

Abstract
Many features of gene transcription in human cells remain unclear, mainly due to a lack of quantitative approaches to follow genome transcription with nucleotide precision in vivo. Here we present a robust genome-wide approach to study RNA polymerase (Pol) II-mediated transcription in human cells at single-nucleotide resolution by native elongating transcript sequencing (NET-seq). Elongating RNA polymerase and the associated nascent RNA is prepared by cell fractionation, avoiding immunoprecipitation or RNA labeling. The 3′-ends of nascent RNAs are captured through barcode linker ligation and converted into a DNA sequencing library. The identity and abundance of the 3′-ends are determined by high-throughput sequencing, revealing the exact genomic locations of Pol II. Human NET-seq can be applied to study the full spectrum of Pol II transcriptional activities, including the production of unstable RNAs and transcriptional pausing. Using the protocol described here, a NET-seq library can be obtained from human cells in 5 days.
INTRODUCTION
Elucidating how genome transcription is regulated in human cells requires genomic tools that reveal the exact location of transcribing RNA polymerases. The application of genome-wide methods that map RNA polymerase II (Pol II) density along genomes in vivo has uncovered new general features of gene transcription and has changed the view of how genomes are transcribed in living cells1–3. Originally, it was thought that the recruitment of Pol II to the gene promoter and transcription initiation were the major regulatory steps during RNA synthesis4. However, genomic methods that map Pol II occupancy in vivo have shown that Pol II is predominantly located in a region 50–100 nt downstream of the transcription start site (TSS) at the majority of active mammalian genes indicating promoter-proximal pausing2,3,5,6. These genome-wide studies provided strong evidence that transcription elongation by Pol II is a key regulatory step in RNA production. Additionally, genomic approaches also found that Pol II transcription in the sense orientation is usually accompanied by divergent antisense transcription at the majority of promoters of active mammalian genes7–9. Genome-wide Pol II profiling studies revealed that promoter-proximal pausing of Pol II in the sense direction typically co-occurs with promoter-proximal pausing in the upstream antisense orientation in mammalian cells uncovering new complexities of genome transcription6,10–14.
Until now, two genome-wide strategies have mainly been applied to map RNA polymerase density across mammalian genomes as described in more detail in the ‘Comparison with other approaches’ section below: (i) chromatin immunoprecipitation (ChIP) based approaches5,15–20 and (ii) transcription run-on based methods8,21,22. Here, we provide a step-by-step protocol for native elongating transcript sequencing (NET-seq), an alternative method to map Pol II genome transcription with single-nucleotide precision and DNA strand-specificity in unperturbed human cells. Human NET-seq is a robust Pol II profiling approach that does not require any genetic modification or labeling of the nascent RNA and its application to different human cell types is straightforward. We have successfully applied the NET-seq approach to 4 different human cell types as described in detail below.
Development and overview of human NET-seq
Native elongating transcript sequencing (NET-seq) allows RNA polymerase transcription elongation to be studied with single-nucleotide precision in vivo. The NET-seq methodology was originally developed for budding yeast grown to mid-log phase23,24. In the original yeast NET-seq approach native Pol II transcription elongation complexes are quantitatively purified by immunoprecipitation using a C-terminally 3× FLAG-tagged version of the Pol II subunit Rpb323,24. A very similar approach has recently been applied to bacterial cells, mapping the RNA polymerase density across the bacterial genome25,26. Although the NET-seq strategy was recently applied to Drosophila S2 and Kc cells27,28, a NET-seq method was not available for mammalian cells. To provide a simple and robust genomic tool for visualizing Pol II transcription with single-nucleotide precision in human cells, we developed a human NET-seq approach29. Human NET-seq revealed new general aspects of Pol II transcription such as widespread transcriptional pausing across the expressed human genome and a new promoter-proximal transcriptional activity in the antisense orientation29.
Human NET-seq is a quantitative genomic approach. This requires a strategy to quantitatively (>95% as determined by western blot analysis) purify transcriptionally engaged RNA polymerase from human cells as well as an efficient sequencing library preparation method (Fig. 1).
Figure 1.

Schematic overview of the human NET-seq protocol. Each panel describes a key experimental step of the human NET-seq approach.
In contrast to the yeast NET-seq method, where an epitope-tagged version of Pol II is purified by immunoprecipitation23,24, we found that the most efficient and simple way to quantitatively purify transcribing Pol II from human cells is by cellular fractionation. Here, we exploit the high degree of stability of the Pol II transcription elongation complex, which is very stable even in the presence of high amounts of salts, detergents and urea30–33. Upon fractionation, the cytoplasm and the nucleoplasm are removed, and elongating Pol II associated with the nascent RNA is greatly enriched in the chromatin fraction (Figs. 1 and 2). The removal of the cytoplasmic and nucleoplasmic fractions eliminates the bulk of mature mRNA whereas the nascent RNA is captured in the chromatin fraction. Briefly, human cells are lysed in the presence of mild detergents. Nuclei are separated from the cytoplasm by centrifugation through a sucrose cushion. Next, nuclei are washed to remove cytoplasmic remnants. The nucleoplasm is separated from the chromatin in the presence of urea, salt and mild detergents. Urea removes chromatin bound proteins except the very stable elongating RNA polymerase and histone proteins32,34,35. Since the histone proteins are not detached upon urea treatment, the native chromatin can be collected by low-speed centrifugation using a refrigerated benchtop centrifuge. No ultracentrifugation is required. The chromatin-associated nascent RNA is purified and remaining DNA is degraded by DNase treatment.
Figure 2.
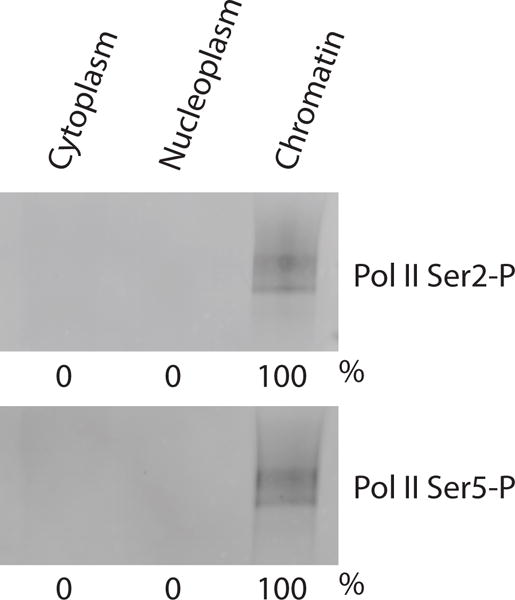
Representative western blot showing the quantitative purification of elongating RNA polymerase II by cell fractionation. Subcellular fractions have been obtained from ~1× 107 HeLa S3 cells. Sample volumes have been adjusted to the cytoplasmic fraction (largest volume) so that the western blot signals between the different fractions can be compared. The subcellular fractions have been probed with antibodies directed against the Pol II CTD Ser2-phosphorylated (first panel; 3E10 antibody) and CTD Ser5-phosphorylated form (second panel; 3E8 antibody). The numbers below the panels represent the western blot signals in percent as obtained by image quantification using ImageJ 1.47v software68.
The cell fractionation approach described here is based on protocols published by the Ueli Schibler and Douglas Black laboratories32,34,36. However, the fractionation procedure has been modified and extensively optimized to ensure that:
>95% of elongating Pol II together with the nascent RNA is captured in the chromatin fraction. This was achieved by a systematic optimization of cell lysis conditions as well as of the buffers and solutions used for subcellular fractionation.
cross-contamination between the subcellular fractions is minimized. This was realized by optimizing buffer and washing conditions during subcellular fractionation.
the risk of transcriptional run-on and RNA/protein degradation during sample preparation is reduced. This was accomplished by performing the cell fractionation in the presence of α-amanitin (a strong inhibitor of elongating Pol II), RNase and protease inhibitors, and by conducting all experimental steps on ice or at 4°C. Furthermore, a fast and efficient cell lysis method is used, leading rapid depletion of the nuclear NTP pool and further reducing the risk of run-on transcription during cell fractionation.
Apart from its simplicity and robustness, other major advantages of this purification approach are that it avoids restarting Pol II in vitro to label and purify nascent RNA and that it evades possible biases from epitope masking and cross-reactivity when antisera is used for Pol II purification.
The 3′-ends of the purified nascent RNA are then converted into a DNA sequencing library (Fig. 1). The human NET-seq library generation is based on the library preparation method that was originally developed for budding yeast23,24, but includes two important modifications to minimize biases and to increase the efficiency by which the human nascent RNA is converted into a NET-seq library:
Barcode DNA linker ligation. In contrast to the original yeast NET-seq library preparation method23,24, human NET-seq employs a barcoded DNA linker with a random hexameric sequence at the 5′-end (Table 1). The use of this new DNA linker pool has two main advantages. First, mispriming artifacts that can arise during reverse transcription are strongly reduced29. We observed that due to the length of human nascent RNA, the RT primer anneals to stretches of complementarity within the nascent RNA that are as short as six nucleotides leading to frequent mispriming events29. By ligating a DNA linker pool that possesses a random hexameric sequence at the 5′-end mispriming events during RT are dramatically decreased29. Second, the random hexameric sequence serves as a molecular barcode that allows PCR duplicates and residual mispriming RT events to be identified and bioinformatically removed29.
Depletion of abundant chromatin-associated mature RNAs. Specific depletion of the most abundant chromatin-associated mature RNAs increases the fraction of sequencing reads that originate from nascent Pol II transcripts29. Specific depletion is performed by subtractive hybridization using a set of 20 biotinylated DNA oligos (Table 2) that anneal to the 3′-ends of the original mature RNA and are removed with streptavidin coupled magnetic beads (Fig. 1). The specific subtractive hybridization strategy described here is based on the tailored rRNA depletion protocol of the ribosome profiling approach37.
TABLE 1.
DNA and RNA oligos required for NET-seq library preparation (5′ to 3′)
| DNA oligos | ||
| Barcode DNA Linker | /5rApp/(N)6CTGTAGGCACCATCAAT/3ddC | |
| oLSC007 (RT primer) | /5Phos/ATCTCGTATGCCGTCTTCTGCTTG/iSp18/CACTCA/iSp18/TCCGACGATCATTGATGGTGCCTACAG | |
| oNTI231 (reverse primer) | CAAGCAGAAGACGGCATACGA | |
| oLSC006 (sequencing primer) | TCCGACGATCATTGATGGTGCCTACAG | |
| RNA oligo | ||
| oGAB11 (control oligo) | agucacuuagcgauguacacugacugug | |
|
| ||
| 5rApp: | 5′-Riboadenylate | |
| (N)6: | random hexameric sequence (handmixed by Integrated DNA Technologies) | |
| 3ddC: | 3′-Dideoxycytidine | |
| 5Phos: 5′-Phosphate | ||
| iSp18: internal 18-atom hexa-ethylenglycol spacer | ||
| CRITICAL Please note that the 5′-Adenine nucleotide of the pre-adenylated Barcode DNA Linker is a ribonucleotide (“rA”). | ||
TABLE 2.
Biotinylated depletion DNA oligos
| Gene | Transcript | DNA sequence (5′ to 3′) |
|---|---|---|
| RNVU1-1 | snRNA | CAGGGGAAAGCGCGAACGCAGTCCCCCACTACC |
| RNVU1-7 | snRNA | CAGGGGAAAGCGCGGACGCAGTCCCCCACT |
| RNU2-1 | snRNA | TGCACCGTTCCTGGAGGTACTGCAATACCAGGTCGATG |
| RNU4-1 | snRNA | GCCTACAGCAGTCTCCGTAGAGACTGTCAAAAATTGCCA |
| RNU5B-1 | snRNA | AGCCTTGTCGGAACAAGGCCTCAAAAAATTAGCTTAAGAC |
| RNU5E-1 | snRNA | AGCCTTGCCAAAGCAAGGCCTCAAAAAATTGGGTTAAG |
| SNORD3D | snoRNA | CCACTCAGACCGCGTTCTCTCCCTCTCACTCCCCAA |
| SNORD12B | snoRNA | CAGACAGAAACTGGCTTAGAAAAGTTAGATCAACATAGTCGATC |
| SNORD12C | snoRNA | CTAACTGGCAAAATATAAGACGTCAGCATTGTCGATCTGATG |
| SNORD27 | snoRNA | CTCAGTAGTAAGATGACATCACTTGAAAGTTCAGCCATATGC |
| SNORD29 | snoRNA | CTCAGGTGTTCATGTATTTTCACTGTCGGTCATAGTGAGC |
| SNORD31 | snoRNA | TACCTTTCAGTCACACATTGATCAGACTGGGGCGGTAT |
| SNORD80 | snoRNA | CATCAGATAGGAGCGAAAGACTTAATATTGCTCATCAGCG |
| SNORD81 | snoRNA | TTGTCATCAAGTAATCAGTGAGAGAGTTCAAGTTGG |
| SNORD118 | snoRNA | GGAGCAATCAGGGTGTTGCAAGACCTGATTACGCAGAG |
| RNA5S1 | rRNA | CACCCGGTATTCCCAGGCGGTCTCCCATCCAAGTAC |
| RNA5-8S5 | rRNA | AGCGACGCTCAGACAGGCGTAGCCCCGGGAGG |
| RNA28S5 | rRNA | ACAAACCCTTGTGTCGAGGGCTGACTTTCAATAGATCGCA |
| MT-TM (ID: 4569) | mitochondrial tRNA | GTACGGGAAGGGTATAACCAACATTTTCGGGGTATGGG |
| MT-TV (ID: 4577) | mitochondrial tRNA | TGGTCAGAGCGGTCAAGTTAAGTTGAAATCTCCTAAGTG |
DNA oligos possess a 5′-biotin-TEG and are HPLC purified.
