

Abstract
Background
Transitions into conscious states are partially mediated by inactivation of sleep networks and activation of arousal networks. Pharmacologic hastening of emergence from general anesthesia has largely focused on activating subcortical monoaminergic networks, with little attention on antagonizing the GABA type A receptor (GABAAR). As the GABAAR mediates clinical effects of many common general anesthetics, we hypothesized that negative GABAAR modulators would hasten emergence, possibly via cortical networks involved in sleep.
Methods
We investigated the capacity of the benzodiazepine rescue agent, flumazenil, recently shown to promote wakefulness in hypersomnia patients, to alter emergence. Using an in vivo rodent model and an in vitro GABAAR heterologous expression system, we measured flumazenil’s effects on behavioral, neurophysiological, and electrophysiological correlates of emergence from isoflurane anesthesia.
Results
Animals administered intravenous flumazenil (0.4 mg/kg, n=8) exhibited hastened emergence compared to saline-treated animals (n=8) at cessation of isoflurane anesthesia. Wake-like electroencephalographic (EEG) patterns occurred sooner and exhibited more high-frequency EEG power following flumazenil administration (median latency ± MAD: 290±34 s) compared to saline administration (473±186 s, p=0.042). Moreover, flumazenil-treated animals exhibited decreased impact on post-anesthesia sleep. In vitro experiments in HEK293T cells demonstrated that flumazenil inhibited isoflurane-mediated GABA current enhancement (n = 34 cells, 88.7±2.42% potentiation at 3 µM). Moreover, flumazenil exhibited weak agonist activity on the GABAAR (n=10 cells, 10.3±3.96% peak GABA EC20 current at 1 μM).
Conclusions
Flumazenil can modulate emergence from isoflurane anesthesia. We highlight the complex role GABAARs play in mediating consciousness and provide mechanistic links between emergence from anesthesia and arousal.
Introduction
Anesthesiologists can pharmacologically reverse many components of the anesthetized state (e.g., neuromuscular blockade). However, restoring wakefulness following general anesthesia has proven more challenging. Recent pre-clinical work has demonstrated a role for hastening arousal via increasing circulating catecholamines1–3 and cyclic AMP levels4 but less attention has been placed on antagonizing the main molecular target of modern anesthetics5. Activity through the GABA type A receptor (GABAAR) is considered a critical part of the cellular mechanisms involved in unconsciousness and a principal central nervous system target of anesthetics6. We reasoned that negative modulation of the GABAAR could expedite recovery from general anesthesia, and focused on flumazenil as a candidate anesthetic reversal agent.
Flumazenil is best known as a competitive antagonist at the benzodiazepine-binding site on the GABAA receptor, and has long-standing clinical use as an emergency treatment for benzodiazepine overdose. Its potential for improving recovery from anesthesia/sedation using benzodiazepines has been examined in clinical studies with mixed results7. One study found that patients that were administered flumazenil upon emergence from isoflurane anesthesia had a faster recovery, improved vigilance, and a subjective rating of recovery as more pleasant8. Conversely, studies have shown that flumazenil can potentiate the sedative qualities of propofol in patients undergoing minor surgery9. Our previous work demonstrated that flumazenil can promote wakefulness in hypersomnic patients10. Here, we explore the possibility that the effect of flumazenil on promoting wakefulness translates to the post-anesthetized state.
Transitions between conscious and unconscious states (i.e. sleep and anesthesia) involve similar brain networks that mediate arousal11,12. Despite the heavy influence of cortical electroencephalography (EEG) on sleep categorization13, the manipulation of subcortical networks for anesthetic emergence has received more research attention. This might be because many of the brain stem nuclei involved in arousal are fairly well-characterized and the pharmacology of these exhibits regional anatomic specificity14,15. For example, pharmacologic or electrical stimulation of the ventral tegmental area (rich in dopaminergic neurons) has successfully reversed the effects of general anesthesia2,3,16. Similarly, excitation of the thalamus through microinfusion of potassium channel antagonists17 or nicotine18 resulted in behaviors indicative of arousal. The cholinergic system has also been manipulated to reverse volatile anesthesia globally via intracerebraventricular injection of neostigmine19.
We hypothesized that flumazenil can reverse the actions of isoflurane anesthesia on the GABAA receptor and expedite emergence. To test these hypotheses, we used a rodent model of anesthesia emergence to test the effect of flumazenil on several physiological correlates of emergence, as well as a heterologous expression system to test the effect of flumazenil on GABAA receptor function. We predicted that animals treated with flumazenil would exhibit hastened behavioral and neurophysiological markers of emergence post isoflurane anesthesia. At the cellular level, we further predicted that flumazenil would antagonize the actions of isoflurane at the GABAA receptor.
Materials & Methods
Experimental design
Figure 1 summarizes our experimental paradigm. Briefly, adult male Sprague-Dawley rats (Charles River) recovered from EEG implantation surgery (see EEG/EMG recording: surgery section below) for 7–10 days before receiving isoflurane anesthesia. Baseline EEG data were collected in activity chambers for 48 hours prior to anesthesia. After 60 minutes of isoflurane anesthesia, rats were given an intravenous injection of either saline or flumazenil, returned to their activity chamber, and passively observed through emergence from anesthesia. During emergence, we used qualitative behavioral measures and quantitative EEG (qEEG) to characterize time to wakeful behaviors and neurophysiologic effects on brain activity. Animals remained connected to EEG and electromyography (EMG) electrodes for 48 hours after anesthesia. During this time, animals continued their normal active/inactive cycle, and sleep periods were analyzed.
Figure 1.
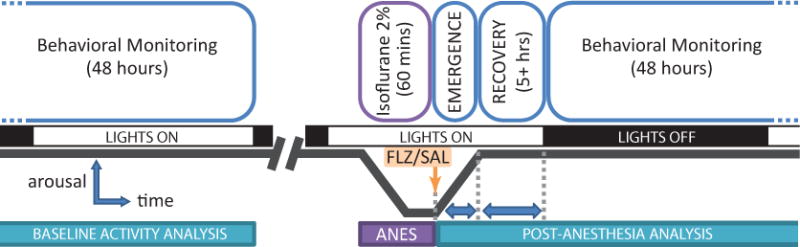
Experimental paradigm. Adult male rats were outfitted with EEG/EMG electrodes and EEG/EMG was recorded for a 48-hour period prior to anesthesia induction for sleep scoring. Animals were then administered an induction dose of isoflurane, followed by a maintenance dose of 2% isoflurane in oxygen for 60 minutes. At the cessation of isoflurane, intravenous flumazenil (0.4 mg/kg) or an equivalent volume of saline was administered, and the animal was placed into the long-term EEG/EMG recording chamber. Emergence characteristics (EEG wake, eye blink, ambulation) were measured. EEG/EMG were recorded for at least 48 hours postoperatively for qEEG analysis and sleep scoring.
A total of 26 animals were used in the study: 14 animals in the saline control group, and 12 animals in the flumazenil group. All protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University and the Atlanta VA Medical Center. Throughout the protocol, animals were housed under a 12:12 hour light:dark cycle with food and water provided ad libitum. Note that the anesthesia challenge and recovery occurred during the inactive portion of the animal’s circadian cycle.
