

Abstract
IL-6 inhibition has been unsuccessful in treating psoriasis, despite high levels of tissue and serum IL-6 in patients. Additionally, de novo psoriasis onset has been reported following IL-6 blockade in rheumatoid arthritis patients. To explore mechanisms underlying these clinical observations, we backcrossed an established psoriasiform mouse model (IL-17C+ mice) with IL-6 deficient mice (IL-17C+KO) and examined the cutaneous phenotype. IL-17C+KO mice initially exhibited decreased skin inflammation, however this decrease was transient and reversed rapidly, concomitant with increases in skin Tnf, Il36α/β/γ, Il24, Epgn and S100a8/a9 to levels higher than those found in IL-17C+ mice. Comparison of IL-17C+ and IL-17C+KO mouse skin transcriptomes with that of human psoriasis skin, revealed significant correlation among transcripts of psoriasis patient skin and IL-17C+KO mouse skin, and confirmed an exacerbation of the inflammatory signature in IL-17C+KO mice that aligns closely with human psoriasis. Transcriptional analyses of IL-17C+ and IL-17C+KO primary keratinocytes confirmed increased expression of proinflammatory molecules, suggesting that in the absence of IL-6, keratinocytes increase production of numerous additional proinflammatory cytokines. These preclinical findings may provide insight into why arthritis patients being treated with IL-6 inhibitors develop new onset psoriasis and why IL-6 blockade for the treatment of psoriasis has not been clinically effective.
Keywords: IL-17, IL-36, IL-6, psoriasis, mouse model
The factors responsible for the initiation of a psoriatic skin lesion have yet to be conclusively determined, but involve a tightly orchestrated series of events involving T cells, innate immune cells, dermal fibroblasts, endothelial and epithelial cells, together with chemokines and cytokines that work in concert to exacerbate keratinocyte proliferation giving rise to visible skin plaques (Lowes et al., 2014). The importance of cytokine-mediated inflammation as a driving factor for psoriasis, in particular that of IL-17A and TNFα, is highlighted by the efficacy of TNFα- and IL-17-targeted inhibitors for psoriasis (Crow, 2012, Gordon et al., 2014, Langley et al., 2014, Papp et al., 2014, Papp et al., 2012, Silva et al., 2010).
Psoriasis patients also exhibit high levels of IL-6 in lesional skin and serum, although IL-6 inhibition has thus far been unsuccessful in mitigating human psoriasis (Castells-Rodellas et al., 1992, Grossman et al., 1989, Mease et al., 2016, Neuner et al., 1991). Paradoxically, psoriasis onset following IL-6 inhibition in rheumatoid arthritis patients has also been reported (Grasland et al., 2013, Laurent et al., 2010), although the mechanisms underlying this remain unclear.
We recently engineered and characterized a novel transgenic mouse model of psoriasiform skin inflammation that models the cutaneous overexpression of IL-17C observed in human psoriasis lesional skin (K5-promoter driven IL-17C overexpression, hereafter called IL-17C+) which produced skin-specific elevated levels of TNFα, IL-17A and IL-6 (Johnston et al., 2013). IL-17C is derived primarily from epithelial cells and exerts context-specific inflammatory effects in the skin and gut and has been hypothesized to regulate Th17 cell differentiation via Iκβζ (Chang et al., 2011, Johnston et al., 2013, Ramirez-Carrozzi et al., 2011, Song et al., 2011). IL-17C, like IL-17A, synergizes with TNFα to elicit inflammatory responses similar to those induced by IL-17A combined with TNFα (Chang et al., 2011, Johnston et al., 2013, Pappu et al., 2011, Ramirez-Carrozzi et al., 2011, Song et al., 2011) including increases in IL-6; and IL-17C+ mice treated with TNFα blocking antibodies have an improved skin phenotype and decreased IL-6.
To determine if IL-6 contributes directly to the skin pathogenesis associated with IL-17C, IL-17C+ mice were backcrossed with IL-6-deficient (KO) animals (IL-17C+KO) and the skin phenotype examined at the histological, cellular and molecular levels.
RESULTS
IL-17C+KO mice have delayed skin phenotype
IL-17C+ animals were backcrossed to IL-6KO mice and the skin phenotype assessed. Initial examination revealed that young adult (~10-weeks of age) IL-17C+KO mice had less skin inflammation than age-matched IL-17C+ animals, determined by a composite severity score based on visual analysis (Figure 1a–b). However, this difference reversed by 14-weeks of age (Figure 1a–b). At the histological level, involved lesional skin from both mouse lines developed similar levels of acanthosis regardless of age (Figure 1c–d), with the epidermal thickness being ~5–6 fold greater than wildtype (WT) C57Bl/6 control animals (indicated by the dashed line), consistent with previous observations (Johnston et al., 2013). Similar levels of dermal angiogenesis, T cell and myeloid cell infiltration in lesional skin of IL-17C+ and IL-17C+KO mice were observed regardless of age (data not shown).
Figure 1. IL-17C+KO mice develop a delayed inflammatory skin phenotype.
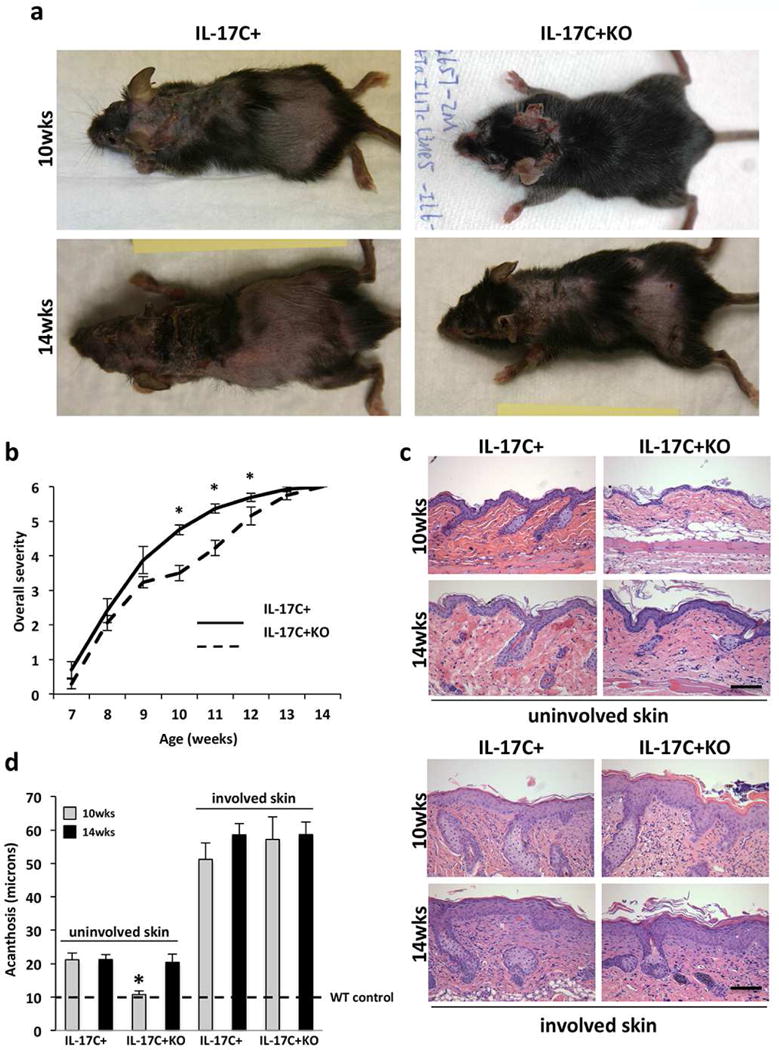
(a) Representative images of IL-17C+ and IL-17C+KO mice at 10- and 14-weeks of age. (b) Mouse overall severity scale between 7- and 14-weeks of age. (c) Representative images of H&E stained uninvolved and involved skin from IL-17C+ and IL-17C+KO mice at 10- and 14-weeks of age. (d) Mean epidermal thickness (acanthosis; μm) measures (± SEM) of involved and uninvolved dorsal skin of IL-17C+ (n=8 at 10 weeks; n=5 at 14 weeks) and IL-17C+KO (n=8 at 10 weeks, n=8 at 14 weeks) mice at 10-weeks and 14-weeks of age. The dashed line indicates wildtype mouse levels. Scale bars in c = 100 μm; *P<0.05.