Finally, the identity and the abundance of the 3′-ends of the original nascent RNA are determined by high-throughput single-end sequencing. This identifies the last nucleotide that was incorporated into the nascent RNA chain and thus the exact genomic position of the RNA polymerase active site. Following these main experimental steps, NET-seq provides a quantitative measure of Pol II density across the human genome with single-nucleotide resolution.
Here we present a step-by-step protocol for performing NET-seq in cultured human cells, from cell lysis through to the quality control measures of the final NET-seq library (Fig. 1). The human NET-seq protocol was originally applied to HeLa S3 and HEK293T cells29;. We have since successfully performed NET-seq on two other human cell types (K562 and MOLT-4 cells, data not shown) suggesting that the technique is robust and works for different human cell types. The detailed description of the experimental steps from cell lysis to the quality control measures of the final NET-seq library will allow our colleagues to adapt this approach in their own laboratories.
Advantages and applications of human NET-seq
NET-seq can be applied to address a broad range of biological questions. The most basic applications of the NET-seq approach can be derived from its main attributes. NET-seq maps Pol II density DNA-strand specifically and therefore can be used to investigate how transcriptional directionality (sense and antisense transcription) is regulated in vivo. Due to the single-nucleotide resolution, the human NET-seq approach provides a quantitative measure of Pol II occupancy at each nucleotide that is transcribed. Prominent peaks of Pol II density at specific nucleotides or within narrow regions reveal pausing sites, locations where Pol II is detected with a higher probability. Thus, NET-seq is a powerful approach to study how transcriptional pausing is regulated in the promoter-proximal region as well as throughout gene-body regions of genes. Additionally, the high spatial resolution allows Pol II transcriptional activities (e.g. sense and different types of antisense transcription29) that arise in close proximity to each other, to be distinguished. Human NET-seq now provides the opportunity to investigate how these activities are co-regulated in normal cells and upon disease conditions or treatments.
Furthermore, NET-seq captures the RNA as it is being produced. Therefore, NET-seq can be applied to study unstable RNA species, such as promoter upstream transcripts (PROMPTs)7 and upstream antisense RNAs (uaRNAs)38 as well as enhancer derived non-coding RNAs (eRNAs)39,40, before these transcripts are turned over by cellular RNA degradation pathways41–43. Human NET-seq detects these unstable RNA species29. Standard RNA-seq methods mainly capture steady-state mRNA levels of fully processed mature RNAs and thus struggle to detect unstable transcripts.
Application of human NET-seq revealed strong Pol II pauses at the boundaries of exons that are retained during splicing but not at exons that are subsequently removed by the spliceosome29. This indicates that Pol II recognizes exons with different processing fates where Pol II slows down at exons that are retained in the final mRNA. The determinants of Pol II pausing at exon boundaries and how Pol II transcription and splicing are coupled in vivo remain to be determined. NET-seq will help to elucidate these links and will shed new light on the regulatory mechanisms that underlie these fundamental biological processes.
Although the NET-seq approach has been established and optimized for the analysis of Pol II transcription the adaptation of the protocol to the RNA polymerase I (Pol I) and RNA polymerase III (Pol III) transcriptional systems should be straightforward. Following the human NET-seq protocol described here, we detect Pol I and Pol III transcription29. However, the NET-seq library preparation may require further adaptations to study Pol I and Pol III transcription in an unbiased way. For example, the extended secondary structures in Pol I and Pol III transcripts may pose a problem for the reverse transcription reaction.
Limitations of human NET-seq
To fully comprehend the potential applications of NET-seq and to extract meaningful conclusions, it is important to be cognizant of its limitations. NET-seq reveals the exact position of Pol II by determining the 3′-ends of Pol II-associated nascent RNA. While Pol II involved in elongation can be localized with NET-seq, the exact genomic positions of Pol II that is part of the pre-initiation complex (PIC) at promoter regions or of Pol II involved in transcription initiation around the transcriptional start site (TSS) of genes cannot be mapped. PIC formation occurs before nascent RNA is being produced by Pol II. The nascent transcripts produced by Pol II in close proximity to the TSS are too short to produce uniquely mappable sequencing reads; minimum read lengths of ~18 nt are required for unique mapping to the human reference genome.
Purification of transcriptionally engaged RNA polymerase will co-purify nascent RNA and RNA processing intermediates from splicing and 3′-end cleavage. Thus, the NET-seq signal at the last nucleotide of introns and exons can either be due to elongating Pol II or splicing intermediates. Similarly, it is not clear whether the NET-seq signal that is obtained at the polyadenylation (pA) site, where the transcript is cleaved and polyadenylated, relates to transcriptionally engaged Pol II or to a 3′-end processing intermediate. Therefore, NET-seq signals at these single-nucleotide positions are computationally removed by discarding the corresponding sequencing reads (Box 4, steps 7–8).
Box 4. Guidance to the bioinformatics analysis of human NET-seq data.
The following protocol describes the general steps for the computational analysis of human NET-seq data. For more information please see Mayer et al.29. The bioinformatics analysis of human NET-seq data also employs custom scripts that will be shared upon request.
-
1|
Use sequencing reads that pass the default Illumina quality filter (BCL2FASTQ2).
-
2|
Remove the six 5′-end nucleotides that correspond to the random molecular barcode from sequencing reads.
CRITICAL STEP The information of the barcode sequence should remain associated with the corresponding sequencing read. This is important for the identification of reads that arise from RT mispriming events (step 4).
-
3|
Align both sets of sequencing reads (containing and not containing the barcode sequence) to the human reference genome using the STAR aligner (v.2.4.0)65.
CRITICAL STEP Perform the alignment without providing the transcriptome information to avoid any biases favoring annotated regions.
-
4|
Identify and remove reads that arise from mispriming events during RT. Reads that originate from RT mispriming can be identified by aligning the reads still containing the barcode sequence and that map without mismatches in the six 5′-end nucleotides to the human reference genome. Therefore, these reads appear to have “barcode sequences” that perfectly match the genomic sequence adjacent to the aligned read and are discarded.
-
5|
Record the position of the 5′-end of the sequencing read that corresponds to the 3′-end of the original nascent RNA, with a custom script applying the HTSeq package66.
-
6|
Identify and remove sequencing reads due to PCR duplication i.e. reads that align to the same genomic position and contain the same barcode sequence. These reads are computationally removed using a custom script.
-
7|
Identify and remove sequencing reads due to splicing intermediates i.e. reads that align exactly to the 3′-end nucleotide of introns and 3′-end nucleotide of exons. These reads are computationally removed using a custom script.
-
8|
Optional: Identify and remove sequencing reads that arise from chromatin-associated mature RNAs. Sequencing reads that arise from polyadenylated mature RNAs do not align to the human reference genome and are discarded. Sequencing reads that originate from non-polyadenylated mature RNAs or chromatin-associated RNA that is cleaved but not yet polyadenylated align exactly to the polyadenylation (pA) site of the corresponding gene i.e. the site where the transcript is cleaved and polyadenylated. Reads that align exactly to the pA site can be computationally removed using a custom script.
-
9|
Visualize human NET-seq data in a genome browser such as the Integrative Genomics Viewer (IGV)67.
Purification of transcriptionally engaged Pol II will also co-purify some fully processed mature RNAs. Abundant non-coding mature RNAs, such as those associated with the spliceosome, are removed by specific depletion (Procedure steps 71–80). Although the vast majority of mature mRNAs are experimentally removed by discarding the cytoplasmic and nucleoplasmic fractions (Procedure steps 8–18), as determined by western blot analysis and subcellular RNA sequencing[add citation to the cell paper], some mature mRNAs are likely captured in the final NET-seq library. These mature mRNAs can be polyadenylated or non-polyadenylated at their 3′ ends. As this protocol is designed to sequence the 3′ ends of RNAs, the polyadenylated transcripts are not a problem. First, the long polyadenyated tracks are technically challenging for reverse transcription reactions, PCR and sequencing platforms to capture. Second, any sequencing reads that arise from polyadenylated mature RNAs do not align to the human reference genome due to polyadenine stretches and are removed bioinformatically. NET-seq reads that arise from non-polyadenylated mature mRNAs align exactly to sites corresponding to the polyadenylation site for the transcript (see the ‘Experimental design’ section for more information). The identification of this subset of mature mRNAs and the subsequent removal of the corresponding sequencing reads depends on the annotation of the pA sites, which has been strongly improved over the last years and comprehensive maps are available58,59. For genes where pA site information is lacking, these mature mRNAs might introduce low levels of background signals. These unspecific signals are restricted to a few single-nucleotide positions. Therefore, with the focus by NET-seq on 3′ end sequencing, we expect that these unspecific signals occur at a much lower level as compared to background signals that are usually observed in IP based genomic methods for which the unspecific pull-down of RNAs represents an inherent problem when the entire molecule is fragmented and sequenced.
Our conclusion that mature mRNAs have negligible impact, if any, on NET-seq results, is based on two experimental results. First, we performed human NET-seq upon conditions where productive Pol II transcription elongation is blocked, by inhibiting the Cdk9 subunit of the positive transcription elongation factor b (P-TEFb) [cite the Cell paper to make it clear that the data aren’t presented here]. P-TEFb represents a key player in the release of promoter-proximally paused Pol II60,61. In agreement with previous studies5,38, Cdk9 inhibition resulted in a genome-wide loss of productive elongating Pol II in both sense and antisense orientation29 indicating that NET-seq quantitatively captures transcribing Pol II. If mature mRNAs were influencing the NET-seq signal, we would not expect a complete reduction in productive elongating Pol II levels upon one hour of P-TEFb inhibition. Second, key observations of mammalian Pol II transcription made by human NET-seq, such as widespread promoter-proximal pausing and divergent transcription originating from bidirectional promoters are consistent with observations by many laboratories using a broad range of genomic methods5–9,18,62–64. Nevertheless, mature RNAs likely introduce some background signal, which is important to consider when inspecting NET-seq data.
The human NET-seq strategy described here requires 1× 107 cells as an input and therefore works best for cells that can be obtained in larger quantities. Current efforts aim to reduce the amount of cells that are required per NET-seq experiment.
Comparison with other approaches
Prior to the development of NET-seq, two main strategies existed to determine the genome-wide location of Pol II and to study gene transcription in vivo; Pol II chromatin immunoprecipitation (ChIP) and transcription run-on. The Nicholas Proudfoot and Maria Carmo-Fonseca laboratories have also recently developed a variant of the NET-seq approach for mammalian cells, called mNET-seq44.
Pol II chromatin immunoprecipitation
In this approach, chromatin is crosslinked and Pol II bound DNA is purified by immunoprecipitation (IP) typically using antibodies directed against Pol II. The DNA is converted into a library that is either hybridized to microarrays (ChIP-chip;15,16,45–51) or applied to high-throughput sequencing (ChIP-seq;5,17,18). Although, these approaches have revealed the genomic Pol II density in various model organisms including yeast and humans, the spatial resolution is usually limited to >200 bp. In conventional genomic ChIP experiments crosslinked chromatin is typically fragmented into the range of 200–500 bp limiting the spatial resolution to >200 bp52. ChIP data usually suffers from high levels of background signals due to unspecific pulldown of DNA that can arise from cross-reactive antibodies or the unspecific binding of DNA to beads used in IP experiments. Furthermore, these methods lack DNA-strand specificity and the transcriptional state of Pol II remains unclear. To clarify whether Pol II is transcriptionally engaged, ChIP methods need to be complemented by other approaches such as permanganate footprinting assays53. Recent improvements of the genome-wide ChIP approach could greatly increase the spatial resolution and decrease non-specific background signals19,20. Although these new ChIP approaches lack single-nucleotide resolution and DNA strand-specificity, they provide an opportunity to determine the location of Pol II during pre-initiation and initiation of transcription complementing NET-seq and transcription run-on data.
