

Abstract
Adamantanes such as amantadine (1) and rimantadine (2) are FDA-approved anti-influenza drugs that act by inhibiting the wild-type M2 proton channel from influenza A viruses, thereby inhibiting the uncoating of the virus. Although adamantanes have been successfully used for more than four decades, their efficacy was curtailed by emerging drug resistance. Among the limited number of M2 mutants that confer amantadine resistance, the M2-V27A mutant was found to be the predominant mutant under drug selection pressure, thereby representing a high profile antiviral drug target. Guided by molecular dynamics simulations, we previously designed first-in-class M2-V27A inhibitors. One of the potent lead compounds, spiroadamantane amine (3), inhibits both the M2-WT and M2-V27A mutant with IC50 values of 18.7 and 0.3 μM, respectively, in in vitro electrophysiological assays. Encouraged by these findings, in this study we further examine the in vitro and in vivo antiviral activity of compound 3 in inhibiting both amantadine-sensitive and -resistant influenza A viruses. Compound 3 not only had single to submicromolar EC50 values against M2-WT- and M2-V27A-containing influenza A viruses in antiviral assays, but also rescued mice from lethal viral infection by either M2-WT- or M2-V27A-containing influenza A viruses. In addition, we report the design of two analogs of compound 3, and one was found to have improved in vitro antiviral activity over compound 3. Collectively, this study represents the first report demonstrating the in vivo antiviral efficacy of inhibitors targeting M2 mutants. The results suggest that inhibitors targeting drug-resistant M2 mutants are promising antiviral drug candidates worthy of further development.
Keywords: Influenza virus, M2 proton channel, V27A, drug resistance, spiroadamantane, antiviral
Graphical abstract

1. Introduction
Influenza virus infections are a persistent public health concern. Despite the existence of influenza vaccines and antiviral drugs, influenza viruses claim 250,000–500,000 lives per year worldwide (Thompson et al., 2010). In the unpredictable event of influenza pandemics, the death toll is normally several orders higher (Monto and Webster, 2013). Although influenza vaccines are generally effective in preventing influenza infection, they are less effective for the elderly, children, and immunocompromised persons (Houser and Subbarao, 2015). As a result, they are the most vulnerable populations for influenza virus infection. Moreover, it normally takes at least six months to produce and manufacture sufficient quantities of influenza vaccines for public use; thus influenza vaccines are unlikely to be an option to confine the first wave of an emerging influenza pandemic (Houser and Subbarao, 2015). In contrast, small molecule antiviral drugs can be stockpiled and readily distributed in bulk quantities; thus they are not an alternative, but rather an indispensable complement to vaccines. Currently there are two classes of anti-influenza drugs. M2 inhibitors such as amantadine (1) and rimantadine (2) inhibit the viral M2 proton channel and are viral entry inhibitors. Neuraminidase inhibitors such as oseltamivir, zanamivir, and peramivir inhibit the viral neuraminidase and are viral egress inhibitors. The emergence of drug-resistant influenza strains now necessitates the development of the next generation of antiviral drugs. Resistance to adamantanes is so widely spread that the CDC no longer recommends the use of adamantanes in prophylaxis and the treatment of influenza infection. This leaves neuraminidase inhibitors as the last FDA-approved option. The alarming fact is that oseltamivir-resistant strains have been continuously reported among human and avian influenza viruses (Govorkova et al., 2013; Hurt et al., 2009; Hurt, 2014). In fact, the 2008–2009 seasonal influenza viruses with the H275Y NA mutation were completely resistant to the only orally bioavailable drug, oseltamivir (Hurt, 2014; Renaud et al., 2011). Therefore, the potential of resistance evolution, coupled with the lack of an alternative antiviral drug with a distinct mechanism of action, calls for the development of the next generation of antiviral drugs (Lee and Yen, 2012; Loregian et al., 2014).
With the aim of developing novel anti-influenza drugs, we chose the M2 proton channel as the drug target because M2 is one of the most conserved viral proteins (Dong et al., 2015; Wang, 2016; Wang et al., 2015; Wang et al., 2011c). Although many M2 mutants emerged under drug selection pressure in cell cultures (Abed et al., 2005; Brown et al., 2010), animals (Herlocher et al., 2003), and human patients (Hayden and Hay, 1992; Iwahashi et al., 2001), only a few of them acquired the fitness to transmit among humans (Dong et al., 2015; Furuse et al., 2009a; Furuse et al., 2009b). Genetic studies have shown that the predominant M2 mutants circulating among humans are S31N, V27A, and L26F (Dong et al., 2015). Among this limited number of mutants, V27A was shown to be the predominant mutation under drug selection pressure (Furuse et al., 2009a; Furuse et al., 2009b). We therefore are interested in designing the second generation of M2 channel blockers by targeting the V27A mutant (Balannik et al., 2009; Rey-Carrizo et al., 2013; Wang et al., 2011a; Wang et al., 2011b).
M2 is a homo-tetrameric proton channel that is embedded in the viral membrane (Hong and DeGrado, 2012). The proton conductance function is encoded by the transmembrane domain spanning residues 25–46. The V27 side-chain faces the centroid of the channel and forms the first gate (Pielak and Chou, 2010). H37 and W41 are conserved residues that are critical for proton conductance and selectivity (Balannik et al., 2010). Guided by molecular dynamics simulations, we previously generated a homology model of the V27A mutant (Wang et al., 2011a). The model showed an enlarged channel cavity at the N-terminus compared with M2-WT. This prediction was later confirmed by the NMR investigation of M2-V27A (Pielak and Chou, 2010) as well as molecular dynamics simulations from another group (Yi et al., 2008). On the basis of this observation, we designed a 3D-shaped spiroadamantane amine molecule (3) that has improved shape complementarity to the V27A mutant compared to amantadine (Wang et al., 2011a). Compound 3 was found to have potent channel blockage against V27A mutant in electrophysiological assays, with an IC50 of 0.3 ± 0.1 μM. It also inhibits the M2-WT channel with an IC50 of 18.7 ± 1.4 μM. The mechanism of this dual inhibition phenomenon was revealed by molecular dynamics simulations. It suggested that compound 3 binds deeper in the M2-WT channel and that there is only one layer of water between the ammonium from compound 3 and the H37 (Wang et al., 2011a). In the case of the M2-V27A mutant channel, compound 3 shifted up toward the N-terminus, and there are two layers of water between the ammonium from compound 3 and the H37.
Although a few compounds were reported to inhibit the M2-V27A mutant channel (Rey-Carrizo et al., 2014; Rey-Carrizo et al., 2013; Wang et al., 2011a), there has been no report on their antiviral activity against the M2-V27A-containing influenza A virus. Given the promising channel blockage efficacy of compound 3 in inhibiting both the M2-WT and the M2-V27A mutant, we took a step further to evaluate the in vitro and in vivo antiviral efficacy of compound 3. Specifically, the antiviral activity of compound 3 against M2-WT and the M2-V27A-containing influenza A viruses was tested in plaque assays. For in vivo studies, the non-toxic dose of compound 3 was determined, then the effect of compound 3 on body weight and survival rate of mice infected with a lethal dose of influenza A viruses was observed. Furthermore, we designed and synthesized two analogs of compound 3 with the aim of further improving the antiviral efficacy.
2. Materials and methods
2.1. Compounds
Compound 3 was synthesized as previously reported (Wang et al., 2011a). Novel synthesis procedures and compound characterization by NMR and MS can be found in Fig. 5 and the Supplementary Data. Amantadine was purchased from Sigma (St. Louis, MO). Oseltamivir (as Tamiflu®) was obtained from a local pharmacy as 75 mg capsules. The compounds were prepared in sterile water for oral gavage (p.o.) administration. For oseltamivir, this required using one or more entire capsules, since excipients were also present.
Fig. 5. Synthesis procedures of compound 3 analogs.