In contrast to these findings, acanthosis of uninvolved skin of young adult (~10-week old) IL-17C+KO mice was significantly reduced in uninvolved skin compared to age-matched IL-17C+ mice (Figure 1d). 10-week old IL-17C+KO mice also had significantly less dermal angiogenesis (Figure 2a–b), fewer skin infiltrating CD4+, CD8+, and F4/80+ cells (Figure 2c; Figure S1) and decreased expression of phospho-STAT3 (Figure 2D) compared to age matched IL-17C+ mice. These observations suggest a role for IL-6 in initiating skin inflammation given the differences in non-lesional uninvolved skin. In contrast, once skin inflammation has occurred (in involved lesional skin) deletion of IL-6 does not alter the cutaneous histological phenotype. The differences observed at 10-weeks of age in IL-17C+ and IL-17C+KO mice were eliminated by 14-weeks of age (Figure 1b–c), such that uninvolved skin of IL-17C+KO mice increased in thickness, contained more inflammatory cells and dermal vasculature and resembled uninvolved skin of IL-17C+ animals. These observations suggest that alternative proinflammatory cells and/or cytokines increase in the absence of IL-6, promoting skin of IL-17C+KO mice to develop a phenotype more similar to that observed for IL-17C+ mice.
Figure 2. Cutaneous inflammation, angiogenesis and phospho-Stat3 increase in IL-17C+KO uninvolved mouse skin mice between 10 and 14 weeks.
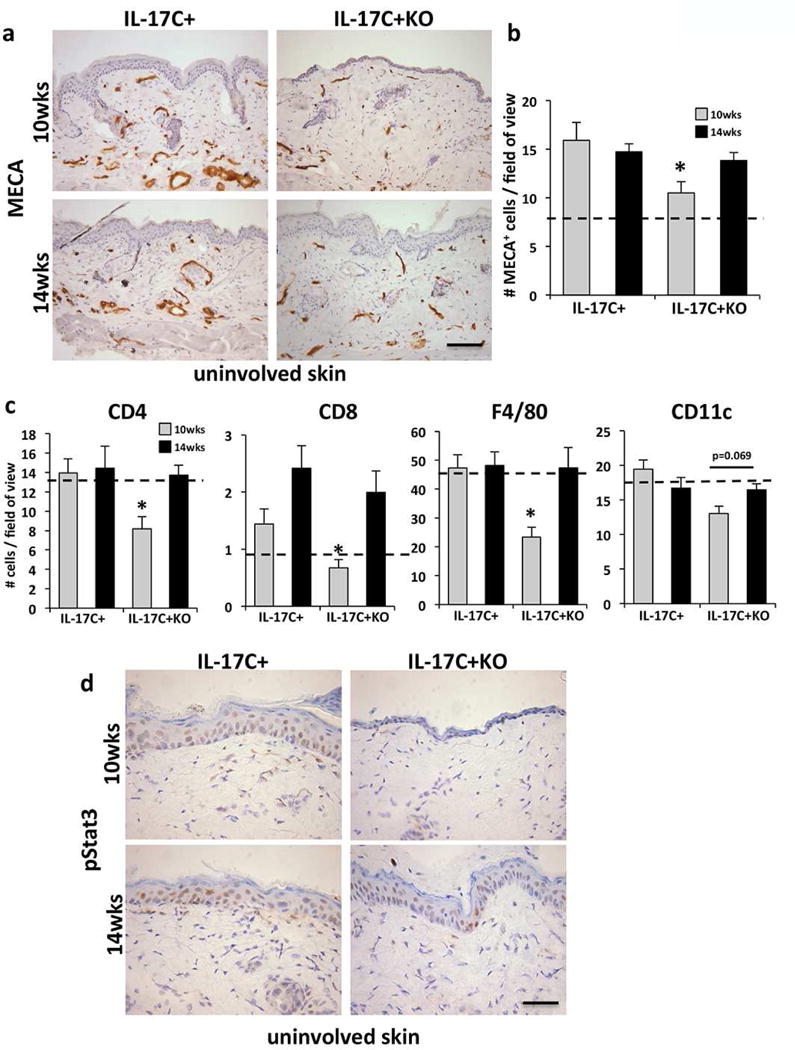
(a) MECA-stained uninvolved skin of 10- and 14-week old IL-17C+ and IL-17C+KO mice. (b) Quantification of MECA+ cells (mean cell number/field of view ± SEM) in IL-17C+KO (n=8) and IL-17C+ mice (n=7) at 10- and 14-weeks of age (n=5 each). (c) Immune cell quantification (mean # cells/field of view ± SEM) for CD4+, CD8+ T cells, CD11c+ myeloid cells and F4/80+ macrophages in IL-17C+KO vs. IL-17C+ uninvolved skin at 10- and 14-weeks of age. (d) Phospho-Stat3 immunostaining of uninvolved skin from 10- and 14-week old IL-17C+KO and IL-17C+ mice. The dashed line in B and C indicates wildtype mouse levels. Scale bars in a and d = 100 μm. *P<0.05.