Transcription run-on
To date, two transcription run-on approaches have been developed to map genomic Pol II density: global run-on-sequencing (GRO-seq)8 and precision nuclear run-on and sequencing (PRO-seq)21,22. In both methods transcription is halted and then restarted in vitro in the presence of modified nucleotides that enable the purification of nascent RNA. The purified nascent RNA is then converted into a DNA sequencing library and subjected to high-throughput sequencing. These methods provide a DNA-strand specific quantitative measure of transcriptionally engaged Pol II and have provided new insights into gene regulation in various model organisms including human cells and Drosophila cells. Similar to human NET-seq, GRO-seq and PRO-seq require nascent RNAs of a minimum length (at least ~18 nt) so that the corresponding sequencing reads can uniquely align to the human reference genome. Therefore, these methods do not provide any information about the localization of Pol II during pre-initiation or transcription initiation. Furthermore, transcription run-on based methods are strongly dependent on an efficient restart of transcription under non-physiological conditions, a process that is sensitive to the experimental setup and the transcriptional status of Pol II54. The pool of Pol II enzymes that are recovering from transcriptional pauses where the nascent RNA is misaligned in the polymerase active site cannot be restarted in vitro and thus escape detection by nuclear run-on based methods27. Thus comparison of transcription run-on data with NET-seq data that maps all Pol II – paused, recovering and not paused – enables the identification of where Pol II is in the process of pause recovery [cite the Henikoff, 3′NT-seq paper].
mNET-Seq
mNET-seq uses immunoprecipitation (IP) to purify Pol II (and its different CTD-phosphorylated forms) with the associated nascent RNA from HeLa cells44, and thus is capable of mapping subpopulations of Pol II genome-wide. The ability to analyze Pol II complexes in a phosphorylation-dependent manner comes at the cost of additional steps. mNET-seq requires that Pol II transcription complexes are released from the chromatin by a micrococcal nuclease (MNase) digest, a process that is inefficient and may release a biased subset of transcriptionally engaged Pol II44. Subsequently, the purified nascent RNA must be phosphorylated at its 5′-end before entering a sequencing library protocol. In contrast, the human NET-seq protocol quantitatively purifies human Pol II elongation complexes by cellular fractionation, and uses an optimized library preparation method to reduce experimental biases (Fig. 1).
Experimental design
Each experimental step of the protocol as described in the ‘PROCEDURE’ section has been optimized and we recommend to always including controls to monitor the success of key steps prior to sequencing a NET-seq library. Below we describe the steps that need particular attention before adapting the human NET-seq methodology. It is important that each step of the library construction works with high efficiency so that the DNA sequencing library quantitatively reflects the original pool of nascent RNA molecules.
Cell fractionation
The cell fractionation approach used in this protocol has been optimized for an input of 1× 107 cells. If more input is required, we recommend that the fractionation is performed on parallel samples rather than increasing the amount of cells per sample. Using more cells per sample reduces the cell lysis efficiency and increases the risk of cross-contamination between the different subcellular fractions.
To avoid run-on transcription the fractionation is performed in the presence of α-amanitin, a very strong inhibitor of elongating Pol II55,56. Furthermore, all steps are performed on ice or at 4°C. To avoid the risk of RNA and protein degradation the fractionation is conducted in the presence of RNase and protease inhibitors.
For RNA purification it is important to use the miRNeasy kit instead of the RNeasy kit (Qiagen) to minimize RNA size biases.
Quality controls for cell fractionation
Cell fractionation represents the first fundamental step of the human NET-seq approach and monitoring its success is critical before proceeding with the NET-seq library preparation. We recommend performing western blot analysis, probing the subcellular fractions with antibodies directed against the elongating form of Pol II. We suggest using the well characterized antibodies directed against the CTD Ser2- (3E10) and CTD Ser5 (3E8)-phosphorylated froms of Pol II, that have been originally developed in the laboratories of Dirk Eick and Elisabeth Kremmer57. ≥95% of elongating Pol II should be captured in the chromatin fraction (Fig. 2).
When applying the cell fractionation approach to a new cell-type we also recommend performing the following control experiments. To check for potential cross-contamination between the subcellular fractions we suggest probing the different fractions on a western blot with antibodies raised against subcellular marker proteins (chromatin fraction: FL-126 directed against Histone 2B; nucleoplasmic fraction: C-18 raised against U1 snRNP70; cytoplasmic fraction: 6C5 recognizing GAPDH; please also see ‘MATERIALS’ section for more information). The cytoplasmic and nucleoplasmic protein markers should be absent in the chromatin fraction. Additionally, the enrichment of nascent intron-containing RNAs in the chromatin fraction compared to the nucleoplasmic and cytoplasmic fractions can be monitored by subcellular RNA-seq or by reverse transcription coupled with quantitative real-time PCR (RT-qPCR) using specific primer pairs directed against intronic regions of the nascent RNA. We performed subcellular RNA-seq for HeLa S3 and HEK293T cells revealing that intron-containing nascent RNA is greatly enriched in the chromatin fraction29. The quantity and quality of the purified RNA can be monitored using a NanoDrop spectrophotometer.
Barcode DNA linker ligation
Although other adaptor and/or barcoding strategies can be used, we strongly recommend using the Barcode DNA linker design described here and the truncated version of the T4 RNA ligase 2 (from NEB) for NET-seq library construction. High-throughput sequencing requires libraries that contain specific sequences, such as a binding site for the sequencing primer. In the NET-seq protocol the DNA linker provides the sequence where the RT primer anneals, which in turn introduces the binding site for the sequencing primer. Human NET-seq uses a barcode DNA linker that is pre-adenylated and possesses a random hexameric sequence at its 5′-end (Table 1). The Barcode DNA linker is ligated to the 3′-hydroxyl group of nascent RNA molecules by a truncated version of the T4 RNA ligase 2. This truncated enzyme cannot adenylate the 5′-ends of the RNA substrate thus reducing background ligation. Additionally, the DNA linker possesses a dideoxy cytosine at the 3′-end to prevent its extension by the reverse transcriptase during the first strand cDNA synthesis. This keeps the reverse transcription directional and is important for the strand specificity of the NET-seq library.
RNA fragmentation
RNA fragmentation by partial alkaline hydrolysis should result in a homogenous RNA pool with most of the molecules being in the range of 35 to 100 nt (Fig. 3a). We have observed that the optimal fragmentation time can vary with different batches of alkaline fragmentation solution, with the preferred fragmentation time usually between 10 and 40 min. We recommend that the optimal RNA fragmentation time is determined for each new batch of fragmentation solution. Even when the same batch of fragmentation solution is used, we suggest validating the fragmentation time every four months to minimize the risk of over- or under-fragmentation of the RNA pool and to maximize reproducibility of human NET-seq data.
Figure 3.

Representative gels showing the size selections during NET-seq library preparation. (a) Optimization of RNA fragmentation time and size selection of fragmented RNA. The RNA was obtained from HeLa S3 cells. 1 μg of RNA was used per fragmentation reaction. The fragmented RNA was separated on a 15% (wt/vol) TBE-Urea gel. The optimal fragmentation time was 30 min. The blue bracket indicates the region that was excised from the gel. The fragmented RNA between 35–100 nt was selected. (b) Size selection of the cDNA after reverse transcription (RT). The RT product was separated on two lanes of a 10% (wt/vol) TBE-Urea gel. The blue bracket indicates the region that was excised from the gel. The cDNA in the range of 85–160 nt was selected. (c) Optimization of final PCR amplification reaction. PCR reactions were stopped after 6, 8, 10 and 12 amplification cycles. The PCR products obtained from four different test amplifications were separated on an 8% (wt/vol) TBE gel. The PCR product that corresponds to the NET-seq library runs at around 150 nt. Note that PCR products in the higher molecular range were obtained by ten amplification cycles or higher, indicating over-amplification. For this NET-seq library, the optimal number of amplification cycles was eight. The lower band (~120 nt, brown asterisk) corresponds to a PCR product obtained from empty circles that arise from unextended RT primers (d) Purification of PCR products. The PCR products of four final PCR reactions using eight amplification cycles were separated on an 8% (wt/vol) TBE gel. The excised band is indicated by a blue bracket. The PCR product that arises from empty circles is labeled by a brown asterisk as in (c).
Specific depletion of highly abundant mature RNAs
Although the vast majority of fully spliced mature RNAs are removed with the cytoplasmic and nucleoplasmic fractions, some mature RNA species are present in the chromatin fraction. These RNAs include chromatin-associated mature RNAs such as small nuclear RNAs (snRNAs) that are an integral part of spliceosomes. Although, the fraction of chromatin-associated mature RNAs is further reduced by the Urea treatment during the separation of the chromatin from the nucleoplasm, some fully processed RNAs are not completely removed and are part of the final NET-seq library, as described in the Limitations section. NET-seq reads due to abundant non-coding mature RNA species, such as spliceosomal-associated RNAs, markedly reduce the amount of informative sequencing data and it is therefore preferable to remove them prior to sequencing. Sequencing of our first NET-seq libraries generated from HeLa S3 cells identified the most abundant mature RNAs in the chromatin fraction and biological replicates identified the same RNAs as being the most abundant in different HeLa S3 NET-seq libraries. Based on these sequencing results we designed biotinylated DNA oligos complementary to the 3′-regions of the 20 most abundant mature RNAs in our NET-seq libraries (Table 2). To deplete abundant mature RNAs, these DNA oligos are hybridized to first-strand cDNA during library preparation; duplexes formed between the biotinylated oligos and their targets are removed by streptavidin coupled magnetic beads. The same set of depletion oligos has been successfully applied during NET-seq library preparations from HEK293T cells. The set of depletion oligos might need to be amended for other cell types since chromatin-associated mature RNAs can vary in different cell- and tissue-types.
PCR amplification
Before high-throughput sequencing on an Illumina platform, the NET-seq library is amplified by a limited number of PCR cycles using an Illumina Index forward primer and a NET-seq specific reverse primer (oNTI231, Table 1). To avoid over-amplification, the minimal number of PCR cycles that are required to obtain a NET-seq library needs to be determined for each library preparation (see step 89 of the ‘PROCEDURE’ section and legend of Fig. 3c for more information). We typically test between 6 and 12 PCR amplification cycles (Fig. 3c). The NET-seq library is then amplified with the minimal amount of PCR cycles (Fig. 3d).
Quality controls for library preparation
The human NET-seq library construction requires several enzymatic reactions, such as ligation, reverse transcription, circularization and PCR amplification (Fig. 1). We have observed variations between different enzyme lots and so we suggest keeping track of the lot numbers for each enzyme. We also include a purified RNA oligonucleotide (oGAB11, Table 1)24 sample, which is processed in parallel to the library sample (steps 25–28, 31–70 and 81–89 of the ‘PROCEDURE’ section), as a control to monitor the success of each enzymatic reaction. In particular, we recommend using this control RNA oligonucleotide to determine the ligation efficiency; ligation of the DNA linker to the 3′-end of the nascent RNA is one of the most critical steps in the NET-seq protocol as only nascent RNA molecules that ligate with the DNA linker are captured in the NET-seq library. To ensure a high sensitivity of the NET-seq approach and to minimize biases, the ligation efficiency should be at least 90% (Supplementary Fig. 1). After the DNA linker has been ligated to oGAB11, the ligation product can be used to monitor the success of first-strand cDNA synthesis by SuperScript III reverse transcriptase; if successful, a distinct band should be observed at approximately 100 nt. This oGAB11 cDNA can then be used to determine the circularization efficiency by the CircLigase. Alternatively, circularization efficiency can be monitored by circularizing the RT primer oLSC007 (Table 1). The circularized product migrates more slowly than the linear molecule and can be visualized by gel electrophoresis. oGAB11 cDNA also serves as a positive control during PCR amplification by the Phusion DNA polymerase.
MATERIALS
REAGENTS
-
HeLa S3 cells (ATCC, cat. no. CCL-2.2) or HEK293T cells (ATCC, cat. no. CRL-11268)
CAUTION It is important to regularly check cell lines to ensure they are authentic and are not infected with mycoplasma. Handle human cell lines according to the manufacturer’s instructions. Work in a fume hood, use sterile equipment and wear gloves to minimize the risk of contamination.
