

Abstract
Soft tissue calcification is associated with aging, common conditions such as diabetes or hypercholesterolemia and with certain genetic disorders. ABCC6 is an efflux transporter primarily expressed in liver facilitating the release of ATP from hepatocytes. Within the liver vasculature, ATP is converted into pyrophosphate (PPi), a major inhibitor of ectopic calcification. ABCC6 mutations thus lead to reduced plasma PPi levels resulting in the calcification disorder pseudoxanthoma elasticum (PXE) and some cases of generalized arterial calcification of infancy (GACI). Most mutations in ABCC6 are missense and many retain/preserve transport activity but are retained intracellularly. We have previously shown that the chemical chaperone 4-phenylbutyrate (4-PBA) promotes the maturation of ABCC6 mutants to the plasma membrane. In a humanized mouse model of PXE, we investigated whether 4-PBA treatments could rescue the calcification inhibition potential of selected ABCC6 mutants. We used the dystrophic cardiac calcification (DCC) phenotype of Abcc6−/− mice as an indicator of ABCC6 function to quantify the effect of 4-PBA on human ABCC6 mutants transiently expressed in the liver. We showed that 4-PBA administrations restored the physiological function of ABCC6 mutants resulting in enhanced calcification inhibition. This study identifies 4-PBA treatments as a promising strategy for allele-specific therapy of ABCC6-associated calcification disorders.
INTRODUCTION
Ectopic calcification is commonly associated with hypercholesterolemia, diabetes, chronic renal insufficiency and certain rare genetic diseases. ABCC6 is an organic anion transporter primarily present in the basolateral plasma membrane of hepatocytes (Pomozi et al., 2013). ABCC6 deficiency is linked to several mineralization pathologies: pseudoxanthoma elasticum (PXE, OMIM 264800) is characterized by late onset and progressive calcifications in the skin leading to prominent dermal manifestations as well as in vascular and ocular tissues (Le Saux et al., 2000); β-thalassemia (OMIM 187550), which may be related to reduced levels of ABCC6 protein in the liver (Martin et al., 2011); and a subset of generalized arterial calcification of infancy (GACI, OMIM 208000), that is typically associated with ENPP1 mutations and results in early patient mortality (Nitschke et al., 2012). In several inbred strains of mice, including C3H/HeJ, ABCC6 deficiency causes an acute calcification phenotype affecting the myocardium and the media of large arteries (Aherrahrou et al., 2008; Doehring et al., 2006; Meng et al., 2007). Because this peculiar phenotype occurs in response to a tissue injury, it is referred to as dystrophic cardiac calcification or DCC (Brampton et al., 2014).
A recent metabolomics analysis revealed that ABCC6 facilitates the cellular efflux of nucleotide triphosphates, including ATP, which is rapidly converted into inorganic pyrophosphate (PPi) by the ectonucleotidase ENPP1 (Jansen et al., 2013). PPi is a potent inhibitor of ectopic calcification. ABCC6 is responsible for the majority of PPi release from the liver, and accounts for ~60% of PPi plasma levels in mice and humans (Jansen et al., 2014).
4-phenylbutyrate (4-PBA) is a short-chain fatty acid used in several clinical applications, including the treatment of urea cycle disorders, where it works as a nitrogen-scavenging molecule. 4-PBA also functions as a chemical chaperone and can correct the trafficking defects of misfolded proteins (Iannitti and Palmieri, 2011) including many transmembrane proteins associated with human diseases such as ABCC7, ABCA1 or ABCB11 (Gonzales et al., 2015; Hayashi et al., 2016; Rubenstein and Zeitlin, 2000; Sorrenson et al., 2013). As the majority of ABCC6 mutations are missense, we recently developed a methodology to investigate the functional consequences of these mutations on the human protein. In these studies a majority of ABCC6 missense mutants presented a transport activity similar to the wild type protein as determined by in vitro assays. However, most of these mutants presented abnormal cellular localization, failing to reach the plasma membrane both in cultured cells and mouse liver (Le Saux et al., 2011; Pomozi et al., 2014). The localization of the transmembrane ABCC6 protein into the basolateral plasma membrane is very relevant to its physiological function as an efflux transporter (Ilias et al., 2002; Le Saux et al., 2011; Pomozi et al., 2014; Pomozi et al., 2013). Treatment with 4-PBA significantly increased the plasma membrane localization for several transport-competent ABCC6 missense mutants in vitro and in vivo (Le Saux et al., 2011; Pomozi et al., 2014).
In the present study, we addressed whether calcification inhibition could be restored in Abcc6−/− mice expressing human ABCC6 mutants in their liver and treated with 4-PBA.