IL-6 intradermal injections into IL-17C+KO mouse skin returns the phenotype to that of IL-17C+ mice
Given that IL-6 depletion appeared to decrease the severity of skin lesions early in development we asked whether exogenous recombinant IL-6 introduced into uninvolved skin of young IL-17C+KO mice (beginning at 8 weeks of age) would result in an enhanced skin phenotype. IL-6 injected IL-17C+KO skin histologically resembled IL-17C+ uninvolved skin, including similar levels of acanthosis and T cell and macrophage skin infiltration (Figure 3a–b). IL-6 injections also increased phospho-STAT3 activity (Figure 3c). Further analyses of the IL-6 vs. BSA injected skin identified increases in inflammatory proteins including S100A8/A9, IL-36α and TNFα. These findings suggest that introduction of exogenous IL-6 into skin genetically overexpressing IL-17C (in the absence of IL-6) promotes immune cell recruitment and increases proinflammatory cytokines and innate defense proteins that potentiate keratinocyte proliferation and promote the development of a skin phenotype that phenocopies uninvolved skin of IL-17C+ mice.
Figure 3. Intradermal IL-6 injection into IL-17C+KO mouse skin elicits a IL-17C+ skin phenotype.
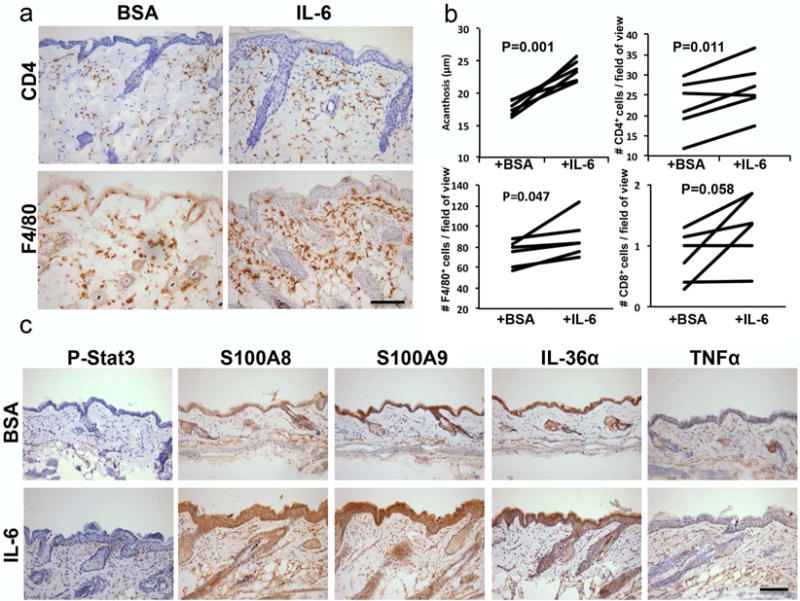
(a) CD4+ and F4/80+ immunohistochemistry on IL-17C+KO mice skin intradermally injected with BSA or recombinant IL-6 every other day for 16 days. (b) Quantification of epidermal thickness (acanthosis, μm) and infiltrating immune cells (CD4+, CD8+ and F4/80+; mean number of cells/field of view) in IL-6 and BSA injected IL-17C+KO (n=6). Significance values are as indicated (b) Representative images from IL-17C+KO skin injected with either BSA or IL-6 immunostained for phospho-Stat3, S100A8/A9, IL-1F6 and TNFα. Scale bar in a and c = 100 μm.
Taking advantage of the KC-Tie2 psoriasis mouse which has elevated cutaneous IL-17C and TNFα (Ward N. L. et al., 2011, Wolfram et al., 2009)(Figure S2a), and when backcrossed with IL-6KO mice has a sustained skin phenotype (Golden, 2015), we validated the findings observed in IL-17C+KO mice and determined that intradermal injections of IL-6 or TNFα into the skin of KC-Tie2-IL-6KO mice also lead to an exacerbation of skin inflammation (Figure S2b), demonstrating in a second psoriasiform model (one where IL-17C is not genetically manipulated) that similar synergy between IL-17C, IL-6 and TNFα may contribute to skin phenotype. Further evidence of this was seen in primary keratinocytes stimulated with combinations of IL-17C, IL-6 and TNFα where increases in expression of proinflammatory transcripts, including S100A8/A9, IL-36γ and TNFα were observed (Figures S3–4).
IL-17C+KO mice have exacerbated inflammation that closely models human psoriasis
We were very interested in the mechanisms underlying the rapid reversal of the delayed phenotype in IL-17C+KO mice. We performed RNAseq on involved and uninvolved skin isolated from IL-17C+ and IL-17C+KO mice and compared changes in identified transcripts with established datasets of psoriasis patient lesional (PP) and nonlesional (PN) skin (n=44 patients) (Swindell et al., 2016). Genes altered in involved skin of IL-17+KO mice were more similar to those altered in human psoriasis lesions (rs = 0.312), as compared with those genes altered in involved skin of IL-17C+ mice (rs = 0.125; Figure 4a). Genes increased in mouse phenotypes and psoriasis lesions included Il19, S100a8/a9, Cxcl5, Cxcl1 and Klk6/13 (Figure 4b), while decreased genes included Wnt2, Trim55, and Myoc (Figure 4c). To determine why expression shifts in IL17C+KO involved skin were more similar to those in human psoriasis lesions, we evaluated subsets of genes responsive to IL-17C-, TNFα-, IL-22-, IFNγ- and IL-36-family cytokines. Interestingly, such genes were differentially correlated between mouse phenotypes, with stronger correlations in IL-17C+KO involved skin (PP/PN vs. IL17C+KO/CTL, with CTL= uninvolved skin) compared with IL-17C+ involved skin (PP/PN vs. IL17C+KO/CTL) (Figure 4d). Taken together, these results suggest that the absence of IL-6 in IL-17C+ animals leads to compensatory increases of proinflammatory transcripts that more closely model human psoriasis.
Figure 4. Gene expression changes in IL-17C+ and IL-17C+KO skin and comparison to human psoriasis.
