

Abstract
Aim
We showed previously that ELF3 protein levels are increased in osteoarthritic (OA) cartilage, that ELF3 accounts for inflammatory cytokine-driven MMP13 gene expression, and that, upon induction by IL-1β, ELF3 binds to the COL2A1 promoter and suppresses its activity in chondrocytes. Here, we aimed to further investigate the mechanism/s by which ELF3 represses COL2A1 transcription in chondrocytes.
Methods and Results
We report that ELF3 inhibits Sox9-driven COL2A1 promoter activity by interfering with the activator functions of CBP/300 and Sox9. Co-transfection of the pGL2B-COL2A1 (−577/+3428 bp) reporter construct with Sox9 and with Sox5 and/or Sox6 increased COL2A1 promoter activity, and ELF3 overexpression significantly reduced the promoter transactivation. Co-transfection of ELF3 with the pLuc 4×48 enhancer construct, containing the 89-bp COL2A1 promoter and lacking the previously defined ELF3 binding sites, decreased both basal and Sox9-driven promoter activity. Co-transfection of ELF3 with a Gal4 reporter construct also inhibited Gal4-Sox9-driven transactivation, suggesting that ELF3 directly interacts with Sox9. Using truncated Sox9 fragments, we found that ELF3 interacts directly with the HMG domain of Sox9. Importantly, over-expression of ELF3 significantly decreased Sox9/CBP-dependent HAT activity. Finally, we show evidence that increased ELF3 mRNA expression in OA chondrocytes correlates with hypermethylation of the proximal promoter, suggesting that ELF3 transcription is subjected to epigenetic control in OA disease.
Conclusion
Our results highlight the contribution of ELF3 to transcriptional regulation of COL2A1 and its potential role in OA disease, and uncover epigenetic mechanisms at play in the regulation of ELF3 and its downstream targets in articular chondrocytes.
Keywords: methylation, transcription, osteoarthritis, collagen, chondrocytes
INTRODUCTION
The E74-like factor 3 (ELF3) is an epithelial-specific member of the ELF subset of the ETS family of transcription factors (1, 2). ELF3 is expressed in non-epithelial tissues in response to inflammatory cytokines, including interleukin (IL)-1β, due to transactivation of the ELF3 promoter via a high affinity NF-κB binding site (3). Following activation by MAPKs, ELF3 directly activates transcription of NOS2 (4), COX2 (5), and MMP13 (6). In previous studies, we reported that ELF3 protein levels are increased in articular cartilage isolated from patients with osteoarthritis (OA) (6), and that ELF3 acts as a transactivator of the MMP13 transcription (6) and as a potent suppressor of the COL2A1 transcriptional activity (7). These results suggested that the transcriptional dysregulation of ELF3 in OA disease might lead to not only increased cartilage catabolism, but also abnormal anabolism.
During skeletal development, COL2A1 expression is regulated in a coordinated fashion by growth and differentiation factors, which modulate a series of transcriptional events requiring elements within both the promoter and first intron regions (8–10). The high mobility group (HMG) protein Sox9 plays an essential role during sequential steps of chondrocyte differentiation and the Sox9-binding intronic enhancer in the COL2A1 gene is required for chondrogenesis in transgenic mice in vivo (10–13). In mouse chimeras generated with Sox9 knockout embryonic stem cells, the mesenchymal progenitors lacking Sox9 are excluded from cartilage tissue and are unable to promote transcription of the COL2A1 gene (13). L-Sox5 and Sox6 are co-expressed with Sox9 in differentiated chondrocytes and cooperate with Sox9 in the binding to the first intron enhancer to permit COL2A1 transcription both in vitro and in vivo (12, 14–16). Several studies suggest that the proximal COL2A1 promoter may operate at specific times during development when negative regulation is required via promoter regions distinct from those required for positive regulation (10, 17).
Sox9 also regulates the transcription of genes encoding type IX collagen (Col9a1) (18), type XI collagen (Col11a1) (19, 20), and aggrecan (Agc) (21). The cooperative effects of L-Sox5 and Sox 6, however, are not observed among all cartilage-specific genes. For example, L-Sox5 and Sox6 are not required for Sox9-dependent lineage commitment and prechondrocyte differentiation in embryos (22), and the Col9a1 enhancer can be activated by Sox9 dimers in the absence of L-Sox5 and Sox6 (23). In adult human articular cartilage, the collagen turnover and associated COL2A1 mRNA levels are very low and Sox9 does not co-localize with COL2A1 mRNA (24); however decreased levels of Sox9 mRNA are detected near the cartilage lesions in OA disease (25). Importantly, Sox9 utilizes a cAMP-response element-binding protein (CREB)-binding protein (CBP)/p300 to control cartilage-specific gene expression (26), and Furumatsu and colleagues (27) showed that the Sox9-induced COL2A1 expression requires interaction between Sox9 and CBP/p300, and that intrinsic CBP/p300 histone acetyltransferase (HAT) activity is fundamental for the COL2A1 promoter activation.
In the present study, we found that the dysregulated ELF3 expression in OA cartilage correlates with increased methylation of the proximal promoter. We also demonstrated that ELF3 overexpression inhibits the COL2A1 transactivation driven by Sox9/Sox6 overexpression. We showed direct protein-protein interactions between ELF3 and Sox9 in GST pull-down assays, which also revealed that ELF3 interacts with the HMG domain of Sox9. Furthermore, ELF3 overexpression inhibited Sox9/CBP-driven HAT activity. Together, these results indicate that ELF3 transcription may be controlled by epigenetic events in OA disease, and suggest that ELF3 may serve to sequester Sox9 to prevent maintenance of an open chromatin network and CBP to block its interaction with the basal transcriptional machinery.
MATERIALS AND METHODS
Cell culture
Human immortalized C-28/I2 chondrocytes were cultured in Dulbecco’s Modified Eagle’s medium (DMEM)/Ham’s F12 containing 10% fetal bovine serum (FBS), as described previously (6, 7).
Human primary chondrocyte isolation
Human articular cartilage was obtained after hemiarthroplasty following femoral neck fracture or total hip arthroplasty for OA with patient consent and prior approval of the local Institutional Review Board. Cartilage was dissected within 6 hours of surgery, and non-OA fractured neck-of-femur and OA human primary chondrocytes were isolated as previously described (28). Briefly, samples were obtained from the deep zone of cartilage from patients with femoral neck fracture for isolation of non-OA/healthy chondrocytes, whereas cartilage pieces adjacent to weight-bearing areas of OA femoral heads (lacking surface zones) were harvested to obtain OA chondrocytes. Cartilage samples were then cut into small fragments and digested with 10% trypsin (Lonza) in phosphate-buffered saline (PBS) for 30 minutes, followed by sequential digestions in 1 mg/ml of hyaluronidase (Sigma-Aldrich) in PBS for 15 minutes, and in 10 mg/ml of collagenase B (Roche) in α-modified Eagle’s medium (α-MEM; Sigma-Aldrich) for 12 to 15 hours at 37°C. Following isolation, genomic DNA and total RNA were extracted simultaneously from the harvested chondrocytes for bisulfite sequencing and RT-qPCR analyses, respectively, as described previously (29).