(A) Synthesis of the guanidine analog 7. (B) Synthesis of the dithiane analog 16.
2.2. Viruses
M2-WT-expressing A/Udorn/72 (H3N2), A/WSN/33 (M2-N31S) (H1N1), A/Solomon Islands/3/2006 (H1N1), and M2-V27A-expressing A/WSN/33 (M2-N31S + V27A) (H1N1) were used to infect MDCK cells (American Type Culture Collection, Manassas, VA) in the presence or absence of compounds to evaluate their antiviral activity. Influenza A/WSN/33 (M2-N31S) (H1N1) and A/WSN/33 (M2-N31S + V27A) (H1N1) viruses were generated by reverse genetics of the A/WSN/33 (H1N1) virus. The M genes of the resulting recombinant viruses were sequenced and the sequences were confirmed as expected. The virus was later titrated in BALB/c mice for lethality. Influenza A/Udorn/72 (H3N2) was obtained from Dr. Robert Lamb's laboratory at the Northwestern University. Influenza A/Solomon Islands/3/2006 (H1N1) virus was obtained from the Centers for Disease Control and Prevention (Atlanta, GA). The virus was passaged seven times in mice to enhance its virulence, and later titrated in BALB/c mice for lethality. Approximately a 100% lethal dose of each H1N1 virus was selected for use in this strain of mouse.
2.3. Plaque assays
Plaque assay were performed according to previous reports (Ma et al., 2016a; Ma et al., 2016b). M2-WT-expressing A/Udorn/72 (H3N2), A/WSN/33 (M2-N31S) (H1N1), A/Solomon Islands/3/2006 (H1N1), and M2-V27A-expressing A/WSN/33 (M2-N31S + V27A) (H1N1) were used to infect MDCK cells in the presence or absence of compounds to evaluate their antiviral activity. Briefly, a confluent monolayer of MDCK cells was incubated with ~100-pfu virus samples in DMEM with 0.5% bovine serum albumin for 1 h at 4 °C, then 37 °C for 1 h. The inoculums were removed, and the cells were washed with phosphate-buffered saline (PBS). The cells were then overlaid with DMEM containing 1.2% Avicel microcrystalline cellulose (FMC BioPolymer, Philadelphia, PA) and N-acetyl trypsin (2.0 μg/ml). To examine the effect of the compounds on plaque formation, the overlay media were supplemented with compounds at testing concentrations. At day 2 after infection, the monolayers were fixed and stained with crystal violet dye solution (0.2% crystal violet, 20% methanol). Plaque area was quantified using ImageJ (Guzman et al., 2014) and the fifty percent virus-inhibitory concentration (EC50) values were subsequently determined.
2.4. Cytotoxicity assays
Evaluation of the cytotoxicity of compounds was carried out using the neutral red uptake assay (Repetto et al., 2008). Briefly, 80,000 cells/ml MDCK cells in DMEM medium, which was supplemented with 10% FBS and 100 U/ml of Penicillin-Streptomycin solution, were dispensed into clear 96-well cell culture plates (Corning Cat #: CLS3362) at 100 μL/well. Twenty-four hours later, the growth medium was removed and washed with 100 μL PBS buffer; then 200 μL fresh DMEM (no FBS) medium containing serial diluted compounds was added to each well. After incubation for 24 h at 37 °C with 5% CO2 in a CO2 incubator, the medium was removed and replaced with 100 μl DMEM medium containing 40 mg/ml neutral red for 4 h at 37 °C. The amount of retained neutral red was determined by absorbance at 540 nm using a Multiskan FC Microplate Photometer (Fisher Scientific). The 50% cytotoxic concentration (CC50) values were calculated from best-fit dose response curves with variable slope in Prism 5.
2.5. Electrophysiological assays
All compounds were initially tested in a TEVC assay using Xenopus laevis frog oocytes microinjected with RNA expressing either the WT or the V27A mutant of A/Udorn/72-M2, as described in a previous report (Balannik et al., 2009). The potency of the inhibitors was expressed as the percent inhibition of M2 current observed after 2 min of incubation with 100 μM compounds.
2.6. Animals
Female 18–20 g BALB/c mice were obtained from Charles River Laboratories (Wilmington, MA) for the antiviral study and for toxicity determinations. The animals were maintained on standard rodent chow and tap water ad libitum. They were housed 5 per cage in corn cob bedding and quarantined for 48 h prior to use. Mice were block randomized so that approximately the same average initial body weight was in each group. Before and during the infection and treatment, mice are observed for general well-being (activity, physical appearance, and unusual behaviors following treatment). This was in addition to monitoring body weight.
2.7. Experiment design of animal studies
BALB/c mice were anesthetized by intraperitoneal injection of ketamine/xylazine (50/5 mg/kg), and the animals were infected intranasally with a 90-μl suspension of influenza virus (day 0). The virus challenge was approximately 3–4 50% mouse lethal infectious doses, equating to about 1-2 × 104 50% cell culture infectious doses (CCID50) of virus per mouse. Oral treatments were given twice a day (at 12 h intervals) for 5 days starting 30 min before virus challenge. Parameters for assessing the infection were survival, mean day of death, body weight changes, and lung infection parameters (hemorrhage score, weight, and virus titer). Animals were weighed every other day through day 21 of the infection. Mice whose body weights fell below 30% of initial weight were humanely euthanized and deaths were noted. During infections, there were 10 compound-treated and 20 placebos per group held for determining survival. Five uninfected mice per group were treated with compound 3 or placebo were assessed with 10 normal controls (uninfected, untreated) for drug toxicity in one experiment.
To assess lung infection parameters, lungs from sacrificed animals (5 mice/group, held for this purpose) were harvested. Lung hemorrhage score was assessed in a blinded manner by visual inspection for color changes from pink to plum. This occurs regionally in the lungs, not by a gradual color change of the whole lung. Hemorrhage scores ranged from 0 (normal) to 4 (total lung showing plum color) in 0.5 unit increments averaged over the whole lung. The scoring range correlates to 0 to 100% lung involvement. The lungs were weighed and then homogenized in cell culture medium, followed by freezing at −80 °C. Later, thawed lungs were serially diluted and titrated by the endpoint dilution method in 96-well plates of MDCK cells (Smee et al., 2008), using 4 microwells per dilution, and by diluting homogenates out to a 108 dilution. Cultures were incubated at 37 oC for 6 days and wells scored for presence or absence of viral cytopathic effect in order to determine the 50% viral titer endpoints. Virus titers were reported as log10 cell culture infectious doses per gram of lung tissue (log10 CCID50/g).
2.8. Statistical analysis of animal data
Kaplan–Meier plots for multiple group comparisons were analyzed by the Mantel–Cox log-rank test to determine statistical significance. Subsequently, pairwise comparisons were made by the Gehan–Breslow–Wilcoxon test. Percent survival results were analyzed by the two-tailed Fishers exact test. Mean day of death and lung hemorrhage data were not normally distributed. Analysis of these parameters was made by Kruskal-Wallis test followed by pairwise comparisons analyzed by the two-tailed Mann-Whitney U-test. Mean lung weight and lung viral titer data were normally distributed, and were analyzed by one-way analysis of variance (ANOVA) followed by Tukey–Kramer multiple comparisons test. All comparisons were made between drug-treated and placebo groups, and were analyzed using InStat® and Prism® software (GraphPad Software, San Diego, CA).
2.9. Ethics regulation of laboratory animals
The animal experiments were conducted in accordance with the approval of the Institutional Animal Care and Use Committee of Utah State University. They were performed in the AAALAC-accredited Laboratory Animal Research Center of Utah State University. The U. S. Government (National Institutes of Health) approval (PHS Assurance No. A3801-01) and conduct of the work were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
3. Results
3.1. In vitro antiviral efficacy of compound 3
As compound 3 was confirmed already to be a potent blocker of both M2-WT and M2-V27A proton channels (Table 1) (Wang et al., 2011a), we then wanted to confirm its antiviral efficacy against both M2-WT- and M2-V27A-containing influenza A viruses. The A/Udorn/72 (H3N2) virus and the A/WSN/33 (M2-N31S + V27A) (H1N1) virus, which contain M2-WT and M2-V27A, respectively, were chosen to assess the drug sensitivity of compound 3. The A/WSN/33 (M2-N31S + V27A) (H1N1) virus was generated by reverse genetics. The asparagine residue in position 31 was mutated to serine, and the A/WSN/33-N31S (H1N1) was amantadine-sensitive (Wang et al., 2013), similar to the M2-WT. In addition, valine in position 27 was mutated to alanine. As shown in Fig. 1A, compound 3 had a similar antiviral activity as amantadine (1) in inhibiting the M2-WT-containing virus A/Udorn/72 (H3N2), with an EC50 value of 0.3 ± 0.1 μM. When tested against another M2-WT-containing virus A/WSN/33 (M2-N31S) (H1N1) virus, both amantadine (1) and compound 3 showed potent antiviral activity with EC50 values of 0.2 ± 0.01 μM, and 0.1 ± 0.01 μM, respectively (Table S1). In contrast, the A/WSN/33 (M2-N31S + V27A) (H1N1) virus was nearly completely resistant to amantadine (1) (Fig. 1B), and no plaque reduction was observed up to 1 μM. Further increasing the concentration of amantadine (1) to 50 μM also failed to significantly inhibit the replication of the A/WSN/33 (M2-N31S + V27A) (H1N1) virus (plaque reduction less than 10 %). For this reason, oseltamivir carboxylate (4) was used as a positive control, and it showed complete inhibition at 200 nM. Gratifyingly, compound 3 showed dose-dependent inhibition of the M2-V27A mutant–containing A/WSN/33 (M2-N31S + V27A) (H1N1) virus with an EC50 of 1.8 ± 0.2 μM.
Table 1.
Channel blockage activity of amantadine (1), rimantadine (2), and spiroadamantane amine (3) in inhibiting the A/Udorn/72 (M2-WT) and A/Udorn/72 (M2-V27A) proton channels.
| Electrophysiological Assay IC50 (μM) | ||
|---|---|---|
| M2-WT | M2-V27A | |