RESULTS
We and others have previously reported that 4-PBA administration rescued the plasma membrane targeting of several human ABCC6 mutants in wild type mice (Aranyi et al., 2013; Jin et al., 2015; Le Saux et al., 2011; Pomozi et al., 2014). In this study, we transiently expressed four ABCC6 missense mutants in the liver of Abcc6−/− mice and confirmed that these variants were also successfully routed back to the plasma membrane after 4-PBA injections (Fig. 1) using a methodology we previously described (Le Saux et al., 2011; Pomozi et al., 2014). Furthermore, we verified that 4-PBA redirected a representative ABCC6 mutant (p.Q1347H) to the basolateral compartment of the plasma membrane of hepatocytes, which is the normal location of both human and mouse ABCC6 (Pomozi et al., 2013; Scheffer et al., 2002; Sinko et al., 2003). For these control experiments, we used immunofluorescent staining to examine the co-localization of the p.Q1347H mutant and the wild type ABCC6 (as a control) with basolateral (Na-taurocholate co-transporting polypeptide (NTCP) and cadherin) and apical markers, (ABCB11) with and without 4-PBA treatments. Co-localization was quantified on immunofluorescent images using ImageJ2 (Schindelin et al., 2012) by calculating Pearson’s correlation coefficients between mutant and wild type ABCC6 and both basolateral markers. We found that, in the absence of 4-PBA treatment, the p.Q1347H mutant showed a predominant intracellular staining with a clear demarcation from NTCP and cadherin. After 4-PBA treatment however, the immunofluorescent staining pattern of the p.Q1347H mutant became similar to that of both basolateral markers with a significant increase of the Pearson’s correlation coefficients (Fig. 2A, B, D, E), indicating that the wild type and 4-PBA-treated mutant primarily localize in the basolateral plasma membrane compartment. Moreover, we observed very little evidence, if any, of co-localization of the wild type and the p.Q1347H mutant (with and without 4-PBA treatment) with the ABCB11 in the apical compartment of the plasma membrane and the corresponding Pearson’s correlation coefficients values were close to 0 (Fig. 2C, F). Collectively, these data indicate that upon 4-PBA treatment, the ABCC6 mutant p.Q1347H assumes a normal and physiological plasma membrane localization.
Figure 1. Intracellular localization of human ABCC6 variants expressed in Abcc6−/− mice.

Hydrodynamic tail vein injections led to the successful expression of human wild type and mutant ABCC6. ABCC6 was detected on liver frozen sections by immunofluorescence using the M6II-31 monoclonal antibody (green) to verify the plasma membrane localization with or without 4-PBA treatments. Co-labeling with the S-20 polyclonal antibody (red) was used to control for the absence of the endogenous mouse Abcc6 and wild type mice (Abcc6+/+) were used as positive controls. DAPI was used as counter staining (blue). With 4-PBA treatments, ABCC6 mutant proteins were predominantly located in the plasma membrane. Scale bar = 50μm.
Figure 2. The human ABCC6 variant p.Q1347H localizes to the basolateral membrane of Abcc6−/− mice treated with 4-PBA.

Liver frozen sections were used to perform immunofluorescent co-labeling between ABCC6 (green) and the basolateral markers (B) cadherin and (B) Na-taurocholate co-transporting polypeptide (NTCP), as well as (C) the apical marker (ABCB11) (red) to determine the plasma membrane localization. DAPI was used as counter staining (blue). To estimate co-localization between the normal and mutant ABCC6 and cadherin (D), NTCP (E) and ABCB11 (F), a Pearson’s correlation coefficient was calculated (ImageJ2) from sets of individual immunofluorescence images from animals with or without 4-PBA treatment. With 4-PBA treatments, the representative ABCC6 mutant p.Q1347H showed enhanced co-localization with the basolateral markers while no evidence of co-localization is seen with ABCB11 in the apical membrane. Scale bar = 20μm. Results for panels D–F are shown +/− SEM. * p<0.05, ** p<0.01, *** p<0.001.
Several years ago, Abcc6 was linked to DCC in several inbred strain of mice carrying a naturally occurring Abcc6 mutation (Aherrahrou et al., 2008; Meng et al., 2007). Although, 3 additional loci affecting the penetrance and expression of DCC were mapped to chromosomes 4, 12, and 14 (Ivandic et al., 2001), ABCC6 is the major determinant of this cardiac calcification phenotype (Aherrahrou et al., 2007; Korff et al., 2006a; Korff et al., 2006b). Moreover, we have shown that in Abcc6−/− mice the hepatic expression of ABCC6 has a predominant influence on the development of DCC (Brampton et al., 2014). Therefore, to determine if 4-PBA could restore the calcification inhibition potential of ABCC6 mutants, we quantified DCC as a measure of ABCC6 function. With this approach we first determined that the expression of the unrelated LacZ gene (Fig. 3A) or 4-PBA treatment by itself (not shown) had no significant influence on DCC in Abcc6−/− mice. After having established a baseline of DCC reduction (−66%, p=0.0001) using the wild type human ABCC6, we evaluated the effects of 4-PBA treatments in mice expressing 4 human disease-causing ABCC6 variants (Fig. 3A), which were chosen based on our previous results (Le Saux et al., 2011; Pomozi et al., 2014). The hepatic expression of the mutants alone provided no calcification inhibition. However, the concomitant administration of 4-PBA, led to significant DCC reduction for 3 of the variants: p.R1314W (−62.8%, p=0.0045), p.Q1347H (−64.2%, p=0.0005), and p.S1121W (−45.0%, p=0.045). Note that p.R1314W is an ABCC6 mutation common to PXE and GACI. The reduction in dystrophic calcification associated with these mutants was comparable to the wild-type human ABCC6 protein (p=0.62). The 4-PBA treatment of the p.R1114P mutant resulted in some calcification inhibition that did not reach statistical significance (p=0.10). Because most PXE patients are diagnosed during the second decade of life, with a calcification phenotype already developed, we addressed the question of whether established ABCC6-dependent calcification could also be reversed after restoring ABCC6 function with 4-PBA treatment. For this purpose, we have injected the p.Q1347H mutant in Abcc6−/− mice and started 4-PBA administration 36 hours after induction of DCC by cryoinjury, at a time when calcification is well-developed (Korff et al., 2006a). The results, shown on Fig. 3B, indicated that the restoration of ABCC6 function after cryionjury could not reverse existing calcification.
