

Abstract
Background
The ‘obesity paradox’ of critical illness refers to better survival with a higher body mass index. We hypothesized that fat mobilized from excess adipose tissue during critical illness provides energy more efficiently than exogenous macronutrients and could prevent lean tissue wasting.
Methods
In lean and premorbidly obese mice, the effect of 5 days of sepsis‐induced critical illness on body weight and composition, muscle wasting, and weakness was assessed, each with fasting and parenteral feeding. Also, in lean and overweight/obese prolonged critically ill patients, markers of muscle wasting and weakness were compared.
Results
In mice, sepsis reduced body weight similarly in the lean and obese, but in the obese with more fat loss and less loss of muscle mass, better preservation of myofibre size and muscle force, and less loss of ectopic lipids, irrespective of administered feeding. These differences between lean and obese septic mice coincided with signs of more effective hepatic fatty acid and glycerol metabolism, and ketogenesis in the obese. Also in humans, better preservation of myofibre size and muscle strength was observed in overweight/obese compared with lean prolonged critically ill patients.
Conclusions
During critical illness premorbid obesity, but not nutrition, optimized utilization of stored lipids and attenuated muscle wasting and weakness.
Keywords: Critical illness, Obesity, Skeletal muscle, Metabolism
Introduction
Critical illness is defined as any acute medical condition necessitating vital organ support without which death would be imminent. Whether evoked by sepsis, trauma, or major surgery, hypercatabolism is a hallmark of critical illnesses. Hypercatabolism in the acute phase of critical illness is presumed to be an adaptive response providing the essential fuel for energy production in vital organs.1, 2 However, when hypercatabolism persists it may result in muscle wasting and weakness, a clinical manifestation referred to as intensive care unit (ICU) acquired weakness (ICUAW).3, 4 Prevalence of ICUAW varies according to the study population, but up to 80% of ICU patients appear to suffer from muscle wasting and/or muscle weakness.5, 6 ICUAW is associated with impaired weaning from mechanical ventilation, delayed rehabilitation and prolonged hospitalization, late death, and greater impaired functional outcome of survivors.5, 7 Muscle wasting during critical illness is explained by reduced myofibrillary protein synthesis and by increased protein breakdown via the ubiquitin–proteasome and cathepsin pathways, whereas insufficient autophagy activation may contribute to reduced myofibre quality.8, 9, 10, 11, 12 Parenteral provision of macronutrients during acute critical illness does not prevent muscle wasting and weakness and may in fact exert deleterious effects via further suppression of autophagic myofibre quality control.10, 13
Several large cohort studies and meta‐analyses have demonstrated a lower risk of death for critically ill patients who are overweight or obese as compared with those with a normal body mass index (BMI), an association referred to as the ‘obesity paradox’.14, 15, 16, 17, 18 Although this paradox remains incompletely understood, it has been suggested that having a larger nutritional reserve, stored in the excess adipose tissue, may be beneficial.19 This mechanism has also been suggested to underlie a similar obesity paradox observed in patients suffering from chronic diseases such as heart and kidney failure.20, 21, 22, 23 Nevertheless, the underlying pathways through which being overweight or obese prior to becoming critically ill could be beneficial, have not yet been clarified.
We hypothesized that during critical illness, fat mobilized from excess adipose tissue can provide energy to vital organs more efficiently than exogenous macronutrients, and that this might prevent lean tissue wasting. We tested this hypothesis in a centrally catheterized mouse model of cecal ligation and puncture (CLP)‐induced septic critical illness and in a human study. In lean and premorbidly obese mice, the effect of 5 days of critical illness on body weight and composition, muscle wasting, and weakness was compared, each with fasting and parenteral feeding. Additionally, in matched lean and overweight/obese critically ill patients, we compared markers of muscle wasting in muscle biopsies of two muscle groups [musculus (m.) vastus lateralis and m. rectus abdominis] as well as muscle strength, quantified by the Medical Research Council (MRC) sum score.
Materials and methods
Setup of the animal model
Diet‐induced obesity
Male, 12‐week‐old C57BL/6J mice (Janvier SAS Chassal, France) received ad libitum standard chow (10% fat, E15745‐04, ssniff, Soest, Germany), or ad libitum high‐fat diet (45% fat, E15744‐34, ssniff) for 12 weeks. Body weight was quantified weekly. Only animals placed on the high‐fat diet that reached a body weight above 30 g but below 45 g (to avoid morbid obesity‐associated metabolic alterations) within the 12 weeks of diet were included in the study.24 Tail blood glucose measurements indicated that all mice remained normoglycemic during the obesity‐inducing diet (Accu‐check, Roche, Basel, Switzerland).
Mice experiment 1
At 24 weeks of age, lean and obese animals were randomly allocated to ‘healthy control’ (lean healthy mice (n = 8) and obese healthy mice (n = 9)) or to ‘CLP’. CLP groups were randomly subdivided into a fasted‐CLP or fed‐CLP group. The CLP‐induced septic critical illness model and nutritional protocols of the animals have previously been described in detail.25 Total body, fat, and fat‐free mass were quantified with DEXA scans at the start (day −2) and at the end of the 5 day experiment, as previously described.25 After 5 days of critical illness, animals were sacrificed by decapitation, vital organs were removed, snap frozen in liquid nitrogen and stored at −80°C, or preserved in paraformaldehyde. In lean CLP animals, 9/15 fasted and 7/11 fed mice survived until day 5. In obese CLP animals, 9/10 fasted and 10/18 fed mice survived until day 5.
Mice experiment 2
We examined muscle force ex vivo in lean and diet‐induced obese CLP mice (Supplemental methods). The experimental setup was comparable to that of mice experiment 1. Mice were randomly allocated to ‘healthy control’ or ‘CLP’. As in experiment 1 we had observed that muscle wasting occurred irrespective of feeding, we now only compared parenterally fed with pair‐fed lean (n = 17) and obese (n = 15) healthy mice. Until day 5, 15/18 lean and 15/18 obese CLP mice survived. After 5 days, animals were deeply anaesthetized and the m. extensor digitorum longus (EDL) was carefully dissected from both hind limbs for ex vivo muscle force measurements (Supplemental methods).
Mice body and tissue composition and mass
To control for potential illness‐ or resuscitation‐related changes in fluid content, dry weight of isolated tissues was obtained by a freeze‐drying process. Myofibre cross‐sectional area (CSA) was quantified on digital microscopy images of haematoxylin and eosin stained paraffin sections with in‐house designed algorithms, as described earlier.26 In addition, the presence of myofibre degeneration, necrosis, and inflammation was histologically evaluated as described earlier.10 Triglyceride content of tissues was determined with a commercial colorimetric assay (triglyceride quantification kit, Abcam, Cambridge, UK).
Mice circulating fatty acids, glycerol, and ketone bodies
Serum concentrations of free fatty acids, glycerol, and 3‐hydroxybutyrate (3‐HB) were determined with commercially available assay kits (free fatty acid fluorometric assay kit, Cayman Chemical Company, Ann Arbor, MI, USA; glycerol assay kit, Sigma‐Aldrich, Saint Louis, MO, USA; EnzyChrom ketone body assay kit, BioAssay Systems, Hayward, CA, USA).
Mice tissue gene expression and protein expression analyses
Messenger RNA of skeletal muscle and liver was isolated, and cDNA was quantified in real‐time as previously described with commercial TaqMan® Assays (Applied Biosystems, Carlsbad, CA, USA) (Supplemental methods).27 Data were normalized to ribosomal 18S (Rn18s) gene expression and expressed as fold change of the mean of the controls. Microtubule‐associated protein 1A/1B‐light chain 3 (LC3)‐I, LC3‐II (Ab from Sigma‐Aldrich), and p62 protein (Ab from Novus, Littleton, CO, USA) levels were quantified in m. gastrocnemius with Western blots as previously described.27 Data were expressed relative to the means of the controls. Commercial kits were used to measure proteasome (20S‐proteasome activity assay, InnoZyme) and cathepsin B/L activities (Cathepsin L activity kit, Millipore, Merck KGaA, Darmstadt, Germany) in m. tibialis anterior homogenates.