EEG/EMG recording: surgery
EEG electrodes were surgically implanted into male rats weighing 370 – 450 g as described previously20. Animals were anesthetized with chloral hydrate (400 mg/kg intraperitoneal) and placed into a stereotaxic frame. Four holes were drilled in the skull and two pairs of sterile 0–80 × 3/32 screw electrodes (Plastics One, Roanoke VA) inserted. With reference to bregma, the first electrode pair was placed AP −1.5 mm, ML 3.0 mm and AP −6.3 mm, ML 3.5 mm. These lateral coordinates overlie primary forelimb somatosensory cortex as well as primary visual cortex; thus, this allows recording of cortical areas (referred to as the “cortical” lead). The second electrode pair was placed contralaterally at AP +2.5 mm, ML 1.5 mm and AP −3.6 mm, ML 1.5 mm, and overlaid primary and secondary motor cortex. These medial coordinates allow recording of hippocampal areas (referred to as the “hippocampal” lead). Note that this is not a true hippocampal lead, nor can the electrical activity measured from this lead be explicitly linked to the hippocampus. Neck EMG was recorded with intramuscular electrode pairs inserted into the left and right nuchal muscles. EEG and EMG electrodes were attached to a micro-connector (Winchester Electronics, Norwalk, CT) and a dental acrylic compound (Plastics One, Roanoke, VA) was applied to ensure fixation of the assembly to the skull. Once the acrylic skullcap had dried, the free ends of the incision were sutured, leaving the micro-connector pins exposed for recurrent, minimally invasive recording.
EEG/EMG recording: isoflurane emergence protocol and testing
EEG-outfitted animals were placed in an anesthesia induction chamber pre-charged with 4% isoflurane in oxygen and maintained for 2 minutes; during this time, all animals had loss of righting reflex. Animals were then moved to an absorbent covered heating pad and fitted with a nose cone for anesthesia, a rectal temperature probe, and a pulse oximetry clip. Animals were maintained at 2% isoflurane for 60 minutes. During steady state anesthesia delivery, body temperature, respiratory rate, heart rate, SpO2, and expired gases were measured at 5-minute intervals. At 56 minutes of steady state, animals were fitted with a 24 gauge lateral tail vein catheter and from 58–60 minutes, animals were randomized to receive an intravenous injection of either 0.9% sterile saline or 0.4 mg/kg flumazenil (West-Ward Pharmaceuticals, Eatontown, NJ) a dose that roughly corresponds to 5 mg flumazenil in a 70 kg human (the maximum recommended intravenous dosing for treatment of benzodiazepine overdose)21. Animals were then fitted with an EEG recording cable and the isoflurane was stopped. Animals were immediately placed in the EEG recording chamber, tethered to the commutator, visually monitored and timed for eye blink and ambulation as behavioral signs of emergence from anesthesia. Ambulation was defined as coordinated activity involving fore and hind limbs, in a diagonal cross-matched pattern (not ataxic). Observers were blinded to the animal’s treatment group for all behavioral studies and sleep scoring. The positioning of the EEG recording cable precludes measurement of a righting reflex, so first signs of ambulation were recorded as a replacement metric along with the ongoing EEG/EMG monitoring. EEG wake was defined empirically from EEG traces as the transition from high-amplitude, low-frequency oscillations to low-amplitude, high-frequency oscillations. Note that ambulation always occurred after EEG wake.
A subset of animals was used for quantifying sleep characteristics (8 in each group). In these animals, EEG and EMG signals were pre-amplified by Grass model 12A5 amplifiers. Signals were then processed for sleep stage scoring via Somnologica Science (Embla, Buffalo, NY); EEG signals were high-pass filtered at 1 Hz and low-pass filtered at 30 Hz while EMG signals were high-pass filtered at 10 Hz and low-pass filtered at 40 Hz. The amplified and filtered signals were outputted to a National Instruments analog-to-digital converter (PCI-MIO-16E-4), digitally processed, and viewed on a real-time basis. Data were parsed into 10-second epochs for sleep stage scoring using standard criteria; each epoch was scored by one blinded observer and categorized as wake, non-rapid-eye movement sleep (NREM), or rapid-eye movement sleep (REM).
To evaluate changes in sleep behavior, we compared total sleep time, NREM, and REM sleep for 24-hour periods before isoflurane anesthesia (baseline; BL) and after isoflurane anesthesia (post-anesthesia day 1; PAD1). For both periods, analysis began at the beginning of the animal’s active phase (6PM). This time was chosen as it minimized disturbances of the animal and was most convenient for cage maintenance. Note that the PAD1 period began more than 5 hours after anesthetic emergence, at which point it is estimated that 95% of isoflurane has been eliminated22.
EEG spectral analysis
For a subset of animals (12 in each group), re-amplified raw EEG traces from emergence and recovery periods were imported into MATLAB (MathWorks, Natick, MA) for spectral analysis using custom functions. Two saline animals were not included as their EEG headcap had fallen off shortly after anesthesia. In order to preserve frequency content throughout alpha, delta, and theta spectral bands, raw EEG traces were low-pass filtered at 40 Hz and high-pass filtered at 0.25 Hz. Spectrograms were computed for individual animals in MATLAB (spectrogram.m) to show the change of spectral EEG properties during anesthesia emergence. Spectrograms were calculated using 10 s windows with a 1 s shift, and covered the entire emergence period, beginning at cessation of isoflurane anesthesia to three minutes after EEG wake. Note that spectrograms were used for qualitative analyses only and no statistical analyses were performed on these data.
To quantitatively compare differences in EEG between flumazenil and saline groups, we calculated power spectral density (PSD) from 20 s EEG episodes during early emergence and late emergence. We defined early emergence as the 20-second epoch beginning 30 s after cessation of isoflurane and late emergence as the 20-second epoch beginning at EEG wake. In two out of 24 cases, the late emergence episode was distorted by artifacts; in these animals, we used the first artifact-free 20-second episode after EEG wake.
PSDs were calculated in MATLAB using the pwelch function with default settings. The presented results are based on the relative power in the 0.5 to 30 Hz range, normalized by division through the total power in this range.
in vitro electrophysiology
Cell Culture and transfection
Human (wild-type: α1, β2, γ2) GABAARs and GFP cDNAs were subcloned into the pCIS2 vector and transfected into human embryonic kidney (HEK) 293 cells (American Type Culture Collection, Manassas, VA) using X-tremeGENE 9 transfection reagent (Roche Diagnostics, IN, USA). These cells were maintained in culture on poly-D-lysine-treated glass coverslips in a solution containing Eagle minimum essential medium supplemented with 5% fetal bovine serum (Hyclone, Logan, UT), L-glutamine (0.292 μg/mL), penicillin G sodium (100 U/mL), and streptomycin sulfate (100 mg/mL).
Recording
For whole-cell patch clamp recording, coverslips were transferred 36–72 hours after transfection and cDNA removal to a recording chamber and continuously superfused with extracellular solution (145 mM NaCl, 3 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 6 mM D-glucose, and 10mM HEPES–NaOH adjusted to pH 7.4). GABAAR-expressing cells were identified by GFP fluorescence and voltage clamped at −60 mV using either a Multiclamp 700A or Axopatch −1D amplifier (Molecular Devices, Sunnyvale, CA). The resistance of the patch pipette was 4–6 MΩ when filled with intracellular solution (145 mM N-methyl-D-glucamine hydrochloride, 5 mM dipotassium ATP, 1.1 mM EGTA, 2 mM MgCl2, 5 mM HEPES–KOH, and 0.1 mM CaCl2 adjusted to pH 7.2). Responses were low-pass filtered (100 Hz, −3 dB, four-pole Bessel) and digitized with a 1322A interface (Molecular Devices) using pCLAMP and stored for off-line analysis. All experiments were performed at room temperature (21–24°C).
Rapid application of drugs to activate ligand-gated chloride currents was performed as described previously23. Solutions including GABA and/or modulators were perfused onto the cell using a motor-driven solution exchange device (Rapid Solution Changer RSC-160; Molecular Kinetics, Indianapolis, IN). Solutions were exchanged within approximately 50 ms. Laminar flow out of the rapid solution changer head was achieved by driving all solutions at identical flow rates (1.0 mL/min) via multichannel infusion pump (KD Scientific, Holliston, MA). The solution changer was driven by protocols in the acquisition program pCLAMP 9.2 (Molecular Devices).