Figure 3. 4-PBA treatments rescue ABCC6 function.

A: The wild type ABCC6 protein, four PXE/GACI mutants and LacZ were transiently expressed in Abcc6−/− liver prior to inducing dystrophic cardiac calcification (DCC). Abcc6−/− mice were exposed to 4-PBA for 7 days (controls received saline injections) with a cumulative dose of 1,000 mg/kg/day. The level of calcification (DCC phenotype) was measured as total heart calcium content and normalized to tissue weight. B: The p.Q1347H variant was transiently expressed in Abcc6−/− liver prior to inducing dystrophic cardiac calcification (DCC). Abcc6−/− mice were administered 4-PBA 36 hrs after DCC induction (controls received saline injections) until the harvesting of hearts. The level of calcification (DCC phenotype) was measured as total heart calcium content and normalized to tissue weight. Results are +/− SEM. * p<0.05, ** p<0.01.
To confirm DCC quantification data, which only measured excess calcium in heart tissues, visual and histological characterization of cardiac calcification was assessed at the time of dissection by the presence of white deposits at the site of injury (Fig. 4A) and by Alizarin Red staining on paraffin-embedded sections (Fig. 4B). Results showed reduced mineralization in mice expressing the wild type ABCC6 and a comparable reduction for the representative p.Q1347H mutant after 4-PBA treatment which is consistent with calcification quantifications.
Figure 4. Histological evaluation of dystrophic cardiac calcification (DCC) of Abcc6−/− mice.

After excision, hearts were fixed in formalin and paraffin embedded. Sections were stained with Alizarin Red S and imaged. A: representative images obtained with Abcc6−/− mice illustrate the macroscopic appearances of the surface lesions outlined with a dashed circle. The calcification is visible as white deposits. B: microscopic images of heart cross sections of Abcc6−/− mice show the reduction of dystrophic cardiac calcification (Red deposits and white arrows) after expression of the p.Q1347H mutant and 4-PBA treatment. The expression of the gene LacZ and the wild type ABCC6 were used as negative and positive controls respectively. Scale bars = 200 μm.
As ABCC6 function facilitates the release of nucleosides triphosphates, and particularly ATP, which is quickly converted into PPi in the liver vasculature (Jansen et al., 2014; Jansen et al., 2013), we assayed plasma PPi levels of mice expressing the p.Q1347H mutant, which produced the most significant DCC rescue (Fig. 3). After several attempts in two different laboratories using various methodologies previously described (Jansen et al., 2014; Jansen et al., 2013), we were unable to detect significant change in plasma PPi levels (not shown). Therefore, we evaluated the effect of 4-PBA treatment on PPi production by the p.Q1347H mutant in HEK293 cells. Interestingly, the transfection with the mutant protein led to an increase in PPi concentration in the supernatant over baseline levels (empty vector). This was not surprising as we previously found that ABCC6 missense mutants translocate to the plasma membrane more efficiently in non-polarized cells in culture than in vivo (Pomozi et al., 2014). The addition of 5mM of 4-PBA in the culture medium had little effects on HEK293 cells transfected with an empty vector whereas it triggered a 64% increase (p=0.03) in PPi release into the supernatant of transfected cells (Fig. 5). This result confirmed that 4-PBA restored the function of the p.Q1347H and probably that of the others mutant (Fig. 3, 4), and provides support to our speculation that the chaperone effects of this drug rescued some of the ABCC6-mediated PPi production.
Figure 5. Effect of 4-PBA on PPi production in vitro.