DMEM (Life Technologies, cat. no. 11995)
Fetal Bovine Serum (FBS) (Life Technologies, cat. no. 16000)
-
Penicillin-Streptomycin (10,000 U/ml; Life Technologies, cat. no. 15140)
CAUTION Penicillin-Streptomycin is toxic. Handle media containing these antibiotics with care and dispose of waste according to institutional regulations.
RNase/DNase-free H2O (Life Technologies, cat. no. 10977–015)
1× PBS (Life Technologies, cat. no. 10010–023)
10× TBE buffer (Life Technologies, cat. no. 15581–044)
-
α-amanitin (Sigma, cat. no. A2263)
CAUTION α-amanitin is toxic. Handle solutions containing α-amanitin with care and dispose of waste according to institutional regulations.
-
DTT (0.1 M; part of the SuperScript III First-Strand Synthesis System, Life Technologies, cat. no. 18080–051)
CAUTION DTT is toxic and corrosive and is an irritant. Handle solutions containing DTT with care and dispose of waste according to institutional regulations.
-
NP-40, molecular biology grade (Life Technologies, cat. no. 28324)
CAUTION NP-40 is an irritant. Handle solutions containing NP-40 with care and dispose of waste according to institutional regulations.
-
Triton X-100, molecular biology grade (Sigma, cat. no. T9284)
CAUTION Triton X-100 is harmful and is an irritant. Triton X-100 is hazardous to the environment. Handle solutions containing Triton X-100 with care and dispose of waste according to institutional regulations.
Tween 20, molecular biology grade (Sigma, cat. no. 274348)
Sucrose, molecular biology grade (Sigma, cat. no. S0389)
Glycerol, molecular biology grade (Sigma, cat. no. G5516)
Urea, molecular biology grade (Sigma, cat. no. U6504)
-
Sodium carbonate anhydrous, proteomics grade (VWR, cat. no. M138)
CAUTION Sodium carbonate is an irritant. Handle solutions containing Sodium carbonate with care and dispose of waste according to institutional regulations.
Sodium bicarbonate (VWR, cat. no. 3509)
Sodium acetate, RNase-free (3 M; Life Technologies, cat. no. AM9740)
NaCl, RNase-free (5 M; Life Technologies, cat. no. AM9760G)
-
EDTA, RNase-free (0.5 M; Life Technologies, cat. no. AM9260G)
CAUTION EDTA is an irritant. Handle solutions containing EDTA with care and dispose of waste according to institutional regulations.
Tris-HCl, RNase-free (1 M, pH 7.0; Life Technologies, cat. no. AM9850G)
Tris-HCl, RNase-free (1 M, pH 8.0; Life Technologies, cat. no. AM9855G)
HEPES, RNase-free (1 M, pH 7.5; Teknova, cat. no. H1035)
-
NaOH solution (1.0 N; Sigma, cat. no. S2770)
CAUTION NaOH containing solutions are corrosive. Handle solutions containing NaOH with care and dispose of waste according to institutional regulations.
-
HCl, Hydrochloric acid concentrate (1.0 mol for 1L standard solution 1.0 N; Sigma, cat. no. 38282)
CAUTION HCl containing solutions are corrosive and is an irritant. Handle solutions containing HCl with care and dispose of waste according to institutional regulations
SUPERase. In (Life Technologies, AM2696)
-
Protease inhibitor mix cOmplete, EDTA-free (Roche, 11873580001)
CAUTION This mix is an irritant. Handle solutions containing the Protease inhibitor mix with care and dispose of waste according to institutional regulations.
Benzonase Nuclease (Sigma, cat. no. E1014)
GlycoBlue (15 mg/ml; Life Technologies, cat. no. AM9515)
-
Isopropanol, molecular biology grade (Sigma, cat. no. 278475)
CAUTION Isopropanol is highly flammable and volatile. Isopropanol is an irritant. Handle solutions containing Isopropanol with care and dispose of waste according to institutional regulations.
-
Ethanol, molecular biology grade (VWR, cat. no. V1016)
CAUTION Ethanol is highly flammable and volatile. Ethanol is an irritant. Handle solutions containing Ethanol with care and dispose of waste according to institutional regulations.
-
Sodium dodecyl sulfate (SDS; Sigma, cat. no. L3771)
CAUTION SDS is corrosive and flammable. SDS is an irritant. Handle solutions containing SDS with care and dispose of waste according to institutional regulations.
-
Pol II CTD Ser2-P antibody (3E10; Active Motif, cat. no. 61083)
CRITICAL: We highly recommend using this well characterized Pol II CTD Ser2-P antibody for western blot control experiments (Please see step 19 of the “PROCEDURE” section and Box 1 for more details.).
-
Pol II CTD Ser5-P antibody (3E8; Active Motif, cat. no. 61085)
CRITICAL: We highly recommend using this well characterized Pol II CTD Ser5-P antibody for western blot control experiments (Please see step 19 of the “PROCEDURE” section and Box 1 for more details.).
Histone 2B antibody (FL-126; Santa Cruz Biotechnology, cat. no. sc-10808)
GAPDH antibody (6C5; Life Technologies, cat. no. AM4300)
U1 snRNP70 antibody (C-18; Santa Cruz Biotechnology, cat. no. sc-9571)
-
Chloroform, molecular biology grade (Sigma, cat. no. 288306)
CAUTION Chloroform is volatile and toxic. Chloroform is an irritant. Handle solutions containing chloroform with care and dispose chloroform waste according to the institutional regulations.
-
miRNeasy Mini Kit (50) (Qiagen, cat. no. 217004)
CAUTION The RWT buffer is corrosive and is an irritant. The Qiazol Lysis Reagent is toxic and corrosive. Use personal protective equipment when handling this kit and dispose waste according to the institutional regulations.
CRITICAL: It is critical to use the miRNeasy Mini Kit instead of the RNeasy Mini Kit to avoid RNA length biases.
RNase-Free DNase Set (50) (Qiagen, cat. no. 79254)
-
PEG8000, molecular biology grade (part of T4 RNA Ligase 2, truncated, NEB, cat. no. M0242S)
CRITICAL: Store PEG8000 solution at −20°C.
Dimethyl sulfoxide (DMSO), molecular biology grade (Sigma, cat. no. D8418)
T4 RNA Ligase Buffer (part of T4 RNA Ligase 2, truncated, NEB, cat. no. M0242S)
-
T4 RNA Ligase 2, truncated (NEB, cat. no. M0242S)
CRITICAL: We recommend to keep track of the enzyme lot since the ligase activity can vary between different batches.
Orange G (Sigma, cat. no. O3756)
-
SYBR Gold Nucleic Acid Gel Stain (10,000× concentrate; Life Technologies, cat. no. S-11494)
CAUTION SYBR Gold Nucleic Acid Gel Stain is flammable. Nucleic acid stains are usually mutagenic. Use personal protective equipment when handling nucleic acid gel stains and dispose waste according to the institutional regulations.
RNA control ladder (0.1–2 kb; Life Technologies, cat. no. 15623-100)
DNA control ladder (10 bp; Life Technologies, cat. no. 10821-015)
TBE-Urea Sample Buffer, 2× (Life Technologies, cat. no. LC6876)
dNTP mix (10 mM; Life Technologies, cat. no. 18427-013)
5× First-Strand Buffer (part of SuperScript III First-Strand Synthesis System, Life Technologies, cat. no. 18080-051)
SuperScript III First-Strand Synthesis System (Life Technologies, cat. no. 18080-051)
CircLigase ssDNA Ligase (100 U/μl; Epicentre, cat. no. CL4111K)
CircLigase Reaction Buffer, 10× (part of CircLigase ssDNA Ligase, Epicentre, cat. no. CL4111K)
ATP, 1 mM (part of CircLigase ssDNA Ligase, Epicentre, cat. no. CL4111K)
-
MnCl2, 50 mM (part of CircLigase ssDNA Ligase, Epicentre, cat. no. CL4111K)
CAUTION MnCl2 is toxic and hazardous to the environment. Handle solutions containing MnCl2 with care and dispose of waste according to institutional regulations.
Dynabeads MyOne Streptavidin C1 (Life Technologies, cat. no. 65001)
20× SSC (Life Technologies, cat. no. AM9763)
Phusion High-Fidelity DNA Polymerase (2,000 U/ml; NEB, cat. no. M0530S)
Phusion HF Buffer, 5× (part of Phusion High-Fidelity DNA Polymerase, NEB, cat. no. M0530S)
Qubit dsDNA HS Assay Kit (Life Technologies, cat. no. Q32851)
High Sensitivity DNA Analysis Kit (Agilent Technologies, cat. no. 5067-4626)
Box 1. Checking cell fractionation by western blotting TIMING 5 h (plus overnight and ~2.5 h the next day).
-
1|
Adjust the volumes of the nucleoplasmic (step 18, “PROCEDURE” section) and chromatin fractions (step 19, “PROCEDURE” section) to the volume of the cytoplasmic fraction (step 12, “PROCEDURE” section) by adding 1× PBS. The volume of the cytoplasmic fraction is typically ~500 μl. Mix well.
CRITICAL STEP It is important to completely resuspend the sticky chromatin pellet. We found that this can be accomplished in the presence of 250 U Benzonase.
-
2|
Use 50 μl of each subcellular fraction and add 50 μl of 2× SDS buffer to each fraction, and boil the samples at 95°C for 5 min.
PAUSE POINT Samples can be stored up to months at −20°C.
-
3|
Load 10 μl of sample (from step 2: cytoplasmic, nucleoplasmic and chromatin fractions) per lane and separate by standard SDS-polyacrylamide gel electrophoresis (PAGE).
-
4|
Probe the membrane overnight at 4°C with antibodies directed against transcribing RNA polymerase II (Pol II): Ser2 CTD phosphorylated form (3E10, 1:1000 dilution); Ser5 CTD phosphorylated form (3E8, 1:1000 dilution) (Fig. 2). We recommend using these anti-Pol II antibodies since their CTD specificities have been extensively characterized57.
Optionally the membrane can also be probed with antibodies raised against the subcellular marker proteins GAPDH (cytoplasm), U1 snRNP70 (nucleoplasm) and Histone 2B (chromatin). We recommend probing the subcellular fractions with these antibodies especially in pilot experiments and in cases where a new cell type is used.
TROUBLESHOOTING
EQUIPMENT
15 cm × 2.5 cm Tissue Culture Dish (VWR, cat. no. 25383-103)
Cell Scrapers (GeneMate, cat. no. T-2443-2)
-
Scalpels (Electron Microscopy Sciences, cat. no. 72042-11)
CAUTION Handle sharps with care. Dispose sharps according to the institutional regulations.
0.2 ml RNase/DNase-free PCR tubes (Corning, cat. no. 3745)
0.5 ml RNase/DNase-free microfuge tubes (Life Technologies, cat. no. AM12350)
-
1.5 ml RNase/DNase-free microfuge tubes (Life Technologies, cat. no. AM12450)
Microfuge tube filter: Costar Spin-X centrifuge tube filters (Sigma, cat. no. CLS8162-96EA)
15 ml RNase/DNase-free centrifuge tubes (VWR, cat. no. 89039-670)
20G Needle (BD, cat. no. 305175)CAUTION Handle sharps with care. Dispose of sharps according to institutional regulations.
22G Needle (BD, cat. no. 305155)CAUTION Handle sharps with care. Dispose of sharps according to institutional regulations.
1 ml Syringe (BD, cat. no. 309628)
Cell counter Countess (Life Technologies, cat. no. C10227)
Refrigerated centrifuge 5810R (VWR, cat. no. 89305-180)
Refrigerated microcentrifuge 5424R (VWR, cat. no. 97058-914)
TBE-Urea gels, 15% (Life Technologies, cat. no. EC68852BOX)
TBE-Urea gels, 10% (Life Technologies, cat. no. EC68752BOX)
TBE gels, 8% (Life Technologies, cat. no. EC62152BOX)
Mini-Cell polyacrylamide gel box, XCell SureLock (Life Technologies, cat. no. EI0001)
Electrophoresis power supply (VWR, cat. no. 93000-744)
Black gel box (Licor, cat. no. 929-97301)
Shaker (BioExpress, cat. no. S-3200-LS)
Vortexer (VWR, cat. no. 58816-121)
Magnetic Rack (Thermo Scientific, cat. no. 21359)
Thermal cycler (Fisher Scientific, cat. no. E950030020)
Thermomixer (Core Life Sciences, cat. no. H5000-HC)
NanoDrop 2000 UV-Vis Spectrophotometer (Thermo Scientific, cat. no. ND-2000)
2100 BioAnalyzer (Agilent Technologies, cat. no. G2940CA)
Qubit 2.0 Fluorometer (Life Technologies, cat. no. Q32866)
MiSeq, HiSeq or NextSeq next-generation sequencing platform (Illumina)
REAGENT SETUP
50× Protease inhibitor mix
Dissolve one tablet of Protease inhibitor cocktail (Roche, see ‘REAGENTS’ section) in 1 ml of pre-cooled RNase-free H2O. Prepare before use and store aliquots up to one year at −20°C. CAUTION The Protease inhibitor mix is an irritant.