Pharmacology
To study the effect of modulators on GABAAR currents, cells were superfused with extracellular saline before switching solutions or concentrations for 2 seconds followed by a return to saline for at least 8 seconds before any subsequent drug application. GABA was diluted in extracellular solution shortly before use. Flumazenil was dissolved in dimethyl sulfoxide (DMSO), and when used, equivalent amounts of DMSO were added to the other solutions for control comparisons. Care was taken to remove all air bubbles from the perfusion setup when working with isoflurane. Previous dose-response studies of GABA on evoked current in our laboratory consistently demonstrated that responses evoked by 100 μM GABA or less did not desensitize, and the amplitude of the responses to 100 μM and above typically declined by 10–15% in the continued presence of agonist. For flumazenil dose-response studies, flumazenil or GABA doses were individually infused through dedicated syringes to avoid chemical contamination. To study anesthetic-reversal effects of flumazenil, we measured peak currents evoked by the EC20 of GABA (10 μM), by the EC20 of GABA plus 280 μM isoflurane (approximately equivalent to 1 MAC24–26), or both plus increasing doses of flumazenil. Flumazenil doses for in vivo studies were chosen based on approximate plasma and CSF concentrations10 adjusted for the higher metabolic rate of rats21.
Analysis
To quantify dose-response relationships, peak current amplitudes were extracted from raw data using custom MATLAB functions and organized in Microsoft Excel (Redmond, WA). To fit a nonlinear dose-response curve and derive standard Hill coefficients, current peaks were fitted to a Hill equation of the form
where I is the peak of each current, Imax is the maximum whole cell current amplitude, [GABA] is the GABA concentration, EC50 is the GABA concentration eliciting a current equal to half of Imax, and nH is the Hill coefficient.
To measure flumazenil reversal of isoflurane-induced potentiation of GABAAR function, isoflurane potentiation was first calculated as the percentage increase in the peak response to the application of 280 μM isoflurane and GABA, relative to the 10μM GABA response, or
Reversal by isoflurane was calculated as the difference between this potentiation and the potentiation observed in the presence of an ascending dose of flumazenil (0.1, 1.0, 3.0, 10, or 30 μM), or
Statistical analysis
To compare latencies to emergence, we used the Mann-Whitney-U test (unpaired data using MATLAB function ranksum); to compare differences in baseline and post-anesthesia sleep behavior, we used the Wilcoxon signed-rank test (paired data using MATLAB function signrank). Center estimations are presented as median ± median absolute deviation (MAD) for these non-parametric tests. For emergence times and sleep times, we presented measures of effect size together with 95% confidence intervals derived through 10000-fold bootstrapping27 in addition to the p-values, allowing us to quantify the magnitude of an effect. The effect size of choice was the area under the receiver-operating characteristic curve (AUC) for the independent emergence times and Hedges’ g for the dependent sleep behavior. The Hedges’ g is a modification of Cohen’s d that makes it better applicable to small sample sizes. We consider values of |g| > 0.8 a strong effect and |g| < 0.5 a medium effect28. It must be noted that these boundaries follow no strict rules, but may help to give an impression regarding the strength of an effect. We only present the g values if the corresponding 95% CIs do not contain 0. Consistent with the statistical comparison of EEG spectra used by other groups29 we compared PSDs between saline and flumazenil groups; we calculated the 95% confidence interval using the jackknife method and evaluated significant differences using the MATLAB two_group_test function with a significance level set to p<0.001 using methods from the Chronux toolbox30,31. For in vitro electrophysiological studies, statistical significance for dose-response relationships was assessed using one-way ANOVA with Dunnett’s post-hoc comparisons (α = 0.05). Data are presented as mean ± SEM.
Results
Intravenous flumazenil hastens EEG and behavioral correlates of emergence from isoflurane anesthesia
Representative EEG/EMG traces during EEG emergence and recovery are shown for a saline-treated animal (Figure 2A and 2B) and a flumazenil-treated animal (Figure 2C and 2D). Arrows indicate EEG wake (left), eye blink (middle) and ambulation (right). Note that nuchal EMG activity precedes ambulation as the animal is making subtle movements while tonic muscle activity is returning. The corresponding bar plots of the emergence times are presented in Figure 3. Time to EEG wake was significantly shorter for the flumazenil group (median latency ± MAD 290±34 s) than for the saline group (473±186 s; p=0.042; AUC: 0.74 [0.55 0.93]). A similar trend was noted with other markers of emergence. Flumazenil-treated animals had shorter latencies to eye blink (293±25 s) compared to the saline-treated animals (482±193 s; p=0.054; AUC 0.73 [0.53 0.92]), and shorter latencies to ambulation (flumazenil 348±109 s, saline 646±169 s; p=0.117; AUC of 0.68 [0.45 0.89]) that did not achieve statistical significance. The corresponding bar plots of the emergence times are presented in Figure 3. There were no differences in the latency from EEG wake to ambulation across groups (p=0.643; AUC: 0.44 [0.21 0.67]). Although changes in ventilatory parameters are a major contributor to isoflurane elimination and consequently latency to end emergence, we recorded respiratory parameters in pilot studies and did not observe any statistically significant differences among the groups (Table 1).
Figure 2.
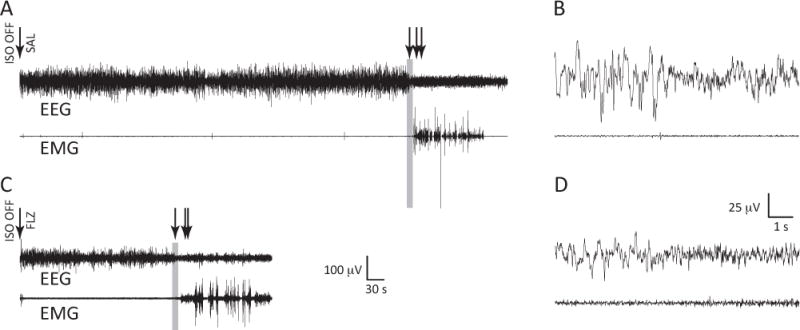
EEG/EMG recordings of the transition to arousal from anesthesia. A, Representative waveforms from an animal given saline (SAL) via tail vein catheter at the cessation of isoflurane anesthesia. EEG from the medial (hippocampal) lead is shown. Arrows designate (from left to right): isoflurane cessation and administration of saline, EEG wake (defined as a transition to lower amplitude, faster oscillatory activity), eye blink, and ambulation. B, Time-expanded 10-second epoch capturing the transition to a wakeful EEG (before the onset of movement). The gray rectangle in A corresponds to this expansion in B. C, Representative waveforms from an animal given flumazenil (FLZ) via tail vein catheter at the cessation of isoflurane anesthesia. Arrows designate (from left to right): isoflurane cessation and administration of flumazenil, EEG wake, eye blink, and ambulation. D, Time-expanded 10-second epoch capturing the transition to a wakeful EEG (before the onset of movement). The gray rectangle in C corresponds to this expansion in D.
Figure 3.
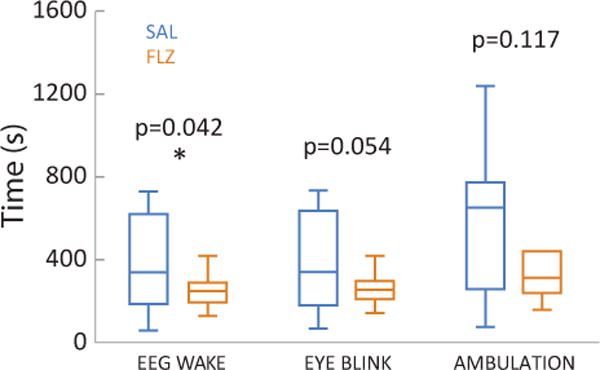
Flumazenil hastens EEG markers of emergence from isoflurane. Boxplots reflecting median, quartiles, and range are shown for EEG wake, eye blink, and ambulation in animals treated with saline (SAL – blue; n = 14) versus flumazenil (FLZ – orange; n = 12) at the cessation of anesthesia. * p=0.042; AUC: 0.74 [0.55 0.93] using Mann-Whitney-U test and AUC.