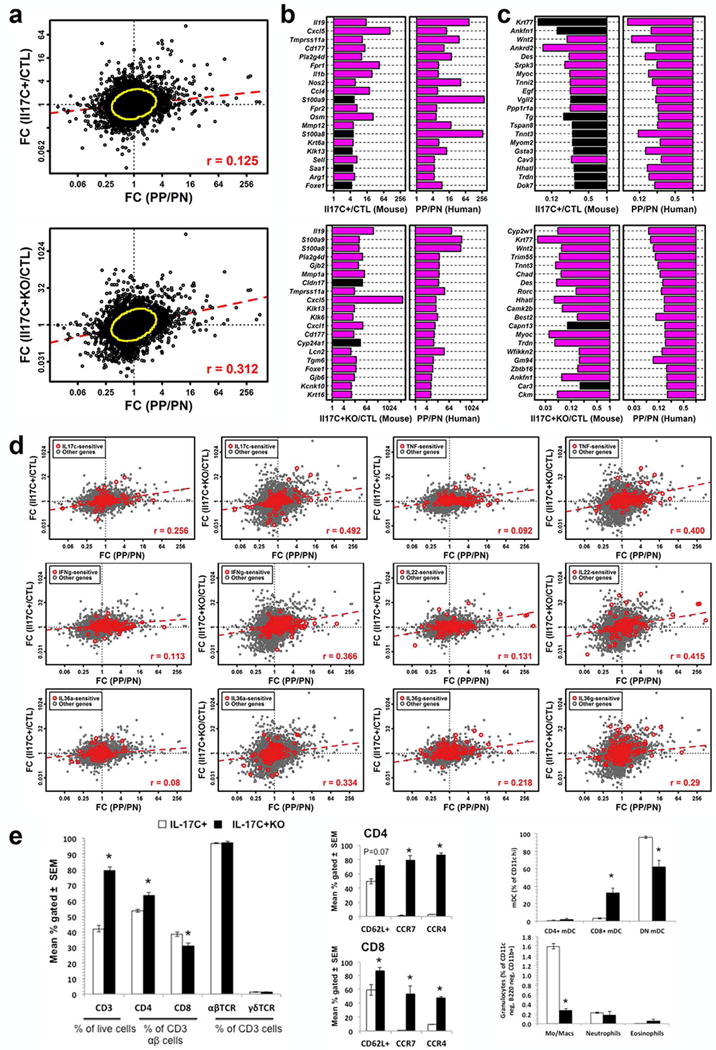
(a) Comparison of fold-changes in IL-17C+ and IL-17C+KO involved and uninvolved skin (CTL; n = 3/group) and human psoriasis (involved (PP)/uninvolved (PN); n = 44 patients). Yellow circles encompass the 90% of genes closest to the bivariate centroid (Mahalanobis distance). Fold-changes for genes (b) increased and (c) decreased in IL-17C+ and IL-17C+KO mice and human psoriasis (magenta bars, FDR < 0.10). (d) Cytokine-sensitive genes and associations among IL-17C+ and IL-17C+KO involved skin and human psoriasis. Red symbols represent cytokine-sensitive genes identified from in vitro experiments performed using cultured keratinocytes (100 cytokine-increased + 100 cytokine-decreased; grey symbols: all other genes). (e) Flow cytometric analysis of skin-draining lymph node (SDLN) immune cells from IL-17C+ and IL17C+KO mice examining CD3+ and CD4+ T cell populations and homing molecule expression of CD62L, CCR7 and CCR4 on CD4+ and CD8+ T cells. Analysis of SDLN myeloid cells in IL-17C+ and IL-17C+KO mice. * P<0.05.
To further explore the cellular mechanisms mediating these bioinformatics results, we analyzed immune cells isolated from skin draining lymph nodes of IL-17C+ and IL17C+KO mice and identified increases in CD3+ and CD4+ T cell populations in IL-17C+KO mice compared to IL-17C+ mice, and increases in T cell-derived CD62L+, CCR7+ and CCR4+ expression. CD8+ myeloid dendritic cells increased, however macrophages decreased (Figure 4e). These observations demonstrate an exacerbation of inflammation in IL-17C+KO mice and support the idea that in the absence of IL-6, alternative inflammatory responses occur that can elicit an exacerbated or at the very least, a similar skin phenotype to IL-17C+ mice.
To further identify and validate alternative inflammatory mediators in IL-17C+KO mice, psoriasis-related immune cell-, endothelial cell- and keratinocyte-derived cytokines were examined. TNFα levels were evaluated by ELISA, as our prior work had identified critical synergy between IL-17C and TNFα (Johnston et al., 2013). Uninvolved skin of 10-week old IL-17C+KO mice had significantly higher TNFα protein than age-matched IL-17C+ mice (7.3±0.6 vs. 4.8±0.7pg/ml; P=0.01). Moreover, further increases (~3-fold) in TNFα were seen in uninvolved skin of IL-17C+KO mice as the mice aged (14-weeks old), and the skin phenotype worsened toward a phenotype that resembled IL-17C+ mice (7.3±0.6 vs. 21.2 ±4.9pg/ml; P=0.003; Figure 5a). Paradoxically, serum TNFα levels were significantly reduced in 10-week old IL-17C+KO mice (7.07±1.4 vs. 16.7±3.7pg/ml; P=0.03; Figure 5a), consistent with, and likely reflective of, less overall skin inflammation in IL-17C+KO mice (i.e., less body surface area with lesional skin; Figure 1a). The difference in serum TNFα levels was eliminated by 14-weeks of age, as IL-17C+KO mice exhibited increased skin inflammation.
Figure 5. Compensatory proinflammatory molecules increase in uninvolved skin of IL-17C+KO mice between 10 and 14 weeks.
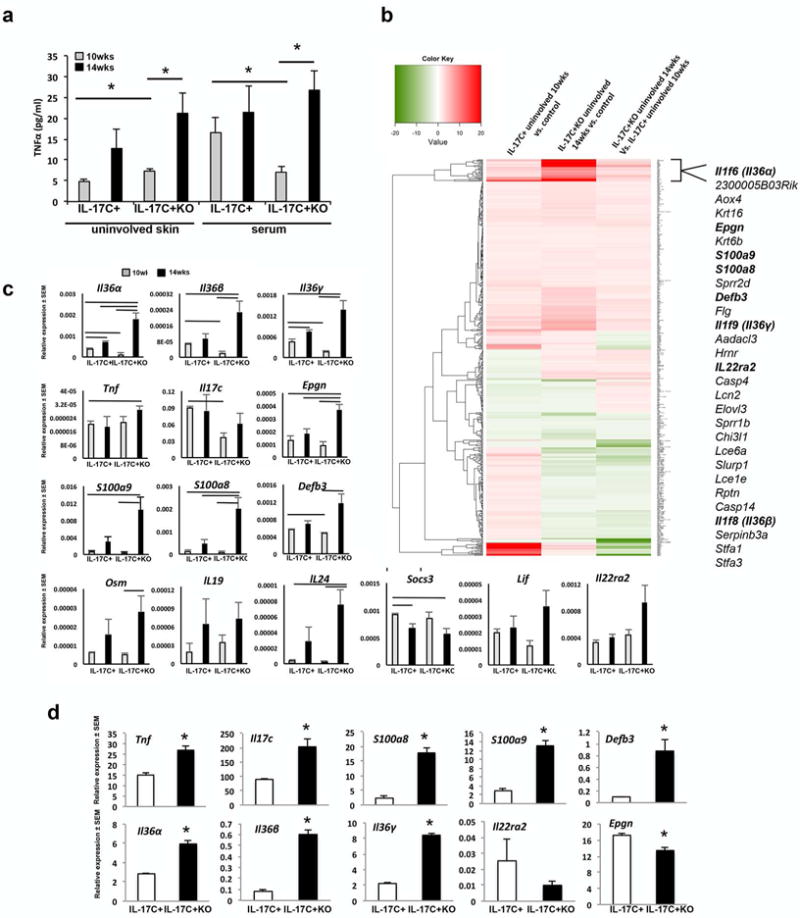
(a) Uninvolved skin and serum TNFα levels in IL-17C+ and IL-17C+KO mice at 10 and 14 weeks (n=6 each). (b) Heat map of changing transcripts between IL-17C+ and control mice at 10-weeks of age compared to genes altered in 14-week old IL-17C+KO mice vs. controls (n=4–5/group). (c) qRT-PCR validation of transcripts identified in 10- and 14-week old uninvolved skin of IL-17C+ and IL-17C+KO mice (n=4/group). Solid lines indicate P<0.05 between groups. (d) qRT-PCR on primary neonatal mouse keratinocytes from IL-17C+ and IL-17C+KO mice examining expression levels of Tnf, Il17c, S100a8/a9, Defb3, Il36α/β/γ, Il22ra2 and Epgn (n= 3/group performed in triplicate). *P<0.05. Lines indicate significant differences between groups with P<0.05.