Patient studies
Myofibre cross‐sectional area
In vivo skeletal muscle needle biopsies were obtained from the m. vastus lateralis of the m. quadriceps femoris of ICU patients on day 8 ± 1 of ICU stay.13, 28 As healthy references, in vivo m. vastus lateralis needle biopsies (n = 20) were available from healthy individuals undergoing minor urological intervention or surgery for inguinal herniation. Healthy volunteers had comparable demographics as ICU patients (Supplemental table 1). From 122 patients of whom myofibre CSA were available,13 we selected 51 lean patients (BMI ≤25 kg/m2) and 51 overweight/obese patients (BMI >25 kg/m2), matched with use of propensity scores obtained by logistic regression (one‐to‐one nearest neighbour matching without replacement and with a caliper of 0.2).29 For this propensity score matching, the following baseline characteristics were used: gender, age, presence of malignancy, diabetes, APACHE II score on admission, and randomization (Supplemental table 1). Next, we investigated postmortem m. rectus abdominis skeletal muscle biopsies, harvested immediately after death, from which myofibre CSA was available.10 From 148 available biopsies, 43 lean and 43 overweight/obese patients were propensity score matched similarly as the first set. As healthy references, in vivo m. rectus abdominis biopsies (n = 11) were available from non‐critically ill individuals with comparable demographics (Supplemental table 2).
Muscle weakness
In fully cooperative patients, who were still in the ICU on day 8 ± 1, muscle strength was quantified with the MRC sum score.13, 28 To correct for a possible bias by an effect of the randomized intervention on ICU stay, a random sample of patients discharged from the ICU was assessed in the regular hospital ward on post‐randomization day 8 ± 1. Clinically relevant weakness was diagnosed when the MRC sum score was lower than 48. Of the 352 patients that were tested on post‐admission‐day 8 ± 1, 139 lean and 139 overweight/obese patients were propensity score matched, similarly as the first sets (Supplemental table 3). Of the 139 lean patients, 74 patients were tested on the regular hospital ward, 65 in the ICU. Of the 139 overweight/obese patients, 76 patients were tested on the ward, 63 in the ICU.
Statistics
Normally distributed data were compared with one‐way analysis of variance (ANOVA) with post hoc Fisher's LSD test for multiple comparisons. Not‐normally distributed data were analysed with parametric tests after log‐ or (double) square root‐transformation if this resulted in a normal distribution. Comparison of proportions was performed using Fisher's exact tests. Continuous non‐normally distributed data were compared with non‐parametric Mann–Whitney U‐tests. Two‐sided P‐values ≤0.05 were considered statistically significant (Statview 5.0.1, JMP 8.0.1 and SPSS 22 were used). Data are presented as box plots with median, interquartile ranges, and 10th and 90th percentiles or as bars with whiskers, representing means and standard error of the mean (SEM). ANOVA or Mann–Whitney P‐values are presented in figure legends. Post‐hoc P‐values <0.1 are plotted on the figures.
Study approval
All animals were treated according to the Principles of Laboratory Animal Care (U.S. National Society for Medical Research) and the Guide for Care and Use of Laboratory Animals (National Institutes of Health). The protocols for these animal studies had been approved by the Institutional Ethical Committee for Animal Experimentation (project number P051/2010 and P050/2015). The study protocol of the human studies had been approved by the Institutional Review Board of the KU Leuven (ML1820, ML2707, ML4190, ML1094). Written informed consent was obtained from the patients' closest family member and from healthy volunteers.
Results
Mice study—body composition
Prior to CLP, body weight was significantly higher in obese than in lean mice (35.3 ± 0.5 g vs. 29.4 ± 0.6 g, P < 0.0001). This was attributable to a higher fat mass in obese compared with lean mice (9.0 ± 0.6 g vs. 4.7 ± 0.2 g, P < 0.0001), whereas fat‐free mass was equal in obese and lean mice (23.5 ± 0.4 g vs. 22.7 ± 0.4 g, P = 0.2).
After 5 days of CLP‐induced critical illness, all animals lost a comparable amount of body weight; hence body weight remained higher in obese vs. lean CLP mice (Figure 1a–b). Critical illness also resulted in loss of fat mass, but obese CLP mice lost more than double the amount of fat mass over 5 days of illness than lean CLP mice (Figure 1c–d). In both lean and obese CLP mice, the loss of body weight and fat mass was unaffected by parenteral feeding (Figure 1).
Figure 1.
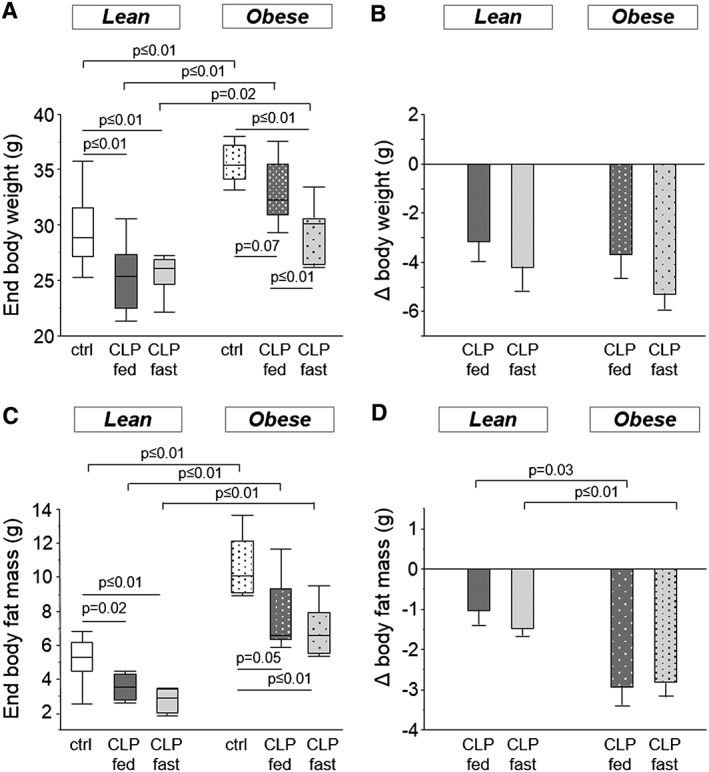
Mice body composition. (a) Body weight after 5 days of CLP‐induced critical illness (ANOVA P ≤ 0.01). (b) Loss of body weight during the 5 day experiment (ANOVA P = 0.4). (c) End body fat mass, measured by DEXA (ANOVA P ≤ 0.01). (d) Loss in body fat mass during 5 days of critical illness (ANOVA P ≤ 0.01). White, healthy lean mice (n = 8); dark gray, fed lean CLP mice (n = 7); light gray, fasted lean CLP mice (n = 9); white dotted, healthy obese mice (n = 9); dark gray dotted, fed obese CLP mice (n = 10); light gray dotted, fasted obese CLP mice (n = 9). [DEXA: dual‐energy‐X‐ray‐absorptiometry, CLP: cecal ligation and puncture, ctrl: healthy control animals, fed: parenterally fed, fast: fasted]
Critical illness resulted in a loss of lean tissue in lean, but not in obese mice, as demonstrated by the reduction in dry weight of isolated m. tibialis anterior and m. soleus (Figure 2a–b). This coincided with a reduced myofibre CSA of the m. tibialis anterior in the lean, but not in obese CLP mice (Figure 2c). Consequently, mean muscle myofibre size was larger in obese CLP mice than lean CLP mice (Figure 2c). Fasting during critical illness tended to further reduce the dry muscle weight (Figure 2a–b), whereas the occurrence of smaller myofibres was present in both fed and fasted animals (Figure 2c).
Figure 2.