Bisulfite modification and determination of the percentage of methylation
Genomic DNA was modified with MethylDetector (Active Motif), according to the manufacturer’s instructions. Fifteen CpG sites located between −811 and −123 bp of the ELF3 proximal promoter sequence were investigated with 6 pairs of nested PCR primers (Table 1), as described (29). The percentage of methylation was calculated for each CpG site by RTqPCR, also as described (29).
Table 1.
| Primers for nested PCR | ||
|---|---|---|
| Primer sequence (5’>3’) | Range (bp) | |
| Forward | TGG TTA GAG TTT TGG GTG TTT TG | −481/−87 |
| Reverse | CCC CAA ATA ATT TAC ATA TTC AAT ACC AC | |
| Forward | GAA TTT TTG GAA TTT AGG GGT TAG TA | −405/−87 |
| Reverse | CCC CAA ATA ATT TAC ATA TTC AAT ACC AC | |
| Forward | TTT TGG TTA GAG TTT TGG GTG TTT | −484/−17 |
| Reverse | CCC TAC AAT AAC CTA AAC TAA TAT CTC ATT | |
| Forward | TTT TGT TTG GTT TAT TTT TGG TTT G | −329/−82 |
| Reverse | CCT AAC CCC AAA TAA TTT ACA TAT TCA | |
| Forward | TTT TGA TTA TTA GAG GGT AGT AGA AGG T | −854/−383 |
| Reverse | TAA CCC CTA AAT TCC AAA AAT TCA AC | |
| Forward | TAG AGG GTA GTA GAA GGT ATT GGA AAA | −844/−458 |
| Reverse | CCA AAA CAC CCA AAA CTC TAA CC | |
| Sequencing primers | ||
| Forward | GTA AAA CGA CGG CCA G | |
| Reverse | CAG GAA ACA GCT ATG AC | |
Quantitative reverse transcription-polymerase chain reaction (RT-qPCR)
Amplifications were carried out using SYBR Green I-based RT-PCR on the Opticon 2 Real Time PCR Detector System (BioRad), as described (6, 28), using the following sets of primers: ELF3 (NM_004433): 5’-CAACTATGGGGCCAAAAGAA-3’ (forward), 5’-TTCCGACTCTGGAGAACCTC-3’ (reverse); GAPDH (NM_002046): 5’-CAAAGTTGTCATGGATGACC-3’ (forward), 5’-CCATGGAGAAGGCTGGGG-3’ (reverse); HPRT1 (NM_000194): 5’-AAAGGACCCCACGAAGTGTT-3’ (forward), 5’-TCAAGGGCATATCCTACAACAA-3’ (reverse). The data were calculated as the ratio of each gene to GAPDH, and HPRT1 was utilized as an additional housekeeping gene control.
Luciferase reporter constructs and expression plasmids
Luciferase reporter plasmids and expression vectors were prepared for transfection using the EndoFree plasmid maxi kit (Qiagen). The COL2A1 sequence spanning –577 to +3428 bp was cloned into the pGL2-basic (pGL2-B) luciferase reporter gene vector (Promega,) to generate the pGL2B-COL2–4.0, as described (7). The expression vectors encoding ELF3, CBP, and p300 were described previously (5–7, 28, 30, 31). The expression vectors encoding L-Sox 5, Sox6, and Sox9 were obtained from Drs. Veronique Lefebvre and Benoit deCrombrugghe (11, 12). Expression plasmids encoding Sox9 fragments comprising amino acids 1–507, 1–327, 1–423, and 182–507, the Gal TK-luc reporter construct, and expression vectors encoding wild-type Gal4-Sox9 were obtained from Dr. Hiroshi Asahara and described elsewhere (26). The sequences of all constructs were confirmed by DNA sequencing performed at the Beth Israel Deaconess Medical Center DNA sequencing facility or at the Weill Cornell Medicine Core Laboratories Center.
Transfections and reporter assays
Transient transfection experiments were carried out in C-28/I2 cells using LipofectAMINE PLUS™ Reagents (Invitrogen). At 24 h before transfection, the cells were plated at 2.5 × 104 cells/cm2 in DMEM/F12 containing 10% FBS. Transfections were carried out in serum-free and antibiotic-free medium using no more than a total of 450 ng of plasmid DNA, including 350 ng of reporter constructs. Cell lysates were prepared by extraction with Reporter Lysis Buffer (Promega) and the protein content was determined using the Coomassie Plus Protein Assay Reagent. Luciferase activities were normalized to the protein content, and the activity of the pRL-SV40 Renilla luciferase control vector was used to assess transfection efficiencies (Promega).
Preparation of fusion proteins and GST pull-down assay
Vectors containing full-length cDNAs encoding whole open reading frames were used to obtain 35S-methionine-labeled recombinant proteins employing the TNT T7 Coupled Reticulocyte Lysate System (Promega), according to the manufacturer’s instructions. Empty vectors were also subjected to in vitro translation to provide control lysates. Briefly, each reaction mixture was composed of 40 µl of TNT T7 Quick Master Mix, 2 µl of 35S-methionine (1,000 Ci/mmol) at 10 µCi/ml, and 1 µg of the DNA template and nuclease-free water to a final volume of 50 µl. The reaction mixture was incubated at 30°C for 90 min. SDS-PAGE analyses were performed to confirm that all proteins were well translated and preparations were stored at −80 °C. The glutathione S-transferase (GST) control (pGEX-4T-2) and fusion (pGEX-4T-2-ELF3) vectors were prepared as described (31). Glutathione-Sepharose beads (Pharmacia) were incubated with 1000 to 1500 µg of GST fusion protein and washed to prepare GST-bound beads. The GST fusion protein beads were incubated with 35S-labeled, in vitro translated protein at 4 °C for 1 h. The beads were washed three times in NETN buffer, and the bound proteins were eluted by boiling the beads for 5 min in SDS-PAGE loading buffer (50 mM Tris, pH 6.8, 30% glycerol, 0.4% SDS, 0.1% bromophenol blue) containing β-mercaptoethanol. Proteins were separated by SDS-PAGE on a 10% polyacrylamide gel, which was then soaked in Amplify fluorographic reagent (Amersham Life Science) and exposed to Kodak X-Omat film.
HAT activity assay
To examine the regulation of the Sox9/CBP-driven HAT activity by ELF3, the C-28/I2 cells were transfected with plasmids expressing ELF3, CBP, and Sox9 alone or in combination, incubated for 24 h, and collected by centrifugation at 600 × g for 5 min at 4 °C. Nuclear extracts were prepared using the nuclear/cytosol fractionation kit (BioVision), according to the manufacturer’s protocol. The HAT activity assay was performed using a kit from BioVision (K332-100) in a 96-well plate with 50 µg of nuclear extract in 40 µl water (final volume) per well. After addition of 65 µl of assay mix (50 µl of 2× HAT assay buffer, 5 µl HAT substrate I, 5 µl HAT substrate II, 5 µl NADH generating enzyme), incubations were carried at 37 °C for 1 h and read in a plate reader at 440 nm.