Table 1.
Flumazenil does not change respiratory parameters in a rodent model of emergence. Average respiratory rate for animals is represented as mean ± SD (Saline n = 6, flumazenil n = 5).
| AGENT | −10′ | −5′ | 0′ | 1′ | 2′ | 3′ | 4′ | 5′ |
|---|---|---|---|---|---|---|---|---|
| SALINE | 51.2 ± 5.0 | 49.7 ± 4.5 | 48.7 ± 4.7 | 63.2 ± 8.7 | 66.8 ± 6.9 | 72.8 ± 5.2 | 73.6 ± 8.3 | 78.0 ± 8.5 |
| FLUMAZENIL | 55.0 ± 3.6 | 52.6 ± 4.2 | 52.6 ± 5.1 | 60.0 ± 6.2 | 62.8 ± 7.2 | 69.0 ± 10.5 | 72.0 ± 10.6 | 80.0 ± 11.3 |
Quantitative EEG reveals neurophysiologic effects of flumazenil administration
Similar to raw EEG traces, all animals regardless of group showed a transition of EEG frequency content. Specifically, we observed a shift in EEG from predominance in delta power (0.5 – 4 Hz) during early emergence toward a more uniform distribution of power among higher frequencies during late emergence. Representative spectrograms for an animal in each group are shown in Figure 4. In general, both cortical (Figure 4A) and hippocampal (Figure 4B) leads exhibited this shift in frequency profiles from emergence to recovery. Qualitatively these two examples also demonstrate more beta activity (~14 – 30 Hz) pre- and post-EEG wake and a more modest theta power on the spectrogram from the hippocampal lead (Figure 4B) in the flumazenil treated example.
Figure 4.
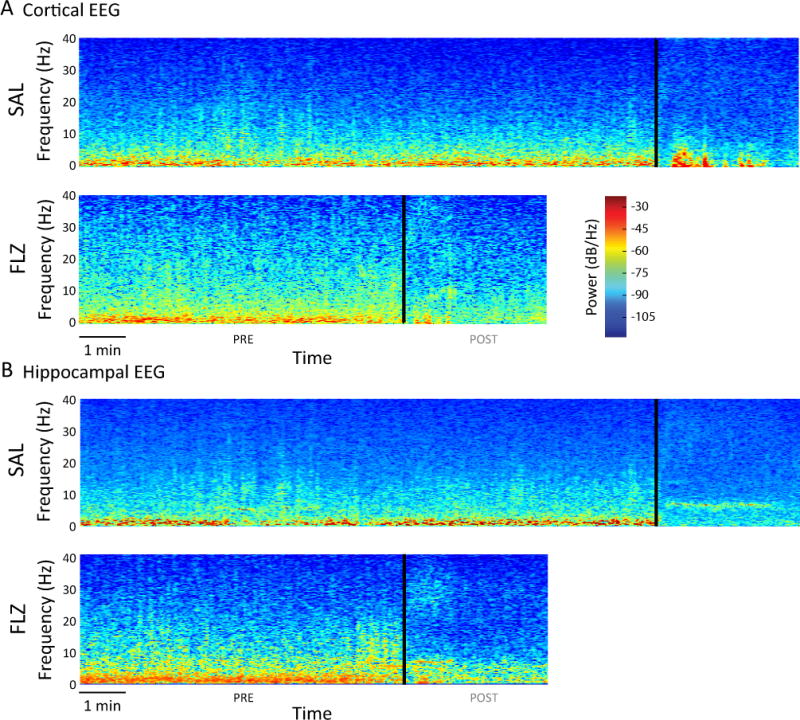
Spectrograms during emergence from anesthesia. Spectrograms were computed from cessation of isoflurane anesthesia to three minutes after EEG wake (black vertical line). Warmer colors (i.e. red) indicate higher power at a given frequency, while cooler colors (i.e. blue) indicate lower power at a given frequency. Data are shown for a representative animal treated with saline or flumazenil. A, Cortical lead. B, Hippocampal lead. Before EEG wake, there is a predominance of power in low (<4 Hz) frequencies with less power in higher frequencies. After EEG wake, power decreases in low frequencies and begins to appear in higher frequencies.
Figure 5 shows averaged PSDs in both leads for all animals by treatment group. During early emergence, spectral content is dominated by delta waves in both groups. This frequency distribution becomes more uniform during late emergence, with an increase in beta power. The spectral properties of the corresponding EEG differed between flumazenil and saline groups. During late emergence, flumazenil-treated animals had significantly increased power over a broad band of frequencies within the beta range in the cortical lead (Figure 5B, top). We did not observe such a phenomenon in the hippocampal lead (Figure 5B, bottom).
Figure 5.
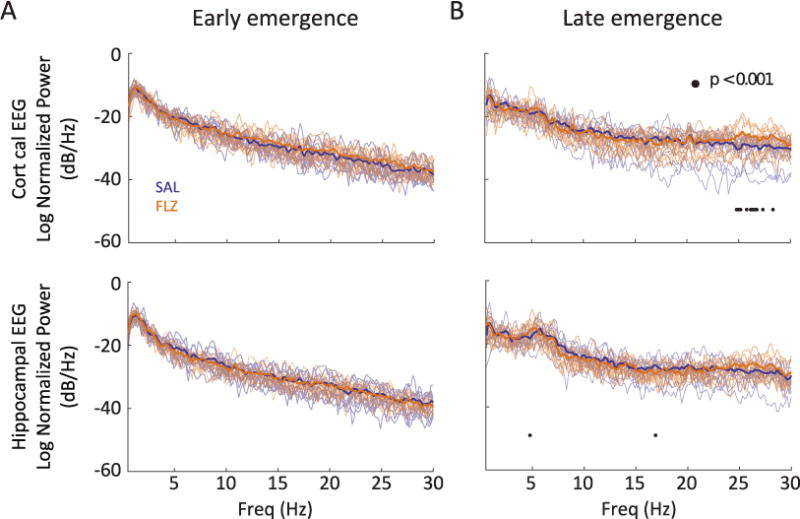
Effect of flumazenil on power spectra during emergence and recovery. Solid lines represent the average normalized power spectral density (PSD) estimates for animals following cessation of anesthesia for early emergence (A) and late emergence (B). Transparent lines designate the raw PSD traces for individual animals in each group. Blue: saline-treated animals; n = 12; Orange: flumazenil-treated animals; n = 12. Significance at p < 0.001 is designated by • as described in Methods section.
Flumazenil administration mitigates increase in sleep times post-anesthesia challenge
Although there was no significant difference in baseline total sleep time between the saline and flumazenil groups (p=0.383), total sleep time for the saline group was significantly increased from 11.1 ± 0.5 h (median ± MAD) at baseline (BL) to 12.7±0.2 h on post-anesthesia day 1 (PAD1) (p=0.008). Hedges’ g was −1.98 [−3.42 −0.54] and indicated a strong effect on sleeping behavior caused by isoflurane. There were no significant differences in sleep time after anesthesia compared to baseline in flumazenil-treated animals (p=0.195). The total sleep time changed from 11.8±1 h to 12.5±0.3 h. Hedges’ g was −0.55 [−1.57 0.46].
Saline-treated animals exhibited significantly more time spent in NREM sleep compared to flumazenil-treated animals. Figure 6A shows the change in total sleep time due to isoflurane anesthesia and the contribution of NREM (6B) and REM (6C) sleep to the total sleep time. For saline-treated animals, isoflurane had a strong effect on the time spent in NREM sleep (BL 9.4±0.3 h; PAD1 10.8±0.3 h; p=0.008; g= −1.69 [−4.06 −1.12]) as well as the time spent in REM sleep (BL 1.5±0.3 h; PAD1 2.0±0.1 h; p=0.008; g=−0.96 [−2.36 −0.66]). With flumazenil these effects disappeared for NREM (BL 10.0±0.7 h; PAD1 10.4±0.3 h; p=0.461; g=−0.39 [−1.53 0.50]). The time spent in REM sleep changed from 1.8±0.3 h to 2.0±0.3 h; p=0.008; g=−0.77 [−1.77 0.06].