Next, we used gene expression microarray analyses coupled with Ingenuity Pathway Analysis (IPA) and qRT-PCR to validate critical gene expression changes between IL-17C+ and IL-17C+KO mice (Figure 5b and Figures S5–7). At ten weeks of age, when IL-17C+KO mice had a less severe phenotype, a number of key psoriasis signature genes, including Il36α/β/γ, Defb3 and Il17c were decreased compared to age-matched IL-17C+ mice. However at 14 weeks, these transcripts, as well as Epgn, S100a8/a9 (Mrp8/14), Il24, and the IL-6 receptor cytokine, Osm, all increased significantly in IL-17C+KO mice (Figure 5b–c). In fact, Epgn, Il36α and S100a8 all increased to levels beyond those observed in IL-17C+ mice at 14-weeks of age; and finally, Tnf, Il36α/β/γ, Il24, Epgn and S100a8/a9 levels all significantly increased in IL-17C+KO mice at 14 weeks compared to IL-17C+ mice at 10 weeks. These changes suggest that the transient, less severe skin phenotype observed in 10-week old IL-17C+KO mice may result from delays in elevation of these signaling molecules, and that by 14-weeks of age these transcripts increase to levels beyond that observed in skin of IL-17C+ mice, allowing the skin phenotype to rapidly resemble the skin of IL-17C+ animals and leading to a transcript signature that more closely resembles human psoriasis lesional skin (Figure 4).
Finally, primary keratinocytes isolated and expanded from IL-17C+ and IL-17C+KO mouse skin confirmed increases in Il17c (2.3-fold; P=0.02), Tnf (1.8-fold; P= 0.006), S100a8 (7-fold; P<0.001); S100a9 (4.5-fold; P=0.002), Defb3 (9.4-fold; P=0.02), and IL-36 family members (Il36α/Il36β/Il36γ; 2-7-fold; P<0.001) in IL-17C+KO keratinocytes compared to IL-17C+ keratinocytes (Figure 5d).
DISCUSSION
We show in the absence of IL-6, the psoriasiform skin phenotype in IL-17C+KO mice is initially delayed, but this delay is transient and rapidly reversed as a result of increases in compensatory proinflammatory factors. This enhanced inflammatory response was confirmed in primary keratinocytes isolated and expanded from IL-17C+KO mice demonstrating that IL-6-deficient keratinocytes increase production of alternative proinflammatory chemokines and cytokines (Figure 5d). Of particular interest was the higher correlation with human psoriasis transcript changes observed between involved and uninvolved skin for the IL-17C+KO mice, providing additional evidence of an exacerbation of skin inflammation that is relevant to human psoriasis. These preclinical findings provide insight into why patients being treated with IL-6 inhibitors may develop new onset psoriasis and why IL-6 blockade for the treatment of psoriasis has not been clinically effective.
IL-6 inhibitors have failed to improve psoriasis skin manifestation despite efficacy in rheumatoid arthritis (RA) and psoriatic arthritis (PsA)(Hartung et al., 2013, Mease et al., 2016). The paradoxical elicitation of psoriasis in patients undergoing treatment with the IL-6 inhibitor tocilizumab (Grasland et al., 2013, Wendling et al., 2012), suggests that inhibition of IL-6 signaling may not improve skin outcomes. Indeed, inhibition of the IL-6 receptor (IL-6R) may allow increases in circulating IL-6. Our data suggest that an alternative reason why IL-6 therapeutic blockade fails to resolve skin inflammation is the ability of the skin to readily and efficiently compensate for the loss of IL-6.
IL-36 family members (Il36α/β/γ) along with S100a8/9, Epgn, Defb3, Osm, and Il24 transcripts all increase significantly between 10 and 14 weeks in IL-17C+KO skin, concomitant with increases in angiogenesis, skin infiltrating T cells and macrophages. Moreover, the transcript changes occurring between involved and uninvolved skin in IL-17C+KO mice more closely correspond with changes identified between human psoriasis lesions and peri-lesional skin. Significant increases in IL-17C-, TNF-, IFNγ-, IL-22- and IL-36-sensitive genes were also observed in IL-17C+KO mice when compared with IL-17C+ mice. These occurred at the same time increases in CD3+ and CD4+ T cells in the skin draining lymph nodes of IL-17C+KO mice were observed. Together, these observations support the idea that alternative cellular and molecular changes compensate for the loss of IL-6 in IL-17C+KO mice and allow skin severity to increase to similar levels as IL-17C+ mice.
Paradoxically, IL-17C+KO animals initially showed a delay in the development of the skin phenotype, potentially a result of decreases in skin IL-17C and IL-36. Exogenous introduction of recombinant IL-6 returned the skin phenotype to levels of IL-17C+ mice concomitant with increases in cutaneous phospho-Stat3, TNFα, IL-36α and S100A8/A9, suggesting a potential capacity of IL-6 to synergize both with IL-17C, TNFα and IL-36α. IL-17C is known to synergize with TNFα (Johnston et al., 2013, Ramirez-Carrozzi et al., 2011), IL-1β (Ramirez-Carrozzi et al., 2011), IL-22 (Song et al., 2014) and most recently with IL-36γ (IL-1F9)(Friedrich et al., 2014) to elicit the production of inflammatory signals from epithelial cells. Thus we also demonstrated that IL-6 synergizes with IL-17C alone and in combination with TNFα to drive a psoriasis-like signature from primary mouse and human keratinocytes, including increases in S100a8/a9, Defb3, Il36α/β/γ, Tnf and additional Il17c (Figures S3–4). Interestingly, recombinant IL-6 and TNFα, together, but not alone, injected into uninvolved skin of K5-IL-17C mice also significantly increases acanthosis and skin infiltrating CD4+ and F4/80+ immune cells (Figure S8). These in vitro and in vivo observations support the ability of IL-17C, IL-6 (and TNFα) to promote skin inflammation and identify another IL-17C-synergistic signaling partner in addition to IL-1β, IL-22 and IL-36γ.