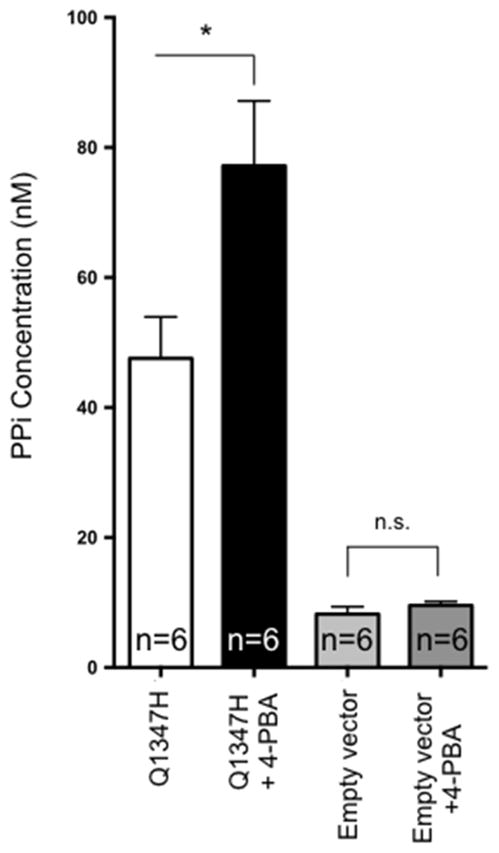
HEK293 cells were transfected with a plasmid carrying a cDNA encoding the ABCC6 mutant p.Q1347H or with an empty vector as transfection control. The presence of 5mM of 4-PBA in the culture medium resulted in a significant increase in PPi production suggesting the restoration of p.Q1347H cellular function. Results are +/− SEM, * p<0.05, n.s. = not significant.
DISCUSSION
Although palliative treatment exists for PXE patients (Finger et al., 2011), both PXE and GACI are still incurable diseases. PPi has a short half-life in plasma and there is currently no administration protocol available for this molecule for long-term therapy. Bisphosphonates, non-hydrolysable analogs of PPi, were tested in GACI (Otero et al., 2013) and in Abcc6−/− mice (Li et al., 2015). Early treatments improve GACI outcome but Etidronate has undesirable side-effects (Otero et al., 2013). Moreover, bisphosphonates have pleiotropic effects on cellular functions and the large doses of Etidronate necessary in Abcc6−/− mice to achieve modest effects (Li et al., 2015) suggest these analogs may not be the best treatment option. Another approach at rescuing the common ABCC6 R1141X variant using the drug PTC-124 has been tested in mice (Zhou et al., 2013) but because this premature stop codon is recoded as amino acids other than the original arginyl residue, its translational value remains unclear.
There is now a growing interest in developing and testing a pharmacological correction for mutated transmembrane proteins. Developing new drugs for humans is a long and costly process, and generally unattractive for orphan diseases. One current and very promising trend to circumvent this problem is to re-purpose existing drugs and using them towards pathologies not initially identified for these drugs. 4-PBA has been used for many years for the clinical treatment of urea cycle disorders and thalassemia (Dover et al., 1992; Maestri et al., 1996; Perrine et al., 1993). 4-PBA also influences the transcription of endoplasmic reticulum chaperones (Iannitti and Palmieri, 2011) and this physiological property has been used to treat patients with familial intrahepatic cholestasis type 2 caused by ABCB11mutations that result in intracellular retention of an otherwise functional protein (Gonzales et al., 2012; Gonzales et al., 2015; Hayashi et al., 2016; Naoi et al., 2014). In the present study, the doses of 4-PBA used in our humanized mouse model of PXE were similar to those used clinically (Maestri et al., 1996) and successfully restored the calcification inhibition potential of 3 selected ABCC6 missense mutants. However, for one of the 4 mutants (p.R1114P), the rescue of calcification inhibition was only minimal despite normal transport activity and plasma membrane localization after 4-PBA treatment (Pomozi et al., 2014). This suggested that the arginine to proline substitution might have compromised other essential functions of the protein, such as substrate specificity, and illustrates some limitations to the applicability of 4-PBA to rescue ABCC6 missense mutations.
Although we evidenced the positive effect of 4-PBA on the correct plasma membrane targeting of ABCC6 mutants and PPi production in cell culture, we could not detect a similar rise in pyrophosphate in murine plasma samples. This may be explained by the methodologies employed in our study. Unlike human PXE patients who express mutant ABCC6 in all of their hepatocytes, the hydrodynamic tail vein injections used in our study only results in partial and transient transfection (Brampton et al., 2014; Jiang et al., 2010). Considering the inherent difficulties of measuring PPi in biological fluids (O’Neill et al., 2010; Russell et al., 1971) and its short half-life (O’Neill et al., 2011), it is therefore, possible that the peak of PPi production was below our detection levels. Furthermore, a facet of ABCC6 physiological function could not be evaluated in this study as the substrate(s) that ABCC6 (potentially) efflux is still unknown (Jansen et al., 2013) and there may be alternative molecular mechanism(s) other than pyrophosphate by which ABCC6 prevents ectopic calcification. Another limitation of our experimental model is that the functional rescue (inhibition of calcification) was not complete. This could be due to the fact that we have restored the cellular localization and the physiological activity of the protein only in liver. The liver has the highest levels of ABCC6 expression but several other tissues and cells also express the protein (Beck et al., 2005; Beck et al., 2003; Douet et al., 2007) and these could contribute significantly to calcification inhibition. However, the effect of 4-PBA on calcification inhibition in human would most likely surpass that obtained with our humanized mouse model as PXE patient express ABCC6 in all liver hepatocytes and other tissues.