|
15.7 ± 1.2 | > 100 |

|
10.8 ± 0.2 | > 100 |

|
18.7 ± 1.4 | 0.3 ± 0.1 |
Fig. 1. Antiviral efficacy of amantadine (1) and compound 3 in inhibiting M2-WT- and M2-V27A-containing influenza A viruses.

(A) Plaque assay results of amantadine (1) and compound 3 in inhibiting M2-WT-containing A/Udorn/72 (H3N2) virus. The EC50 values for amantadine (1) and compound 3 were 0.3 ± 0.1 μM and 0.3 ± 0.1 μM, respectively. (B) Plaque assay results of amantadine (1) and compound 3 in inhibiting M2-V27A-containing A/WSN/33 (M2-N31S + V27A) (H1N1) virus. Oseltamivir carboxylate (4) was used as a control. The EC50 value for compound 3 was 1.8 ± 0.2 μM.
To assess the genetic barrier to resistance for compound 3, serial viral passage experiments were performed. Specifically, the M2-WT-containing virus A/Udorn/72 (H3N2) was amplified for several rounds in the presence of increasing concentrations of compound 3, and the antiviral EC50 value was determined at each passage. At passage 1, compound 3 was applied at 300 nM and the EC50 was 0.9 ± 0.1 μM, which confers a 3-fold increase. At passage 2, compound 3 was applied at 600 nM and the EC50 was more than 10 μM, indicating complete resistance. In parallel, amantadine (1) was also subjected to the serial viral passage experiments. At passage 1, amantadine (1) was applied at 300 nM and the EC50 was more than 10 μM, indicating complete resistance. Thus, compound 3 appears to have a similar genetic barrier to resistance as amantadine.
In summary, compound 3 not only had potent channel blockage activity against the A/Udorn/72 M2-WT and M2-V27A channels with IC50 values of 18.7 ± 1.4 μM and 0.3 ± 0.1 μM, respectively, it also had potent antiviral activity against both M2-WT and M2-V27A mutant–containing influenza A viruses.
3.2. Assaying the in vivo non-toxic dose of compound 3
The in vivo non-toxic dose of compound 3 was assessed with uninfected BALB/c mice. Compound 3 was prepared in sterile water and given to mice by oral gavage (p.o.) administration. The compound was given twice a day (at 12 h intervals) for five days at either 30 mg/kg/day or 100 mg/kg/day. As shown in Fig. 2, a dose of compound 3 up to 100 mg/kg/day was well tolerated. Although mice treated with compound 3 exhibited a 3% body weight difference compared to placebos and normal control animals on day 3, this difference was not statistically significant (P>0.05). For cumulative toxicity to occur, body weights should have been at nadir on day 5 (which was 12 hours after the final treatment). Instead weight gain was seen in all groups between days 3 and 5. No apparent differences in physical activity or behavior were noted among groups during the treatment period or thereafter. On the basis of this result, the maximum dose was set as 100 mg/kg/day for the following efficacy studies.
Fig. 2. Effects of compound 3 on body weight in uninfected mice.