Statistical analysis
Data are reported as mean ± S.E.M. (error bars) of at least three independent experiments. Statistical analysis was performed by ANOVA followed by Student’s t-test with p values of <0.05 considered significant.
RESULTS
ELF3 inhibits the Sox9-driven promoter transactivation
To determine whether overexpression of SOX proteins could enhance the activity of the pGL2B-COL2-4.0 construct, which contains the intronic enhancer, we performed cotransfections with combinations of the three SOX family members. As shown in Figure 1A, cotransfection with sub-optimal concentrations of L-Sox5, Sox6, or Sox9 individually had no significant effect on the activity of the pGL2B-COL2-4.0 construct. In contrast, overexpression of sub-optimal concentrations of Sox9 together with L-Sox5 or Sox6, or all three proteins together, increased the reporter activity by 3- to 4-fold. In agreement with our previous results showing that ELF3 inhibits COL2A1 transcription (7), overexpression of ELF3 prevented the increase in Sox9/Sox6-driven pGL2B-COL2–4.0 activity (Figure 1B). In order to determine whether the inhibitory effect of ELF3 was dependent upon its binding to the previously reported ETS binding sites (7), we transfected the C-28/I2 cells with the pLuc 4×48 enhancer construct containing the 89-bp COL2A1 promoter attached to four 48-bp enhancer elements, but lacking other promoter regions that bind to Elf3. In cotransfection experiments, we found that the pLuc 4×48 basal activity was almost completely abolished by ELF3 overexpression (Figure 2A). Importantly, while cotransfection of Sox9 and Sox6 dramatically increased the pLuc 4×48 transactivation, overexpression of ELF3 significantly reduced the Sox9/Sox6-driven pLuc 4×48 activity (Figure 2B). These results indicate that forced expression of ELF3 is sufficient to repress the constitutive Sox9 actions, and suggest that ELF3 inhibitory actions on Sox9 do not require binding of ELF3 to distal promoter sites. To further assess the interactions of ELF3 that inhibit Sox9-driven promoter transactivation, we performed additional transfection assays using a Gal TK-luc reporter construct, cotransfected with expression vectors encoding Gal4-Sox9 and ELF3. As shown in Figure 2C, we found that ELF3 overexpression also inhibited the Gal4-Sox9-driven transactivation, suggesting that ELF3 directly interacts with Sox9.
Figure 1. ELF3 overexpression inhibits the Sox9,6-driven COL2A1 promoter transactivation.

Luciferase reporter assays performed in C-28/I2 cells were cotransfected with a pGL2B(−577/+3428)COL2A1 reporter construct (pGL2B-4.0-Luc) together with (A) expression vectors encoding sub-optimal concentrations of L-Sox5, Sox6, or Sox9, alone or in combination, or (B) expression vectors encoding Sox9 and Sox6, alone or in combination with expression vectors encoding ELF3. *indicates p<0.001 vs. Sox5, Sox6, Sox9 and Sox5,6; #indicates p<0.05 vs. Sox5,9; ***indicates p<0.001.
Figure 2. The inhibitory effect of ELF3 on Sox9-driven transcription does not depend upon binding of ELF3 to ETS binding sites.
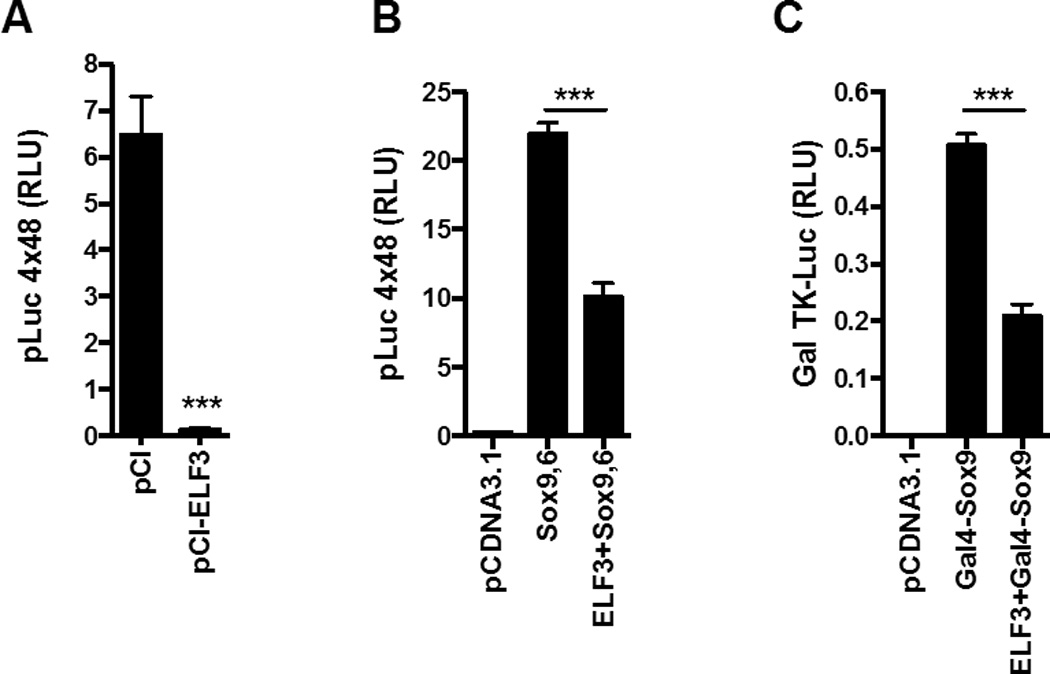
The C-28/I2 cells were co-transfected with the pLuc 4×48 enhancer construct containing the 89-bp COL2A1 promoter with four 48-bp enhancer elements and with (A) expression vectors encoding ELF3 or (B) expression vectors encoding Sox9 and Sox6 alone or in combination with ELF3. (C) The C-28/I2 cells were co-transfected with the Gal TK-luc reporter construct and expression vectors encoding Gal4-Sox9 alone or together with ELF3. ***indicates p<0.001.