In conclusion, our study demonstrated the viability of using 4-PBA to rescue the functional defects of certain ABCC6 missense mutations and restore their calcification inhibition potential. This interpretation is restricted to the ABCC6 variants studied and the restoration of their physiological function as determined by injury-induced DCC, which is distinct from the typical PXE phenotype; we could not assess the PXE-like spontaneous calcification of Abcc6−/− mice because of our experimental settings. However, based on our results, we propose that 4-PBA treatments could serve as an allele-specific therapy for PXE/GACI. Approximately 75% of PXE patients harbor at least one missense allele and many of these patients carrying missense mutants with residual transport activity could be eligible for 4-PBA treatment. As PXE and GACI are recessive diseases, the rescue of single missense allele should be sufficient. Therefore, if 4-PBA treatment is eventually used to treat PXE patients, one will have to define which clinical criteria to use to assess treatment efficacy. Moreover, because we found that 4-PBA treatment of ABCC6 mutations prevents but does not reverse mineralization, the sooner treatment is initiated, the better.
MATERIAL AND METHODS
The experiments described in this manuscript were conducted at the University of Hawaii and Hungarian Academy of Sciences following identical experimental protocols so that results from one laboratory were systematically duplicated and verified in the other facility and later integrated. The results shown on Figure 1 and 4 were obtained at the University of Hawaii. The results of Figure 2 and 5 were primarily generated at Hungarian Academy of Sciences. Figure 3 was developed at the University of Hawaii with contribution from the Hungarian Academy of Sciences.
Animals
C57BL/6J mice, designated here as wild type were derived from mice purchased from The Jackson Laboratory. Abcc6tm1Aabb mice were generated on 129/Ola background (Gorgels et al., 2005) and backcrossed into a C57BL/6J >10 times. These mice are herein designated Abcc6−/−. Both male and female, 3 to 4 month-old age-matched Abcc6−/− and wild type mice were used, as gender had no significant influence on our previous results. All animals were housed in approved animal facilities at the University of Hawaii School of Medicine and the Research Center for Natural Sciences, Hungarian Academy of Sciences. Mice were kept under routine laboratory conditions with 12-hours light-dark cycle, a constant temperature of 23°C and with ad libitum access to water and chow in similar facilities at the University of Hawaii and Hungarian Academy of Sciences. The University of Hawaii and Hungarian Academy of Sciences Institutional Animal Care and Use Committees approved this study. Experiments have been conducted according to national guidelines.
Immunodetection
Liver-specific expression of ABCC6 variants in mice and immunohistochemical staining were performed as described in a previous study (Le Saux et al., 2011). Briefly, after mouse euthanasia, multiple liver lobes were quickly harvested, placed in Optimum Cutting Temperature (OCT) compound and stored at −80°C. Immunofluorescent staining was performed on 6 μm-thick frozen sections. The rat monoclonal anti-ABCC6 M6II-31 antibody (sc-59618) was used to specifically detect the human ABCC6. The anti-Abcb11 antibody and anti-Ntcp antibody were gifts from Dr. Bruno Stieger (Univ. Zurich, Switzerland). The rabbit polyclonal anti-Cadherin antibody was purchased from Abcam (Cambridge, MA). The rabbit polyclonal S-20 anti-Abcc6 antibody (sc-5787) was used to verify the absence of the endogenous protein in Abcc6−/− mice as we have already demonstrated the specificity of this antibody on frozen liver sections and isolated primary hepatocytes from Abcc6−/− mice, (Le Saux et al., 2011; Pomozi et al., 2013). These primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The secondary antibodies were Alexafluor 488 and 568 (Life Technologies, CA). Immunofluorescent images were acquired with a Zeiss LSM 710 confocal laser scanning microscope. For evaluation of co-localization, individual images of cells for the ABCC6 Q1347H mutant were collected and processed with Photoshop CS6 (Adobe, San Jose, CA). The Pearson’s correlation coefficient was calculated using the ImageJ2 software (Schindelin et al., 2012) using the Coloc 2 tool.
Liver-specific expression of ABCC6 cDNA in mice
Inducing the transient expression of a specific cDNA by hydrodynamic tail vein injection has been described previously (Alino et al., 2003; Le Saux et al., 2011; Liu et al., 1999; Zhang et al., 1999; Zhang et al., 2004). Briefly, cDNAs encoding the wild type and several variants of the human ABCC6 or the β-galactosidase were sub-cloned into the pLIVE vector carrying the mouse albumin promoter and a fetoprotein enhancer that ensured a liver-specific expression (Mirus Bio, Madison, WI). Plasmid DNA constructs were delivered to 3-month old Abcc6−/− mice by hydrodynamic tail vein injection. The injections were performed with a 27-gauge needle with a volume of 1.5 to 2ml of DNA in a solution of TransIT EE® according to the manufacturer’s instruction (Mirus Bio Madison, WI). Mice were injected with the optimal dosage of 30 to 60 μg of plasmid, as previously defined (Le Saux et al., 2011).
Myocardial cryoinjury to induce DCC
Cardiac injury was instilled through trans-diaphragm cryoinjury as previously described (Brampton et al., 2014). Mice were sacrificed by CO2 asphyxiation 7 days after injury to ensure full DCC development. Hearts were quickly removed, rinsed in phosphate buffered saline, imaged and either minced and placed in a 0.15N HCl solution to quantify calcium content (see below) or paraffin-embedded for histology analysis with Alizarin Red staining.