α-amanitin solution (1 mM)
Dissolve 1 mg α-amanitin in 1 ml RNase-free H2O. Prepare before use and store aliquots up to one year at −20°C. CAUTION α-amanitin is toxic. Handle α-amanitin solution with care and dispose of waste according to institutional regulations.
Cytoplasmic lysis buffer
0.15% (vol/vol) NP-40, 10 mM Tris-HCl (pH 7.0), 150 mM NaCl, 25 μM α-amanitin, 50 U SUPERase. In and 1× Protease inhibitor mix. For one reaction mix 3.8 μl of 10% (vol/vol) NP-40, 2.5 μl of 1 M Tris-HCl (pH 7.0), 7.5 μl of 5 M NaCl, 5 μl of 1× Protease inhibitor mix (50×), 6.2 μl of 1 mM α-amanitin, 0.6 μl of SUPERase. In (20 U/μl) and 224.4 μl of RNase-free H2O. Prepare this solution freshly before use with RNase-free reagents and keep on ice. CAUTION α-amanitin is toxic. Handle solutions containing α-amanitin with care and dispose of waste according to institutional regulations. NP-40 and the Protease inhibitor mix are irritants.
Sucrose buffer
10 mM Tris-HCl (pH 7.0), 150 mM NaCl, 25% (wt/vol) sucrose, 25 μM α-amanitin, 50 U SUPERase. In and 1× Protease inhibitor mix. For one reaction mix 5 μl of 1 M Tris-HCl (pH 7.0), 15 μl of 5 M NaCl, 250 μl of 50% (wt/vol) filter-sterilized sucrose, 10 μl of 1× Protease inhibitor mix (50×), 12.5 μl of 1 mM α-amanitin, 1.2 μl of SUPERase. In (20 U/μl) and 206.3 μl of RNase-free H2O. Prepare this solution freshly before use with RNase-free reagents and keep on ice. CAUTION α-amanitin is toxic. Handle solutions containing α-amanitin with care and dispose of waste according to institutional regulations. The Protease inhibitor mix is an irritant.
Nuclei wash buffer
0.1% (vol/vol) Triton X-100, 1 mM EDTA, in 1× PBS, 25 μM α-amanitin, 50 U SUPERase. In and 1× Protease inhibitor mix. For one reaction mix 2 μl of 0.5 M EDTA solution, 10 μl of 10% (vol/vol) Triton X-100, 20 μl of 1× Protease inhibitor mix (50×), 25 μl of 1 mM α-amanitin, 2.5 μl of SUPERase. In (20 U/μl) and 940.5 μl of 1× PBS. Prepare this solution freshly before use with RNase-free reagents and keep on ice. CAUTION α-amanitin is toxic. Handle solutions containing α-amanitin with care and dispose of waste according to the institutional regulations. Triton X-100 is harmful and is an irritant. Triton X-100 is hazardous to the environment. Handle solutions containing Triton X-100 with care and dispose of waste according to institutional regulations. The Protease inhibitor mix is an irritant.
Glycerol buffer
20 mM Tris-HCl (pH 8.0), 75 mM NaCl, 0.5 mM EDTA, 50% (vol/vol) glycerol, 0.85 mM DTT, 25 μM α-amanitin, 50 U SUPERase. In and 1× Protease inhibitor mix. For one reaction mix 5 μl of 1 M Tris-HCl (pH 8.0), 3.8 μl of 5 M NaCl, 0.5 μl of 0.25 M EDTA, 125 μl of 100% (vol/vol) filter-sterilized glycerol, 2.1 μl of 0.1 M filter-sterilized DTT, 5 μl of 1× Protease inhibitor mix (50×), 6.2 μl of 1 mM α-amanitin, 0.6 μl of SUPERase. In (20 U/μl) and 101.8 μl of RNase-free H2O. Prepare this solution freshly before use with RNase-free reagents and keep on ice. CAUTION α-amanitin is toxic. Handle solutions containing α-amanitin with care and dispose of waste according to institutional regulations. DTT is toxic and corrosive. DTT, EDTA and the Protease inhibitor mix are irritants.
Nuclei lysis buffer
1% (vol/vol) NP-40, 20 mM HEPES (pH 7.5), 300 mM NaCl, 1M Urea, 0.2 mM EDTA, 1 mM DTT, 25 μM α-amanitin, 50 U SUPERase. In and 1× Protease inhibitor mix. For one reaction mix 25 μl of 10% (vol/vol) NP-40, 5 μl of 1 M HEPES (pH 7.5), 0.5 μl of 0.1 M EDTA, 15 μl of 5 M NaCl, 25 μl of 10 M filter-sterilized Urea, 2.5 μl of 0.1 M filter-sterilized DTT, 5 μl of 1× Protease inhibitor mix (50×), 6.2 μl of 1 mM α-amanitin, 0.6 μl of SUPERase. In (20 U/μl) and 165.2 μl of RNase-free H2O. Prepare this solution freshly before use with RNase-free reagents and keep on ice. CAUTION α-amanitin is toxic. Handle solutions containing α-amanitin with care and dispose of waste according to institutional regulations. DTT is toxic and corrosive. DTT, EDTA, NP-40 and the Protease inhibitor mix are irritants.
Chromatin resuspension solution
25 μM α-amanitin, 50 Units SUPERase. In and 1× Protease inhibitor mix Roche in 1× PBS. Mix 5 μl of 1 mM α-amanitin, 4 μl of of 1× Protease inhibitor mix (50×), 0.5 μl of SUPERase. In (20 U/μl) and 190.5 μl of 1× PBS. This volume is sufficient for 3 reactions. Prepare this solution freshly before use with RNase-free reagents and store on ice. CAUTION α-amanitin is toxic. Handle solutions containing α-amanitin with care and dispose of waste according to institutional regulations. The Protease inhibitor mix is an irritant.
Alkaline fragmentation solution (2×)
For a 5 ml stock solution mix 0.6 ml 0.1 M Na2CO3 and 4.4 ml 0.1 M NaHCO3. Store 500 μl aliquots in air-tight screw cap tubes at room temperature (22°C) up to months. CAUTION Sodium carbonate is an irritant.
RNA precipitation solution
For one reaction (562 μl) mix 60 μl 3 M sodium acetate (pH 5.5), 2 μl GlycoBlue (15 mg/ml) and 500 μl RNase-free H2O. Prepare freshly before use and store on ice.
SUPERase. In/DTT mix
For one reaction (1.3 μl) mix 0.5 μl SUPERase. In (20 U/μl) and 0.8 μl 0.1 M DTT. Prepare freshly before use and store on ice. CAUTION DTT is toxic and corrosive. DTT is an irritant.
DNA soaking buffer
For one reaction (668 μl) mix 40 μl 6.7 μl 1 M Tris-HCl (pH 8.0), 40 μl 5 M NaCl, 1.3 μl 0.5 M EDTA and 620 μl RNase-free H2O. Prepare freshly before use and store at room temperature. CAUTION EDTA is an irritant.
HCl (1N)
Dilute Hydrochloric acid concentrate (Sigma, see ‘REAGENTS’ section) by adding DNase-free H2O to a final volume of 1L. Store 1N HCl up to one year at room temperature. CAUTION HCl containing solutions are corrosive and cause irritations. Handle solutions containing HCl with care and dispose of waste according to institutional regulations.
SDS buffer (2×)
100 mM Tris-HCl (pH 7.0), 4% (wt/vol) SDS and 20% (vol/vol) glycerol. Mix 5 ml of 1 M Tris-HCl (pH 7.0), 10 ml of 20% (wt/vol) SDS, 10 ml of 100% (vol/vol) filter-sterilized glycerol and 25 ml filter-sterilized and deionized H2O. The volume will be enough to process at least 50 subcellular fractions. Prepare before use and store at room temperature for several months. CAUTION SDS is corrosive and flammable. SDS is an irritant.
Depletion oligo mix
Resuspend each of the 20 biotinylated DNA oligos (Table 2) with 10 mM Tris-HCl (pH 8.0) so that the final concentration of each depletion oligo solution will be 200 μM. Next, combine 5 μl of each depletion oligo solution and mix. The final concentration of each DNA oligo in the Depletion oligo mix will be 10 μM. The volume of the Depletion oligo mix will be 100 μl. This volume will be enough for 50 depletion experiments when two depletion reactions are performed per sample. Prepare before use and store indefinitely at −20°C.
Bind/wash buffer (2×)
5 mM Tris-HCl (pH 7.0), 2 M NaCl, 1 mM EDTA and 0.2% (vol/vol) Triton X-100. Mix 5 μl of 1 M Tris-HCl (pH 7.0), 400 μl of 5 M NaCl, 2 μl of 0.5 M EDTA, 20 μl of 10% Triton X-100 and 573 μl of RNase-free H2O. The volume will be enough to perform seven specific depletion experiments using two depletion reactions per sample (see Procedure steps 76–85 and Box 3). Store at room temperature for several months. CAUTION EDTA and Triton X-100 are irritants. Triton X-100 is harmful and hazardous to the environment. Handle solutions containing Triton X-100 with care and dispose of waste according to institutional regulations.
Box 3. Preparation of Streptavidin coupled magnetic beads for specific depletion TIMING 10 min.
The following protocol describes how to prepare the Dynabeads MyOne Streptavidin C1 (10 mg/ml) for specific depletion of the 20 most abundant chromatin-associated mature RNAs that are captured in NET-seq libraries obtained from HeLa S3 and HEK293T cells.
-
1|
Resuspend beads by gentle vortexing.
-
2|
Transfer 37.5 μl beads per depletion reaction to a DNase-free 1.5 ml microfuge tube.
-
3|
Place the tube on a magnetic rack for 1 min and withdraw all the supernatant from the tube.
-
4|
Remove the tube from the magnetic rack and resuspend beads in 37.5 μl 1× bind/wash buffer.
-
5|
Repeat this washing procedure (steps 3 and 4) two more times.
-
6|
Place tube on a magnetic rack for 1 min and withdraw the supernatant from the tube.
-
7|
Remove tube from the magnetic rack and resuspend beads in 15 μl 2× bind/wash buffer.
-
8|
Transfer 10 μl of the resuspended and washed beads to a new tube.
-
9|
Place tube in a thermomixer at 37°C to equilibrate for 15–30 min.
6× DNA loading buffer
Dissolve 6 g Sucrose and 30 mg Orange G in 20 ml RNase-free H2O. Bring to a final volume of 25 ml with RNase-free H2O. Store light-protected at room temperature for up to a year.
Gel staining solution
To stain one TBE or TBE-Urea gel, add 5 μl of SYBR Gold Nucleic Acid Gel Stain to 50 ml 1× TBE buffer and mix. Prepare before use and store light-protected at room temperature. CAUTION SYBR Gold Nucleic Acid Gel Stain is flammable. Nucleic acid stains are usually mutagenic. Use personal protective equipment when handling nucleic acid gel stains and dispose ofwaste according to the institutional regulations.
PROCEDURE
Quantitative purification of RNA polymerase by cell fractionation TIMING 45 min
CRITICAL Cell fractionation has been optimized for HeLa S3 and HEK293T cells. If using other cell types, protocol optimizations might be required.
CRITICAL Cell fractionation is performed on ice or at 4°C with buffers freshly prepared on the same day. All buffers are pre-cooled on ice before use. Use RNase-free reagents and equipment.
-
1|
Grow HeLa S3 or HEK293T cells in a 15 × 2.5 cm dish in DMEM containing 10% (vol/vol) FBS, 100 U/ml penicillin and 100 μg/ml streptomycin until 90% confluent.
CRITICAL STEP Grow human cells according to the Cell Culture Guidelines of the ENCODE project (https://www.encodeproject.org/documents/60b6b535-870f-436b-8943-a7e5787358eb/). This includes keeping track of the growth time, passage number and cell density.
-
2|
Wash cells twice with 10 ml PBS buffer. Perform washes on ice. A typical wash takes less than 1 min.
-
3|
Scrape cells into 1 ml PBS buffer.
-
4|
Determine the cell number by counting using a cell counter (Please see “EQUIPMENT” section for details.). Use 1× 107 cells as input for each cell fractionation. In addition to the experimental sample, also process in parallel a control for western blot analysis (described in Box 1; Fig. 1).
-
5|
Wash each sample of 1× 107 cells with 10 ml PBS.