ELF3 binds to the HMG domain of Sox9 and inhibits the Sox9/CBP-mediated HAT activity in vitro
To determine potential protein-protein interactions between ELF3 and Sox9, we performed GST pull-down assays using bacterially expressed GST-ELF3 and in vitro translated wild type and truncated Sox9 fragments, which had been used previously to study interactions with CBP/p300 (26). Western blotting confirmed that all proteins were well translated (not shown). As shown in Figure 3A, GST-ELF3 interacted with the wild-type Sox9 protein (1–507) and with the truncated Sox9 fragments containing amino acids 1–423 and 1–327, all of which contain the HMG box (amino acids 102–182). On the other hand, the Sox9 truncations without the HMG box (182–507 and 328–507) did not bind to GST-ELF3 (Figure 3A), compared to the strong binding to either CBP or p300 (not shown). These results demonstrate that the ELF3 protein interacts directly with the HMG box domain of Sox9, in contrast to p300, which interacts with the P/Q/S-rich region, as reported by Tsuda et al. (26). The Sox9-driven cartilage-specific gene expression relies on direct protein-protein interactions between Sox9 and CBP/p300 and the intrinsic CBP/p300-HAT activity (26, 27). Thus, considering the inhibitory role of ELF3 in Sox9-driven transcription and the direct protein-protein interaction between ELF3 and Sox9 shown here, it is conceivable that the inhibitory effect of ELF3 on Sox9 involves disruption of the Sox9 and CBP/p300-mediated HAT activity. To test this hypothesis, we transfected C-28/I2 cells with plasmids expressing ELF3, CBP, and Sox9 alone or in combination, and we measured HAT activity at 24 h after transfection. Cotransfection of Sox9 and CBP increased HAT activity, whereas ELF3 overexpression, which had little effect alone, decreased the HAT activity induced by Sox9 and CBP overexpression (Figure 3B). Interestingly, overexpression of ELF3 showed a non statistically significant trend towards reduced CBP-driven HAT activity (Figure 3C), which suggests that ELF3 could potentially repress CBP activity via direct protein-protein interactions (31) independent of Sox9. Together, these results suggest that ELF3 inhibits Sox9-driven transcription by decreasing the Sox9/CBP-dependent HAT activity via protein-protein interactions with the HMG domain of Sox9.
Figure 3. ELF3 interacts with the HMG domain of Sox9 and inhibits the Sox9/CBP-dependent HAT activity in vitro.

(A) GST pull-down assays were performed using GST-ELF3 and in vitro translated fragments of Sox9, previously reported (26). The schematic illustration below shows the fragments of Sox9 expressed by the deletion mutants, and the positive (+) or negative (−) interactions of Sox9 fragments with ELF3 are shown on the right. The C-28/I2 cells were transfected with plasmids expressing (B) ELF3, CBP, and Sox9, alone or in combination, or (C) CBP and ELF3, alone or in combination. HAT activity was measured in nuclear extracts at 24 h after transfection. ***indicates p<0.001.
The increased ELF3 mRNA expression in human OA chondrocytes correlates with increased CpG methylation of the proximal ELF3 promoter
Recent evidence has shown that DNA methylation, in combination with other means of epigenomic control, is a central contributing factor to OA disease progression (reviewed in (32)). Hence, a complete analysis of epigenomic alterations is essential for fully understanding the consequences of altered signaling networks in OA cartilage. We have shown that ELF3 interacts with chromatin-modifying factors to regulate transcription in chondrocytes (Figures 1–3), that the activity of ELF3 in chondrocytes depends upon its activation by MAPKs (6), and that ELF3 transcription is induced by inflammatory cytokines in chondrocytes and dysregulated in OA disease (6), suggesting its involvement in the cartilage degradative processes. To begin to better understand the contribution of dysregulated ELF3 expression to cartilage degradation in OA, we analyzed ELF3 mRNA expression levels and the methylation status of the ELF3 proximal promoter in OA and non-OA chondrocytes. Bisulfite sequencing showed increased methylation of CpG sites between −811 and −123 bp of the ELF3 proximal promoter in OA chondrocytes (Figure 4A). RT-qPCR analysis revealed increased ELF3 mRNA levels in OA chondrocytes compared to non-OA chondrocytes (Figure 4B). These results suggest that hypermethylation of the proximal promoter permits enhanced ELF3 transcription in articular chondrocytes in vivo. However, we found that in vitro methylation treatment of −622/+163bp and −322/+163bp CpG-free ELF3 luciferase reporter constructs, generated as described (28, 33), leads to a significant suppression of the ELF3 promoter activities (not shown). The latter suggests that the contribution of DNA methylation to the ELF3 promoter transcriptional control may depend upon the methylation status of specific CpG site/s, and that the same events that may block ELF3 transcriptional repressors may also affect the actions of activators of the ELF3 promoter. These results also highlight the differential contribution of 5-hydroxymethylcytosine (5hmC) as opposed to 5-methylcytosine (5mC), which cannot be distinguished using the methodology that we applied.
Figure 4. The increased ELF3 mRNA levels in OA chondrocytes correlate with hypermethylation of the proximal promoter.
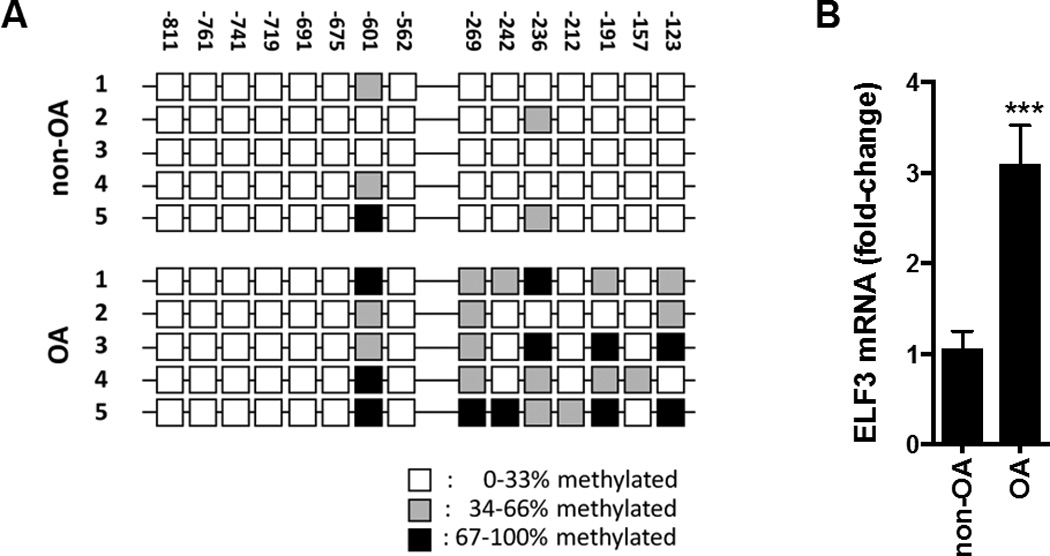
(A) Schematic representation of the proximal human ELF3 promoter sequence (−811bp to −123bp), indicating CpG site positions and percentage of methylation in non-OA and OA human primary chondrocytes, assessed by bisulfite sequencing and (B) ELF3 mRNA levels, assessed by RTqPCR in total RNA isolated from the same cell populations. ***indicates p<0.001.
Taken together, our results show that ELF3 interacts with chromatin-modifying factors to regulate transcription in chondrocytes and that ELF3 transcription is subject to epigenetic control and is altered in OA disease. These results indicate that ELF3 belongs to a complex network of transcriptional mechanisms that in concert drive transcription in chondrocytes, and underline that a complete analysis of epigenomic alterations is essential for fully understanding the consequences of altered signaling networks in joint disease.