Oral gavage treatments were administered twice a day for 5 days starting on day 0. The normal control (uninfected, untreated) group contained 10 mice. The other groups contained 5 mice each. Mean values ± SEM are shown at each time point. This assessment was performed in parallel with the infection experiment reported in Figs. 3 and 4.
3.3. In vivo antiviral efficacy of compound 3 in treatment of an M2-WT-containing influenza A/Solomon Islands/3/2006 (H1N1) virus infection in mice
As the amantadine-sensitive A/Udorn/72 (H3N2) virus was not mouse-adapted and 100% lethality could not be achieved, another strain of M2-WT-containing influenza A virus, A/Solomon Islands/3/2006 (H1N1), was chosen for the in vivo efficacy study. A/Solomon Islands/3/2006 (H1N1) became lethal to mice after seven serial passages in the lungs of infected animals (Murray et al., 2012). To confirm that the mouse-adapted A/Solomon Islands/3/2006 (H1N1) virus was sensitive to amantadine (1) and compound 3, plaque assays were performed. The EC50 values of amantadine (1) and compound 3 in inhibiting the M2-WT-containing A/Solomon Islands/3/2006 (H1N1) virus were 0.1 ± 0.01 μM and 0.1 ± 0.02 μM (Table S1), respectively. Thus the choice of A/Solomon Islands/3/2006 (H1N1) for the in vivo studies was justified. As shown in Fig. 3A, no mice in the placebo group survived beyond day 8 after infection with A/Solomon Islands/3/2006 (H1N1). Oral treatment of compound 3 at a doses of 40 and 100 mg/kg/day or above significantly improved the survival rate to 70% (Fig. 3A, Table 2). In comparison, amantadine (1) was more potent than compound 3, with all doses providing some protection, and doses of 20, 40, and 100 mg/kg/day providing 100% protection (Fig. 3C, Table 2).
Fig. 3. Survival curves and body weight changes for compound 3 (A and B), amantadine (1) (C and D), and oseltamivir (4) in p.o. treatment of an influenza A/Solomon Islands/3/2006 (H1N1) virus infection in mice.

(A) Survival curves for compound 3. (B) Body weight changes for compound 3. (C) Survival curves for amantadine (1). (D) Body weight changes for amantadine (1). Oral gavage treatments were given twice a day for 5 days starting 30 min before virus exposure at day 0. *** P<0.001 compared to placebo, determined by the Mantel-Cox log-rank test with pairwise comparisons made by the Gehan-Breslow-Wilcoxon test. Groups of 10 compound-treated mice and 20 placebos were used. Groups of 10 compound-treated mice and 20 placebos were used. Mean values are shown at each time point. Error bars are not shown because animals were weighed as groups instead of individually in this experiment.
Table 2. Effects of compound 3 and amantadine (1) on an influenza A/Solomon Islands/3/2006 (H1N1) virus infection in mice.
Oral treatments were administrated twice a day (at 12 h intervals) for 5 d starting 30 min before virus exposure on day 0.
| Compound (mg/kg/day) | Survivors/Totala | MDDb ± SD |
|---|---|---|
| 3 (100) | 7/10*** | 10.3 ± 3.1 |
| 3 (40) | 7/10*** | 15.0 ± 6.0* |
| 3 (20) | 1/10 | 11.9 ± 4.6*** |
| 3 (10) | 0/10 | 7.7 ± 1.2 |
| 3 (5) | 0/10 | 7.0 ± 1.1 |
| Amantadine (1) (100) | 10/10*** | - |
| Amantadine (1) (40) | 10/10*** | - |
| Amantadine (1) (20) | 10/10*** | - |
| Amantadine (1) (10) | 6/10*** | 13.0 ± 6.4 |
| Amantadine (1) (5) | 5/10** | 9.4 ± 1.9* |
| Placebo p.o. | 0/20 | 7.1 ± 0.8 |
Statistically evaluated by two-tailed Fisher's exact test.
Mean day of death of mice that died prior to day 22, with pairwise comparisons statistically evaluated by two-tailed Mann-Whitney U-Test.
P<0.05
P<0.001 compared to placebo.
Body weight changes during the infection are shown in Figs. 3B and 3D. Oral treatment with compound 3 was effective in reducing the extent of weight loss at the 40 and 100 mg/kg/day doses (Fig. 3B). In a similar manner, amantadine (1) treatments of 20, 40, and 100 mg/kg/day reduced weight loss during the acute infection (Fig. 3D). Each compound performed in a dose-responsive manner.
The results of this experiment established that compound 3 possessed in vivo antiviral activity against a M2-WT-containing influenza A virus, A/Solomon Islands/3/2006 (H1N1). Compound 3 was effective in reducing mortality and weight loss at 40 and 100 mg/kg/day when given by oral gavage route; however, its potency was less than that of amantadine (1).
3.4. In vivo antiviral efficacy of compound 3 in treatment of a M2-V27A-containing influenza A/WSN/33 (M2-N31S + V27A) (H1N1) virus infection in mice
As shown in Fig. 4A and Table 3, compound 3 at 100 mg/kg/day was highly protective but lower doses were not effective. The positive control drug oseltamivir (4) was 100% protective at a dose of 10 mg/kg/day. Amantadine (1) was not effective in preventing death, as A/WSN/33 (M2-N31S + V27A) (H1N1) was amantadine-resistant in vitro (Fig. 1). High significance was seen in groups treated with compound 3 (100 mg/kg/day) and oseltamivir (4). A considerably less but statistically significant benefit (primarily in delaying the time to death) was afforded by treatment with compound 3 (30 mg/kg/day, 10.3 days MDD) or with amantadine (1) (100 mg/kg/day, 11.1 days MDD). The results of antiviral treatment on lung infection parameters during this same infection are shown in Table 3. On day 6 of the infection the mean lung scores in the compound 3 100 mg/kg/day group were normal, as were lungs from the oseltamivir (4) group (Table 3). Certain other treatments with compound 3 or amantadine (1) also reduced lung hemorrhage scores significantly. Significant reductions in lung weight were found in a few of the compound 3 and amantadine (1) groups, but were not as great as in the oseltamivir (4) group. Lung virus titers were determined from animals sacrificed on day 6. Compound 3 at 100 mg/kg/day and oseltamivir (4) significantly reduced virus titers compared to placebo. Amantadine (1) at 100 mg/kg/day also reduced virus titers, but the results did not achieve statistical significance (P>0.05).
Fig. 4. Survival curves and body weight changes for compound 3, amantadine (1), and oseltamivir (4) on an influenza A/WSN/33 (M2-N31S + V27A) (H1N1) virus infection in mice.