Calcification measurements
The level of mineralization in the whole ventricular area of the hearts minus the atria was quantified with a colorimetric assay (McGee-Russell, 1958) that measures directly the amount of excess calcium, which is then normalized to the weight of the excised tissues, as previously described (Brampton et al., 2014). Briefly, hearts were minced and decalcified for 48 hours in 0.15N HCl and the total calcium content of the supernatant was assessed using the Calcium (CPC) Liquicolor kit (Stanbio). Calcium content was expressed in μg/dL and per milligram of tissue.
4-PBA administration
The procedure for intraperitoneal injections and oral dosage of 4-PBA followed a method adapted from previously described protocol (Pomozi et al., 2014). Mice typically received intraperitoneal injections of 4-PBA (1000 mg/kg/day) 24 hours before hydrodynamic tail vein injections, however, for the calcification reversal experiments, mice were administered 4-PBA 36 hours after cryoinjury. Injection were continued daily thereafter until euthanasia. In addition, drinking water contained 6.25 mg/ml 4-PBA to ensure continuous exposure to the drug.
Cell culture
HEK 293H cells were stably transfected with a plasmid carrying a cDNA encoding the ABCC6 Q1347H mutant or with an empty vector as transfection controls using standard techniques. Transfected cells were seeded onto 12-well plates and grown to confluency. Cells were then incubated with 800μl fresh DMEM medium (containing or not 5mM 4-PBA) for 24 hrs. PPi concentration was determined in harvested media.
Pyrophosphate measurements
PPi in plasma (harvested 24 and 48 hours post hydrodynamic tail vein injections) and culture supernatant were determined as previously described in (Jansen et al., 2013) and (Jansen et al., 2014) respectively.
Data analysis
Data were compared by the Student t test. Values are expressed as mean +/− standard error of the mean (SEM). A p value <0.05 was considered statistically significant. Animal numbers used for individual set of data varied and is shown on the figures. The authors take full responsibility for the integrity of the data and all authors have read and agree to the manuscript as written.
Acknowledgments
We thank Drs. B. Sarkadi, G. Szakács and P. Borst for critical comments on the manuscript and Dr. B. Stieger for his generous gift of antibodies. Financial support came from National Institutes of Health HL108249, G12 MD007601, P30 GM103341, RR003061, RR016453 and 15ADVC-74403 from the Ingeborg v.F. McKee Fund of the Hawaii Community Foundation (in support of OLS) and R01 AR055225 (in support to AV). Additional support was received from the Hungarian grants OTKA 114136, 104227 and OTKA K111625 (to AV).
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aherrahrou Z, Doehring LC, Ehlers EM, Liptau H, Depping R, Linsel-Nitschke P, et al. An alternative splice variant in Abcc6, the gene causing dystrophic calcification, leads to protein deficiency in C3H/He mice. J Biol Chem. 2008;283:7608–7615. doi: 10.1074/jbc.M708290200. [DOI] [PubMed] [Google Scholar]
- Aherrahrou Z, Doehring LC, Kaczmarek PM, Liptau H, Ehlers EM, Pomarino A, et al. Ultrafine mapping of Dyscalc1 to an 80-kb chromosomal segment on chromosome 7 in mice susceptible for dystrophic calcification. Physiol Genomics. 2007;28:203–212. doi: 10.1152/physiolgenomics.00133.2006. [DOI] [PubMed] [Google Scholar]
- Alino SF, Crespo A, Dasi F. Long-term therapeutic levels of human alpha-1 antitrypsin in plasma after hydrodynamic injection of nonviral DNA. Gene Ther. 2003;10:1672–1679. doi: 10.1038/sj.gt.3302065. [DOI] [PubMed] [Google Scholar]
- Aranyi T, Bacquet C, de Boussac H, Ratajewski M, Pomozi V, Fulop K, et al. Transcriptional regulation of the ABCC6 gene and the background of impaired function of missense disease-causing mutations. Frontiers in genetics. 2013;4:27. doi: 10.3389/fgene.2013.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck K, Hayashi K, Dang K, Hayashi M, Boyd CD. Analysis of ABCC6 (MRP6) in normal human tissues. Histochem Cell Biol. 2005;123:517–528. doi: 10.1007/s00418-004-0744-3. [DOI] [PubMed] [Google Scholar]
- Beck K, Hayashi K, Nishiguchi B, Le Saux O, Hayashi M, Boyd CD. The distribution of Abcc6 in normal mouse tissues suggests multiple functions for this ABC transporter. J Histochem Cytochem. 2003;51:887–902. doi: 10.1177/002215540305100704. [DOI] [PubMed] [Google Scholar]