The ability of biologic therapies to affect skin changes is largely dependent upon direct blockade of signaling molecules and/or their cognate receptors. Despite the success of biologic therapies patients with recalcitrant disease emerge and show incomplete drug action, or a propensity for disease recurrence despite therapy. In the case of psoriasis, our data suggests that key combinations of signaling molecules namely IL-6, TNFα, and IL-17C, can combine to elicit skin inflammation and that blocking of any single agent may be temporarily effective, but over time, becomes compensated for by other non-targeted inflammatory molecules. It is likely that combination therapies addressing two or more possible signals may have higher efficacy in psoriasis for patients that become refractory to single agent therapies. An example of this is the bi-specific ABT-122 (AbbVie) antibody currently under development that targets both TNFα and IL-17A. A combined approach may prove useful for patients that fail to respond or stop responding to one targeted drug for multiple reasons: TNFα inhibitors are highly effective for treatment of skin (psoriasis) and joint inflammation (psoriatic arthritis) and work quickly on targeting joint inflammation; in contrast, IL-17A inhibitors clear skin inflammation rapidly and appear to be effective for psoriatic arthritis, but take more time to reach comparable protection afforded by TNFα inhibitors. Therefore, using TNFα and IL-17A inhibitors simultaneously may offer a unique solution to clearance of skin involvement while protecting the joints from further irreversible damage.
Determining the compensatory pathways upregulated upon inhibition of signaling by currently employed therapeutics may inform potential alternative (complementary) targets that must also be inhibited to achieve optimal efficacy of particular inhibitors. Our data suggests that a combination of anti-IL-6 with either TNF or IL-17 inhibitors may improve the efficacy of IL-6 inhibitors for treating psoriatic skin disease.
METHODS
Mice
K5tTA, TetosIL-17C and TetosTie2 engineering have been previously described (Diamond et al., 2000, Johnston et al., 2013, Wolfram et al., 2009). Individual mouse lines were back-crossed to IL-6KO mice (JAX cat# 002650; www.Jax.org) to generate either K5tTA-, Tetos-IL-17C- homozygous IL-6KO mice (IL-17C+KO), or Tetos-Tie2- homozygous IL-6KO mice. These were then mated with each other in the presence of doxycycline (200mg/kg; Bio-Serve, Beltsville, MD) to repress transgene expression until birth (IL-17C+ mice) or for the first week of gestation (KC-Tie2 mice); at which time mice were switched to regular P3000 diet. K5tTA and TetosIL-17C on a C57Bl/6 background were mated using the same approach to provide K5-IL-17C-C57Bl/6 (IL-6 proficient; IL-17C+) controls. C57Bl/6 and IL-6KO mice served as additional background strain controls. Male and female mice were used for all experimental analyses.
IL-17C+ and IL-17C+KO mice and controls were euthanized at 10-weeks or 14-weeks of age. Involved and uninvolved skin from each mouse was harvested for histology, immunohistochemistry and protein and RNA analyses as previously described (Johnston et al., 2013). Although alopecia developed in both IL-17C+ and IL-17C+KO mice this criteria was not used as a measure to identify skin involvement, and was consistent with what we previously reported. Involved and uninvolved areas occur routinely on the mouse body and samples from each were harvested from the same geographic location across mice. Involved skin was defined as skin with well-demarcated scale and erythema, whereas uninvolved demonstrated no phenotypic difference from control skin.
ELISA (R&D, Minneapolis, MN) detected an average ~13-fold increase in IL-6 protein in involved IL-17C+ mouse skin (51.3±11.5pg/ml, range 20-93pg/ml; n=6) and nearly undetectable levels of IL-6 in involved IL-17C+KO mouse skin (4.1±1.9pg/ml, range 0–13pg/ml; n=8). Assay range of the ELISA was 7.8–500pg/ml.
Histological and immunostaining analyses
Formalin-fixed paraffin-embedded skin was sectioned and stained with H&E as described previously (Wolfram et al., 2009). Fresh frozen skin was sectioned and stained using protocols and antibodies previously published (Foster et al., 2014, Johnston et al., 2013, Wolfram et al., 2009) and using antibodies targeting S100A8/A9 and TNFα (all R&D Systems, Minneapolis, MN).
Epidermal thickness (acanthosis) measurements and immune cell quantification of skin was done as previously described (Ward Nicole L. et al., 2011, Wolfram et al., 2009).
ELISA
Sera or protein isolated from skin adjacent to that used for histological and immunostaining analyses using standard protocols as previously published (Wolfram et al., 2009), were used for ELISA according to manufacturer’s instructions.
Intradermal cytokine experiments
Purified recombinant cytokine (IL-6, TNFα, or both; R&D Systems) or BSA (500ng of each protein in 100μl volume) was intradermally injected into the dorsum of uninvolved skin of ~8 week old IL-17C+, IL-17C+KO, KC-Tie2-IL-6KO and/or C57Bl/6 mice every other day for 16 days. Each animal had uninvolved skin injected with both BSA and cytokine at separate sites, and skin from each site was harvested ~2 days after the last injection and used for histological and immunostaining analyses.
Primary mouse keratinocyte culture, stimulation and gene expression analyses
Mouse keratinocytes were isolated and cultured as described (Dlugosz et al., 1995) and stimulated with recombinant mouse IL-17C, TNFα and IL-6 (all 10ng/ml, R&D Systems) for 8h and then RNA isolated and qRT-PCR performed as above.
Primary human keratinocyte cultures, stimulation and gene expression analyses
Normal human keratinocyte cultures were established from sun-protected adult human skin as described (Elder et al., 1991). Keratinocytes were stimulated with recombinant human IL-17C (100 ng/ml), TNFα (20 ng/ml) and IL-6 (20 ng/ml) or combinations thereof and then RNA isolated and qRT-PCR performed as described (Johnston et al., 2011).