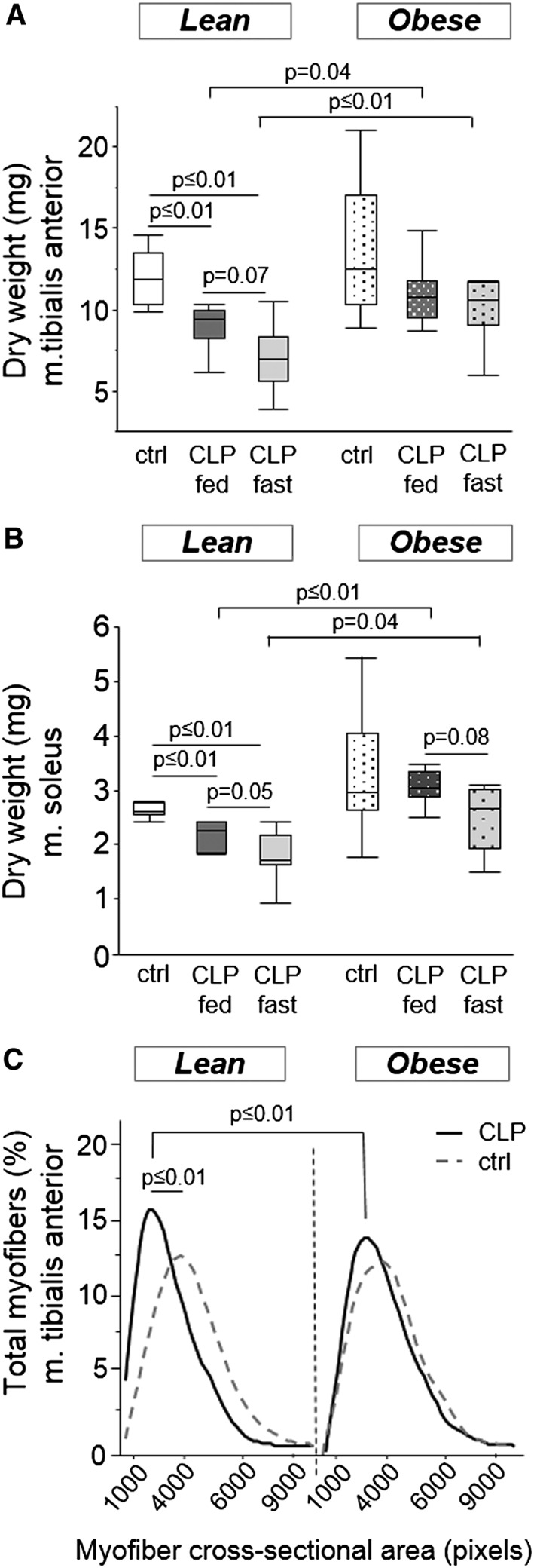
Mice skeletal muscle mass and cross‐sectional area. (a) Dry weight of the m. tibialis anterior (ANOVA P ≤ 0.01). (b) Dry weight of the m. soleus (ANOVA P ≤ 0.01). White, healthy lean mice (n = 8); dark gray, fed lean CLP mice (n = 7); light gray, fasted lean CLP mice (n = 9); white dotted, healthy obese mice (n = 9); dark gray dotted, fed obese CLP mice (n = 10); light gray dotted, fasted obese CLP mice (n = 9). (c) Skeletal muscle myofibre cross‐sectional area (ANOVA P ≤ 0.01). Cross‐sectional area is categorized in blocks of 1000 pixels for each animal. The graph displays smoothed curves of the percentage of myofibres in each category, gray dotted line, healthy animals; black line, CLP mice. Statistical difference reflects mean myofibre cross‐sectional area. Fed and fasted CLP mice were grouped as they were similar (P = 0.3 in lean CLP; P = 0.4 in obese CLP). [CLP: cecal ligation and puncture, ctrl: healthy control animals, fed: parenterally fed, fast: fasted]
Mice study—markers of skeletal muscle atrophy and autophagy
We investigated whether less activation of atrophy pathways could explain the observed preservation of muscle mass and muscle fibre size in obese CLP mice. Compared with lean healthy control mice, muscle protein content tended to be reduced in lean fed CLP mice (83.9 ± 8.3 µg/mg vs. 55.1 ± 10.1 µg/mg, P = 0.06) and was reduced in lean fasted CLP mice (50.4 ± 11.1 µg/mg, vs. lean healthy controls, P = 0.01). In contrast, obese CLP mice preserved their muscle protein content (108.1 ± 20.8 µg/mg vs. 76.1 ± 14.8 µg/mg in fed CLP mice P = 0.8, and 63.5 ± 6.6 µg/mg in fasted CLP mice P = 0.1). Gene expression of markers of the ubiquitin–proteasome system, Fbxo32 and Trim63, were up‐regulated in lean and obese CLP mice (Figure 3a–b). Fasted lean CLP mice displayed a further increase in Fbxo32 and Trim63 expression (Figure 3a–b). Activity of the proteolytic enzyme 20S‐proteasome was unaffected by critical illness (Figure 3c). In contrast, cathepsin activity was increased in lean CLP mice, whereas this increase tended to be attenuated in obese CLP mice (Figure 3d). Compared with lean and obese healthy control animals, fasting further increased the cathepsin activity both in lean and obese CLP mice (Figure 3d).
Figure 3.
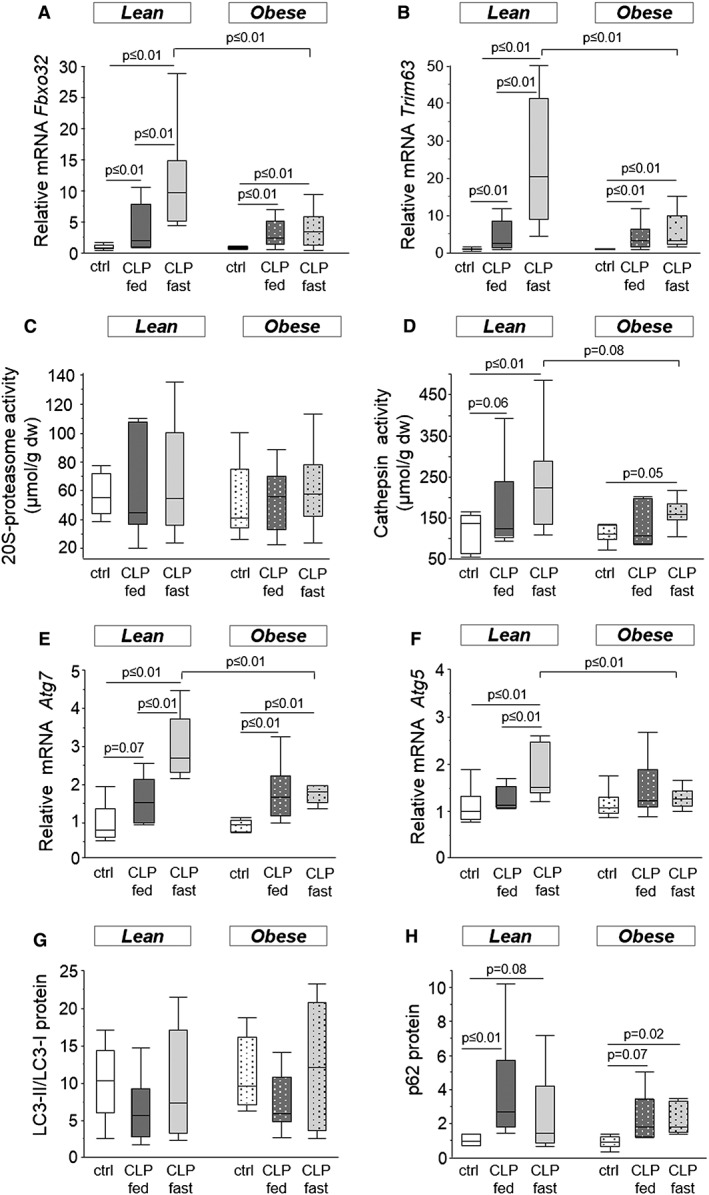
Mice skeletal muscle atrophy and autophagy. (a) Relative mRNA expression of Fbxo32 (ANOVA P ≤ 0.01). (b) Relative mRNA expression of Trim63 (ANOVA P ≤ 0.01). (c) Activity of the 20S‐proteasome (ANOVA P = 0.9). (d) Cathepsin activity (ANOVA P = 0.4). (e) Relative mRNA expression of Atg7 (ANOVA P ≤ 0.01). (f) Relative mRNA expression of Atg5 (ANOVA P ≤ 0.01). (g) LC3‐II/LC3‐I protein ratio, as detected with western blot (ANOVA P = 0.4). (h) Protein level of p62, measured with western blot (ANOVA P ≤ 0.01). Gene expression data are expressed normalized to Rn18s gene expression and as a fold change of the mean of the lean healthy controls. Protein levels are presented as fold change of the mean of lean healthy controls. White, healthy lean mice (n = 8); dark gray, fed lean CLP mice (n = 7); light gray, fasted lean CLP mice (n = 9); white dotted, healthy obese mice (n = 9); dark gray dotted, fed obese CLP mice (n = 10); light gray dotted, fasted obese CLP mice (n = 9). [dw: dry weight, CLP: cecal ligation and puncture, ctrl: healthy control animals, fed: parenterally fed, fast: fasted]
Gene expression of Atg7 was elevated in both lean and obese CLP mice, but most pronounced in fasted lean CLP mice (Figure 3e). In contrast, Atg5 gene expression was only increased in fasted lean CLP mice (Figure 3f). The LC3 protein ratio (LC3‐II/LC3‐I), a marker of autophagosome formation, appeared unaffected after 5 days of critical illness (Figure 3g). Protein levels of p62, used as a marker of insufficiently activated autophagy, were elevated in CLP mice, irrespective of obesity or whether the mice were fasted or received parenteral feeding (Figure 3h).