(A) Survival curves for compound 3, amantadine (1), and oseltamivir (4). (B) Body weight changes for compound 3. (C) Body weight change for amantadine (1). Oral gavage treatments were administered twice a day for 5 days starting 30 min pre-infection on day 0. ** P<0.01, *** P<0.001, compared to placebo, determined by the Mantel-Cox log-rank test with pairwise comparisons made by the Gehan-Breslow-Wilcoxon test. Groups of 10 compound-treated mice and 20 placebos were used. Groups of 10 compound-treated mice and 20 placebos were used. Mean values ± SEM are shown at each time point.
Table 3. Effects of compound 3, amantadine (1), and oseltamivir (4) on an influenza A/WSN/33 (M2-N31S + V27A) (H1N1) virus infection in BALB/c mice.
Oral treatments were administered twice a day (at 12 h intervals) for 5 d starting 30 min before virus exposure on day 0.
| Mean Lung Parameters ± SD (Day 6), N=5/group | |||||
|---|---|---|---|---|---|
| Compound (mg/kg/day) | Survivors/Totala | MDDb,c ± SD | Hemorrhage Scorec | Weightd (mg) | Virus Titerd (Log10 CCID50/g) |
| 3 (100) | 7/8e, * | 11.0 | 0.0 ± 0.0** | 246±21* | 6.2 ± 0.5** |
| 3 (30) | 2/10 | 10.3 ± 1.0 | 1.0 ± 1.1* | 288 ± 22 | 7.4 ± 0.4 |
| 3 (10) | 0/9e | 8.7 ± 0.9 | 0.9 ± 0.2* | 246 ± 36* | 7.4 ± 0.4 |
| 3 (3) | 0/10 | 8.4 ± 0.5 | 1.7 ± 1.0 | 284 ± 46 | 7.2 ± 0.4 |
| 3 (1) | 0/10 | 9.3 ± 0.9 | 1.2 ± 0.8* | 302 ± 53 | 7.7 ± 0.4 |
| Amantadine (1) (100) | 2/10 | 11.1 ± 3.4 | 1.7 ± 1.2 | 282 ± 5 | 6.6 ± 0.5 |
| Amantadine (1) (30) | 0/10 | 10.1 ± 1.9 | 1.4 ± 0.9 | 224±38** | 7.3 ± 0.4 |
| Amantadine (1) (10) | 1/10 | 9.3 ± 1.4 | 3.4 ± 0.5 | 346 ± 65 | 7.3 ± 0.3 |
| Oseltamivir (4) (10) | 10/10*** | - | 0.0 ± 0.0** | 178±28*** | 2.5 ± 0.2*** |
| Placebo | 1/20 | 9.0 ± 0.8 | 3.0 ± 1.2 | 336 ± 32 | 7.3 ± 0.3 |
Statistically evaluated by two-tailed Fisher's exact test.
Mean day of death of mice that died prior to day 22. None of the compound treatment results were significantly different than placebo (P>0.05).
Pairwise comparisons were statistically evaluated by two-tailed Mann-Whitney U-Test.
Statistically analyzed by Tukey-Kramer multiple comparisons test.
Group sizes were originally at 10. Deaths occurred during the treatment phase (which was likely due to p.o. treatment trauma), and these mice were excluded from the study.
P<0.05
P<0.01
P<0.001 compared to placebo.
Body weights during the infection are depicted in Figs. 4B and 4C. Minimal weight loss occurred in the oseltamivir-treated group, followed by the compound 3 (100 mg/kg/day) (Fig. 4B) and the amantadine (1) (100 mg/kg/day) (Fig. 4C) groups. Rebound in body weight occurred faster in the compound 3 (100 mg/kg/day) group than in the comparable amantadine group.
In conclusion, the 100-mg/kg/day dose of compound 3 was effective in terms of survival outcome, protection from body weight loss, and improvement in lung hemorrhage score. In contrast, amantadine (1) showed minimum effectiveness at 100 mg/kg/day in treating the M2-V27A virus infection.
3.5. Design, synthesis, and in vitro activity of analogs of compound 3
Encouraged by the promising in vitro and in vivo antiviral efficacy of compound 3, we then designed two analogs of compound 3 (Fig. 5) with the aim to further improve antiviral efficacy. Compound 7 is the guanidine analog of 3, and it was synthesized through a two-step synthesis procedure with an overall yield of 68%. Compound 16 is the dithiane analog of compound 3. Synthesis of compound 16 started from 2-amino-1,3-propanediol (8). The amine was first protected with benzyl chloroformate (CbzCl) to give the intermediate 10 with 85% yield, then the alcohol in 10 was converted to thioester 12 through the Mitsunobu reaction with 72% yield. The ester 12 was hydrolyzed and the dithiol intermediate 13 was condensed with 2-adamantone 14 to give the protected dithiane 15 in 65% overall yield. Finally, the Cbz protecting group was cleaved by Pd/C and ammonium formate, and the hydrochloride salt of the amine (16) was formed by treatment of HCl in methanol. The overall yield for the last two steps was 78%.
The channel blockage activity of the newly synthesized compounds 7 and 16 was assayed against both M2-WT and M2-V27A proton channels using the two-electrode voltage clamp (TEVC) assays (Table 4). For the M2-WT channel inhibition, compound 7 had a similar potency as compound 3, reaching 94.5 ± 2.0 % current inhibition at 100 μM. Compound 16 had reduced channel blockage with 64.0 ± 1.2 % at 100 μM. For M2-V27A channel inhibition, all compounds nearly completely inhibited the current conductance at 100 μM, and the IC50 values for compounds 3, 7, and 16 in inhibiting the A/Udorn/72 (M2-V27A) were 0.3 ± 0.1 μM, 0.5 ± 0.2 μM, and 0.4 ± 0.1 μM, respectively (Table 4). Collectively, the two analogs, 7 and 16, had confirmed channel blockage activity against both M2-WT and M2-V27A mutant channels. When tested against the M2-S31N channel, none of the compounds (3, 7, and 16) showed more than 12 % channel inhibition at 100 μM. These results were expected since compounds 3, 7, and 16 were designed to target the M2-V27A mutant, not the M2-S31N mutant.
Table 4.
Channel blockage activity of compound 3 and its two analogs, 7 and 16, in inhibiting the A/Udorn/72 (M2-WT) and A/Udorn/72 (M2-V27A) proton channels.
| Electrophysiological Assay % current inhibition at 100 μM and IC50 (μM) | ||
|---|---|---|
| M2-WT | M2-V27A | |
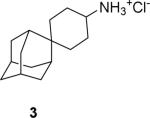
|
89.1 ± 0.4 % at 100 μM IC50 = 18.7 ± 1.4 μM |
95.2 ± 0.3 % at 100 μM IC50 = 0.3 ± 0.1 μM |
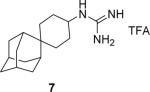
|
94.5 ± 2.0 % at 100 μM IC50 = 8.5 ± 2.3 μM |
93.3 ± 1.3 % at 100 μM IC50 = 0.5 ± 0.2 μM |
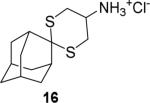
|
64.0 ± 1.2 % at 100 μM IC50 N.T.* |
94.4 ± 1.0 % at 100 μM IC50 = 0.4 ± 0.1 μM |
N.T. = not tested.
We then tested the antiviral activity of compounds 7 and 16 against both M2-WT-containing A/Udorn/72 (M2-WT) (H3N2) virus and the M2-V27A-containing A/WSN/33 (M2-N31S + V27A) (H1N1) virus in plaque assays (Fig. 6). Compounds 7 and 16 each had potent antiviral activity against the M2-WT-containing A/Udorn/72 (H3N2) virus, with EC50 values of 6.4 ± 0.4 μM and 0.07 ± 0.01 μM, respectively. In contrast, only compound 16 had potent antiviral activity against the M2-V27A-containing A/WSN/33 (M2-N31S + V27A) (H1N1) virus, with an EC50 value of 1.0 ± 0.1 μM. Compound 7 had no effect on the replication of the M2-V27A-containing A/WSN/33 (M2-N31S + V27A) (H1N1) virus up to 30 μM, despite its potent channel blockage, as confirmed by the TEVC assay (Table 4). The cytotoxicity of compounds 3, 7, and 16 were tested against the MDCK cells using the neutral red method, and the two analogs, 7 and 16, were less cytotoxic than compound 3 (Table 5).
Fig. 6. Antiviral efficacy of compounds 7 and 16 in inhibiting M2-WT and M2-V27A-containing influenza A viruses.