DISCUSSION
In the current work, we show that the dysregulation of ELF3 transcription correlates with epigenetic changes. We also show that ELF3 directly interacts with the HMG domain of Sox9 and represses COL2A1 promoter activity by inhibiting Sox9- and CBP/p300-mediated HAT activity, in addition to its direct binding to ETS binding sites in the distal region of the COL2A1 promoter, as we showed previously (7). ELF3 is not normally expressed by non-epithelial cells, but is induced by the inflammatory cytokines IL-1β and TNFα via induction of NF-κB, which binds to and activates the ELF3 promoter (3). Our previous work showed that the altered ELF3 levels in OA cartilage correlated with increased MMP13 protein levels and activity (6), and in vitro studies showed that ELF3 accounts for IL-1β- and TNFα-induced MMP13 expression (6) and mediates IL-1β-driven COL2A1 repression (7) in chondrocytes, by binding to the respective promoters.
In differentiated chondrocytes at a post-developmental stage of low matrix turnover, the COL2A1 promoter in vivo is not active and the negative response to cytokines cannot be observed unless its transcription is activated. The nuclear transcription factor, Sox9, is one of earliest developmental markers expressed in chondroprogenitors undergoing condensation and it is required for the subsequent stages of chondrogenesis characterized by the deposition of matrix containing collagens II, IX and XI and aggrecan in the cartilage anlagen (9). L-Sox5 and Sox6, which are not present in early mesenchymal condensations, but are co-expressed later with Sox9 and required for chondrocyte differentiation, have a high degree of sequence identity with each other, but have no sequence homology with Sox9 except in the HMG box (22). They can form homo- or heterodimers, which bind more efficiently to pairs of HMG box sites than to single sites, but unlike Sox9, they contain no transcriptional activation domain. In cytokine-activated genes, the assembly of higher order nucleoprotein complexes orchestrated by high mobility group (HMG)-I(Y) factors may be important for integrating the responses to the induced signaling pathways (34). Similarly, Sox9 and related HMG factors are architectural proteins that act to maintain the nucleosomes in an open configuration, thereby exposing the endogenous, chromatin-integrated COL2A1 promoter to factors that interact directly with the promoter.
The interaction between Sox9 and CBP/p300, modulated in part by Smad2/3, is essential for the COL2A1 expression (27, 35, 36). In addition to affecting chromatin-DNA structure, HMG proteins act to enhance transcription by recruiting transcriptional regulators such as NF-κB, ATF-2, c-Jun, and interferon regulatory factor-1 that bind directly to DNA. ELF3 has two DNA binding domains, a classical ETS domain, which can bind to the ETS sites in the COL2A1 promoter, and an A/T hook domain, which is found also in HMG proteins and recognizes the A/T-rich region of double-stranded DNA (1). In our study, GST-ELF3 pull-down assays showed that ELF3 interacts with Sox9, as well as with L-Sox5 and Sox6 (not shown), all of which contain HMG domains. Using truncated Sox9 fragments we demonstrated that ELF3 interacts with the HMG box toward the N-terminus of Sox9, which is distinct from the CBP/p300-interacting site in the C-terminus reported by Tsuda et al. (26).
Similar to other ETS factors such as ETS-1, which binds to two cysteine-histidine rich regions of CBP (37), ELF3 can also interact with CBP/p300 (31). CBP/p300 acts as a positive regulator of chondrocyte-specific gene expression by interacting with the P/Q/S-rich region in the carboxy-terminus of Sox9 (26) and by binding to and sequestering negative factors such as C/EBP (38). CBP/p300 has intrinsic HAT activity by which it relaxes the chromatin and enhances the function of its associated transcription factors. Indeed, Sox9-induced COL2A1 transcription is dependent upon the interaction between Sox9 and CBP/p300, showing that the intrinsic CBP/p300-HAT activity is fundamental for COL2A1 promoter activation (36). Whereas interactions with CBP generally enhance the activities of DNA-bound transcription factors such as CREB and Stat1 (39), our experiments show that the interaction with ELF3 significantly attenuates endogenous HAT activity. In contrast to the CBP/p300-Ets-1 complex, which exhibits functional HDAC activity and promotes chromatin remodeling and modification of transcription factors and adaptor proteins (40), our results indicate that ELF3 interaction with both Sox9 and CBP/p300 interferes with HAT activity in the context of the COL2A1 promoter. The potential capacity of IL-1β to upregulate p300 expression, as shown in endothelial cells (31), may account partially for the less effective inhibition of COL2A1 promoter activity by IL-1β, compared to the potent inhibition by overexpressed ELF3 (7).
Here, we report another mechanism by which ELF3 levels are dysregulated in OA cartilage and may account for the catabolic role of this ETS transcription factor. We found that OA chondrocytes express increased levels of ELF3 mRNA compared to chondrocytes isolated from non-OA cartilage, and its increased expression correlates with hypermethylation of the proximal promoter. DNA methylation is one of the principal mechanisms by which cells maintain dominant phenotypes and stable chromatin configurations (41, 42) by the actions of DNA methyltransferases (DNMTs), which can maintain stable methylation patterns during cell division and can also result in de novo methylation of unmethylated DNA (43–45). DNA methylation or de-methylation can alter the ability of specific transcription factors to bind to their DNA target sequences, thereby activating or repressing the expression of specific genes (46–50). In human chondrocytes, treatment with inflammatory cytokines decreases the percentage of CpG methylation associated with downregulation of DNMT1 (29) in manner dependent upon NF-κB transcriptional activity (51).
Altered DNA methylation is associated with abnormal gene expression in several pathologies (52), including OA (46, 53). Genome-wide DNA methylation studies have identified epigenomic changes in OA cartilage (54) along with differences between mild and severe OA (55), different epigenomic landscapes in hip and knee cartilage (56) and distinct clusters of OA patients (57, 58). Numerous studies have revealed the importance of DNA methylation in controlling the expression of OA-related genes, including Phlpp1 (59), IL8 (60), and genes encoding proteinases, including MMPs and ADAMTSs (28, 53, 61). DNA hypomethylation correlates with aberrant expression of several catabolic genes in OA chondrocytes, including IL1B, MMP3, 9, and 13, ADAMTS4, and NOS2 (28, 29, 47, 53, 62). Together, these studies represent strong functional evidence that pathological alterations in methylation status do occur, are subsequently maintained, and have important roles in OA disease.
