

Abstract
Aims: Among the putative mechanisms proposed to be common factors in Down syndrome (DS) and Alzheimer's disease (AD) neuropathology, deficits in protein quality control (PQC) have emerged as a unifying mechanism of neurodegeneration. Considering that disturbance of protein degradation systems is present in DS and that oxidized/misfolded proteins require polyubiquitinylation for degradation via the ubiquitin proteasome system, this study investigated if dysregulation of protein polyubiquitinylation is associated with AD neurodegeneration in DS.
Results: Postmortem brains from DS cases before and after development of AD neuropathology and age-matched controls were analyzed. By selectively isolating polyubiquitinated proteins, we were able to identify specific proteins with an altered pattern of polyubiquitinylation as a function of age. Interestingly, we found that oxidation is coupled with polyubiquitinylation for most proteins mainly involved in PQC and energy metabolism.
Innovation: This is the first study showing alteration of the polyubiquitinylation profile as a function of aging in DS brain compared with healthy controls. Understanding the onset of the altered ubiquitome profile in DS brain may contribute to identification of key molecular regulators of age-associated cognitive decline.
Conclusions: Disturbance of the polyubiquitinylation machinery may be a key feature of aging and neurodegeneration. In DS, age-associated deficits of the proteolytic system may further exacerbate the accumulation of oxidized/misfolded/polyubiquitinated proteins, which is not efficiently degraded and may become harmful to neurons and contribute to AD neuropathology. Antioxid. Redox Signal. 26, 280–298.
Keywords: : Alzheimer disease, Down syndrome, proteasome, proteomics, trisomy21, ubiquitin
Introduction
Down syndrome (DS) is the most frequent genetic cause of intellectual disability caused by the presence of three copies of chromosome 21 (Chr21). Several studies reveal that DS and Alzheimer's disease (AD) neuropathology have many common features that include deposition of senile plaques and neurofibrillary tangles, together with cellular dysfunction such as mitochondrial defects, increased oxidative stress, and impairment of protein quality control (PQC) (4, 27, 51, 53, 57). Around two-thirds of individuals with DS develop dementia in their 50s, but the severity of neuropathology and dementia varies significantly among DS population. Triplication of APP is considered the major pathological event to which converge both AD-DS and normal AD, but it is clear that several other genes contribute to the neurodegenerative process. Moreover, understanding the cross-talk between genetic and environmental factors could provide insights into the molecular mechanisms responsible of early onset AD in DS. Thus, DS offers a unique model to investigate the early molecular changes that disturb neuronal homeostasis and that with age lead slowly, but irreversibly, to neuronal death [reviewed in Wiseman et al. (72)].
Innovation.
Ubiquitinylation is a post-translation modification that controls several intracellular processes, among which regulation of protein half-life is the most well recognized. Recent evidence suggests that genetically defined shifts in the global ubiquitinylation pattern of the proteome might initiate age-specific response programs. Innovation of the present study is the finding that disturbance of polyubiquitinylation may contribute to neurodegeneration in Down syndrome (DS) brain and transition to Alzheimer's disease (AD) phenotype. Determination and better understanding of the onset of the altered ubiquitome profile in DS brain may contribute to identification of key molecular regulators of age-associated cognitive decline in DS, which ultimately culminates in the development of AD.
The ability of neurons to compensate for the increasing burden of age-dependent stresses largely relies on the efficiency of proteolytic systems (32). A wide set of proteases regulate cell functions, but the main proteolytic systems responsible for the removal of proteins and organelles are the proteasome (ubiquitin-proteasome system [UPS]) and the endosomal–lysosomal system (24). The UPS regulates the levels of cytosolic and nuclear proteins, while the endosomal–lysosomal system largely is responsible for the degradation of secretory and internalized proteins.
Several studies demonstrate that UPS integrity is required for normal life span. Identifying ubiquitin-dependent degradation pathways that specifically control the steady-state levels of life span regulators further highlights a key role of the UPS during aging and in the development of age-related diseases (68). In line with these observations, recent evidence suggests that a genetically defined alteration in the ubiquitinylation profile of the proteome might promote age-specific response programs (39). In accord with this view, results obtained in worms suggested proteostasis failure as a possible programmed event that causes accumulation of damaged/misfolded proteins at an early time of life and might represent the first event presaging aging (3).