(A) Plaque assay results of compounds 7 and 16 in inhibiting M2-WT containing A/Udorn/72 (H3N2) virus. The EC50 values for 7 and 16 were 6.4 ± 0.4 μM and 0.07 ± 0.01 μM, respectively. (B) Plaque assay results of compounds 7 and 16 in inhibiting M2-V27A-containing A/WSN/33 (M2-N31S + V27A) (H1N1) virus. Compound 7 was not active and compound 16 had an EC50 of 1.0 ± 0.1 μM. Oseltamivir carboxylate (4) was used as a control.
Table 5.
Antiviral efficacy and cytotoxicity of compounds 3, 7, and 16.
| Compounds | Antiviral Efficacy EC50 (μM) | Cytotoxicity CC50 (μM) | |
|---|---|---|---|
| A/Udorn/72 (H3N2) M2-WT | A/WSN/33 (M2-N31S + V27A) (H1N1) M2-V27A | ||

|
0.3 ± 0.1 | 1.8 ± 0.2 | 27.6 ± 0.5 |

|
6.4 ± 0.4 | > 30 | 150.5 ± 3.2 |

|
0.07 ± 0.01 | 1.0 ± 0.1 | 74.8 ± 1.2 |
As shown, the correlations between the channel blockage results and the antiviral efficacy were not linear. Compound 16 had reduced M2-WT channel blockage compared to compound 3 (64.0 ± 1.2 % versus 89.1 ± 0.4 %); however, the antiviral activity of compound 16 in inhibiting the M2-WT-containing A/Udorn/72 (H3N2) virus was four-fold greater than compound 3 (EC50 = 0.07 ± 0.01 μM versus 0.3 ± 0.1 μM). Similarly, compound 7 had very poor antiviral (EC50 > 30 μM) potency against M2-V27A-containing A/WSN/33 (M2-N31S + V27A) (H1N1) virus despite its potent channel blockage (93.3 ± 1.3 % at 100 μM). The discrepancy promoted us to take a further look at the drug binding kinetics. It is known that the adamantane class of drugs bind with slow-on and very slow-off rates to the M2 channel, and the rate of recovery varies significantly between individual compounds (Balannik et al., 2009; Ma et al., 2016b; Tu et al., 1996; Wang et al., 1993). Examination of the current traces from electrophysiological recordings revealed interesting findings (Fig. 7). In these experiments, the pH is first dropped to 5.5 to allow measurement of channel conduction. Once a steady-state value is approached, the compound is added, leading to rapid inhibition. After the full extent of inhibition is achieved free drug is removed by perfusion with drug-free pH 8.5 buffer (the high pH avoids membrane leakage from prolonged exposure to low pH). After a delay of 1 min, the buffer is again returned to pH 5.5, and the extent of reversal of channel block assessed from the fractional change in proton flux relative to the value prior to drug treatment. In the case of compound 3, as shown by the second activation trace (Fig. 7A), the M2-V27A channel remained partially occluded even with perfusion with compound-free solution (pH = 8.5). The channel conductance current at the second activation event was 50.4 ± 7.5% of the maximum current. In contrast, the M2-V27A channel inhibition by compound 7 was nearly completely reversible (Fig. 7B), reaching 93.9 ± 1.7% of the maximum current at the second activation event. This observation provides an attractive explanation for the lack of antiviral activity of compound 7. The dithiane compound 16, on the other hand, showed essentially irreversible inhibition of the M2-V27A channel on this time scale, and only 12.2 ± 1.3 % current was recovered at the second activation event (Fig. 7C). Taken together, it became apparent that the kinetics of drug inhibition is a critical parameter for channel inhibition: a very slow rate of reversal appears to correlate with potent antiviral efficacy, while more rapidly reversible channel blockers were less active. As such, caution has to be taken when predicting the compound's antiviral activity based on its percent channel blockage in electrophysiological assays.
Fig. 7. Electrophysiological assays of compounds 3, 7, and 16 in inhibiting the current conductance of the A/Udorn/72 (M2-V27A) proton channel.