Figure 6.
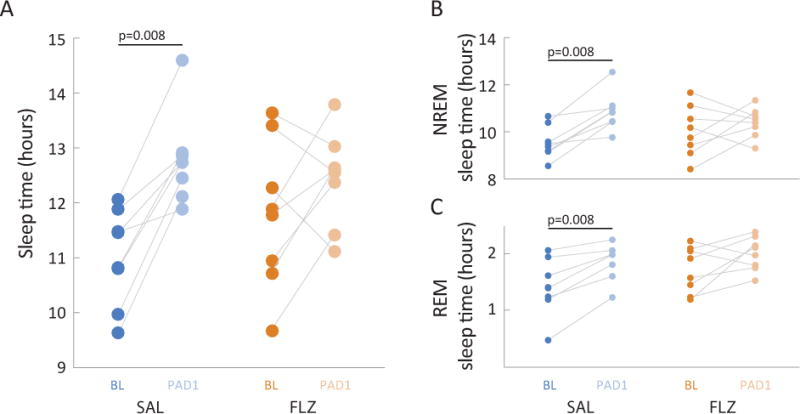
A single dose of flumazenil results in near baseline sleep characteristics following isoflurane anesthesia and mitigates an increase in REM sleep. For both saline (SAL – blue) and flumazenil (FLZ – orange) treated animals, sleep characteristics were evaluated for a 24-hour period before (baseline – BL) and after (post-anesthesia day 1 – PAD1) isoflurane anesthesia. A, Total sleep times. Saline-treated animals were asleep for significantly longer following anesthesia compared to baseline. There were no significant differences in baseline sleep times between the two groups (p=0.383, Mann-Whitney U test). Connected pairs of dots represent corresponding sleep times for individual animals. * p=0.008, Wilcoxon signed rank; strong effect. B, C, Amount of time in NREM (B) and REM (C) sleep. Connected pairs of dots represent time spent in NREM sleep for saline-treated (blue) and flumazenil-treated (orange) animals. Saline-treated animals spent significantly more time in both NREM and REM sleep following anesthesia compared to baseline. BL – darker shaded dots; PAD1 – lighter shaded dots. n = 8 animals per group; * Wilcoxon signed rank; strong effect for both REM (p = 0.008) and NREM (p=0.008) sleep.
Flumazenil is a competitive antagonist of GABAARs in absence and presence of isoflurane
The dose-response relationship of applied GABA concentration with and without co-applied flumazenil was tested using α1β2γ2s GABAA receptors heterologously expressed in HEK-293 cells. Electrophysiological recordings of GABAAR-mediated chloride current amplitudes were measured using whole-cell patch clamp recordings. Figure 7A shows representative traces from a single cell of a chloride current evoked by increasing concentrations of GABA in the absence (black trace) and presence of flumazenil (orange trace). These experiments were done pairwise: with each cell, flumazenil lowered the maximal peak GABA current without shifting the dose-response relation, consistent with a competitive rather than non-competitive antagonist. The summative dose-response curve (Figure 7B) demonstrates that this effect is most consistently seen at higher GABA concentrations (GABA > 30 μM).
Figure 7.
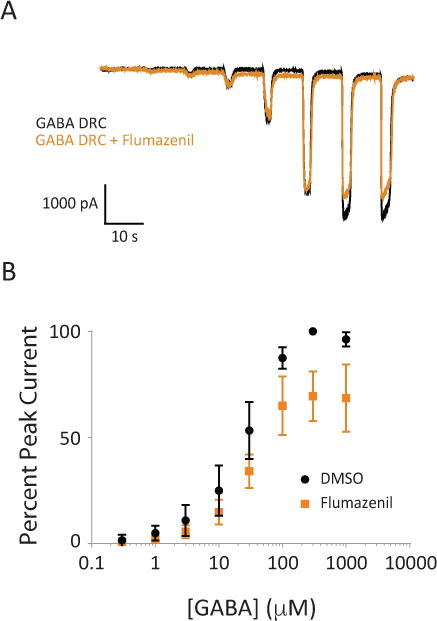
Flumazenil can act as a competitive antagonist at high GABA concentrations. A, Representative traces of GABA-evoked chloride currents in the presence and absence of 4 μM flumazenil. Black traces are GABA alone, orange traces are co-application of GABA and flumazenil. B, Summative dose-response curves for application of GABA and GABA plus flumazenil to α1β2γ2s receptors (n = 8 cells). Error bars represent SEM.
Flumazenil also partially reversed the isoflurane enhancement of GABAAR currents. Figure 8A shows representative traces from a single cell of a chloride current evoked by EC20 GABA (black bars), and isoflurane (purple bars). Addition of isoflurane enhanced chloride currents through GABAARs. However, in the presence of increasing concentrations of flumazenil (orange bars), peak chloride currents decreased slightly (Figure 8A inset). Figure 8B represents population data as a flumazenil inhibitory dose-response curve. In the absence of flumazenil, percent potentiation of GABA currents by 280 μM isoflurane was 82.0 ± 7.5 (SEM), on par with the expected magnitude of isoflurane enhancement. The magnitude of reversal was dose-dependent, and statistically significant at higher dosages of flumazenil (one-way repeated measures ANOVA with Dunnett’s test, 3μM p = 0.007; 10μM p = 0.013; 30 μM p = 0.009).
Figure 8.
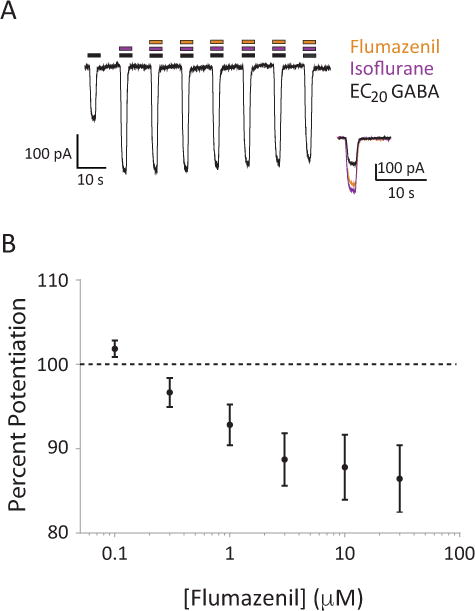
Flumazenil can inhibit the enhancement of GABA currents by isoflurane. A, Representative traces of GABA receptor activation in multiple treatment conditions. Black bars represent co-application of isoflurane; orange bars represent co-application of increasing flumazenil concentrations (from left to right: 0.1, 0.3, 1.0, 3.0, 10, 30 μM). Inset is an overlay of EC20 GABA response (black), EC20 GABA response potentiated by isoflurane (280 μM), and the flumazenil antagonized EC20 GABA response potentiated by isoflurane. B, Summative dose-response curve for the effect of increasing doses of flumazenil on isoflurane enhanced EC20 response (n = 34 cells). Error bars represent SEM.
Flumazenil has intrinsic agonist activity on the GABAA receptor
Flumazenil modulated GABAA receptor function in the absence of GABA or any other agonist or receptor modulator. Figure 9A shows that flumazenil elicited chloride currents in a sigmoidal, dose-dependent manner. In comparison to an EC20 GABA response elicited from the same cell, the maximal flumazenil evoked-current is relatively small suggesting weak intrinsic activity. Figure 9B represents an agonist dose-response relationship for flumazenil’s effects on measured chloride current, scaled to the within-cell EC20 GABA current value. These effects suggest that flumazenil is a weak agonist of the GABAAR when GABA itself is low or nonexistent, and competes with GABA at high doses. This competitive antagonist property also allows flumazenil to incompletely reverse the enhancement of the GABAAR by isoflurane even at moderate (EC20) doses of GABA.