RNA Sequencing and analyses
Total RNA was converted to mRNA by polyA purification and cDNA was generated using random primers. Products were sequenced on a 50 cycle single end HiSeq 2000 system (Illumina; 6 samples/lane; high output mode; version 3 reagents). This generated an average 29.9 million 50 bp reads was generated per sample (range: 23.2 – 38.7 million). Reads were quality-filtered and mapped to the mouse genome (UCSC, GRCm38/mm10) using a workflow described previously (Swindell et al., 2016, Swindell et al., 2014), with reads mapped using tophat2 (Kim et al., 2013) and gene counts and FPKM calculated using HTSeq (Anders et al., 2015) and Cufflinks (Trapnell et al., 2012), respectively. Raw gene counts were normalized using the trimmed mean of M-values method (Robinson and Oshlack, 2010) and differential expression analyses were performed using edgeR (Robinson et al., 2010). Differential expression trends in IL-17C+ and IL-17C+KO mice were compared with those estimated from lesional (PP) and uninvolved (PN) skin from psoriasis patients. Psoriasis differential expression statistics were obtained from a recent meta-analysis study that included 44 psoriasis patients (Swindell et al., 2016) (Gene Expression Omnibus series identifiers GSE41745, GSE54456/GSE63979 and GSE66511). Cytokine-sensitive genes (Figure 4d) were identified from microarray comparisons between cytokine-treated keratinocyte cultures and control-treated keratinocytes (GSE12109, GSE25400, GSE32620, GSE32975 and GSE53751) (Swindell et al., 2016).
RNA, microarray, IPA analyses and real time qRT-PCR validation on murine skin
RNA was isolated from skin adjacent to that used for histology and immunostaining and was used for microarray, Ingenuity Pathways Analysis (Ingenuity Systems Inc., Redwood City, CA) and qRT-PCR analyses as previously described (Wolfram et al., 2009).
Individual genes of interest were validated using an Applied Biosystems StepOne Plus Real-Time PCR system following our established protocol (Johnston et al., 2013) and using mouse-specific probes and primers obtained from Applied Biosystems (Grand Island, NY). Individual values for each animal and each primer/probe set were normalized to GAPDH and then presented as an average/group/time point ± SEM.
RNA-seq and microarray data have been submitted to Gene Expression Omnibus and are available under the accession GSE86140.
Flow cytometry
Mouse skin draining lymph nodes were minced with sterile razor blades then disrupted further in GentleMACS C-tubes (Miltenyi) followed by enzymatic digestion with 0.1mg/ml DNase I, 0.5mg/ml collagenase, 0.1mg/ml hyaluronidase (Sigma) in HBSS at 37°C for 30 minutes with occasional agitation. Cell suspensions were passed through a 40μm cell strainer (Fisher) then placed in red blood cell lysis buffer (155mM NH4Cl, 0.01M KHCO3, 0.01% EDTA). After washing, cellular FC receptors were blocked for 20 minutes at 4°C with anti-CD16/CD32 antibodies (Biolegend) before staining with a viability dye (Invitrogen) and antibodies against CD3e (clone 145-2C11), CD4 (RM4-5), CD8a (5–6.7), TCR-β (H57-597), TCR-γδ (GL3), CD11b (M1/70), CD11c (N418), CD62L (MEL-14), CCR4 (2G12), CCR7 (4B12) or appropriate isotype controls (all FACS antibodies from Biolegend, San Diego, CA) for 30 minutes. Cells were washed then analyzed on a LSR2 flow cytometer (BD Bioscience), gating lymphocyte populations guided by isotype control binding.
Statistics
All data are represented as mean ± SEM. Analysis of between group comparisons were completed using an unpaired, unequal variance, two-tailed Student’s t-test and statistical significance was defined as P≤0.05. For intradermal injections where BSA and recombinant cytokine were injected into the same animal; a paired two-tailed Student’s t-test was used and statistical significance remained defined as P≤0.05.
Study approval
All animal protocols were consistent with guidelines issued by the American Association for Accreditation of Laboratory Animal Care and were approved by the Case Western Reserve University Institutional Animal Care And Use Committee (IACUC; Cleveland, OH). All human subjects protocols were approved by the institutional review boards (IRB) of the University of Michigan (Ann Arbor, MI) or Case Western Reserve University (Cleveland, OH) and written informed consent was received from participants prior to inclusion in the study.
Supplementary Material
Acknowledgments
This work was supported in part by The Lozick Discovery Grant, the National Psoriasis Foundation, the American Skin Association, the Babcock Foundation Endowment, the Murdough Family Center of Psoriasis, the Kenneth and Frances Eisenberg Emerging Scholar Award of the A. Alfred Taubman Medical Research Institute, and the following grants from the National Institutes of Health: K08 grant AR060802, R01 AR069071, P30 AR39750, R01 AR063437, R01 AR062546; R21 AR063852; K01 AR064765. Microarray experiments were supported by the Gene Expression & Genotyping Core Facility of the Case Comprehensive Cancer Center (P30 CA043703). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS USED
- CCL20
chemokine (C-C motif) ligand 20
- IPA
Ingenuity Pathways Analysis
- K5
keratin 5
- KO
knockout
- MECA
mouse endothelial cell antigen
- PN
uninvolved human psoriasis skin
- PP
involved lesional human psoriasis skin
- qRT-PCR
quantitative reverse-transcription PCR
- SDLN
skin draining lymph node
- SEM
standard error of the mean
- Tetos
tetracycline operator sequence
- Th
T helper
- TNF
tumor necrosis factor
- tTA
tetracycline transactivator
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors state no conflict of interest
References
- Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castells-Rodellas A, Castell JV, Ramirez-Bosca A, Nicolas JF, Valcuende-Cavero F, Thivolet J. Interleukin-6 in normal skin and psoriasis. Acta dermato-venereologica. 1992;72(3):165–8. [PubMed] [Google Scholar]
- Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011;35(4):611–21. doi: 10.1016/j.immuni.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JM. Therapeutics: Silencing psoriasis. Nature. 2012;492(7429):S58–9. doi: 10.1038/492S58a. [DOI] [PubMed] [Google Scholar]
- Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol. 2000;115(5):788–94. doi: 10.1046/j.1523-1747.2000.00144.x. [DOI] [PubMed] [Google Scholar]
- Dlugosz AA, Glick AB, Tennenbaum T, Weinberg WC, Yuspa SH. Isolation and utilization of epidermal keratinocytes for oncogene research. Methods Enzymol. 1995;254:3–20. doi: 10.1016/0076-6879(95)54003-2. [DOI] [PubMed] [Google Scholar]