To exclude the involvement of increased fibrosis or myostatin‐associated hypertrophy in the preservation of muscle mass, we quantified gene expression of fibrogenic genes Col1a1 and S100a4, muscle growth inhibiting factor Mstn, and hypertrophy marker Igf1 (Supplemental Fig. 1 ). These markers were largely unaffected by illness. However, fasted lean CLP mice demonstrated lower Col1a1 and higher Mstn gene expression levels than healthy mice and fasted obese CLP mice (Supplemental Fig. 1). Histological analysis of the muscle in lean and obese CLP mice indicated similar signs of fibrosis (63% and 71%, respectively, P = 0.6) and fasciitis (11% and 6%, respectively, P = 0.8). Also structural abnormalities (58% and 65%, respectively, P = 0.6) and signs of necrosis (16% and 6%, respectively, P = 0.3) were equally observed in lean and obese CLP mice.
Mice study—ectopic triglyceride content
To determine whether an effect on lipid content might contribute to the preservation of muscle mass in obese CLP mice, muscle triglyceride content was quantified. Whereas healthy lean and obese mice had comparable muscle triglyceride contents, muscle triglyceride content was decreased in lean CLP mice, irrespective of nutritional intake (Figure 4a). In contrast, triglyceride content of the muscle was preserved in fed and fasted obese CLP mice (Figure 4a). Furthermore, muscle mass of m. tibialis anterior correlated significantly with muscle triglyceride content (R = 0.498, P = 0.0002). Next, we investigated whether a comparable effect was present in the liver. Healthy lean and obese mice had comparable hepatic triglyceride contents (Figure 4b), confirming the absence of potentially adverse features of a morbid obesity‐associated liver steatosis.30 Liver triglyceride content was decreased in lean CLP mice, irrespective of nutritional intake, whereas it was preserved in fed and fasted obese CLP mice (Figure 4b).
Figure 4.
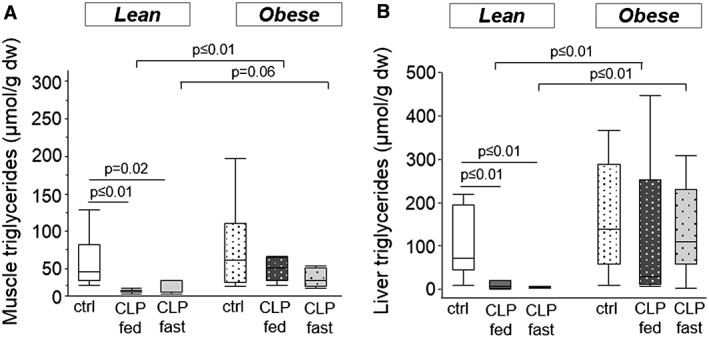
Mice muscle and hepatic triglyceride content. (a) Triglyceride content of skeletal muscle tissue (Mann–Whitney P ≤ 0.01). (b) Hepatic triglyceride content (Mann–Whitney P ≤ 0.01). White, healthy lean mice (n = 8); dark gray, fed lean CLP mice (n = 7); light gray, fasted lean CLP mice (n = 9); white dotted, healthy obese mice (n = 9); dark gray dotted, fed obese CLP mice (n = 10); light gray dotted, fasted obese CLP mice (n = 9). [dw: dry weight, CLP: cecal ligation and puncture, ctrl: healthy control animals, fed: parenterally fed, fast: fasted]
Mice study—markers of fatty acid and glycerol metabolism
The observations of enhanced loss of fat mass (Figure 1b) and preservation of ectopic (muscle and liver) fat (Figure 4) in obese CLP mice suggest that during CLP‐induced critical illness, more fat was mobilized from the adipose tissue of obese CLP mice. Therefore, we subsequently quantified circulating levels of fatty acids and glycerol and markers of hepatic uptake and metabolism of these substrates. Serum fatty acid concentration was not different from controls in lean CLP mice. In contrast, in obese CLP mice, fatty acid serum concentration was decreased (obese healthy control mice 1384.4 ± 221.3 μM vs. obese CLP mice 804.9 ± 76.8 μM; P = 0.007) (Figure 5a). Hepatic gene expression of Cd36, a fatty acid transporter, was markedly increased in obese (healthy, fasted‐CLP, and fed‐CLP) mice compared with lean (healthy, fasted‐CLP, and fed‐CLP) mice (Figure 5b). Hepatic gene expression of Acadl, the first enzyme of β‐oxidation, was comparable in lean, obese, healthy, and CLP mice (data not shown). However, hepatic gene expression of Hmgcs2, encoding for the mitochondrial enzyme that catalyses the first step of ketogenesis, decreased in lean CLP animals (lean healthy control mice 1.0 ± 0.1 vs. lean CLP mice 0.6 ± 0.1; P = 0.01), whereas its expression was unaltered in obese CLP mice (Figure 5c). Thus, obese CLP mice showed higher Hmgcs2 gene expression than lean CLP mice. This difference in gene expression coincided with higher serum ketone body (3‐HB) concentrations in obese CLP mice than in lean CLP mice (Figure 5d).
Figure 5.
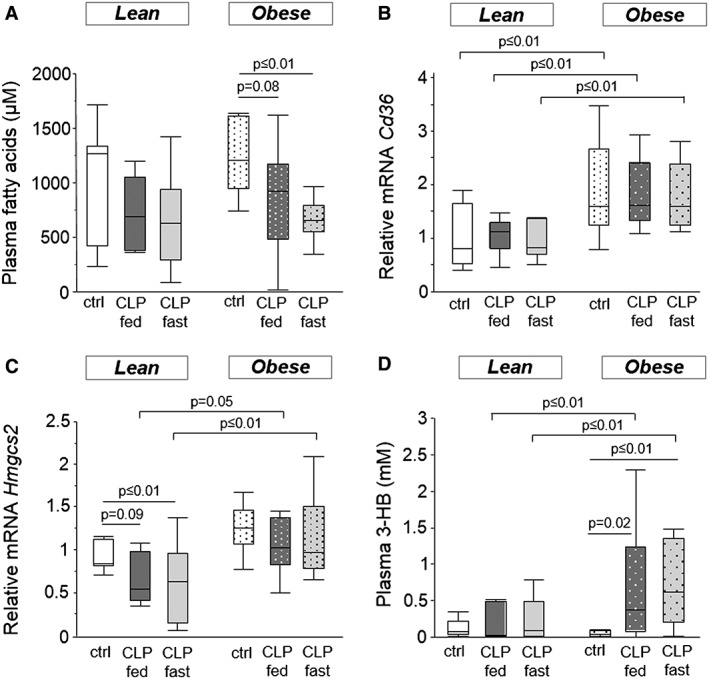
Mice fatty acid metabolism. (a) Serum fatty acid concentration (ANOVA P ≤ 0.01). (b) Relative mRNA expression of Cd36 (ANOVA P ≤ 0.01). (c) Relative Hmgcs2 mRNA expression (ANOVA P ≤ 0.01). (d) Ketone body serum concentration (ANOVA P ≤ 0.01). Gene expression data are expressed normalized to Rn18s gene expression and as a fold change of the mean of the lean healthy controls. White, healthy lean mice (n = 8); dark gray, fed lean CLP mice (n = 7); light gray, fasted lean CLP mice (n = 9); white dotted, healthy obese mice (n = 9); dark gray dotted, fed obese CLP mice (n = 10); light gray dotted, fasted obese CLP mice (n = 9). [CLP: cecal ligation and puncture, ctrl: healthy control animals, fed: parenterally fed, fast: fasted, 3‐HB: 3‐hydroxybutyric acid]
Healthy lean mice had lower serum glycerol concentrations than healthy obese mice. Whereas circulating glycerol was not altered by critical illness in lean mice, glycerol serum concentrations in obese CLP mice were reduced after 5 days of critical illness (Figure 6a). Gene expression of Aqp9, an aquaglyceroporin membrane channel that conducts glycerol into the liver, was unaltered (Figure 6b). However, gene expression of Gk, encoding the rate‐limiting enzyme in the hepatic conversion from glycerol to glucose, was higher in obese compared with lean mice (Figure 6c). Overall, these parameters were not affected by the nutritional intake, neither in lean nor in obese CLP mice (Figures 5 and 6).
Figure 6.