Figure 9.

Flumazenil exhibits weak agonist effects on the GABA receptor in the absence of GABA. A, Representative traces of chloride currents through the GABA receptor evoked by application of increasing flumazenil concentrations (orange bars, from left to right: 0.1, 0.3, 1.0, 3.0, 10, 30 μM) followed by the response of the GABA receptor to a 10 μM application of GABA (EC20) for comparison. B, Summative dose-response curve for flumazenil’s effect on the GABA receptor in the absence of GABA (n = 10 cells). Error bars represent SEM.
Discussion
The present work focused on a potential role of flumazenil in altering emergence from anesthesia. Previous work with hypersomnic patients has demonstrated that modulation of GABA signaling via flumazenil improves vigilance and wakefulness in these patients10. Our work supports the notion that antagonism of GABAergic signaling can contribute to the re-establishment of waking behavior. While this arousal from anesthesia could in part be due to modulation of common subcortical arousal networks involved in sleep32, our work cannot exclude modulation of cortical GABAergic synapses33. We expected that negative modulation of the GABAA receptor could mitigate the hypoactive effects of isoflurane and subsequently expedite emergence. In this study we found that flumazenil had a modest effect on hastening emergence in rats, and this is associated with an increase in high frequency neurophysiologic activity in the cortex. Flumazenil administration prevented prolongation of total sleep time in the first post-anesthesia day and did not significantly change sleep architecture. Taken together, administration of flumazenil during emergence may prevent an adverse effect of isoflurane on normal sleep/wake cycles. The results from pharmacologic experiments of flumazenil on the GABAA receptor suggest that although antagonism of the GABAA receptor is possible with flumazenil, partial agonism can also be exhibited, consistent with the pharmacological profile of a competitive antagonist with weak intrinsic agonist activity. These in vitro findings might help explain some of the variability in emergence characteristics previously shown with flumazenil administration in humans8,34,35.
Flumazenil hastens behavioral and neurophysiological markers of emergence from anesthesia through its interaction with sleep and arousal networks
Our neurophysiologic findings support the notion that flumazenil’s effect on post-anesthesia behavior is related to its effects on sleep and arousal networks. Metrics of emergence such as time to ambulation and EEG wake occurred significantly earlier in flumazenil treated animals. In both saline- and flumazenil-treated animals, EEG wake was readily detected by a transition in the EEG record from predominantly large, slow delta waves to a more uniform distribution of EEG power. This increased EEG power in higher frequency bands (> 14 Hz) is indicative of arousal and occurred significantly earlier in animals treated with flumazenil. Human EEG data gathered during recovery from general anesthesia characteristically exhibits an increase in high-frequency power36–38. Our results are also consistent with those of Dahaba et al., who determined that flumazenil changed the frontal EEG of patients undergoing intravenous anesthesia towards a pattern more indicative of awakening39. In humans, increased power in higher frequency bands is also more indicative of the lighter stages of sleep (such as N1 and REM)40 more common in the early morning in preparation for waking.
Although hypoactivity is expected after general anesthesia, flumazenil administration appeared to mitigate this effect for isoflurane. As compared to their baseline, saline-treated animals spent significantly more time asleep in the 24 hours after anesthesia (PAD1). Conversely, the percentage of time spent asleep for flumazenil-treated animals was more similar to their baseline. The observed differences in post anesthesia sleep may be explained by the differences observed during emergence from isoflurane anesthesia. For example, flumazenil-treated animals exhibited overall higher beta power during the peri-emergence period, whereas theta power was higher in the saline group. This shift in EEG power towards higher frequency bands with flumazenil administration has also been observed in humans41. The higher beta frequencies seen with flumazenil at late emergence may represent a more active cortex. Theta EEG frequencies have been associated with mnemonic processing42 as well with acute stress43. Thus, the increased theta activity seen with saline-treated animals during late emergence may be caused by a more stressful emergence from anesthesia that consequently led to higher need of sleep and a higher proportion of REM sleep in the 24 h post anesthesia. Especially since REM sleep seems to play a major role in dealing with traumatic and stressful experiences44. Our results suggest that increased theta power may be an inconsistent marker of emergence; more evidence will be needed to fully understand the role of increased theta power post-anesthesia.
Evidence in the sleep literature demonstrates that the predominance of lower frequency bands during awakening is more associated with abnormal arousals during slow-wave sleep45. Furthermore, parasomnias such as confusional arousal, night terrors and sleepwalking are more common during these stages46.
The effects of flumazenil on promoting an awake state were not limited to the emergence period, as flumazenil-treated animals did not exhibit significant increases in sleep in the 24 hours after anesthesia as compared to their baseline. In contrast, saline-treated animals exhibited a significant increase in sleep (total sleep time, NREM and REM) in the day following cessation of anesthesia. Whereas propofol anesthesia appears to satisfy some sleep need47, accrual of sleep debt (specifically REM) is known to occur with the administration of isoflurane in rodents48,49. Although sleep deprivation was not a part of our study, the animals that received a single administration of flumazenil prior to emergence did not demonstrate the change in sleep patterns as the saline-treated animals. These results may highlight the importance of the arousal process on subsequent sleep architecture. Similarly, we observed a lasting effect (> 5 hours) on vigilance in hypersomnic patients administered a single dose of flumazenil10. Of course, the possibility remains that an a priori difference in general waking behaviors (from sleep or anesthesia) could exist among our drug/vehicle treated groups. Thus, further experiments characterizing these differences would be warranted.
A dual mechanism of flumazenil explains variability in clinical emergence from anesthesia
Taken in the context of our previous work with flumazenil in patients with hypersomnia10 and clinical case reports of flumazenil in reversing emergence delirium50, our results suggest that flumazenil has a more complex relationship with GABAARs than pure antagonism. Our results are consistent with clinical studies that suggest flumazenil can reverse anesthesia even in the absence of benzodiazepines35. However, Schwieger and Szlam found that flumazenil had no effect on the anesthetic requirements of dogs undergoing inhaled anesthesia51 and other studies show that flumazenil can potentiate the sedative qualities of propofol in patients undergoing minor surgery9. Because of its weak agonist activity, flumazenil has been suggested as having utility in treating anxiety from alcohol withdrawal52. In contrast to pure GABA antagonists with epileptogenic properties, flumazenil actually has anticonvulsant properties in the absence of chronic benzodiazepine use53. These observations fit a mixed or partial agonist action for this drug at the GABAA receptor54. Therefore, it is possible that higher-dose flumazenil administration regimes could augment the unconsciousness produced by general anesthesia via enhancement of GABA signaling. It would be interesting to determine whether similar effects on sleep architecture were observed after a prolongation of anesthetic effects via flumazenil.
Our results suggest that inhibition of GABAA receptors with flumazenil is effective at hastening wakeful states and perhaps can mitigate post-anesthesia sleep disturbances in a rodent model of general anesthesia with inhaled isoflurane. Understanding the role of GABA in transitions to conscious states may help us prevent significant neurological consequences after administration of general anesthesia, such as post-operative delirium. Similarly, by identifying overlapping neurophysiologic features in the transitions to consciousness we can better determine how to capitalize on the beneficial effects of sleep in the peri-operative period.