-
6|
Collect cells by centrifugation at 500g for 2 min at 4°C.
-
7|
Remove the supernatant by aspiration.
CRITICAL STEP It is important to remove the supernatant completely at this step. If the supernatant is not completely removed the Cytoplasmic lysis buffer (step 8) will be diluted which affects the cell lysis efficiency.
-
8|
Add 200 μl Cytoplasmic lysis buffer and transfer to a RNase-free 1.5 ml microfuge tube. Using a cut 1,000 μl pipette tip, pipette the sample up and down 10×.
-
9|
Incubate the cell lysate on ice for 5 min.
-
10|
Using a cut 1,000 μl pipette tip, layer the cell lysate onto 500 μl Sucrose buffer.
-
11|
Collect cell nuclei by centrifugation at 16,000g for 10 min at 4°C.
-
12|
Remove supernatant.
CRITICAL STEP The supernatant represents the cytoplasmic fraction. The cytoplasmic fraction is only retained in case of the western blot control (Box 1).
-
13|
Wash nuclei with 800 μl Nuclei wash buffer.
-
14|
Collect washed nuclei by centrifugation at 1,150g for 1 min at 4°C.
CRITICAL STEP To remove cytoplasmic mature RNAs it is important to remove the supernatant completely.
-
15|
Add 200 μl Glycerol buffer. Using a cut 1,000 μl tip, resuspend washed nuclei by pipetting up and down. Transfer to a new 1.5 ml RNase-free microfuge tube.
-
16|
Add 200 μl Nuclei lysis buffer, mix by pulsed vortexing and incubate on ice for 2 min.
-
17|
Centrifuge at 18,500g for 2 min at 4°C.
-
18|
Remove supernatant completely.
CRITICAL STEP It is important to completely remove the supernatant containing nucleoplasmic RNAs. The nucleoplasmic fraction is only retained in case of the western blot control (Box 1).
-
19|
Resuspend the chromatin in 50 μl Chromatin resuspension solution.
CRITICAL STEP It is important to resuspend the sticky chromatin pellet prior to the preparation of the nascent RNA. This step increases the RNA yield. This step is performed for the experimental sample as well as for the control sample. The experimental sample is used for the preparation of the nascent RNA (step 20). The control sample is employed for the western blot analysis as described in Box 1.
Preparation of nascent RNA TIMING 1 h
-
20|
Add 700 μl QIAzol Lysis Reagent (part of miRNeasy Mini Kit, Qiagen) to the resuspended chromatin from step 19.
-
21|
Mix thoroughly by slowly pipetting up and down using a 1 ml syringe with a 22G needle. Alternatively, the chromatin pellet can also be solubilized by gentle vortexing.
CRITICAL STEP Mix very carefully until the solution is homogenous. Mix slowly to avoid spilling the sample. Be aware that the QIAzol Lysis reagent contains phenol. Handle solutions with care and according to institutional regulations.
-
22|
Prepare RNA using the miRNeasy Mini Kit following the manufacturer’s instructions including a DNase treatment (see following step).
-
23|
Remove DNA from the RNA during RNA preparation (step 22). This is accomplished by performing an on-column DNase digestion using the RNase-Free DNase Set (Qiagen) according to the manufacturer’s instructions.
-
24|
Determine the quantity and the quality of the prepared RNA using a NanoDrop 2000. The RNA yield is usually in the range of 20–30 μg. The expected A260/A280 ratio is 2.1. Please also see the “ANTICIPATED RESULTS” section for more information.
PAUSE POINT The isolated RNA can be stored for months at −80°C.
Barcode DNA linker ligation TIMING 3.5 h
-
25|
Denature the RNA sample from step 24 and 5 μl of the ligation control oligonucleotide oGAB11 (10 μM; Table 1) for 2 min at 80°C in a thermomixer.
-
26|
Prepare DNA linker ligation mix for each RNA sample and for oGAB11 in 0.2 ml RNase-free PCR tubes as described in the table below. Prepare 3 ligation reactions per RNA sample; the RNA input per ligation reaction is 1 μg.
CRITICAL STEP Before adding the truncated T4 RNA Ligase 2 it is important to mix the ligation reaction until it is homogenous. Then add the Ligase and mix again. Incompletely mixed samples will negatively affect the ligation efficiency.
Component Amount per reaction (μl)
Final RNA sample oGAB11 control PEG8000 (50% vol/vol) 8.0 8.0 20% (vol/vol) DMSO 2.0 2.0 10% (vol/vol) T4 RNA Ligase Buffer (10×) 2.0 2.0 1× Barcode DNA Linker (1 μg) 1.0 1.0 RNA sample (1 μg) 6.0 – oGAB11 (10 μM) – 1.0 0.5 μM RNAse-free H2O – 5.0 Truncated T4 RNA Ligase 2 1.0 1.0 200 U
-
27|
Incubate Ligation mixes for 3 h at 37°C in a thermal cycler.
-
28|
Add 0.7 μl EDTA (0.5 M) to each ligation mix to stop the ligation reaction.
RNA fragmentation TIMING 45 min (plus overnight and ~3.5 h the next day)
-
29|
Add 20 μl of 2× Alkaline fragmentation solution to each ligation reaction. Please note that the Procedure steps 29 and 30 are not performed for the oGAB11 ligation control.
-
30|
RNA at 95°C in a thermal cycler for the adjusted time.
CRITICAL STEP The fragmentation time needs to be adjusted whenever a new batch of Alkaline fragmentation solution is applied. An over or under fragmentation of the nascent RNA pool can lead to systematic biases. In a typical experiment RNA is fragmented between 10 and 40 min at 95°C (Fig. 3a). The optimal fragmentation time is when most RNA molecules are in the required size range, usually between 35–100 nt (Fig. 3a, lane 6).
-
31|
Add fragmented RNA sample (from step 30) to 562 μl RNA precipitation solution. Add the non-fragmented oGAB11 ligation control (from step 28) to 562 μl RNA precipitation solution.
-
32|
Precipitate the fragmented RNA and the non-fragmented oGAB11 ligation control by adding 750 μl Isopropanol. Mix well. Precipitate for ≥1 h at −80°C.
PAUSE POINT Precipitation can be left at −80°C overnight.
-
33|
Centrifuge at 20,000g for 30 min at 4°C to pellet the RNA.
-
34|
Remove supernatant and wash pellet with 750 μl 80% (vol/vol) ice-cold Ethanol.
-
35|
Spin sample at 20,000g for 2 min at 4°C for and remove supernatant.
-
36|
Airdry RNA pellet for 10 min at room temperature.
-
37|
Resuspend RNA pellet in 10 μl RNase-free H2O. Add 10 μl 2× TBU Denaturing sample buffer to each RNA sample and mix.
-
38|
Prepare RNA control ladder and the oGAB11 control. Add 1.0 μl RNA control ladder or oGAB11 to 9 μl RNase-free H2O. Add 10 μl 2× TBU Denaturing sample buffer to each control sample and mix.
-
39|
Denature RNA sample and RNA control samples (oGAB11 ligation control, ladder and oGAB11) for 2 min at 80°C. Cool samples on ice for 3 min.
-
40|
Prerun a 15% (wt/vol) polyacrylamide TBE-Urea gel at 200 V for 15 min in 1× TBE.
-
41|
Separate the fragmented RNA samples and the RNA control samples (including the oGAB11 ligation control) by polyacrylamide gel electrophoresis at 200 V for 65 min.
-
42|
Stain the gel in 50 ml Gel staining solution for 5 min at room temperature on a shaker. Protect the gel from light during staining by the use of a black gel box.
-
43|
Visualize the fragmented RNA and the oGAB11 ligation control under blue/UV light (Fig. 3a and Supplementary Fig. 1). For the fragmented RNA excise the region between 35 and 100 nt (Fig. 3a). For the oGAB11 ligation control excise the narrow band at ~55 nt (Supplementary Fig. 1, lane 4).
-
44|
Extract the RNA from the gel slice by rapid gel extraction as described in Box 2.
-
45|
Precipitate RNA by adding 2 μl 15 mg/ml GlycoBlue and 50 μl 3M Sodium acetate (pH 5.5). Mix well. Add 750 μl Isopropanol and mix well. Incubate precipitations at −80°C for ≥1 h.
PAUSE POINT The RNA precipitations can be stored at −80°C overnight.
-
46|
Recover RNA as described in Steps 33–36.
-
47|
Resuspend size-selected RNA and the oGAB11 ligation control in 10 μl pre-cooled RNase-free H2O.
Box 2. Rapid gel extraction TIMING 20 min.
This protocol for rapidly extracting RNA from a polyacrylamide TBE-Urea gel is similar to the gel extraction protocol provided by the Jonathan Weissman laboratory24,37.
-
1|
Pierce the bottom of a 0.5 ml RNase-free microfuge tube with a 21-G needle.
-
2|
Put the pierced 0.5 ml tube in 1.5 ml RNase-free microfuge tube.
-
3|
Place the gel slice (from step 44 or step 63 of the Procedure) into the inner pierced 0.5 ml tube.
-
4|
Centrifuge at 20,000g for 4 min at room temperature.
-
5|
Add 200 μl RNase-free H2O and mix.
-
6|
Incubate the sample for 10 min at 70°C in a thermomixer.
-
7|
Vortex for 30 sec at a medium intensity setting.
-
8|
Cut the tip off of a 1,000 μl pipette tip and transfer the gel slurry into a Microfuge tube filter.
-
9|
Centrifuge at 20,000g for 3 min at room temperature.
-
10|
Combine eluates from 3 original ligation reactions. The expected volume is approximately 600 μl.
Reverse transcription (RT) TIMING 4 h (plus overnight and ~1 h the next day)
-
48|
Prepare the RT reaction mix tabulated below in a 0.2 ml RNase-free PCR tube and store on ice.
Component Amount per reaction (μl) Final
5× First-Strand Buffer 3.3 1× dNTPs (10 mM) 0.8 0.5 mM Reverse primer oLSC007 (10 μM) 0.5 0.3 μM
-
49|
Add 4.6 μl RT reaction mix to 10 μl RNA sample and to the oGAB11 control RNA sample.
-
50|
Incubate for 2 min at 80°C in a thermal cycler, then cool on ice for 3 min.
-
51|
Add 1.3 μl SUPERase. In/DTT mix and mix well.
-
52|
Add 0.8 μl SuperScript III (200 U/μl) and mix.
-
53|
Incubate for 30 min at 48°C in a thermal cycler.
-
54|
Add 1.8 μl 1N NaOH, mix well and incubate for 20 min at 98°C.
-
55|
Neutralize reaction by adding 1.8 μl 1N HCl, mix well and put on ice.
-
56|
Add 20 μl 2× TBU Denaturing sample buffer to each cDNA sample and the oGAB11 cDNA control, then mix.
-
57|
Prepare DNA control ladder. Add 1.0 μl DNA control ladder to 9 μl RNase-free H2O. Add 10 μl 2× TBU Denaturing sample buffer to each cDNA sample and mix.
-
58|
Denature cDNA sample, oGAB11 cDNA control and DNA control ladder for 3 min at 95°C in a thermomixer. Cool samples on ice for 3 min.
-
59|
Prerun a 10% (wt/vol) polyacrylamide TBE-Urea gel at 200 V for 15 min in 1× TBE.
-
60|
Separate the cDNA sample, the oGAB11 cDNA control and the DNA ladder by polyacrylamide gel electrophoresis at 200 V for 65 min.
-
61|
Stain the gel in 50 ml Gel staining solution for 5 min at room temperature on a shaker. Protect the gel from light during staining by the use of a black gel box.
-
62|
Visualize the gel under blue/UV light and excise the cDNA between 85 and 160 nt (Fig. 3b).
TROUBLESHOOTING
-
63|
Extract the cDNA from the gel slice by rapid gel extraction as described in steps 1–9 of Box 2. Combine eluates from 2 lanes (expected volume approximately 400 μl), add 25 μl 5 M NaCl and mix.
-
64|
Precipitate cDNA by adding 2 μl 15 mg/ml GlycoBlue and 750 μl Isopropanol. Mix well. Incubate precipitations at −20°C for ≥1 h.
PAUSE POINT The cDNA precipitation can be stored at −20°C overnight.
-
65|
Recover cDNA as described in Steps 33–36.
-
66|
Resuspend cDNA and oGAB11 control cDNA in 15 μl pre-cooled RNase-free H2O each.
PAUSE POINT The cDNA can be stored indefinitely at −20°C.
Circularization of cDNA TIMING 1.5 h
-
67|
Prepare Circularization mix as tabulated below and store on ice.