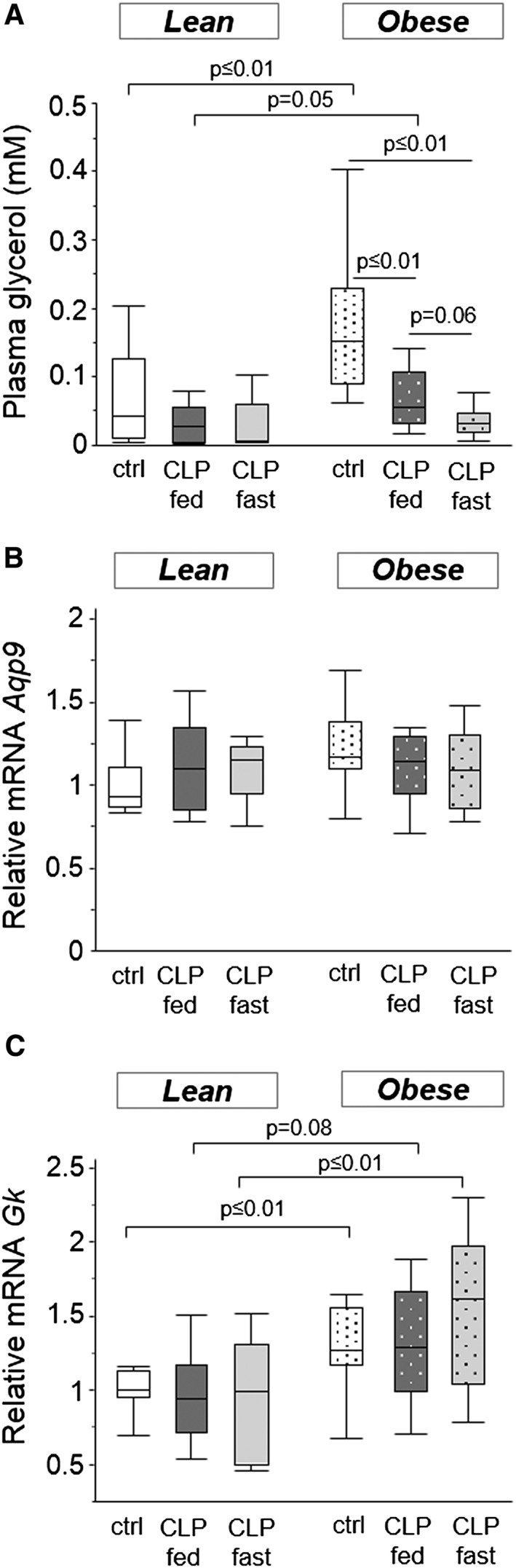
Mice glycerol metabolism. (a) Serum glycerol concentrations (ANOVA P < 0.01). (b) Relative mRNA levels of Aqp9 (ANOVA P = 0.4). (c) Relative mRNA levels of Gk (ANOVA P = 0.04). Gene expression data are expressed normalized to Rn18s gene expression and as a fold change of the mean of the lean healthy controls. White, healthy lean mice (n = 8); dark gray, fed lean CLP mice (n = 7); light gray, fasted lean CLP mice (n = 9); white dotted, healthy obese mice (n = 9); dark gray dotted, fed obese CLP mice (n = 10); light gray dotted, fasted obese CLP mice (n = 9). [CLP: cecal ligation and puncture, ctrl: healthy control mice, fed: parenterally fed, fast: fasted]
Mice study—muscle strength and recovery from fatigue
After 5 days of critical illness, only the lean but not the obese mice lost m. EDL mass (Figure 7a). Lean and obese healthy control mice had comparable twitch tensions (39.2 ± 1.9 mN vs. 43.9 ± 4.6 mN; P = 0.5), tetanic tensions (213.3 ± 9.5 mN vs. 203 ± 15.4 mN; P = 0.4) and fatigue recovery rates (42% vs. 38%; P = 0.2) (Figure 7). Peak twitch tension was unaltered in lean and obese CLP mice (data not shown). However, lean CLP mice demonstrated a lower peak tetanic tension and a lower recovery from fatigue than lean control mice, whereas obese CLP mice maintained peak tetanic tensions and the recovery from fatigue was comparable to obese healthy control mice (Figure 7b–c). Compared with lean CLP mice, obese CLP mice tended to have higher peak tetanic tensions and displayed better recovery from fatigue (Figure 7b–c).
Figure 7.
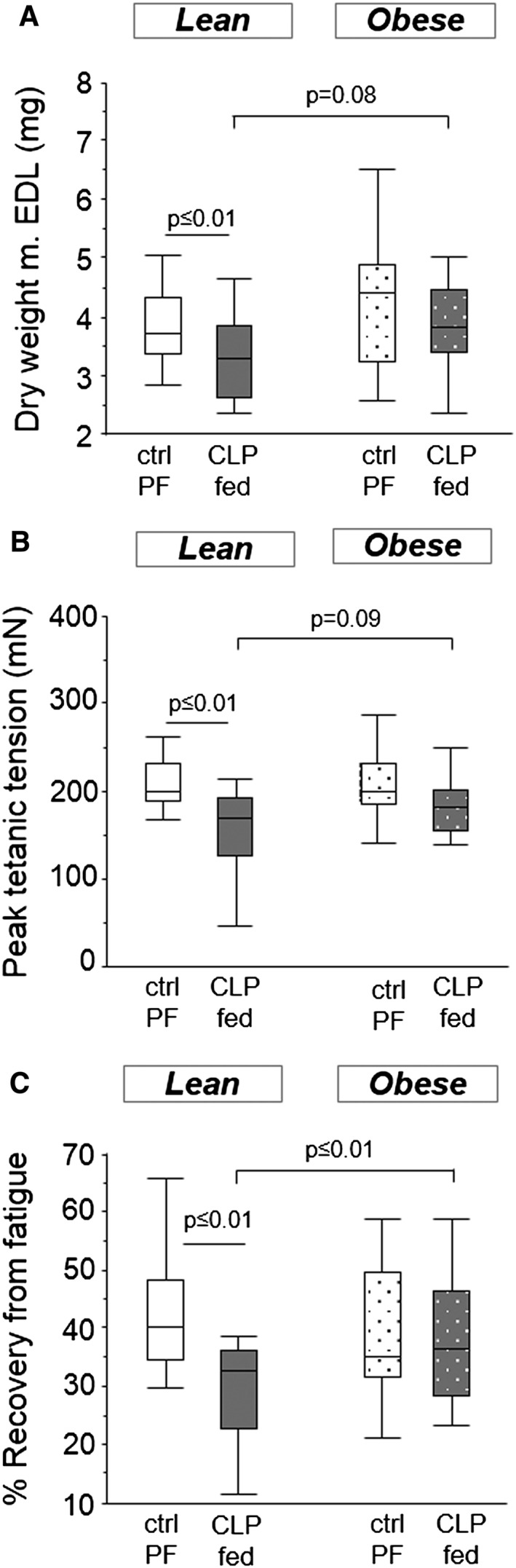
Mice muscle force. Ex vivo force measurements of the m. extensor digitorum longus (EDL). (a) Dry weight (ANOVA P = 0.04). (b) Peak tetanic muscle tensions (ANOVA P ≤ 0.01). (c) Recovery from fatigue after 10 min, as percentage of initial muscle force (ANOVA P ≤ 0.01). White, healthy lean mice, pair‐fed (n = 17); dark gray, fed lean CLP mice (n = 15); white dotted, healthy obese mice, pair‐fed (n = 15); dark gray dotted, fed obese CLP mice (n = 15). [CLP: cecal ligation and puncture, ctrl: healthy control animals, PF, pair‐fed, fed: parenterally fed]
Patient study—muscle wasting and weakness
We next assessed whether attenuation of muscle wasting and muscle weakness was also present in obese/overweight vs. lean prolonged critically ill patients. Myofibre CSA of the m. vastus lateralis was comparable in lean and overweight/obese healthy volunteers. However, myofibres were significantly smaller in lean prolonged critically ill patients than in healthy volunteers, as illustrated by a shift to the left in the histogram of myofibre size distribution (Figure 8a). In contrast, overweight/obese critically ill patients maintained their myofibre size compared with healthy controls, which resulted in larger myofibres compared with lean critically ill patients (Figure 8a). These findings were confirmed in a second set of postmortem m. rectus abdominis biopsies (Figure 8b). Furthermore, fewer overweight/obese prolonged critically ill patients suffered from muscle weakness than lean prolonged critically ill patients (12% vs. 24%: P = 0.004).
Figure 8.