Acknowledgments
We acknowledge technical help from Joey Carlson, BS and Lee Bannister, MD from the laboratory of Professor Jenkins Department of Anesthesiology, Emory University School of Medicine, Atlanta, Georgia, USA. We thank the following members of Dr. García’s Neuroanesthesia Laboratory, Department of Anesthesiology, Atlanta VA/Emory University School of Medicine, Atlanta, Georgia, USA, for critical discussions of preliminary results: Vincent Ciavatta, PhD; Edyta Bichler, PhD; Qiangguo Xu, MD; Sarah Burke, MD; September Hesse, PhD. We thank the following members of Dr. Rye’s Sleep Research Group, Department of Neurology, Emory University School of Medicine, Atlanta, Georgia, USA for discussions of sleep pharmacology: Prabhit Jyoti, BS and Lynn Marie Trotti, MD. This work was supported from Departmental Resources, the James S. McDonnell Foundation (220020346 to PSG), and a VA Career Development Award (BX001677 to PSG). The authors declare no competing interests. Dr. Rye wishes to disclose the following relationship: US Patent Application pending (20110028418) “The use of GABAA receptor antagonists for the treatment of excessive sleepiness and sleep disorders associated with excessive sleepiness”; Jazz Pharmaceuticals; UCB Pharma; Xenoport Inc.
References
- 1.Chemali JJ, Van Dort CJ, Brown EN, Solt K. Active emergence from propofol general anesthesia is induced by methylphenidate. Anesthesiology. 2012;116:998–1005. doi: 10.1097/ALN.0b013e3182518bfc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solt K, Cotten JF, Cimenser A, Wong KF, Chemali JJ, Brown EN. Methylphenidate actively induces emergence from general anesthesia. Anesthesiology. 2011;115:791–803. doi: 10.1097/ALN.0b013e31822e92e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor NE, Chemali JJ, Brown EN, Solt K. Activation of D1 dopamine receptors induces emergence from isoflurane general anesthesia. Anesthesiology. 2013;118:30–9. doi: 10.1097/ALN.0b013e318278c896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q, Fong R, Mason P, Fox AP, Xie Z. Caffeine accelerates recovery from general anesthesia. J Neurophysiol. 2014;111:1331–40. doi: 10.1152/jn.00792.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia PS, Kolesky SE, Jenkins A. General anesthetic actions on GABA(A) receptors. Curr Neuropharmacol. 2010;8:2–9. doi: 10.2174/157015910790909502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–86. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- 7.Ricou B, Forster A, Bruckner A, Chastonay P, Gemperle M. Clinical evaluation of a specific benzodiazepine antagonist (RO 15–1788). Studies in elderly patients after regional anaesthesia under benzodiazepine sedation. Br J Anaesth. 1986;58:1005–11. doi: 10.1093/bja/58.9.1005. [DOI] [PubMed] [Google Scholar]
- 8.Weinbroum AA, Geller E. Flumazenil improves cognitive and neuromotor emergence and attenuates shivering after halothane-, enflurane- and isoflurane-based anesthesia. Can J Anaesth. 2001;48:963–72. doi: 10.1007/BF03016585. [DOI] [PubMed] [Google Scholar]
- 9.Adachi YU, Watanabe K, Higuchi H, Satoh T. Flumazenil reduces the hypnotic dose of propofol in male patients under spinal anesthesia. J Anesth. 2002;16:9–12. doi: 10.1007/s540-002-8087-2. [DOI] [PubMed] [Google Scholar]
- 10.Rye DB, Bliwise DL, Parker K, Trotti LM, Saini P, Fairley J, Freeman A, Garcia PS, Owens MJ, Ritchie JC, Jenkins A. Modulation of vigilance in the primary hypersomnias by endogenous enhancement of GABAA receptors. Sci Transl Med. 2012;4:161ra151. doi: 10.1126/scitranslmed.3004685. [DOI] [PubMed] [Google Scholar]
- 11.Brown EN, Lydic R, Schiff ND. General anesthesia, sleep, and coma. N Engl J Med. 2010;363:2638–50. doi: 10.1056/NEJMra0808281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown EN, Purdon PL, Van Dort CJ. General anesthesia and altered states of arousal: a systems neuroscience analysis. Annu Rev Neurosci. 2011;34:601–28. doi: 10.1146/annurev-neuro-060909-153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silber MH, Ancoli-Israel S, Bonnet MH, Chokroverty S, Grigg-Damberger MM, Hirshkowitz M, Kapen S, Keenan SA, Kryger MH, Penzel T, Pressman MR, Iber C. The visual scoring of sleep in adults. J Clin Sleep Med. 2007;3:121–31. [PubMed] [Google Scholar]
- 14.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 15.Silber MH, Rye DB. Solving the mysteries of narcolepsy: the hypocretin story. Neurology. 2001;56:1616–8. doi: 10.1212/wnl.56.12.1616. [DOI] [PubMed] [Google Scholar]
- 16.Solt K, Van Dort CJ, Chemali JJ, Taylor NE, Kenny JD, Brown EN. Electrical stimulation of the ventral tegmental area induces reanimation from general anesthesia. Anesthesiology. 2014;121:311–9. doi: 10.1097/ALN.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alkire MT, Asher CD, Franciscus AM, Hahn EL. Thalamic microinfusion of antibody to a voltage-gated potassium channel restores consciousness during anesthesia. Anesthesiology. 2009;110:766–73. doi: 10.1097/aln.0b013e31819c461c. [DOI] [PubMed] [Google Scholar]
- 18.Alkire MT, McReynolds JR, Hahn EL, Trivedi AN. Thalamic microinjection of nicotine reverses sevoflurane-induced loss of righting reflex in the rat. Anesthesiology. 2007;107:264–72. doi: 10.1097/01.anes.0000270741.33766.24. [DOI] [PubMed] [Google Scholar]
- 19.Hudetz AG, Wood JD, Kampine JP. Cholinergic reversal of isoflurane anesthesia in rats as measured by cross-approximate entropy of the electroencephalogram. Anesthesiology. 2003;99:1125–31. doi: 10.1097/00000542-200311000-00019. [DOI] [PubMed] [Google Scholar]
- 20.Keating GL, Kuhar MJ, Bliwise DL, Rye DB. Wake promoting effects of cocaine and amphetamine-regulated transcript (CART) Neuropeptides. 2010;44:241–6. doi: 10.1016/j.npep.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. The FASEB Journal. 2007;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 22.Saab BJ, Maclean AJ, Kanisek M, Zurek AA, Martin LJ, Roder JC, Orser BA. Short-term memory impairment after isoflurane in mice is prevented by the alpha5 gamma-aminobutyric acid type A receptor inverse agonist L-655,708. Anesthesiology. 2010;113:1061–71. doi: 10.1097/ALN.0b013e3181f56228. [DOI] [PubMed] [Google Scholar]
- 23.Richardson JE, Garcia PS, O’Toole KK, Derry JM, Bell SV, Jenkins A. A conserved tyrosine in the beta2 subunit M4 segment is a determinant of gamma-aminobutyric acid type A receptor sensitivity to propofol. Anesthesiology. 2007;107:412–8. doi: 10.1097/01.anes.0000278875.36639.2c. [DOI] [PubMed] [Google Scholar]
- 24.Franks NP, Lieb WR. Selective actions of volatile general anaesthetics at molecular and cellular levels. Br J Anaesth. 1993;71:65–76. doi: 10.1093/bja/71.1.65. [DOI] [PubMed] [Google Scholar]
- 25.Franks NP, Lieb WR. Temperature dependence of the potency of volatile general anesthetics: implications for in vitro experiments. Anesthesiology. 1996;84:716–20. doi: 10.1097/00000542-199603000-00027. [DOI] [PubMed] [Google Scholar]
- 26.Jenkins A, Franks NP, Lieb WR. Effects of temperature and volatile anesthetics on GABA(A) receptors. Anesthesiology. 1999;90:484–91. doi: 10.1097/00000542-199902000-00024. [DOI] [PubMed] [Google Scholar]