Component Amount per reaction (μl) Final
CircLigase 10× Reaction Buffer 2.0 1× ATP (1 mM) 1.0 50 μM MnCl2 (50 mM) 1.0 2.5 mM
-
68|
Add 4 μl Circularization mix to 15 μl cDNA sample and the oGAB11 cDNA control sample in a 0.2 ml RNase-free PCR tube, then mix well.
TROUBLESHOOTING
-
69|
Add 1 μl CircLigase (100 U/μl) and mix.
-
70|
Incubate CircLigase reaction for 60 min at 60°C and for 10 min at 80°C in a thermal cycler.
PAUSE POINT Circularized cDNA can be stored indefinitely at −20°C.
Specific depletion of highly abundant mature RNAs TIMING 1.5 h
-
71|
Prepare two specific depletion reactions per sample, but not for the oGAB11 control sample. Prepare depletion reactions in 0.2 ml DNase-free PCR tubes. The Depletion DNA oligo pool that has been successfully used for HeLa S3 and HEK293T cells is given in Table 2. Please note that the oGAB11 control sample is not subjected to steps 71–80. Store the oGAB11 control sample on ice until is required at step 81.
Component Amount per reaction (μl) Final
Circularization reaction (from Step 70) 5.0 Depletion DNA oligo pool 1.0 1 μM (per Depletion oligo) 20× SSC 1.0 2× DNase-free H2O 3.0
-
72|
Perform subtractive hybridization in a thermal cycler as given below.
Temperature Time
Denature 99°C 90 sec Annealing 99°C to 37°C in 0.1°C steps 1 sec (per 0.1°C step) Final Annealing 37°C 15 min
-
73|
Prepare Dynabeads MyOne Streptavidin C1 (10 mg/ml) for specific depletion as described in Box 3.
-
74|
Transfer 10 μl of depletion reaction directly from the 0.2 ml PCR tube in the thermal cycler (from step 72) to the washed and equilibrated beads in the thermomixer. Immediately mix by pipetting.
-
75|
Incubate in the thermomixer for 15 min at 37°C with mixing at 1,000 r.p.m.
-
76|
Transfer tubes from the thermomixer into a magnetic rack and leave for 1 min. Transfer the supernatant into a new 1.5 ml microfuge tube.
CRITICAL STEP The supernatant needs to be transferred carefully. Any remaining magnetic beads in the supernatant will have a negative impact on subsequent steps.
-
77|
Add 2 μl GlycoBlue, 6 μl 5 M NaCl and 74 μl DNase-free H2O to each depletion reaction and mix. Add 150 μl Isopropanol and mix.
-
78|
Incubate circularized and depleted DNA for ≥1 h at −20°C.
PAUSE POINT Precipitation can be left at −20°C overnight.
-
79|
Recover DNA as described in Steps 33–36.
-
80|
Resuspend 2 DNA pellets per sample in 10 μl ice cold H2O.
PAUSE POINT DNA can be stored indefinitely at −20°C.
PCR amplification of the cDNA sequencing library TIMING 4 h (plus overnight and ~1 h the next day)
-
81|
Prepare PCR mix for 4 pilot PCR amplification reactions for both the cDNA sample and the oGAB11 control cDNA sample. Mix well and store on ice.
TROUBLESHOOTING
Component Amount for 4.5 reactions (μl) Final
Phusion HF Buffer, 5× 18 1× dNTPs (10 mM) 1.8 0.2 mM Forward primer (Illumina Index primer, 100 μM) 0.45 0.5 μM oNTI231 (Reverse primer, 100 μM) 0.45 0.5 μM DNase-free H2O 63.9 Phusion DNA Polymerase (2 U/μl) 0.9 1.8 U
-
82|
For each PCR reaction put 19 μl PCR master mix in a 0.2 ml RNase-free PCR tube.
-
83|
Add 1 μl circularized cDNA and mix well.
-
84|
Perform PCR pilot amplifications as tabulated below. Remove one PCR tube for each sample at the end of the extension step after 6, 8, 10 and 12 amplification cycles.
Cycle number Denature Anneal Extend
1 98°C, 30 sec 2–14 98°C, 10 sec 60°C, 10 sec 72°C, 5 sec
-
85|
Add 3.4 μl 6× DNA loading dye to each tube and mix well.
-
86|
Prepare DNA control ladder. Add 1.0 μl DNA control ladder to 9 μl DNase-free H2O. Add 2 μl 6× DNA loading dye and mix well.
-
87|
Separate the PCR products and the DNA control ladder by TBE gel electrophoresis on an 8% (wt/vol) TBE gel at 180 V for 55 min.
-
88|
Stain the gel in 50 ml Gel staining solution for 5 min at room temperature on a shaker. Protect the gel from light during staining by the use of a black gel box.
-
89|
Visualize the gel under blue/UV light and identify the optimal PCR amplification cycle for each cDNA sequencing library (Fig. 3c). The NET-seq library runs at around 150 nt (Fig. 3c). The optimal PCR amplification cycle is characterized by a clear band at ~150 nt and the absence of PCR products at the higher molecular range (Fig. 3c, lane 3).
TROUBLESHOOTING
-
90|
Perform 4 PCR amplification reactions per sample with the adjusted amplification cycle as described in Steps 81–89. The oGAB11 control sample is not subjected to steps 90–106.
TROUBLESHOOTING
-
91|
For each sample and control, excise the band that contains the PCR product from the gel (Fig. 3d).
CRITICAL STEP Excise the broad band around 150 nt. Avoid contamination from the lower band that runs at ~120 nt, representing PCR product from empty circles. Empty circles are circularized cDNA molecules that arise from unextended RT primers and hence do not contain any information about the original nascent RNA.
-
92|
Pierce the bottom of a 0.5 ml DNase-free microfuge tube with a 21-G needle.
-
93|
Put the pierced 0.5 ml tube in 1.5 ml DNase-free microfuge tube.
-
94|
Place the gel slices into the inner pierced 0.5 ml tube.
-
95|
Centrifuge at 20,000g for 4 min at room temperature.
-
96|
Add 670 μl DNA soaking buffer and mix.
-
97|
Incubate at room temperature at 1,500 r.p.m. overnight in a thermomixer.
-
98|
Using a cut 1,000 μl pipette tip, transfer the gel slurry into a Microfuge tube filter.
-
99|
Centrifuge at 20,000g for 3 min at room temperature. Transfer the eluate to a new 1.5 ml microfuge tube.
-
100|
Precipitate the NET-seq library by adding 2 μl 15 mg/ml GlycoBlue and 680 μl Isopropanol. Mix well. Incubate precipitations at −20°C for ≥1 h.
PAUSE POINT The DNA precipitations can be stored at −20°C overnight.
-
101|
Recover DNA as described in Steps 33–36.
-
102|
Resuspend the NET-seq library in 10 μl Tris-HCl (10 mM, pH 8.0).
PAUSE POINT The DNA sequencing library can be stored indefinitely at −20°C.
Quantify and characterize the NET-seq library TIMING 1 h
-
103|
Prepare a 1:5 dilution of the NET-seq library by adding 1 μl of the NET-seq library to 4 μl of Tris-HCl (10 mM, pH 8.0) and mix.
-
104|
Use 1 μl of the diluted NET-seq library for quantification with the Qubit Fluorometer using the Qubit dsDNA HS Assay Kit. Prepare the sample and perform the measurement according to the manufacturer’s protocol.
-
105|
Use 1 μl of the diluted NET-seq library for characterization on the Agilent BioAnalyzer using the High Sensitivity DNA Analysis Kit according to the manufacturer’s instructions.
-
106|
Sequence the human NET-seq library from the 3′-end on the Illumina platform using oLSC006 (Table 1) as a custom sequencing primer. Perform sequencing according to the manufacturer’s instructions. NET-seq libraries are typically sequenced on MiSeq, HiSeq or NextSeq next-generation sequencing platforms. A reasonable coverage is obtained with 100–200 Million reads. A brief guidance to the bioinformatics analysis of human NET-seq data is given in Box 4. A more detailed description of the computational analysis of human NET-seq data can also be found in the Supplemental Information of the original human NET-Seq publication:29.
TROUBLESHOOTING
Troubleshooting advice is provided in Table 3.
TABLE 3.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 4, Box 1 | Western blot signals of the cytoplasmic marker protein GAPDH in the chromatin fraction | Reduced cell lysis efficiency due to an excess of input per cell fractionation reaction | Reduce the amount of input. The amount of cells per fractionation reaction should not exceed 2× 107 HeLa S3 or HEK293T cells. |
| The supernatant after washing the cell nuclei (step 18) is not completely removed. | Completely remove the supernatant after washing the cell nuclei. | ||
| 4, Box 1 | Strong western blot signals for elongating Pol II (CTD Ser5-P and Ser2-P) in the cytoplasmic or nucleoplasmic fractions | Treatment during cell lysis is too harsh leading to dissociation of transcribing Pol II. | Reduce the amount of cell lysis buffer that is added to the cell pellet. Be careful: too little lysis buffer can negatively affect the cell lysis efficiency. |
| 62 | No smear of cDNA is observed after the reverse transcription | The DNA linker ligation is inefficient. | Mix the viscous ligation reaction properly (step 26). Replace components of the ligation reaction every 4 months. |
| The reverse transcriptase enzyme has lost activity. | Replace the SuperScript III enzyme and other reagents of the RT reaction every 4 months and note the enzyme lot. | ||
| 68 | A brown precipitate forms after adding the RT product to the circularization mix. | The reason is unclear. | Replace the isopropanol precipitation (step 64) by an ethanol precipitation to reduce the amount of salt that co-precipitates with the cDNA. Additionally, reduce the amount of MnCl2 to 50%. |
| 89 | A DNA smear that migrates slower in an 8% TBE gel appears (Fig. 3c, 10 and 12 PCR cycles). | PCR over-amplification | Reduce the number of PCR cycles. |
| 90 | Strong PCR band at ~120 nt on an 8% TBE gel (Fig. 3c,d). | PCR product that originates from empty circles. Empty circles arise from unextended RT primers that get circularized by the CircLigase enzyme. | Precisely excise the RT product (step 62) and use less RT primer. Precisely excise the final PCR product at around 150 nt (Fig. 3d). Run the 8% TBE gel longer than 45 min. |
ANTICIPATED RESULTS
RNA extraction (steps 20–24) from the chromatin prepared from 1× 107 HeLa S3 and HEK293T cells typically results in a total RNA yield of 20–30 μg with an A260/A280 ratio of 2.1 as determined by NanoDrop spectrophotometer measurement. The efficiency of the DNA linker ligation is usually >95% as monitored by polyacrylamide gel electrophoresis of a control ligation using the RNA oligonucleotide oGAB11 (Table 1, Supplementary Fig. 1, steps 25–28 and 38–43). Most of the RNA after fragmentation by partial alkaline hydrolysis (steps 29–43) typically runs between 10 and 150 nt as revealed by polyacrylamide gel electrophoresis (Fig. 3a). The cDNA that is obtained upon reverse transcription (steps 48–62) runs between 80–160 nt as observed by polyacrylamide gel electrophoresis (Fig. 3b). Gel electrophoresis of the PCR product after the final amplification (steps 81–90) reveals a broad band around 150 nt (Fig. 3c,d). The total yield of a typical NET-seq library from HeLa S3 and HEK293T cells is usually 20–30 ng as determined by Qubit Fluorometer (step 104) and BioAnalyzer (step 105) measurements. The average fragment length of the human NET-seq library is ~150 nt (Fig. 4).
Figure 4.

BioAnalyzer electropherogram of a representative human NET-seq library. The NET-seq library was generated from 3 μg of RNA obtained from HeLa S3 cells. 1.1 ng of the final NET-seq library were loaded onto a high sensitivity DNA microfluidic chip. The profile shows a prominent peak around 150 nt (violet arrow) and a smaller peak around 125 nt (green arrow). The less prominent peak corresponds to background that arises from unextended RT primer leading to a 119 nt PCR product (see also Fig. 3c,d). This background DNA also results in sequencing data. However, these sequencing reads will contain sequences of the 3′ and 5′ linker regions that are part of the RT primer and are computationally removed. The background DNA should be lower than 10% as compared to the amount of the NET-seq library. The peak at 35 nt represents the manufacturer’s internal standard.
High-throughput sequencing of a typical NET-seq library (step 106 and Box 4) obtained from HeLa S3 cells has the following characteristics: ~50% of the sequencing reads map uniquely to the annotated human reference genome. Of those reads 76% align to Pol II transcribed genes, 15% align to Pol I transcribed genes (also included are reads that map to spacer regions of rRNA genes, indicating nascent Pol I transcripts), 8% align to Pol III transcribed genes and <1% align to mitochondrial genes. At protein-coding genes the highest sequencing coverage is obtained at the promoter-proximal region (between TSS and +1kb): >50% of active genes have a coverage of >1 read per kb per million uniquely aligned reads (RPKM)29. To observe typical NET-seq results please consult Mayer et al. (and especially Figures 2D and S1F therein)29.