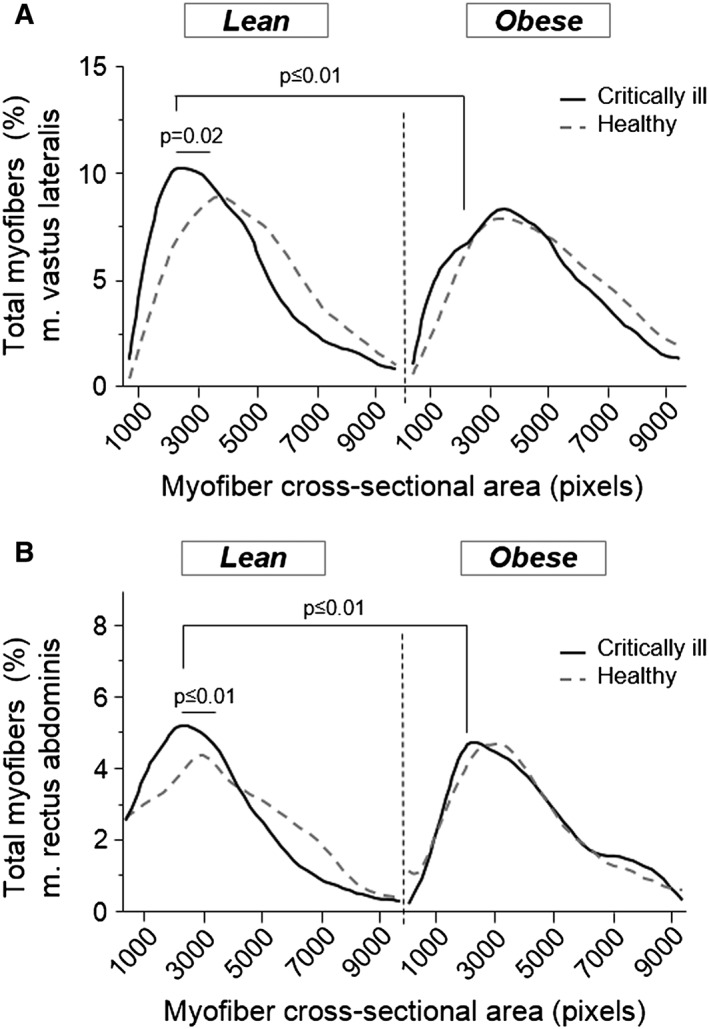
Muscle cross‐sectional area in prolonged critically ill patients. (a) m. vastus lateralis myofibre cross‐sectional area of in vivo biopsies from lean (BMI ≤25; n = 51) and overweight/obese (BMI >25; n = 51) prolonged critically ill patients and lean (n = 11) and overweight/obese (n = 9) healthy controls. (b) m. rectus abdominis myofibre cross‐sectional area of postmortem biopsies from lean (n = 43) and overweight/obese (n = 43) prolonged critically ill patients and lean (n = 4) and overweight/obese (n = 7) healthy controls. Cross‐sectional area is categorized in blocks of 1000 pixels. The graph displays smoothed curves of the percentage myofibres in each category, split up for critically ill patients (black line) and healthy controls (gray dotted line). Statistical difference reflects a change in proportion of small (<2000) myofibres.
Discussion
We here demonstrated in mice and humans that being obese prior to becoming critically ill protected against muscle wasting and weakness. As compared with lean critically ill mice, obese mice showed better preservation of muscle mass and myofibre size, irrespective of whether they were fasted or received parenteral nutrition. Furthermore, obese CLP mice preserved their muscle strength. Obesity, but not nutrition during critical illness, attenuated the loss of lipids and myofibrillary proteins, and increased mobilization and metabolization of fat from adipose tissue. In human muscle biopsies of overweight/obese prolonged critically ill patients, myofibre size appeared more preserved than in lean patients. Moreover, fewer overweight/obese patients suffered from muscle weakness than lean patients, assessed one week after admission to the ICU.
Critical illness is known to induce loss of muscle mass and the development of muscle weakness.8, 10, 11, 31 Although multiple mechanisms underlying muscle wasting and weakness during critical illness have been identified, efficient therapies preventing critical illness‐associated muscle wasting and weakness remain elusive.8, 9, 10, 11 Therefore, the finding that obesity not only protected against lean tissue wasting but also against muscle weakness during critical illness is remarkable.
We observed increased markers of the ubiquitin–proteasome and the autophagy–lysosome pathway in muscle of critically ill mice, in agreement with previous findings in other models.8, 10, 12, 13 Obesity tended to attenuate increased cathepsin activity after 5 days of critical illness. Furthermore, whereas in lean critically ill mice, fasting induced a significant further increase in atrophy markers, this known fasting response32, 33, 34 was absent in obese critically ill mice. In addition to the ubiquitin–proteasome pathway, also insufficiently activated autophagy can play a key role in muscle wasting and the development of muscle weakness.8, 13 Insufficient autophagy activation is characterized by elevated p62 protein levels and an inadequate rise in LC3‐II/LC3‐I ratio.8, 13, 35, 36 We observed p62 accumulation in the presence of an unaltered LC3‐II/LC3‐I ratio and increased expression of autophagy‐related genes Atg5 and Atg7 in muscle of lean and obese CLP mice. Similarly to the findings for the ubiquitin–proteasome pathway, only lean but not obese critically ill animals displayed an additional upregulation in autophagy genes in response to fasting. Histological analysis indicated presence of muscle abnormalities (such as myocyte necrosis, fibrosis, and fasciitis) in our mice model of critical illness consistent with earlier human and rodent observations.37, 38 However, the histological markers as well as gene expression of fibrogenesis and muscle hypertrophy markers were not affected by the presence of obesity during critical illness, suggesting that these pathways did not contribute to the preservation of muscle mass in obese critically ill mice.
The maintenance of muscle proteins and stored ectopic (muscle and liver) triglycerides in obese CLP mice suggests that obese mice, in contrast to lean mice, may use other energy stores during critical illness. The observation that total body fat mass was more reduced during critical illness in obese compared with lean mice, concomitantly with the preservation of ectopic fat, indicates that more lipids were released from adipose tissue stores of obese CLP mice. Theoretically, sufficiently available circulating fatty acids and glycerol for energy consumption would reduce the need for utilization of energy substrates stored in vital organs, such as structural lipids and proteins. However, obese CLP mice did not display higher circulating fatty acids and free glycerol than lean CLP mice. Possibly, we missed a rise in circulating lipids on an earlier time point. On the other hand, obesity has been shown to influence the turnover rate of circulating fatty acids and glycerol, and thus increased metabolization of these substrates by the liver could have decreased their serum concentrations.39, 40 In our mouse study we indeed observed such signs of an elevated fatty acid and glycerol turnover rate in all obese mice, unaffected by nutrition or illness. The increase in circulating ketone bodies in obese CLP mice further indicates enhanced fatty acid metabolism. Obese mice thus appear to have a different metabolic response to prolonged critical illness as compared with lean mice. Together, our findings may suggest that obese critically ill mice preferentially use fat from adipose tissue, while lean critically ill mice may utilize ectopically stored lipids and proteins. Possibly, the combination of mobilizing excess stored triglycerides in adipose tissue, and enhanced metabolism of fatty acids and glycerol from these stores, prevents or decreases the use of stored proteins and triglycerides in muscle tissue during critical illness. Glycerol and fatty acid metabolism can indeed generate vital energy through the production of glucose and ketone bodies.41 In addition, ketone bodies may also directly be involved in the attenuation of muscle wasting, comparable to what has been observed in pancreatic cancer cachexia.42
It was demonstrated earlier that administration of parenteral nutrition to critically ill patients could not attenuate muscle wasting and even aggravated weakness.10, 13, 43 Our observations now suggest that the use of endogenous lipids released from the adipose tissue may counteract muscle wasting. Possibly, infused lipids do not elicit similar metabolic responses as endogenous lipids because of a disturbed uptake of lipids from parenteral nutrition during critical illness. In commercially available parenteral fat emulsions, lipids are organized in artificial chylomicrons that have to be broken down by lipoprotein lipase (LPL) to generate free fatty acids.44, 45 Although the regulation of LPL activity during critical illness is complex, evidence suggests that during sepsis, LPL activity decreases during the initial stages of the disease.46, 47 This could induce inefficient utilization of exogenously administered lipids from parenteral nutrition. Conversely, the utilization of endogenous lipids may be prioritized as the release of lipids stored in the adipose tissue is performed by lipolysis, a process that does not require LPL activity. A study in hypermetabolic septic patients also indicated that parenteral nutrition infusion could not counteract ongoing oxidation of stored fat.48
This study has some limitations. Our mechanistical findings on a shift in mobilization and metabolization of stored fat in obese critically ill mice mainly rely on gene expression data of the enzymes involved and not on flux studies. However, our findings suggest that exogenously administered substrates might not be utilized as efficiently as endogenous substrates during critical illness, which might limit the usability of infusing labelled fatty acid and glycerol tracers to address these questions. Clearly, additional studies are needed to confirm a causal relationship between enhanced mobilization and metabolization of endogenous lipids and the preservation of lean tissue in overweight/obese critically ill patients. Second, we evaluated the effect of premorbid obesity during critical illness in mice, without the presence of obesity‐related comorbidities. The absence of obesity‐related comorbidities was confirmed by comparable skeletal muscle and hepatic triglyceride content, muscle protein content, muscle atrophy markers, muscle mass, and myofibre size in healthy lean and obese mice. However, obesity can induce severe metabolic disturbances which can affect the muscle tissue, as is observed in sarcopenic obesity. Third, one has to be very cautious before extrapolating animal findings to the human setting. Although human muscle mass and function appeared better preserved in overweight/obese than in lean critically ill patients, our findings should be confirmed in other patient cohorts. Furthermore, although human data on muscle mass and weakness were corrected for baseline ICU characteristics, demographics, and presence of diabetes and malignancies, other (unreported) comorbidities might have influenced myofibre size and muscle weakness of the study subjects.