(A) M2-V27A channel conductance traces with and without compound 3. (B) M2-V27A channel conductance traces with and without compound 7. (C) M2-V27A channel conductance trace with and without compound 16.
4. Discussion and conclusions
M2-V27A is one of the most frequent amantadine-resistant mutations among influenza A viruses that are circulating among humans and avian species (Dong et al., 2015; Durrant et al., 2015). More concerning is the fact that M2-V27A mutant is directly correlated with drug selection pressure (Furuse et al., 2009b). Although the most frequent amantadine-resistant mutant M2-S31N remains partially sensitive to amantadine (Wang et al., 2013), the M2-V27A mutant was completely resistant to amantadine (Balannik et al., 2009), which makes it a challenging drug target. Nevertheless, propelled by an integrated approach involving molecular dynamics simulations, NMR, electrophysiology, and medicinal chemistry, we were able to design the first-in-class M2-V27A inhibitors (Wang et al., 2011a; Wang et al., 2011b). In this report, we further evaluated the in vitro and in vivo antiviral activity of the M2-V27A inhibitors. The representative compound, 3, not only had potent in vitro antiviral activity, but also potent in vivo antiviral activity against both M2-WT- and M2-V27A-containing influenza A viruses. This proof-of-concept study demonstrates that drug-resistant M2 mutants are valid antiviral drug targets and should be further pursued.
During the course of drug discovery targeting the M2 proton channels, the percentage of channel blockage or dose-dependent channel blockage (IC50 value) of compounds was normally used to predict their antiviral activity. It is generally assumed that compounds with higher channel blockage activity will display potent antiviral activity. However, as has been demonstrated by compound 7 in inhibiting the M2-V27A channel, this assumption appears incorrect. The results from this study emphasize the role of drug binding kinetics: slow-reversible or irreversible channel blockers correlate with potent antiviral activity, while reversible channel blockers showed weak or no antiviral activity. With such knowledge in mind, priority in lead optimization should be given to compounds with slow or irreversible binding kinetics during the following drug discovery development.
Supplementary Material
Highlights.
Compound 3 displayed potent in vivo antiviral activity against both M2-WT- and M2-V27A-containing influenza A viruses
Dithiane analog (16) had improved in vitro antiviral activity against M2-WT- and M2-V27A-containing influenza A viruses
Potent M2 channel blockers with very slow reversal of channel inhibition kinetics showed potent antiviral activity
Acknowledgements
This work was supported by the startup funds and the NIH AI 119187 from the University of Arizona to J.W. and NIH GM056423 to W.F.D. R.M. was supported by the NIH training grant T32 GM008804. Funding for D.F.S. and for the animal studies was provided by the Respiratory Diseases Branch, DMID, NIAID, NIH under contract HHSN272201000039I. We thank David Bishop for proofreading and editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abed Y, Goyette N, Boivin G. Generation and characterization of recombinant influenza A (H1N1) viruses harboring amantadine resistance mutations. Antimicrob. Agents Chemother. 2005;49:556–559. doi: 10.1128/AAC.49.2.556-559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balannik V, Carnevale V, Fiorin G, Levine B, Lamb R, Klein M, DeGrado W, Pinto L. Functional Studies and Modeling of Pore-Lining Residue Mutants of the Influenza A Virus M2 Ion Channel. Biochemistry. 2010;49:696–708. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, DeGrado WF, Pinto LH. Design and Pharmacological Characterization of Inhibitors of Amantadine-Resistant Mutants of the M2 Ion Channel of Influenza A Virus. Biochemistry. 2009;48:11872–11882. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AN, McSharry JJ, Weng Q, Driebe EM, Engelthaler DM, Sheff K, Keim PS, Nguyen J, Drusano GL. In Vitro System for Modeling Influenza A Virus Resistance under Drug Pressure. Antimicrob. Agents Chemother. 2010;54:3442–3450. doi: 10.1128/AAC.01385-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G, Peng C, Luo J, Wang C, Han L, Wu B, Ji G, He H. Adamantane-Resistant Influenza A Viruses in the World (1902–2013): Frequency and Distribution of M2 Gene Mutations. PloS one. 2015;10:e0119115. doi: 10.1371/journal.pone.0119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant MG, Eggett DL, Busath DD. Investigation of a recent rise of dual amantadine-resistance mutations in the influenza A M2 sequence. BMC Genet. 2015;16:S3. doi: 10.1186/1471-2156-16-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse Y, Suzuki A, Kamigaki T, Oshitani H. Evolution of the M gene of the influenza A virus in different host species: large-scale sequence analysis. Virol. J. 2009a;6:67. doi: 10.1186/1743-422X-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse Y, Suzuki A, Oshitani H. Large-scale sequence analysis of M gene of influenza A viruses from different species: mechanisms for emergence and spread of amantadine resistance. Antimicrob. Agents Chemother. 2009b;53:4457–4463. doi: 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorkova EA, Baranovich T, Seiler P, Armstrong J, Burnham A, Guan Y, Peiris M, Webby RJ, Webster RG. Antiviral resistance among highly pathogenic influenza A (H5N1) viruses isolated worldwide in 2002-2012 shows need for continued monitoring. Antiviral Res. 2013;98:297–304. doi: 10.1016/j.antiviral.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PloS one. 2014;9:e92444. doi: 10.1371/journal.pone.0092444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden FG, Hay AJ. Emergence and transmission of influenza A viruses resistant to amantadine and rimantadine. Curr. Top. Microbiol. Immunol. 1992;176:119–130. doi: 10.1007/978-3-642-77011-1_8. [DOI] [PubMed] [Google Scholar]
- Herlocher ML, Truscon R, Fenton R, Klimov A, Elias S, Ohmit SE, Monto AS. Assessment of development of resistance to antivirals in the ferret model of influenza virus infection. J. Infect. Dis. 2003;188:1355–1361. doi: 10.1086/379049. [DOI] [PubMed] [Google Scholar]