The increased CpG methylation of the proximal promoter of ELF3 and its correlation with increased ELF3 mRNA in OA chondrocytes reported here constitute another example of the sensitivity of gene transcription to altered methylation status. Our previous studies showed that DNA methylation does not affect the binding of ELF3 to the proximal MMP13 promoter, whereas methylation of a specific CpG site nearby prevented HIF-2a binding and MMP13 promoter transactivation (28). OA-related changes in methylation status are also involved in regulating promoter activities of genes associated with anabolism and homeostasis in chondrocytes, including SOD2, SOX9, GDF5, SOST (63–66). In contrast to the hypomethylation of catabolic genes generally observed in OA, hypermethylation of COL9A1 is associated with down-regulation of its expression in OA cartilage. Whereas COL10A1 gene silencing in healthy human articular cartilage is dependent upon the methylation of sparse CpG sites in the COL10A1 promoter (67), COL2A1 mRNA levels do not correlate with changes in the methylation status in the CpG island-rich COL2A1promoter or the Sox9-binding enhancer region (67, 68). Furthermore, DNA methylation is not associated with down-regulation of aggrecan or the replicative factor, p21(WAF1/CIP1), in cartilage due to aging or OA (69, 70).
Our findings on ELF3 indicate that hypermethylation of a given sequence may also drive gene transcription. While DNA methylation of promoter sequences is usually associated with decreased transcriptional activity, methylation of specific CpG sites can also activate transcription, either by enhancing transcription factor binding (71) or perhaps by repressing binding of inhibitors of transcription. The absence of a CpG site within or adjacent to the NF-κB site at −87bp, which is required for ELF3 promoter activity (3, 72), suggests that NF-κB binding would still occur in the presence of the hypermethylated CpG sites studied here. In addition, the methodology utilized in this study did not allow us to assess the relative contribution of 5hmC, which is positively associated with transcription in actively transcribed genes within term placenta (73) and with activation of gene expression during chondrogenic differentiation (74). Thus, 5hmC can mark and perhaps drive enhanced transcription, as opposed to the inhibitory role usually associated with 5mC. Therefore, differential 5hmC could be related with the increased ELF3 expression in OA disease, adding another level of complexity and emphasizing the need for performing comprehensive analyses of DNA methylation status to fully understand its contribution to gene transcription in normal and pathological conditions.
In conclusion, our results indicate that the cytokine-inducible ETS factor, ELF3, is transcriptionally dysregulated in OA chondrocytes via, at least in part, epigenetic mechanisms and that it acts as a repressor of COL2A1 gene transcription not only by binding to the COL2A1 promoter but also by interacting with Sox9 and CBP/p300, decreasing the CBP/p300-dependent HAT activity.
Acknowledgments
This work was supported in whole or in part by National Institutes of Health Grants R01-AG022021, R21-AR054887, and RC4 AR060546 (to M. B. G.).
REFERENCES
- 1.Oettgen P, Alani RM, Barcinski MA, Brown L, Akbarali Y, Boltax J, et al. Isolation and characterization of a novel epithelium-specific transcription factor, ESE-1, a member of the ets family. Mol Cell Biol. 1997;17(8):4419–4433. doi: 10.1128/mcb.17.8.4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oettgen P, Barcinski M, Boltax J, Stolt P, Akbarali Y, Libermann TA. Genomic organization of the human ELF3 (ESE-1/ESX) gene, a member of the Ets transcription factor family, and identification of a functional promoter. Genomics. 1999;55(3):358–362. doi: 10.1006/geno.1998.5681. [DOI] [PubMed] [Google Scholar]
- 3.Grall F, Gu X, Tan L, Cho JY, Inan MS, Pettit AR, et al. Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor alpha in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor kappaB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003;48(5):1249–1260. doi: 10.1002/art.10942. [DOI] [PubMed] [Google Scholar]
- 4.Rudders S, Gaspar J, Madore R, Voland C, Grall F, Patel A, et al. ESE-1 is a novel transcriptional mediator of inflammation that interacts with NF-kappa B to regulate the inducible nitric-oxide synthase gene. J Biol Chem. 2001;276(5):3302–3309. doi: 10.1074/jbc.M006507200. [DOI] [PubMed] [Google Scholar]
- 5.Grall FT, Prall WC, Wei W, Gu X, Cho JY, Choy BK, et al. The Ets transcription factor ESE-1 mediates induction of the COX-2 gene by LPS in monocytes. FEBS J. 2005;272(7):1676–1687. doi: 10.1111/j.1742-4658.2005.04592.x. [DOI] [PubMed] [Google Scholar]
- 6.Otero M, Plumb DA, Tsuchimochi K, Dragomir CL, Hashimoto K, Peng H, et al. E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under proinflammatory stress. J Biol Chem. 2012;287(5):3559–3572. doi: 10.1074/jbc.M111.265744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng H, Tan L, Osaki M, Zhan Y, Ijiri K, Tsuchimochi K, et al. ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes. J Cell Physiol. 2008;215(2):562–573. doi: 10.1002/jcp.21338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies SR, Sakano S, Zhu Y, Sandell LJ. Distribution of the transcription factors Sox9, AP-2, and [delta]EF1 in adult murine articular and meniscal cartilage and growth plate. J Histochem Cytochem. 2002;50(8):1059–1065. doi: 10.1177/002215540205000808. [DOI] [PubMed] [Google Scholar]
- 9.Lefebvre V, Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005;75(3):200–212. doi: 10.1002/bdrc.20048. [DOI] [PubMed] [Google Scholar]
- 10.Leung KK, Ng LJ, Ho KK, Tam PP, Cheah KS. Different cis-regulatory DNA elements mediate developmental stage- and tissue-specific expression of the human COL2A1 gene in transgenic mice. J Cell Biol. 1998;141(6):1291–1300. doi: 10.1083/jcb.141.6.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol. 1997;17(4):2336–2346. doi: 10.1128/mcb.17.4.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefebvre V, Li P, de Crombrugghe B. A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 1998;17(19):5718–5733. doi: 10.1093/emboj/17.19.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22(1):85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 14.Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16(21):2813–2828. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Lloris R, Vinals F, Lopez-Rovira T, Harley V, Bartrons R, Rosa JL, et al. Induction of the Sry-related factor SOX6 contributes to bone morphogenetic protein-2-induced chondroblastic differentiation of C3H10T1/2 cells. Mol Endocrinol. 2003;17(7):1332–1343. doi: 10.1210/me.2002-0254. [DOI] [PubMed] [Google Scholar]