- Elder JT, Fisher GJ, Zhang QY, Eisen D, Krust A, Kastner P, et al. Retinoic acid receptor gene expression in human skin. J Invest Dermatol. 1991;96(4):425–33. doi: 10.1111/1523-1747.ep12469889. [DOI] [PubMed] [Google Scholar]
- Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, et al. IL-36 Promotes Myeloid Cell Infiltration, Activation, and Inflammatory Activity in Skin. J Immunol. 2014 doi: 10.4049/jimmunol.1301481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich M, Tillack C, Wollenberg A, Schauber J, Brand S. IL-36gamma sustains a proinflammatory self-amplifying loop with IL-17C in anti-TNF-induced psoriasiform skin lesions of patients with Crohn’s disease. Inflamm Bowel Dis. 2014;20(11):1891–901. doi: 10.1097/MIB.0000000000000198. [DOI] [PubMed] [Google Scholar]
- Golden JB, Fritz Y, Li Y, Wang Y, Simon DI, McCormick TS, Ward NL. Skin-mediated promotion of thrombosis is abrogated following IL-23/IL-17 inhibition or IL-6 deletion in mouse models of psoriasis. Journal of Investigative Dermatology. 2015;135:S1. [Google Scholar]
- Gordon KB, Leonardi CL, Lebwohl M, Blauvelt A, Cameron GS, Braun D, et al. A 52-week, open-label study of the efficacy and safety of ixekizumab, an anti-interleukin-17A monoclonal antibody, in patients with chronic plaque psoriasis. Journal of the American Academy of Dermatology. 2014;71(6):1176–82. doi: 10.1016/j.jaad.2014.07.048. [DOI] [PubMed] [Google Scholar]
- Grasland A, Mahe E, Raynaud E, Mahe I. Psoriasis onset with tocilizumab. Joint Bone Spine. 2013;80(5):541–2. doi: 10.1016/j.jbspin.2013.03.014. [DOI] [PubMed] [Google Scholar]
- Grossman RM, Krueger J, Yourish D, Granelli-Piperno A, Murphy DP, May LT, et al. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(16):6367–71. doi: 10.1073/pnas.86.16.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung W, Ehrenstein B, Wallisch R, Fleck M. Remission of arthritis but persistent cutaneous lesions following tocilizumab treatment in a RA-patient suffering from concomitant psoriasis. Case Reports in Clinical Medicine. 2013;02(01):3. [Google Scholar]
- Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol. 2013;190(5):2252–62. doi: 10.4049/jimmunol.1201505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston A, Gudjonsson JE, Aphale A, Guzman AM, Stoll SW, Elder JT. EGFR and IL-1 signaling synergistically promote keratinocyte antimicrobial defenses in a differentiation-dependent manner. J Invest Dermatol. 2011;131(2):329–37. doi: 10.1038/jid.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med. 2014;371(4):326–38. doi: 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- Laurent S, Le Parc JM, Clerici T, Breban M, Mahe E. Onset of psoriasis following treatment with tocilizumab. The British journal of dermatology. 2010;163(6):1364–5. doi: 10.1111/j.1365-2133.2010.10005.x. [DOI] [PubMed] [Google Scholar]
- Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of Psoriasis. Annual Review of Immunology. 2014;32(1):227–55. doi: 10.1146/annurev-immunol-032713-120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease P, Gottlieb AB, Berman A, Drescher E, Xing J, Wong R, et al. The Efficacy and Safety of Clazakizumab, an Anti-Interleukin-6 Monoclonal Antibody, in a Phase 2b Study of Adults with Active Psoriatic Arthritis. Arthritis & rheumatology. 2016 doi: 10.1002/art.39700. [DOI] [PubMed] [Google Scholar]
- Neuner P, Urbanski A, Trautinger F, Moller A, Kirnbauer R, Kapp A, et al. Increased IL-6 production by monocytes and keratinocytes in patients with psoriasis. J Invest Dermatol. 1991;97(1):27–33. doi: 10.1111/1523-1747.ep12477880. [DOI] [PubMed] [Google Scholar]
- Papp K, Leonardi C, Menter A, Thompson EH, Milmont CE, Kricorian G, et al. Safety and efficacy of brodalumab for psoriasis after 120 weeks of treatment. Journal of the American Academy of Dermatology. 2014;71(6):1183–90 e3. doi: 10.1016/j.jaad.2014.08.039. [DOI] [PubMed] [Google Scholar]
- Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG, Kricorian G, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366(13):1181–9. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- Pappu R, Ramirez-Carrozzi V, Sambandam A. The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology. 2011;134(1):8–16. doi: 10.1111/j.1365-2567.2011.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. 2011;12(12):1159–66. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LC, Ortigosa LC, Benard G. Anti-TNF-alpha agents in the treatment of immune-mediated inflammatory diseases: mechanisms of action and pitfalls. Immunotherapy. 2010;2(6):817–33. doi: 10.2217/imt.10.67. [DOI] [PubMed] [Google Scholar]
- Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, et al. Alterations in the Microbiota Drive Interleukin-17C Production from Intestinal Epithelial Cells to Promote Tumorigenesis. Immunity. 2014;40(1):140–52. doi: 10.1016/j.immuni.2013.11.018. [DOI] [PubMed] [Google Scholar]
- Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, et al. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol. 2011;12(12):1151–8. doi: 10.1038/ni.2155. [DOI] [PubMed] [Google Scholar]
- Swindell WR, Sarkar MK, Liang Y, Xing X, Gudjonsson JE. Cross-Disease Transcriptomics: Unique IL-17A Signaling in Psoriasis Lesions and an Autoimmune PBMC Signature. J Invest Dermatol. 2016 doi: 10.1016/j.jid.2016.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindell WR, Xing X, Voorhees JJ, Elder JT, Johnston A, Gudjonsson JE. Integrative RNA-seq and microarray data analysis reveals GC content and gene length biases in the psoriasis transcriptome. Physiol Genomics. 2014;46(15):533–46. doi: 10.1152/physiolgenomics.00022.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NL, Hatala DA, Wolfram JA, Knutsen DA, Loyd CM. Cutaneous manipulation of vascular growth factors leads to alterations in immunocytes, blood vessels and nerves: Evidence for a cutaneous neurovascular unit. Journal of dermatological science. 2011;61(1):14–22. doi: 10.1016/j.jdermsci.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NL, Loyd CM, Wolfram JA, Diaconu D, Michaels CM, McCormick TS. Depletion of antigen-presenting cells by clodronate liposomes reverses the psoriatic skin phenotype in KC-Tie2 mice. The British journal of dermatology. 2011;164(4):750–8. [Google Scholar]
- Wendling D, Letho-Gyselinck H, Guillot X, Prati C. Psoriasis onset with tocilizumab treatment for rheumatoid arthritis. The Journal of rheumatology. 2012;39(3):657. doi: 10.3899/jrheum.111166. [DOI] [PubMed] [Google Scholar]
- Wolfram JA, Diaconu D, Hatala DA, Rastegar J, Knutsen DA, Lowther A, et al. Keratinocyte but not endothelial cell specific overexpression of Tie2 leads to the development of psoriasis. Am J Pathol. 2009;174:1443–58. doi: 10.2353/ajpath.2009.080858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.