TIMING
Steps 1–19, Quantitative purification of RNA polymerase by cell fractionation: 45 min.
Steps 20–24, Preparation of nascent RNA: 1 h.
Box 1, steps 1–4, Check cell fractionation by western blotting: 5 h.
Box 1, step 4, Complete western blotting: 2.5 h.
Steps 25–28, Barcode DNA linker ligation: 3.5 h.
Steps 29–32, RNA fragmentation: 45 min, overnight RNA precipitation.
Steps 33–47, Complete RNA fragmentation ~3.5 h.
Steps 48–64, Reverse transcription: 4 h, overnight cDNA precipitation.
Steps 65–66, Complete Reverse transcription: ~1 h.
Steps 67–70, Circularization of cDNA: 1.5 h.
Steps 71–80, Specific depletion of highly abundant mature RNAs: 1.5 h.
Steps 81–97, PCR amplification of the cDNA sequencing library: ~4 h.
Steps 98–102, Complete NET-seq library preparation: ~1 h.
Steps 103–106, Quantify and characterize the NET-seq library: 1 h.
Supplementary Material
Supplementary Figure 1 | Representative gel to monitor the efficiency of DNA linker ligation. The RNA control oligo oGAB11 (lane 2) was ligated to the DNA linker (no barcode, lane 3) and to the Barcode DNA linker (lane 4). The ligation was performed as described in Procedure steps 25 to 28. The samples were separated on a 15% (wt/vol) polyacrylamide TBE-Urea gel as described in Procedure steps 38 to 43. The ligation efficiency is >95% for both ligations (lanes 3 and 4) as determined by image quantification using ImageJ 1.47v software68.
Acknowledgments
We thank Julia di Iulio and Seth Maleri for help developing this approach. We thank Julia di Iulio, Heather Landry and Andrew Snavely for critical comments on the manuscript. This work was supported by US National Institutes of Health NHGRI grants R01HG007173 to L.S.C.; a Damon Runyon Dale F. Frey Award for Breakthrough Scientists (to L.S.C.); and a Burroughs Wellcome Fund Career Award at the Scientific Interface (to L.S.C). A.M. was supported by Long-Term Postdoctoral Fellowships of the Human Frontier Science Program (LT000314/2013-L) and EMBO (ALTF858-2012).
Footnotes
AUTHOR CONTRIBUTIONS
A.M. performed experiments and prepared figures. A.M. and L.S.C. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Margaritis T, Holstege FCP. Poised RNA Polymerase II Gives Pause for Thought. Cell. 2008;133:581–584. doi: 10.1016/j.cell.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 2.Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16:167–177. doi: 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:599–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 5.Rahl PB, et al. c-Myc Regulates Transcriptional Pause Release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jonkers I, Kwak H, Lis JT. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife. 2014;3:e02407. doi: 10.7554/eLife.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Preker P, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- 8.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seila AC, et al. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigova AA, et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. PNAS. 2013;110:2876–2881. doi: 10.1073/pnas.1221904110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seila AC, Core LJ, Lis JT, Sharp PA. Divergent transcription: a new feature of active promoters. Cell Cycle. 2009;8:2557–2564. doi: 10.4161/cc.8.16.9305. [DOI] [PubMed] [Google Scholar]
- 12.Djebali S, et al. Landscape of transcription in human cells. Nature. 2013;488:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14:880–893. doi: 10.1038/nrg3594. [DOI] [PubMed] [Google Scholar]
- 14.Jensen TH, Jacquier A, Libri D. Dealing with Pervasive Transcription. Mol Cell. 2013;52:473–484. doi: 10.1016/j.molcel.2013.10.032. [DOI] [PubMed] [Google Scholar]
- 15.Buck MJ, Lieb JD. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349–360. doi: 10.1016/j.ygeno.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A Chromatin Landmark and Transcription Initiation at Most Promoters in Human Cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raha D, Hong M, Snyder M. ChIP-Seq: a method for global identification of regulatory elements in the genome. Curr Protoc Mol Biol. 2010 doi: 10.1002/0471142727.mb2119s91. Chapter 21, Unit 21.19.1–14. [DOI] [PubMed] [Google Scholar]
- 18.Descostes N, et al. Tyrosine phosphorylation of RNA Polymerase II CTD is associated with antisense promoter transcription and active enhancers in mammalian cells. eLife. 2014;3:e02105. doi: 10.7554/eLife.02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhee HS, Pugh BF. ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol. 2012 doi: 10.1002/0471142727.mb2124s100. Chapter 21, Unit 21.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He Q, Johnston J, Zeitlinger J. ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat Biotech. 2015;33:395–401. doi: 10.1038/nbt.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwak H, Fuda NJ, Core LJ, Lis JT. Precise Maps of RNA Polymerase Reveal How Promoters Direct Initiation and Pausing. Science. 2013;339:950–953. doi: 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang IX, et al. RNA-DNA Differences Are Generated in Human Cells within Seconds after RNA Exits Polymerase II. Cell Reports. 2014;6:906–915. doi: 10.1016/j.celrep.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2012;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Churchman LS, Weissman JS. Native Elongating Transcript Sequencing (NET-seq) Curr Protoc in Mol Biol. 2012 doi: 10.1002/0471142727.mb0414s98. Chapter 4, Unit 4.14.1–4.14.17. [DOI] [PubMed] [Google Scholar]
- 25.Larson MH, et al. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science. 2014;344:1042–1047. doi: 10.1126/science.1251871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vvedenskaya IO, et al. Transcription. Interactions between RNA polymerase and the ‘core recognition element’ counteract pausing. Science. 2014;344:1285–1289. doi: 10.1126/science.1253458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber CM, Ramachandran S, Henikoff S. Nucleosomes Are Context-Specific, H2A.Z-Modulated Barriers to RNA Polymerase. Mol Cell. 2014;53:819–830. doi: 10.1016/j.molcel.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 28.Ferrari F, et al. ‘“Jump Start and Gain”’ Model for Dosage Compensation in Drosophila Based on Direct Sequencing of Nascent Transcripts. Cell Reports. 2013;5:629–636. doi: 10.1016/j.celrep.2013.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer A, di Iulio J, et al. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell. 2015;161:541–554. doi: 10.1016/j.cell.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez-Rucobo FW, Sainsbury S, Cheung AC, Cramer P. Architecture of the RNA polymerase–Spt4/5 complex and basis of universal transcription processivity. EMBO J. 2011;30:1302–1310. doi: 10.1038/emboj.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kireeva ML, Komissarova N, Waugh DS, Kashlev M. The 8-nucleotide-long RNA:DNA hybrid is a primary stability determinant of the RNA polymerase II elongation complex. J Biol Chem. 2000;275:6530–6536. doi: 10.1074/jbc.275.9.6530. [DOI] [PubMed] [Google Scholar]
- 32.Wuarin J, Schibler U. Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol Cell Biol. 1994;14:7219–7225. doi: 10.1128/mcb.14.11.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai H, Luse DS. Transcription initiation by RNA polymerase II in vitro. J Biol Chem. 1987;262:298–304. [PubMed] [Google Scholar]
- 34.Bhatt DM, et al. Transcript Dynamics of Proinflammatory Genes Revealed by Sequence Analysis of Subcellular RNA Fractions. Cell. 2012;150:279–290. doi: 10.1016/j.cell.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khodor YL, et al. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 2011;25:2502–2512. doi: 10.1101/gad.178962.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pandya-Jones A, Black DL. Co-transcriptional splicing of constitutive and alternative exons. RNA. 2009;15:1896–1908. doi: 10.1261/rna.1714509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flynn RA, Almada AE, Zamudio JR, Sharp PA. Antisense RNA polymerase II divergenttranscripts are P-TEFb dependent andsubstrates for the RNA exosome. PNAS. 2011;108:10460–10465. doi: 10.1073/pnas.1106630108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Santa F, et al. A Large Fraction of Extragenic RNA Pol II Transcription Sites Overlap Enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–363. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brannan K, et al. mRNA Decapping Factors and the Exonuclease Xrn2 Function in Widespread Premature Termination of RNA Polymerase II Transcription. Molecular Cell. 2012;46:311–324. doi: 10.1016/j.molcel.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ntini E, et al. Polyadenylation site–induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol. 2013;20:923–928. doi: 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- 44.Nojima T, et al. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell. 2015;161:526–540. doi: 10.1016/j.cell.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayer A, et al. Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol. 2010;17:1272–1278. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- 46.Kim H, et al. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat Struct Mol Biol. 2010;17:1279–1286. doi: 10.1038/nsmb.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeitlinger J, et al. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet. 2007;39:1512–1516. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muse GW, et al. RNA polymerase is poised for activation across the genome. Nat Genet. 2007;39:1507–1511. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinmetz EJ, et al. Genome-Wide Distribution of Yeast RNA Polymerase II and Its Control by Sen1 Helicase. Mol Cell. 2006;24:735–746. doi: 10.1016/j.molcel.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 50.Bataille AR, et al. A Universal RNA Polymerase II CTD CycleIs Orchestrated by Complex Interplays between Kinase, Phosphatase, and Isomerase Enzymes along Genes. Mol Cell. 2012;45:158–170. doi: 10.1016/j.molcel.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 51.Tietjen JR, et al. Chemical-genomic dissection of the CTD code. Nat Struct Mol Biol. 2010;17:1154–1161. doi: 10.1038/nsmb.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mahony S, Pugh BF. Protein–DNA binding in high-resolution. Crit Rev Biochem Mol Biol. 2015;50:269–283. doi: 10.3109/10409238.2015.1051505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gilmour DS, Fan R. Detecting transcriptionally engaged RNA polymerase in eukaryotic cells with permanganate genomic footprinting. Methods. 2009;48:368–374. doi: 10.1016/j.ymeth.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 54.Core LJ, et al. Defining the Status of RNA Polymerase at Promoters. Cell Reports. 2012;2:1025–1035. doi: 10.1016/j.celrep.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lindell TJ, Weinberg F, Morris PW, Roeder RG, Rutter WJ. Specific inhibition of nuclear RNA polymerase II by alpha-amanitin. Science. 1970;170:447–449. doi: 10.1126/science.170.3956.447. [DOI] [PubMed] [Google Scholar]
- 56.Brueckner F, Cramer P. Structural basis of transcription inhibition by α-amanitin and implications for RNA polymerase II translocation. Nat Struct Mol Biol. 2008;15:811–818. doi: 10.1038/nsmb.1458. [DOI] [PubMed] [Google Scholar]
- 57.Chapman RD, et al. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. 2007;318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 58.Ozsolak F, et al. Comprehensive Polyadenylation Site Maps in Yeast and Human Reveal Pervasive Alternative Polyadenylation. Cell. 2010;143:1018–1029. doi: 10.1016/j.cell.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Djebali S, et al. Landscape of transcription in human cells. Nature. 2013;488:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim JB, Sharp PA. Positive Transcription Elongation Factor b Phosphorylates hSPT5 and RNA Polymerase II Carboxyl-terminal Domain Independently of Cyclin-dependent Kinase-activating Kinase. J Biol Chem. 2001;276:12317–12323. doi: 10.1074/jbc.M010908200. [DOI] [PubMed] [Google Scholar]
- 61.Peterlin BM, Price DH. Controlling the Elongation Phase of Transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 62.Scruggs BS, et al. Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Mol Cell. 2015:1–13. doi: 10.1016/j.molcel.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Danko CG, et al. Signaling Pathways Differentially Affect RNA Polymerase II Initiation, Pausing, and Elongation Rate in Cells. Mol Cell. 2013;50:212–222. doi: 10.1016/j.molcel.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laitem C, et al. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II–transcribed genes. Nat Struct Mol Biol. 2015;22:396–403. doi: 10.1038/nsmb.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson JT, et al. Integrative genomics viewer. Nat Biotech. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 | Representative gel to monitor the efficiency of DNA linker ligation. The RNA control oligo oGAB11 (lane 2) was ligated to the DNA linker (no barcode, lane 3) and to the Barcode DNA linker (lane 4). The ligation was performed as described in Procedure steps 25 to 28. The samples were separated on a 15% (wt/vol) polyacrylamide TBE-Urea gel as described in Procedure steps 38 to 43. The ligation efficiency is >95% for both ligations (lanes 3 and 4) as determined by image quantification using ImageJ 1.47v software68.