- 16.Smits P, Dy P, Mitra S, Lefebvre V. Sox5 and Sox6 are needed to develop and maintain source, columnar, and hypertrophic chondrocytes in the cartilage growth plate. J Cell Biol. 2004;164(5):747–758. doi: 10.1083/jcb.200312045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray D, Precht P, Balakir R, Horton WE., Jr The transcription factor deltaEF1 is inversely expressed with type II collagen mRNA and can repress Col2a1 promoter activity in transfected chondrocytes. J Biol Chem. 2000;275(5):3610–3618. doi: 10.1074/jbc.275.5.3610. [DOI] [PubMed] [Google Scholar]
- 18.Zhang P, Jimenez SA, Stokes DG. Regulation of human COL9A1 gene expression. Activation of the proximal promoter region by SOX9. J Biol Chem. 2003;278(1):117–123. doi: 10.1074/jbc.M208049200. [DOI] [PubMed] [Google Scholar]
- 19.Bridgewater LC, Walker MD, Miller GC, Ellison TA, Holsinger LD, Potter JL, et al. Adjacent DNA sequences modulate Sox9 transcriptional activation at paired Sox sites in three chondrocyte-specific enhancer elements. Nucleic Acids Res. 2003;31(5):1541–1553. doi: 10.1093/nar/gkg230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bridgewater LC, Lefebvre V, de Crombrugghe B. Chondrocyte-specific enhancer elements in the Col11a2 gene resemble the Col2a1 tissue-specific enhancer. J Biol Chem. 1998;273(24):14998–15006. doi: 10.1074/jbc.273.24.14998. [DOI] [PubMed] [Google Scholar]
- 21.Sekiya I, Tsuji K, Koopman P, Watanabe H, Yamada Y, Shinomiya K, et al. SOX9 enhances aggrecan gene promoter/enhancer activity and is up-regulated by retinoic acid in a cartilage-derived cell line, TC6. J Biol Chem. 2000;275(15):10738–10744. doi: 10.1074/jbc.275.15.10738. [DOI] [PubMed] [Google Scholar]
- 22.Smits P, Li P, Mandel J, Zhang Z, Deng JM, Behringer RR, et al. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev Cell. 2001;1(2):277–290. doi: 10.1016/s1534-5807(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 23.Genzer MA, Bridgewater LC. A Col9a1 enhancer element activated by two interdependent SOX9 dimers. Nucleic Acids Res. 2007;35(4):1178–1186. doi: 10.1093/nar/gkm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aigner T, Gebhard PM, Schmid E, Bau B, Harley V, Poschl E. SOX9 expression does not correlate with type II collagen expression in adult articular chondrocytes. Matrix Biol. 2003;22(4):363–372. doi: 10.1016/s0945-053x(03)00049-0. [DOI] [PubMed] [Google Scholar]
- 25.Tchetina EV, Squires G, Poole AR. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J Rheumatol. 2005;32(5):876–886. [PubMed] [Google Scholar]
- 26.Tsuda M, Takahashi S, Takahashi Y, Asahara H. Transcriptional co-activators CREB-binding protein and p300 regulate chondrocyte-specific gene expression via association with Sox9. J Biol Chem. 2003;278(29):27224–27229. doi: 10.1074/jbc.M303471200. [DOI] [PubMed] [Google Scholar]
- 27.Furumatsu T, Tsuda M, Yoshida K, Taniguchi N, Ito T, Hashimoto M, et al. Sox9 and p300 cooperatively regulate chromatin-mediated transcription. J Biol Chem. 2005;280(42):35203–35208. doi: 10.1074/jbc.M502409200. [DOI] [PubMed] [Google Scholar]
- 28.Hashimoto K, Otero M, Imagawa K, de Andres MC, Coico JM, Roach HI, et al. Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1beta (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J Biol Chem. 2013;288(14):10061–10072. doi: 10.1074/jbc.M112.421156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hashimoto K, Oreffo RO, Gibson MB, Goldring MB, Roach HI. DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum. 2009;60(11):3303–3313. doi: 10.1002/art.24882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown C, Gaspar J, Pettit A, Lee R, Gu X, Wang H, et al. ESE-1 is a novel transcriptional mediator of angiopoietin-1 expression in the setting of inflammation. J Biol Chem. 2004;279(13):12794–12803. doi: 10.1074/jbc.M308593200. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Fang R, Cho JY, Libermann TA, Oettgen P. Positive and negative modulation of the transcriptional activity of the ETS factor ESE-1 through interaction with p300, CREB-binding protein, and Ku 70/86. J Biol Chem. 2004;279(24):25241–25250. doi: 10.1074/jbc.M401356200. [DOI] [PubMed] [Google Scholar]
- 32.Goldring MB, Marcu KB. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol Med. 2012;18(2):109–118. doi: 10.1016/j.molmed.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klug M, Rehli M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics. 2006;1(3):127–130. doi: 10.4161/epi.1.3.3327. [DOI] [PubMed] [Google Scholar]
- 34.Munshi N, Agalioti T, Lomvardas S, Merika M, Chen G, Thanos D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science. 2001;293(5532):1133–1136. doi: 10.1126/science.293.5532.1133. [DOI] [PubMed] [Google Scholar]
- 35.Furumatsu T, Ozaki T, Asahara H. Smad3 activates the Sox9-dependent transcription on chromatin. Int J Biochem Cell Biol. 2009;41(5):1198–1204. doi: 10.1016/j.biocel.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furumatsu T, Tsuda M, Taniguchi N, Tajima Y, Asahara H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J Biol Chem. 2005;280(9):8343–8350. doi: 10.1074/jbc.M413913200. [DOI] [PubMed] [Google Scholar]
- 37.Yang C, Shapiro LH, Rivera M, Kumar A, Brindle PK. A role for CREB binding protein and p300 transcriptional coactivators in Ets-1 transactivation functions. Mol Cell Biol. 1998;18(4):2218–2229. doi: 10.1128/mcb.18.4.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imamura T, Imamura C, Iwamoto Y, Sandell LJ. Transcriptional Co-activators CREB-binding protein/p300 increase chondrocyte Cd-rap gene expression by multiple mechanisms including sequestration of the repressor CCAAT/enhancer-binding protein. J Biol Chem. 2005;280(17):16625–16634. doi: 10.1074/jbc.M411469200. [DOI] [PubMed] [Google Scholar]
- 39.Korzus E, Torchia J, Rose DW, Xu L, Kurokawa R, McInerney EM, et al. Transcription factor-specific requirements for coactivators and their acetyltransferase functions. Science. 1998;279(5351):703–707. doi: 10.1126/science.279.5351.703. [DOI] [PubMed] [Google Scholar]
- 40.Jayaraman G, Srinivas R, Duggan C, Ferreira E, Swaminathan S, Somasundaram K, et al. p300/cAMP-responsive element-binding protein interactions with ets-1 and ets-2 in the transcriptional activation of the human stromelysin promoter. J Biol Chem. 1999;274(24):17342–17352. doi: 10.1074/jbc.274.24.17342. [DOI] [PubMed] [Google Scholar]
- 41.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson DG, Dent SY. Chromatin: receiver and quarterback for cellular signals. Cell. 2013;152(4):685–689. doi: 10.1016/j.cell.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 44.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19(3):219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 45.Walton EL, Francastel C, Velasco G. Maintenance of DNA methylation: Dnmt3b joins the dance. Epigenetics. 2011;6(11):1373–1377. doi: 10.4161/epi.6.11.17978. [DOI] [PubMed] [Google Scholar]
