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grewal PK. The Ashwell-Morell receptor. Methods Enzymol. 2010;479:223–41. doi: 10.1016/S0076-6879(10)79013-3. [DOI] [PubMed] [Google Scholar]
- 52.Rumjantseva V, et al. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat Med. 2009;15(11):1273–80. doi: 10.1038/nm.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sorensen AL, et al. Role of sialic acid for platelet life span: exposure of beta-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor-expressing liver macrophages and hepatocytes. Blood. 2009;114(8):1645–54. doi: 10.1182/blood-2009-01-199414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grewal PK, et al. Inducing host protection in pneumococcal sepsis by preactivation of the Ashwell-Morell receptor. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1313905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grewal PK, et al. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat Med. 2008;14(6):648–55. doi: 10.1038/nm1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crook M. Platelet sialic Acid and its significance to platelet life-spans. Platelets. 1990;1(3):167. doi: 10.3109/09537109009005484. [DOI] [PubMed] [Google Scholar]
- 57.Crook M. Sialic Acid: its importance to platelet function in health and disease. Platelets. 1991;2(1):1–10. doi: 10.3109/09537109109005496. [DOI] [PubMed] [Google Scholar]
- 58.Reimers HJ, et al. Experimental modification of platelet survival. Adv Exp Med Biol. 1977;82:231–3. doi: 10.1007/978-1-4613-4220-5_48. [DOI] [PubMed] [Google Scholar]
- 59.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112(6):2190–8. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.LaFave LM, Levine RL. JAK2 the future: therapeutic strategies for JAK-dependent malignancies. Trends Pharmacol Sci. 2012;33(11):574–82. doi: 10.1016/j.tips.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 61.Park SO, et al. Conditional deletion of Jak2 reveals an essential role in hematopoiesis throughout mouse ontogeny: implications for Jak2 inhibition in humans. PLoS One. 2013;8(3):e59675. doi: 10.1371/journal.pone.0059675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grisouard J, et al. Selective deletion of Jak2 in adult mouse hematopoietic cells leads to lethal anemia and thrombocytopenia. Haematologica. 2014;99(4):e52–4. doi: 10.3324/haematol.2013.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eulenfeld R, et al. Interleukin-6 signalling: more than Jaks and STATs. Eur J Cell Biol. 2012;91(6–7):486–95. doi: 10.1016/j.ejcb.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 64.Wolber E, Jelkmann W. nterleukin-6 increases thrombopoietin production in human hepatoma cells HepG2 and Hep3B. J Interferon Cytokine Res. 2000;20(5):499–506. doi: 10.1089/10799900050023915. [DOI] [PubMed] [Google Scholar]
- 65.Fallon RJ, Danaher M, Saxena A. The asialoglycoprotein receptor is associated with a tyrosine kinase in HepG2 cells. J Biol Chem. 1994;269(43):26626–9. [PubMed] [Google Scholar]
- 66.Alonzi T, et al. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol Cell Biol. 2001;21(5):1621–32. doi: 10.1128/MCB.21.5.1621-1632.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lesurtel M, et al. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312(5770):104–7. doi: 10.1126/science.1123842. [DOI] [PubMed] [Google Scholar]
- 68.Moh A, et al. Role of STAT3 in liver regeneration: survival, DNA synthesis, inflammatory reaction and liver mass recovery. Lab Invest. 2007;87(10):1018–28. doi: 10.1038/labinvest.3700630. [DOI] [PubMed] [Google Scholar]
- 69.Dalton SR, et al. Carbon tetrachloride-induced liver damage in asialoglycoprotein receptor-deficient mice. Biochem Pharmacol. 2009;77(7):1283–90. doi: 10.1016/j.bcp.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mason K, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 71.Debrincat MA, et al. Mcl-1 and Bcl-xL co-ordinately regulate megakaryocyte survival. Blood. 2012 doi: 10.1182/blood-2011-12-398834. [DOI] [PubMed] [Google Scholar]
- 72.Josefsson EC, et al. Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J Exp Med. 2011;208(10):2017–31. doi: 10.1084/jem.20110750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang H, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14(5):943–51. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 74.Liu ZJ, et al. Expansion of the neonatal platelet mass is achieved via an extension of platelet lifespan. Blood. 2014;123(22):3381–9. doi: 10.1182/blood-2013-06-508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McMillan R. The pathogenesis of chronic immune thrombocytopenic purpura. Semin Hematol. 2007;44(4 Suppl 5):S3–S11. doi: 10.1053/j.seminhematol.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 76■.Li J, et al. Severe platelet desialylation in a patient with glycoprotein Ib/IX antibody-mediated immune thrombocytopenia and fatal pulmonary hemorrhage. Haematologica. 2014;99(4):e61–3. doi: 10.3324/haematol.2013.102897. This report shows that certain anti-GPIbα-antibodies trigger platelet desialylation, a process that deviates platelet clearance from splenic macrophage Fc-receptors to the liver, likely via the AMR, showing that FcγR-independent mechanisms of ITP exist. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li J, et al. Fc-independent phagocytosis: implications for IVIG and other therapies in immune-mediated thrombocytopenia. Cardiovasc Hematol Disord Drug Targets. 2013;13(1):50–8. doi: 10.2174/1871529x11313010006. [DOI] [PubMed] [Google Scholar]
- 78■.Shao L, et al. Successful treatment with oseltamivir phosphate in a patient with chronic immune thrombocytopenia positive for anti-GPIb/IX autoantibody. Platelets. 2014:1–3. doi: 10.3109/09537104.2014.948838. This publication describes an adult chronic ITP patient with an anti-GPIb-IX autoantibody, who was resistant to corticosteroids, IVIG, recombinant human TPO, rituximab, danazol and vindesine (Eldisine®), but has been successfully treated with oseltamivir phosphate, a sialidase inhibitor used to treat influenza. This results show that anti-GPIb-IX autoantibody can induce loss of sialic acid and prevention of sialic acid loss may present an alternative treatment of patients with anti-platelet antibodies resistent to established treatment regimes of ITP. [DOI] [PubMed] [Google Scholar]
- 79.Jansen G, et al. Desialylation accelerates platelet clearance following refrigeration and initiates GPIbα metalloproteinase-mediated cleavage in mice. Blood. 2012;119:1263–73. doi: 10.1182/blood-2011-05-355628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–74. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]