- 27.Hentschke H, Stuttgen MC. Computation of measures of effect size for neuroscience data sets. Eur J Neurosci. 2011;34:1887–94. doi: 10.1111/j.1460-9568.2011.07902.x. [DOI] [PubMed] [Google Scholar]
- 28.Cohen J. Statistical power analysis for the behavioral sciences. Vol. 2. Lawrence Earlbaum Associates; Hillsdale, NJ: 1988. [Google Scholar]
- 29.Akeju O, Westover MB, Pavone KJ, Sampson AL, Hartnack KE, Brown EN, Purdon PL. Effects of sevoflurane and propofol on frontal electroencephalogram power and coherence. Anesthesiology. 2014;121:990–8. doi: 10.1097/ALN.0000000000000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bokil H, Purpura K, Schoffelen JM, Thomson D, Mitra P. Comparing spectra and coherences for groups of unequal size. J Neurosci Methods. 2007;159:337–45. doi: 10.1016/j.jneumeth.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 31.Bokil H, Andrews P, Kulkarni JE, Mehta S, Mitra PP. Chronux: a platform for analyzing neural signals. Journal of neuroscience methods. 2010;192:146–151. doi: 10.1016/j.jneumeth.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedman EB, Sun Y, Moore JT, Hung HT, Meng QC, Perera P, Joiner WJ, Thomas SA, Eckenhoff RG, Sehgal A, Kelz MB. A conserved behavioral state barrier impedes transitions between anesthetic-induced unconsciousness and wakefulness: evidence for neural inertia. PLoS One. 2010;5:e11903. doi: 10.1371/journal.pone.0011903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scharf MT, Kelz MB. Sleep and Anesthesia Interactions: A Pharmacological Appraisal. Curr Anesthesiol Rep. 2013;3:1–9. doi: 10.1007/s40140-012-0007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen AG, Moller JT, Lybecker H, Hansen PA. A random trial comparing recovery after midazolam-alfentanil anesthesia with and without reversal with flumazenil, and standardized neurolept anesthesia for major gynecologic surgery. J Clin Anesth. 1995;7:63–70. doi: 10.1016/0952-8180(94)00005-o. [DOI] [PubMed] [Google Scholar]
- 35.Karakosta A, Andreotti B, Chapsa C, Pouliou A, Anastasiou E. Flumazenil expedites recovery from sevoflurane/remifentanil anaesthesia when administered to healthy unpremedicated patients. Eur J Anaesthesiol. 2010;27:955–9. doi: 10.1097/EJA.0b013e3283398ef9. [DOI] [PubMed] [Google Scholar]
- 36.Chander D, Garcia PS, MacColl JN, Illing S, Sleigh JW. Electroencephalographic variation during end maintenance and emergence from surgical anesthesia. PLoS One. 2014;9:e106291. doi: 10.1371/journal.pone.0106291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ni Mhuircheartaigh R, Warnaby C, Rogers R, Jbabdi S, Tracey I. Slow-wave activity saturation and thalamocortical isolation during propofol anesthesia in humans. Sci Transl Med. 2013;5:208ra148. doi: 10.1126/scitranslmed.3006007. [DOI] [PubMed] [Google Scholar]
- 38.Purdon PL, Pierce ET, Mukamel EA, Prerau MJ, Walsh JL, Wong KF, Salazar-Gomez AF, Harrell PG, Sampson AL, Cimenser A, Ching S, Kopell NJ, Tavares-Stoeckel C, Habeeb K, Merhar R, Brown EN. Electroencephalogram signatures of loss and recovery of consciousness from propofol. Proc Natl Acad Sci U S A. 2013;110:E1142–51. doi: 10.1073/pnas.1221180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dahaba AA, Bornemann H, Rehak PH, Wang G, Wu XM, Metzler H. Effect of flumazenil on bispectral index monitoring in unpremedicated patients. Anesthesiology. 2009;110:1036–40. doi: 10.1097/ALN.0b013e31819db2c4. [DOI] [PubMed] [Google Scholar]
- 40.Rechtschaffen A, Kales A. A manual of standardized terminology, techniques and scoring system for sleep stages of human subjects. 1968 doi: 10.1046/j.1440-1819.2001.00810.x. [DOI] [PubMed] [Google Scholar]
- 41.Pal D, Hambrecht-Wiedbusch VS, Silverstein BH, Mashour GA. Electroencephalographic coherence and cortical acetylcholine during ketamine-induced unconsciousness. Br J Anaesth. 2015;114:979–89. doi: 10.1093/bja/aev095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauseng P, Griesmayr B, Freunberger R, Klimesch W. Control mechanisms in working memory: a possible function of EEG theta oscillations. Neuroscience & Biobehavioral Reviews. 2010;34:1015–1022. doi: 10.1016/j.neubiorev.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Shors TJ, Gallegos RA, Breindl A. Transient and persistent consequences of acute stress on long-term potentiation (LTP), synaptic efficacy, theta rhythms and bursts in area CA1 of the hippocampus. Synapse. 1997;26:209–17. doi: 10.1002/(SICI)1098-2396(199707)26:3<209::AID-SYN2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 44.Mellman TA, Bustamante V, Fins AI, Pigeon WR, Nolan B. REM sleep and the early development of posttraumatic stress disorder. American Journal of Psychiatry. 2014 doi: 10.1176/appi.ajp.159.10.1696. [DOI] [PubMed] [Google Scholar]
- 45.Espa F, Ondze B, Deglise P, Billiard M, Besset A. Sleep architecture, slow wave activity, and sleep spindles in adult patients with sleepwalking and sleep terrors. Clin Neurophysiol. 2000;111:929–39. doi: 10.1016/s1388-2457(00)00249-2. [DOI] [PubMed] [Google Scholar]
- 46.Howell MJ. Parasomnias: an updated review. Neurotherapeutics. 2012;9:753–75. doi: 10.1007/s13311-012-0143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tung A, Bergmann BM, Herrera S, Cao D, Mendelson WB. Recovery from sleep deprivation occurs during propofol anesthesia. Anesthesiology. 2004;100:1419–26. doi: 10.1097/00000542-200406000-00014. [DOI] [PubMed] [Google Scholar]
- 48.Mashour GA, Lipinski WJ, Matlen LB, Walker AJ, Turner AM, Schoen W, Lee U, Poe GR. Isoflurane anesthesia does not satisfy the homeostatic need for rapid eye movement sleep. Anesth Analg. 2010;110:1283–9. doi: 10.1213/ANE.0b013e3181d3e861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pick J, Chen Y, Moore JT, Sun Y, Wyner AJ, Friedman EB, Kelz MB. Rapid eye movement sleep debt accrues in mice exposed to volatile anesthetics. Anesthesiology. 2011;115:702–12. doi: 10.1097/ALN.0b013e31822ddd72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drobish JK, Kelz MB, DiPuppo PM, Cook-Sather SD. Emergence delirium with transient associative agnosia and expressive aphasia reversed by flumazenil in a pediatric patient. A A Case Rep. 2015;4:148–50. doi: 10.1213/XAA.0000000000000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwieger IM, Szlam F, Hug CC., Jr Absence of agonistic or antagonistic effect of flumazenil (Ro 15–1788) in dogs anesthetized with enflurane, isoflurane, or fentanyl-enflurane. Anesthesiology. 1989;70:477–80. doi: 10.1097/00000542-198903000-00018. [DOI] [PubMed] [Google Scholar]
- 52.Knapp DJ, Overstreet DH, Moy SS, Breese GR. SB242084, flumazenil, and CRA1000 block ethanol withdrawal-induced anxiety in rats. Alcohol. 2004;32:101–11. doi: 10.1016/j.alcohol.2003.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scollo-Lavizzari G. The clinical anti-convulsant effects of flumazenil, a benzodiazepine antagonist. Eur J Anaesthesiol Suppl. 1988;2:129–38. [PubMed] [Google Scholar]
- 54.Rusch D, Forman SA. Classic benzodiazepines modulate the open-close equilibrium in alpha1beta2gamma2L gamma-aminobutyric acid type A receptors. Anesthesiology. 2005;102:783–92. doi: 10.1097/00000542-200504000-00014. [DOI] [PubMed] [Google Scholar]