- 46.Barter MJ, Bui C, Young DA. Epigenetic mechanisms in cartilage and osteoarthritis: DNA methylation, histone modifications and microRNAs. Osteoarthritis Cartilage. 2012;20(5):339–349. doi: 10.1016/j.joca.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 47.de Andres MC, Imagawa K, Hashimoto K, Gonzalez A, Roach HI, Goldring MB, et al. Loss of methylation in CpG sites in the NF-kappaB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis Rheum. 2013;65(3):732–742. doi: 10.1002/art.37806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 49.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinson C, Chatterjee R. CG methylation. Epigenomics. 2012;4(6):655–663. doi: 10.2217/epi.12.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imagawa K, de Andres MC, Hashimoto K, Pitt D, Itoi E, Goldring MB, et al. The epigenetic effect of glucosamine and a nuclear factor-kappa B (NF-kB) inhibitor on primary human chondrocytes--implications for osteoarthritis. Biochem Biophys Res Commun. 2011;405(3):362–367. doi: 10.1016/j.bbrc.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 53.Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, Oreffo RO, et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005;52(10):3110–3124. doi: 10.1002/art.21300. [DOI] [PubMed] [Google Scholar]
- 54.Jeffries MA, Donica M, Baker LW, Stevenson ME, Annan AC, Humphrey MB, et al. Genome-wide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis Rheumatol. 2014;66(10):2804–2815. doi: 10.1002/art.38762. [DOI] [PubMed] [Google Scholar]
- 55.Moazedi-Fuerst FC, Hofner M, Gruber G, Weinhaeusel A, Stradner MH, Angerer H, et al. Epigenetic differences in human cartilage between mild and severe OA. J Orthop Res. 2014;32(12):1636–1645. doi: 10.1002/jor.22722. [DOI] [PubMed] [Google Scholar]
- 56.Bomer N, den Hollander W, Ramos YF, Bos SD, van der Breggen R, Lakenberg N, et al. Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernandez-Tajes J, Soto-Hermida A, Vazquez-Mosquera ME, Cortes-Pereira E, Mosquera A, Fernandez-Moreno M, et al. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2012-202783. [DOI] [PubMed] [Google Scholar]
- 58.Rushton MD, Reynard LN, Barter MJ, Refaie R, Rankin KS, Young DA, et al. Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol. 2014 doi: 10.1002/art.38713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bradley EW, Carpio LR, McGee-Lawrence ME, Castillejo Becerra C, Amanatullah DF, Ta LE, et al. Phlpp1 facilitates post-traumatic osteoarthritis and is induced by inflammation and promoter demethylation in human osteoarthritis. Osteoarthritis Cartilage. 2015 doi: 10.1016/j.joca.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takahashi A, de Andres MC, Hashimoto K, Itoi E, Oreffo RO. Epigenetic regulation of interleukin-8, an inflammatory chemokine, in osteoarthritis. Osteoarthritis Cartilage. 2015;23(11):1946–1954. doi: 10.1016/j.joca.2015.02.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bui C, Barter MJ, Scott JL, Xu Y, Galler M, Reynard LN, et al. cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 2012;26(7):3000–3011. doi: 10.1096/fj.12-206367. [DOI] [PubMed] [Google Scholar]
- 62.da Silva MA, Yamada N, Clarke NM, Roach HI. Cellular and epigenetic features of a young healthy and a young osteoarthritic cartilage compared with aged control and OA cartilage. J Orthop Res. 2009;27(5):593–601. doi: 10.1002/jor.20799. [DOI] [PubMed] [Google Scholar]
- 63.Scott JL, Gabrielides C, Davidson RK, Swingler TE, Clark IM, Wallis GA, et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann Rheum Dis. 2010;69(8):1502–1510. doi: 10.1136/ard.2009.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim KI, Park YS, Im GI. Changes in the epigenetic status of the SOX-9 promoter in human osteoarthritic cartilage. J Bone Miner Res. 2013;28(5):1050–1060. doi: 10.1002/jbmr.1843. [DOI] [PubMed] [Google Scholar]
- 65.Reynard LN, Bui C, Syddall CM, Loughlin J. CpG methylation regulates allelic expression of GDF5 by modulating binding of SP1 and SP3 repressor proteins to the osteoarthritis susceptibility SNP rs143383. Hum Genet. 2014 doi: 10.1007/s00439-014-1447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Papathanasiou I, Kostopoulou F, Malizos KN, Tsezou A. DNA methylation regulates sclerostin (SOST) expression in osteoarthritic chondrocytes by bone morphogenetic protein 2 (BMP-2) induced changes in Smads binding affinity to the CpG region of SOST promoter. Arthritis Res Ther. 2015;17:160. doi: 10.1186/s13075-015-0674-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zimmermann P, Boeuf S, Dickhut A, Boehmer S, Olek S, Richter W. Correlation of COL10A1 induction during chondrogenesis of mesenchymal stem cells with demethylation of two CpG sites in the COL10A1 promoter. Arthritis Rheum. 2008;58(9):2743–2753. doi: 10.1002/art.23736. [DOI] [PubMed] [Google Scholar]
- 68.Imagawa K, de Andres MC, Hashimoto K, Itoi E, Otero M, Roach HI, et al. Association of reduced type IX collagen gene expression in human osteoarthritic chondrocytes with epigenetic silencing by DNA hypermethylation. Arthritis Rheumatol. 2014;66(11):3040–3051. doi: 10.1002/art.38774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Poschl E, Fidler A, Schmidt B, Kallipolitou A, Schmid E, Aigner T. DNA methylation is not likely to be responsible for aggrecan down regulation in aged or osteoarthritic cartilage. Ann Rheum Dis. 2005;64(3):477–480. doi: 10.1136/ard.2004.022509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sesselmann S, Soder S, Voigt R, Haag J, Grogan SP, Aigner T. DNA methylation is not responsible for p21WAF1/CIP1 down-regulation in osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2009;17(4):507–512. doi: 10.1016/j.joca.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 71.Rishi V, Bhattacharya P, Chatterjee R, Rozenberg J, Zhao J, Glass K, et al. CpG methylation of half-CRE sequences creates C/EBPalpha binding sites that activate some tissue-specific genes. Proc Natl Acad Sci U S A. 2010;107(47):20311–20316. doi: 10.1073/pnas.1008688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu J, Duan R, Cao H, Field D, Newnham CM, Koehler DR, et al. Regulation of epithelium-specific Ets-like factors ESE-1 and ESE-3 in airway epithelial cells: potential roles in airway inflammation. Cell research. 2008;18(6):649–663. doi: 10.1038/cr.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Green BB, Houseman EA, Johnson KC, Guerin DJ, Armstrong DA, Christensen BC, et al. Hydroxymethylation is uniquely distributed within term placenta, and is associated with gene expression. FASEB J. 2016 doi: 10.1096/fj.201600310R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taylor SE, Li YH, Smeriglio P, Rath M, Wong WH, Bhutani N. Stable 5-Hydroxymethylcytosine (5hmC) Acquisition Marks Gene Activation During Chondrogenic Differentiation. J Bone Miner Res. 2016;31(3):524–534. doi: 10.1002/jbmr.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]