- Hong M, DeGrado WF. Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 2012;21:1620–1633. doi: 10.1002/pro.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser K, Subbarao K. Influenza Vaccines: Challenges and Solutions. Cell Host Microbe. 2015;17:295–300. doi: 10.1016/j.chom.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt A, Holien J, Parker M, Barr I. Oseltamivir Resistance and the H274Y Neuraminidase Mutation in Seasonal, Pandemic and Highly Pathogenic Influenza Viruses. Drugs. 2009;69:2523–2531. doi: 10.2165/11531450-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Hurt AC. The epidemiology and spread of drug resistant human influenza viruses. Curr. Opin. Virol. 2014;8:22–29. doi: 10.1016/j.coviro.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Iwahashi J, Tsuji K, Ishibashi T, Kajiwara J, Imamura Y, Mori R, Hara K, Kashiwagi T, Ohtsu Y, Hamada N, Maeda H, Toyoda M, Toyoda T. Isolation of amantadine-resistant influenza a viruses (H3N2) from patients following administration of amantadine in Japan. J. Clin. Microbiol. 2001;39:1652–1653. doi: 10.1128/JCM.39.4.1652-1653.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Yen HL. Targeting the host or the virus: Current and novel concepts for antiviral approaches against influenza virus infection. Antiviral Res. 2012;96:391–404. doi: 10.1016/j.antiviral.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loregian A, Mercorelli B, Nannetti G, Compagnin C, Palù G. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell. Mol. Life Sci. 2014:1–25. doi: 10.1007/s00018-014-1615-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Li F, Musharrafieh RG, Wang J. Discovery of cyclosporine A and its analogs as broad-spectrum anti-influenza drugs with a high in vitro genetic barrier of drug resistance. Antiviral Res. 2016a;133:62–72. doi: 10.1016/j.antiviral.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Zhang J, Wang J. Pharmacological Characterization of the Spectrum of Antiviral Activity and Genetic Barrier to Drug Resistance of M2-S31N Channel Blockers. Mol. Pharmacol. 2016b;90:188–198. doi: 10.1124/mol.116.105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto AS, Webster RG. Influenza pandemics: History and lessons learned, Textbook of Influenza. John Wiley & Sons, Ltd; 2013. pp. 20–34. [Google Scholar]
- Murray JL, McDonald NJ, Sheng J, Shaw MW, Hodge TW, Rubin DH, O'Brien WA, Smee DF. Inhibition of influenza A virus replication by antagonism of a PI3K-AKT-mTOR pathway member identified by gene-trap insertional mutagenesis. Antivir. Chem. Chemother. 2012;22:205–215. doi: 10.3851/IMP2080. [DOI] [PubMed] [Google Scholar]
- Pielak RM, Chou JJ. Solution NMR structure of the V27A drug resistant mutant of influenza A M2 channel. Biochem. Biophys. Res. Commun. 2010;401:58–63. doi: 10.1016/j.bbrc.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud C, Kuypers J, Englund JA. Emerging oseltamivir resistance in seasonal and pandemic influenza A/H1N1. J. Clin. Virol. 2011;52:70–78. doi: 10.1016/j.jcv.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008;3:1125–1131. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- Rey-Carrizo M, Barniol-Xicota M, Ma C, Frigolé-Vivas M, Torres E, Naesens L, Llabrés S, Juárez-Jiménez J, Luque FJ, DeGrado WF, Lamb RA, Pinto LH, Vázquez S. Easily Accessible Polycyclic Amines that Inhibit the Wild-Type and Amantadine-Resistant Mutants of the M2 Channel of Influenza A Virus. J. Med. Chem. 2014;57:5738–5747. doi: 10.1021/jm5005804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey-Carrizo M, Torres E, Ma C, Barniol-Xicota M, Wang J, Wu Y, Naesens L, DeGrado W, Lamb R, Pinto L, Vazquez S. 3-Azatetracyclo[5.2.1.1(5'8).0(1,5)]undecane Derivatives: From Wild-Type Inhibitors of the M2 Ion Channel of Influenza A Virus to Derivatives with Potent Activity against the V27A Mutant. J. Med. Chem. 2013;56:9265–9274. doi: 10.1021/jm401340p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee DF, Bailey KW, Wong MH, O'Keefe BR, Gustafson KR, Mishin VP, Gubareva LV. Treatment of influenza A (H1N1) virus infections in mice and ferrets with cyanovirin-N. Antiviral Res. 2008;80:266–271. doi: 10.1016/j.antiviral.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MG, Shay DK, Zhou H, Bridges CB, Cheng PY, Burns E, Bresee JS, Cox NJ. Estimates of Deaths Associated With Seasonal Influenza-United States, 1976-2007 (Reprinted from MMWR, vol 59, pg 1057-1062, 2010). JAMA. 2010;304:1778–1780. [Google Scholar]
- Tu Q, Pinto LH, Luo G, Shaughnessy MA, Mullaney D, Kurtz S, Krystal M, Lamb RA. Characterization of inhibition of M2 ion channel activity by BL-1743, an inhibitor of influenza A virus. J. Virol. 1996;70:4246–4252. doi: 10.1128/jvi.70.7.4246-4252.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J. Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. M2 as a target to combat influenza drug resistance: what does the evidence say? Future Virol. 2016;11:1–4. [Google Scholar]
- Wang J, Li F, Ma C. Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses. Biopolymers. 2015;104:291–309. doi: 10.1002/bip.22623. [DOI] [PubMed] [Google Scholar]
- Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Kein ML, DeGrado WF. Molecular Dynamics Simulation Directed Rational Design of Inhibitors Targeting Drug-Resistant Mutants of Influenza A Virus M2. J. Am. Chem. Soc. 2011a;133:12834–12841. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ma C, Wu Y, Lamb RA, Pinto LH, DeGrado WF. Exploring Organosilane Amines as Potent Inhibitors and Structural Probes of Influenza A Virus M2 Proton Channel. J. Am. Chem. Soc. 2011b;133:13844–13847. doi: 10.1021/ja2050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Qiu JX, Soto C, DeGrado WF. Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr. Opin. Struct. Biol. 2011c;21:68–80. doi: 10.1016/j.sbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Ma CL, Wang J, Jo H, Canturk B, Fiorin G, Pinto LH, Lamb RA, Klein ML, DeGrado WF. Discovery of Novel Dual Inhibitors of the Wild-Type and the Most Prevalent Drug-Resistant Mutant, S31N, of the M2 Proton Channel from Influenza A Virus. J. Med. Chem. 2013;56:2804–2812. doi: 10.1021/jm301538e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Cross TA, Zhou H-X. A Secondary Gate As a Mechanism for Inhibition of the M2 Proton Channel by Amantadine. J. Phys. Chem. B. 2008;112:7977–7979. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.