In conclusion, this study showed that being obese when becoming critically ill protects against lean tissue wasting and muscle weakness. Importantly, the administration of parenteral nutrition did not influence muscle wasting. Increased mobilization and metabolization of endogenous fat from the adipose tissue may underlie the preservation of muscle tissue in the obese critically ill mice by preventing the use of ectopically stored lipids and proteins for energy consumption. Furthermore, a less pronounced activation of atrophic pathways in the obese critically ill may also be implicated in the observed preservation of muscle mass and muscle strength.
Author contributions
Conceived and designed the experiments: LL, GVdB, CG, MBM. Conducted the mice experiments: CG, MBM, SD, TD, SET. Acquired data: SVP, CG, MBM, SD, LL, FG, TJ, IV, GH. Contributed materials/analysis tools: KDB, FG, TJ. Analysed data: CG, LL, MBM, IV, GH. Wrote the manuscript: CG, LL, GVdB. All authors read and approved the final manuscript. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia, and Muscle.49
Conflict of interest
The authors have declared that no conflict of interest exists.
Supporting information
Supplemental Fig. 1: Mice skeletal muscle fibrosis and hypertrophy. (A) Relative mRNA expression of fibrogenic gene Col1a1 (ANOVA P ≤ 0.01). (B) Relative mRNA expression of fibrogenic gene S100a4 (Mann–Whitney P = 0.5). (C) Relative mRNA expression of Mstn (ANOVA P ≤ 0.01). (D) Relative mRNA expression of Igf1 (Mann–Whitney P = 0.4). Data are expressed normalized to Rn18s gene expression and as a fold change of the mean of the healthy controls. White, healthy lean mice (n = 8); dark gray, fed lean CLP mice (n = 7); light gray, fasted lean CLP mice (n = 9); white dotted, healthy obese mice (n = 9); dark gray dotted, fed obese CLP mice (n = 10); light gray dotted, fasted obese CLP mice (n = 9). [CLP: cecal ligation and puncture, ctrl: healthy control animals, fed: parenterally fed, fast: fasted]
Supplemental table 1: Baseline and outcome characteristics of matched critically ill patients of whom in vivo muscle biopsies were studied
Supplemental table 2: Baseline and outcome characteristics of matched critically ill patients of whom postmortem muscle biopsies were studied
Supplemental table 3: Baseline and outcome characteristics of matched critically ill patients of whom muscle weakness was assessed by MRC sum score 8 days after ICU admission
Supporting Info Item
Acknowledgements
We thank the control volunteers, patients, and family members for participating in the studies. We thank Annelies Aertgeerts and Inge Derese for excellent technical assistance. GVdB, via the University of Leuven (KU Leuven), receives long‐term structural research support from the Methusalem Program funded by the Flemish Government (METH08/07) and holds a European Research Council Advanced Grant AdvG‐2012‐321670 from the Ideas Program of the European Union seventh framework program. SET received a Research Foundation‐Flanders (FWO) Research Assistant Fellowship.
Goossens, C. , Marques, M. B. , Derde, S. , Vander Perre, S. , Dufour, T. , Thiessen, S. E. , Güiza, F. , Janssens, T. , Hermans, G. , Vanhorebeek, I. , De Bock, K. , Van den Berghe, G. , and Langouche, L. (2017) Premorbid obesity, but not nutrition, prevents critical illness‐induced muscle wasting and weakness. Journal of Cachexia, Sarcopenia and Muscle, 8: 89–101. doi: 10.1002/jcsm.12131.
References
- 1. Vanhorebeek I, Langouche L, Van den Berghe G. Endocrine aspects of acute and prolonged critical illness. Nat Clin Pract Endocrinol Metab 2006;2:20–31. [DOI] [PubMed] [Google Scholar]
- 2. Preiser JC, van Zanten AR, Berger MM, Biolo G, Casaer MP, Doig GS, et al. Metabolic and nutritional support of critically ill patients: consensus and controversies. Crit Care 2015;19:35, doi:10.1186/s13054-015-0737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Puthucheary Z, Harridge S, Hart N. Skeletal muscle dysfunction in critical care: wasting, weakness, and rehabilitation strategies. Crit Care Med 2010;38:S676–S682. [DOI] [PubMed] [Google Scholar]
- 4. Fletcher SN, Kennedy DD, Ghosh IR, Misra VP, Kiff K, Coakley JH, et al. Persistent neuromuscular and neurophysiologic abnormalities in long‐term survivors of prolonged critical illness. Crit Care Med 2003;31:1012–1016. [DOI] [PubMed] [Google Scholar]
- 5. Jolley SE, Bunnell A, Hough CL. Intensive care unit acquired weakness. Chest 2016;doi:10.1016/j.chest.2016.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stevens RD, Dowdy DW, Michaels RK, Mendez‐Tellez PA, Pronovost PJ, Needham DM. Neuromuscular dysfunction acquired in critical illness: a systematic review. Intensive Care Med 2007;33:1876–1891. [DOI] [PubMed] [Google Scholar]
- 7. Schefold JC, Bierbrauer J, Weber‐Carstens S. Intensive care unit‐acquired weakness (ICUAW) and muscle wasting in critically ill patients with severe sepsis and septic shock. J Cachexia Sarcopenia Muscle 2010;1:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Derde S, Vanhorebeek I, Guiza F, Derese I, Gunst J, Fahrenkrog B, et al. Early parenteral nutrition evokes a phenotype of autophagy deficiency in liver and skeletal muscle of critically ill rabbits. Endocrinology 2012;153:2267–2276. [DOI] [PubMed] [Google Scholar]
- 9. Derde S, Vanhorebeek I, Ververs EJ, Vanhees I, Darras VM, Van Herck E, et al. Increasing intravenous glucose load in the presence of normoglycemia: effect on outcome and metabolism in critically ill rabbits. Crit Care Med 2010;38:602–611. [DOI] [PubMed] [Google Scholar]
- 10. Derde S, Hermans G, Derese I, Guiza F, Hedstrom Y, Wouters PJ, et al. Muscle atrophy and preferential loss of myosin in prolonged critically ill patients. Crit Care Med 2012;40:79–89. [DOI] [PubMed] [Google Scholar]
- 11. Puthucheary ZA, Rawal J, McPhail M, Connolly B, Ratnayake G, Chan P, et al. Acute skeletal muscle wasting in critical illness. JAMA 2013;310:1591–1600. [DOI] [PubMed] [Google Scholar]
- 12. Klaude M, Mori M, Tjader I, Gustafsson T, Wernerman J, Rooyackers O. Protein metabolism and gene expression in skeletal muscle of critically ill patients with sepsis. Clin Sci 2012;122:133–142. [DOI] [PubMed] [Google Scholar]
- 13. Hermans G, Casaer MP, Clerckx B, Guiza F, Vanhullebusch T, Derde S, et al. Effect of tolerating macronutrient deficit on the development of intensive‐care unit acquired weakness: a subanalysis of the EPaNIC trial. Lancet Respir Med 2013;1:621–629. [DOI] [PubMed] [Google Scholar]
- 14. Pickkers P, de Keizer N, Dusseljee J, Weerheijm D, van der Hoeven JG, Peek N. Body mass index is associated with hospital mortality in critically ill patients: an observational cohort study. Crit Care Med 2013;41:1878–1883. [DOI] [PubMed] [Google Scholar]
- 15. Fonarow GC, Srikanthan P, Costanzo MR, Cintron GB, Lopatin M, Committee ASA, et al. An obesity paradox in acute heart failure: analysis of body mass index and inhospital mortality for 108,927 patients in the Acute Decompensated Heart Failure National Registry. Am Heart J 2007;153:74–81. [DOI] [PubMed] [Google Scholar]
- 16. Akinnusi ME, Pineda LA, El Solh AA. Effect of obesity on intensive care morbidity and mortality: a meta‐analysis. Crit Care Med 2008;36:151–158. [DOI] [PubMed] [Google Scholar]
- 17. Hogue CW Jr, Stearns JD, Colantuoni E, Robinson KA, Stierer T, Mitter N, et al. The impact of obesity on outcomes after critical illness: a meta‐analysis. Intensive Care Med 2009;35:1152–1170. [DOI] [PubMed] [Google Scholar]