- Brampton C, Aherrahrou Z, Chen LH, Martin L, Bergen AA, Gorgels TG, et al. The level of hepatic ABCC6 expression determines the severity of calcification after cardiac injury. Am J Pathol. 2014;184:159–170. doi: 10.1016/j.ajpath.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehring LC, Kaczmarek PM, Ehlers E, Mayer B, Erdmann J, Schunkert H, et al. Arterial calcification in mice after freeze-thaw injury. Ann Anat. 2006;188:235–242. doi: 10.1016/j.aanat.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Douet V, Heller MB, Le Saux O. DNA methylation and Sp1 binding determine the tissue-specific transcriptional activity of the mouse Abcc6 promoter. Biochem Biophys Res Commun. 2007;354:66–71. doi: 10.1016/j.bbrc.2006.12.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dover GJ, Brusilow S, Samid D. Increased fetal hemoglobin in patients receiving sodium 4-phenylbutyrate. N Engl J Med. 1992;327:569–570. doi: 10.1056/NEJM199208203270818. [DOI] [PubMed] [Google Scholar]
- Finger RP, Charbel Issa P, Schmitz-Valckenberg S, Holz FG, Scholl HN. Long-term effectiveness of intravitreal bevacizumab for choroidal neovascularization secondary to angioid streaks in pseudoxanthoma elasticum. Retina. 2011;31:1268–1278. doi: 10.1097/IAE.0b013e318207d1dc. [DOI] [PubMed] [Google Scholar]
- Gonzales E, Grosse B, Cassio D, Davit-Spraul A, Fabre M, Jacquemin E. Successful mutation-specific chaperone therapy with 4-phenylbutyrate in a child with progressive familial intrahepatic cholestasis type 2. Journal of hepatology. 2012;57:695–698. doi: 10.1016/j.jhep.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Gonzales E, Grosse B, Schuller B, Davit-Spraul A, Conti F, Guettier C, et al. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology. 2015;62:558–566. doi: 10.1002/hep.27767. [DOI] [PubMed] [Google Scholar]
- Gorgels TG, Hu X, Scheffer GL, van der Wal AC, Toonstra J, de Jong PT, et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum Mol Genet. 2005;14:1763–1773. doi: 10.1093/hmg/ddi183. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Naoi S, Hirose Y, Matsuzaka Y, Tanikawa K, Igarashi K, et al. Successful treatment with 4-phenylbutyrate in a patient with benign recurrent intrahepatic cholestasis type 2 refractory to biliary drainage and bilirubin absorption. Hepatol Res. 2016;46:192–200. doi: 10.1111/hepr.12561. [DOI] [PubMed] [Google Scholar]
- Iannitti T, Palmieri B. Clinical and experimental applications of sodium phenylbutyrate. Drugs in R&D. 2011;11:227–249. doi: 10.2165/11591280-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilias A, Urban Z, Seidl TL, Le Saux O, Sinko E, Boyd CD, et al. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6) J Biol Chem. 2002;277:16860–16867. doi: 10.1074/jbc.M110918200. [DOI] [PubMed] [Google Scholar]
- Ivandic BT, Utz HF, Kaczmarek PM, Aherrahrou Z, Axtner SB, Klepsch C, et al. New Dyscalc loci for myocardial cell necrosis and calcification (dystrophic cardiac calcinosis) in mice. Physiol Genomics. 2001;6:137–144. doi: 10.1152/physiolgenomics.2001.6.3.137. [DOI] [PubMed] [Google Scholar]
- Jansen RS, Duijst S, Mahakena S, Sommer D, Szeri F, Varadi A, et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler Thromb Vasc Biol. 2014;34:1985–1989. doi: 10.1161/ATVBAHA.114.304017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RS, Kucukosmanoglu A, de Haas M, Sapthu S, Otero JA, Hegman IE, et al. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci U S A. 2013;110:20206–20211. doi: 10.1073/pnas.1319582110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Dibra F, Lee MD, Oldenburg R, Uitto J. Overexpression of fetuin-a counteracts ectopic mineralization in a mouse model of pseudoxanthoma elasticum (abcc6(−/−)) The Journal of investigative dermatology. 2010;130:1288–1296. doi: 10.1038/jid.2009.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Jiang Q, Wu Z, Shao C, Zhou Y, Yang L, et al. Genetic heterogeneity of pseudoxanthoma elasticum: the Chinese signature profile of ABCC6 and ENPP1 mutations. J Invest Dermatol. 2015;135:1294–1302. doi: 10.1038/jid.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korff S, Riechert N, Schoensiegel F, Weichenhan D, Autschbach F, Katus HA, et al. Calcification of myocardial necrosis is common in mice. Virchows Arch. 2006a;448:630–638. doi: 10.1007/s00428-005-0071-7. [DOI] [PubMed] [Google Scholar]
- Korff S, Schoensiegel F, Riechert N, Weichenhan D, Katus HA, Ivandic BT. Fine mapping of Dyscalc1, the major genetic determinant of dystrophic cardiac calcification in mice. Physiol Genomics. 2006b;25:387–392. doi: 10.1152/physiolgenomics.00010.2006. [DOI] [PubMed] [Google Scholar]
- Le Saux O, Fulop K, Yamaguchi Y, Ilias A, Szabo Z, Brampton CN, et al. Expression and in vivo rescue of human ABCC6 disease-causing mutants in mouse liver. PLoS One. 2011;6:e24738. doi: 10.1371/journal.pone.0024738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25:223–227. doi: 10.1038/76102. [DOI] [PubMed] [Google Scholar]