- 18. Peake SL, Moran JL, Ghelani DR, Lloyd AJ, Walker MJ. The effect of obesity on 12‐month survival following admission to intensive care: a prospective study. Crit Care Med 2006;34:2929–2939. [DOI] [PubMed] [Google Scholar]
- 19. Abhyankar S, Leishear K, Callaghan FM, Demner‐Fushman D, McDonald CJ. Lower short‐ and long‐term mortality associated with overweight and obesity in a large cohort study of adult intensive care unit patients. Crit Care 2012;16:R235, doi:10.1186/cc11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khalid U, Ather S, Bavishi C, Chan W, Loehr LR, Wruck LM, et al. Pre‐morbid body mass index and mortality after incident heart failure: the ARIC Study. J Am Coll Cardiol 2014;64:2743–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mohebi R, Simforoosh A, Tohidi M, Azizi F, Hadaegh F. Obesity paradox and risk of mortality events in chronic kidney disease patients: a decade of follow‐up in tehran lipid and glucose study. J Ren Nutr 2015;doi:10.1053/j.jrn.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 22. Lavie CJ, De Schutter A, Alpert MA, Mehra MR, Milani RV, Ventura HO. Obesity paradox, cachexia, frailty, and heart failure. Heart Fail Clin 2014;10:319–326. [DOI] [PubMed] [Google Scholar]
- 23. Suneja M, Kumar AB. Obesity and perioperative acute kidney injury: a focused review. J Crit Care 2014;29:694 e1–694 e6. [DOI] [PubMed] [Google Scholar]
- 24. Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, et al. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6 J and A/J mice. Metab Clin Exp 1995;44:645–651. [DOI] [PubMed] [Google Scholar]
- 25. Marques M, Perre S, Aertgeerts A, Derde S, Guiza F, Casaer MP, et al. Critical illness induces nutrient‐independent adipogenesis and accumulation of alternatively activated tissue macrophages. Crit Care 2013;17:R193, doi:10.1186/cc12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Janssens T, Antanas L, Derde S, Vanhorebeek I, Van den Berghe G, Guiza Grandas F. Charisma: an integrated approach to automatic H&E‐stained skeletal muscle cell segmentation using supervised learning and novel robust clump splitting. Med Image Anal 2013;17:1206–1219. [DOI] [PubMed] [Google Scholar]
- 27. Langouche L, Vanhorebeek I, Vlasselaers D, Vander Perre S, Wouters PJ, Skogstrand K, et al. Intensive insulin therapy protects the endothelium of critically ill patients. J Clin Invest 2005;115:2277–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Casaer MP, Mesotten D, Hermans G, Wouters PJ, Schetz M, Meyfroidt G, et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med 2011;365:506–517. [DOI] [PubMed] [Google Scholar]
- 29. Gayat E, Pirracchio R, Resche‐Rigon M, Mebazaa A, Mary JY, Porcher R. Propensity scores in intensive care and anaesthesiology literature: a systematic review. Intensive Care Med 2010;36:1993–2003. [DOI] [PubMed] [Google Scholar]
- 30. Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kress JP, Hall JB. ICU‐acquired weakness and recovery from critical illness. N Engl J Med 2014;370:1626–1635. [DOI] [PubMed] [Google Scholar]
- 32. Fuentes EN, Ruiz P, Valdes JA, Molina A. Catabolic signaling pathways, atrogenes, and ubiquitinated proteins are regulated by the nutritional status in the muscle of the fine flounder. PLoS One 2012;7:e44256, doi:10.1371/journal.pone.0044256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015;14:58–74. [DOI] [PubMed] [Google Scholar]
- 34. Jagoe RT, Lecker SH, Gomes M, Goldberg AL. Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J 2002;16:1697–1712. [DOI] [PubMed] [Google Scholar]
- 35. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy‐deficient mice. Cell 2007;131:1149–1163. [DOI] [PubMed] [Google Scholar]
- 36. Kadowaki M, Karim MR. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol 2009;452:199–213. [DOI] [PubMed] [Google Scholar]
- 37. Puthucheary ZA, Phadke R, Rawal J, McPhail MJ, Sidhu PS, Rowlerson A, et al. Qualitative ultrasound in acute critical illness muscle wasting. Crit Care Med 2015;43:1603–1611. [DOI] [PubMed] [Google Scholar]
- 38. Hill NE, Saeed S, Phadke R, Ellis MJ, Chambers D, Wilson DR, et al. Detailed characterization of a long‐term rodent model of critical illness and recovery. Crit Care Med 2015;43:e84–e96. [DOI] [PubMed] [Google Scholar]
- 39. Bjorntorp P, Bergman H, Varnauskas E. Plasma free fatty acid turnover rate in obesity. Acta Med Scand 1969;185:351–356. [DOI] [PubMed] [Google Scholar]
- 40. Karsenty C, Ulmer M, Chanussot F, Ratanasavanh R, Debry G. Paradoxical effect of ethanol on liver lipogenesis in the genetically‐obese Zucker rat. Br J Nutr 1985;54:15–20. [DOI] [PubMed] [Google Scholar]
- 41. Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev 1980;60:143–187. [DOI] [PubMed] [Google Scholar]
- 42. Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab 2014;2:18, doi:10.1186/2049-3002-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vanhorebeek I, Van den Berghe G. Hormonal and metabolic strategies to attenuate catabolism in critically ill patients. Curr Opin Pharmacol 2004;4:621–628. [DOI] [PubMed] [Google Scholar]
- 44. Ferezou J, Bach AC. Structure and metabolic fate of triacylglycerol‐ and phospholipid‐rich particles of commercial parenteral fat emulsions. Nutrition 1999;15:44–50. [DOI] [PubMed] [Google Scholar]
- 45. Carpentier YA, Deckelbaum RJ. In vivo handling and metabolism of lipid emulsions. World Rev Nutr Diet 2015;112:57–62. [DOI] [PubMed] [Google Scholar]
- 46. Gouni I, Oka K, Etienne J, Chan L. Endotoxin‐induced hypertriglyceridemia is mediated by suppression of lipoprotein lipase at a post‐transcriptional level. J Lipid Res 1993;34:139–146. [PubMed] [Google Scholar]
- 47. Lanza‐Jacoby S, Sedkova N, Phetteplace H, Perrotti D. Sepsis‐induced regulation of lipoprotein lipase expression in rat adipose tissue and soleus muscle. J Lipid Res 1997;38:701–710. [PubMed] [Google Scholar]
- 48. Askanazi J, Carpentier YA, Elwyn DH, Nordenstrom J, Jeevanandam M, Rosenbaum SH, et al. Influence of total parenteral nutrition on fuel utilization in injury and sepsis. Ann Surg 1980;191:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6: 315–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1: Mice skeletal muscle fibrosis and hypertrophy. (A) Relative mRNA expression of fibrogenic gene Col1a1 (ANOVA P ≤ 0.01). (B) Relative mRNA expression of fibrogenic gene S100a4 (Mann–Whitney P = 0.5). (C) Relative mRNA expression of Mstn (ANOVA P ≤ 0.01). (D) Relative mRNA expression of Igf1 (Mann–Whitney P = 0.4). Data are expressed normalized to Rn18s gene expression and as a fold change of the mean of the healthy controls. White, healthy lean mice (n = 8); dark gray, fed lean CLP mice (n = 7); light gray, fasted lean CLP mice (n = 9); white dotted, healthy obese mice (n = 9); dark gray dotted, fed obese CLP mice (n = 10); light gray dotted, fasted obese CLP mice (n = 9). [CLP: cecal ligation and puncture, ctrl: healthy control animals, fed: parenterally fed, fast: fasted]
Supplemental table 1: Baseline and outcome characteristics of matched critically ill patients of whom in vivo muscle biopsies were studied
Supplemental table 2: Baseline and outcome characteristics of matched critically ill patients of whom postmortem muscle biopsies were studied
Supplemental table 3: Baseline and outcome characteristics of matched critically ill patients of whom muscle weakness was assessed by MRC sum score 8 days after ICU admission
Supporting Info Item