- Li Q, Sundberg JP, Levine MA, Terry SF, Uitto J. The effects of bisphosphonates on ectopic soft tissue mineralization caused by mutations in the ABCC6 gene. Cell Cycle. 2015;14:1082–1089. doi: 10.1080/15384101.2015.1007809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Song YK, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- Maestri NE, Brusilow SW, Clissold DB, Bassett SS. Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med. 1996;335:855–859. doi: 10.1056/NEJM199609193351204. [DOI] [PubMed] [Google Scholar]
- Martin L, Douet V, Vanwart CM, Heller MB, Le Saux O. A Mouse Model of beta-Thalassemia Shows a Liver-Specific Down-Regulation of Abcc6 Expression. Am J Pathol. 2011;178:774–783. doi: 10.1016/j.ajpath.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee-Russell SM. Histochemical methods for calcium. Journal of Histochemistry & Cytochemistry. 1958;6:22–42. doi: 10.1177/6.1.22. [DOI] [PubMed] [Google Scholar]
- Meng H, Vera I, Che N, Wang X, Wang SS, Ingram-Drake L, et al. Identification of Abcc6 as the major causal gene for dystrophic cardiac calcification in mice through integrative genomics. Proc Natl Acad Sci U S A. 2007;104:4530–4535. doi: 10.1073/pnas.0607620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoi S, Hayashi H, Inoue T, Tanikawa K, Igarashi K, Nagasaka H, et al. Improved liver function and relieved pruritus after 4-phenylbutyrate therapy in a patient with progressive familial intrahepatic cholestasis type 2. The Journal of pediatrics. 2014;164:1219–1227. e1213. doi: 10.1016/j.jpeds.2013.12.032. [DOI] [PubMed] [Google Scholar]
- Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90:25–39. doi: 10.1016/j.ajhg.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill WC, Lomashvili KA, Malluche HH, Faugere MC, Riser BL. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011;79:512–517. doi: 10.1038/ki.2010.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill WC, Sigrist MK, McIntyre CW. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol Dial Transplant. 2010;25:187–191. doi: 10.1093/ndt/gfp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JE, Gottesman GS, McAlister WH, Mumm S, Madson KL, Kiffer-Moreira T, et al. Severe skeletal toxicity from protracted etidronate therapy for generalized arterial calcification of infancy. J Bone Miner Res. 2013;28:419–430. doi: 10.1002/jbmr.1752. [DOI] [PubMed] [Google Scholar]
- Perrine SP, Ginder GD, Faller DV, Dover GH, Ikuta T, Witkowska HE, et al. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N Engl J Med. 1993;328:81–86. doi: 10.1056/NEJM199301143280202. [DOI] [PubMed] [Google Scholar]
- Pomozi V, Brampton C, Fulop K, Chen LH, Apana A, Li Q, et al. Analysis of pseudoxanthoma elasticum-causing missense mutants of ABCC6 in vivo; pharmacological correction of the mislocalized proteins. J Invest Dermatol. 2014;134:946–953. doi: 10.1038/jid.2013.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomozi V, Le Saux O, Brampton C, Apana A, Ilias A, Szeri F, et al. ABCC6 is a basolateral plasma membrane protein. Circ Res. 2013;112:e148–151. doi: 10.1161/CIRCRESAHA.111.300194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am J Physiol Cell Physiol. 2000;278:C259–267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- Russell RG, Bisaz S, Donath A, Morgan DB, Fleisch H. Inorganic pyrophosphate in plasma in normal persons and in patients with hypophosphatasia, osteogenesis imperfecta, and other disorders of bone. J Clin Invest. 1971;50:961–969. doi: 10.1172/JCI106589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer GL, Hu X, Pijnenborg AC, Wijnholds J, Bergen AA, Scheper RJ. MRP6 (ABCC6) detection in normal human tissues and tumors. Lab Invest. 2002;82:515–518. doi: 10.1038/labinvest.3780444. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinko E, Ilias A, Ujhelly O, Homolya L, Scheffer GL, Bergen AA, et al. Subcellular localization and N-glycosylation of human ABCC6, expressed in MDCKII cells. Biochem Biophys Res Commun. 2003;308:263–269. doi: 10.1016/s0006-291x(03)01349-4. [DOI] [PubMed] [Google Scholar]
- Sorrenson B, Suetani RJ, Williams MJ, Bickley VM, George PM, Jones GT, et al. Functional rescue of mutant ABCA1 proteins by sodium 4-phenylbutyrate. Journal of lipid research. 2013;54:55–62. doi: 10.1194/jlr.M027193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- Zhang G, Gao X, Song YK, Vollmer R, Stolz DB, Gasiorowski JZ, et al. Hydroporation as the mechanism of hydrodynamic delivery. Gene Ther. 2004;11:675–682. doi: 10.1038/sj.gt.3302210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Jiang Q, Takahagi S, Shao C, Uitto J. Premature termination codon read-through in the ABCC6 gene: potential treatment for pseudoxanthoma elasticum. J Invest Dermatol. 2013;133:2672–2677. doi: 10.1038/jid.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]