Degradation of a protein via the ubiquitin (Ub) pathway requires two successive steps: (i) covalent binding of multiple ubiquitin moieties to the protein substrate and (ii) proteolytic degradation of the targeted protein by the 26S proteasome system, with the release of free ubiquitin. Moreover, protein post-translational modification with ubiquitin, namely ubiquitinylation, regulates a wide range of processes within cells, not just protein degradation. Ub can be attached to substrate proteins as a single unit or more often as polymeric chains, in which additional ubiquitins are bound through specific isopeptide linkages (41). Ub is tethered to proteins via the action of three proteins: E1 Ub-activating enzymes, E2 Ub-conjugating enzymes, and E3 Ub-ligating enzymes. This enzymatic cascade results in the formation of an isopeptide linkage between the C-terminus of Ub and the ɛ-amino group of a substrate lysine residue. Analogous to protein glycosylation, once Ub is anchored to a substrate protein, additional rounds of conjugation lead to polymeric Ub (poly-Ub) chains.
The conjugation of a poly-Ub chain to a target protein was initially characterized as a signal for protein degradation (30), which still appears to be its dominant role. However, poly-Ub can trigger multiple other functions, including cell cycle, transcription, DNA repair, and membrane trafficking, depending on which lysine residue in the Ub sequence is used to extend the poly-Ub chain. Distinct chain linkages can be formed at all seven internal lysine residues of Ub (K6, K11, K27, K29, K33, K48, and K63) and at its N-terminus. In addition to homotypic Ub chains, with single linkage type, heterotypic chains can also be present containing mixed linkages within the same Ub chain. The most abundant and well-characterized chain linkage is through K48 and is considered to be a key signal for proteasomal degradation, while K63 linkage has a primary role in cell signaling (41, 44). Moreover, recent data also show that the K63 poly-Ub chain may be recognized by the proteasome (42).
The impairment of ubiquitin-dependent proteolysis results in enhanced accumulation of damaged proteins, which can be toxic and become harmful for cellular homeostasis. Recent studies from our group and others demonstrated that disturbance of proteolytic machinery is a key feature of degenerating neurons and appears in several neurodegenerative disorders (4, 8, 15). Proteasome activity was found to be reduced in AD brains than in age-matched controls (38, 50) and also in DS brain (17). In addition, high levels of Ub were detected in brain and cerebrospinal fluid samples from AD patients (43). Moreover, protein inclusions in AD brains generally contain ubiquitinated proteins and are specifically immunoreactive to at least one of the 19S regulatory proteasome subunits, S6b (22).
Understanding the onset of the altered ubiquitome profile in DS brain as a function of age may contribute to the identification of key molecular regulators of age-associated cognitive decline. Recent progress in proteomic approaches has contributed to the identification of targets of ubiquitinylation, but a comprehensive profile of age-related changes of the ubiquitome is still lacking. We analyzed the human postmortem brain from DS cases before and after the development of AD neuropathology (Down syndrome with Alzheimer's disease [DS/AD]). By selectively isolating polyubiquitinated proteins, we identified specific proteins showing an altered pattern of polyubiquitinylation as a function of aging. Considering our previous results showing a disturbance of a key component of the PQC in DS, our findings may contribute to identification of molecular events that may drive the neurodegenerative process in DS brain.
Results
We focused on frontal cortex of human postmortem brain from DS cases before and after the development of AD neuropathology (DS/AD; Table 1). Considering that the development of AD pathology is age dependent in DS and shows characteristic features of AD, identification of common polyubiquitinated proteins by proteomic approaches in both DS and AD can provide novel insights on the overlapping mechanisms that may lead from normal aging to the development of AD. The age of DS cases was under 40 years, while the cases with both DS and AD were over the age of 40 years. Thus, controls were divided into two groups, either less than or equal to 40 years (Ctr Y) or older than 40 years (Ctr O) at death.
Table 1.
Subjects' Demographic Data
| Subjects | PMI | Age | Sex | Race | Cause of death |
|---|---|---|---|---|---|
| Control Y 1 | 5.8 | 39 | Female | Unknown | Unknown |
| Control Y 2 | 12 | 22.8 | Male | African American | Arrhythmia due to hypertrophy cardiomyopathy |
| Control Y 3 | 8 | 33.1 | Male | Caucasian | Cardiac arrhythmia |
| Control Y 4 | 10 | 24.4 | Male | Caucasian | Multiple injuries |
| Control Y 5 | 10 | 10.8 | Female | Caucasian | Asthma |
| Control Y 6 | 14 | 19.8 | Male | Caucasian | Multiple injuries |
| DS 1 | 12 | 19 | Male | Caucasian | Unknown |
| DS 2 | 14 | 15.5 | Male | Caucasian | Chromosome disorder, Trisomy 21 |
| DS 3 | 10 | 40.6 | Male | African American | HCVD |
| DS 4 | 12 | 39.2 | Female | Caucasian | Cancer |
| DS 5 | 13 | 44.5 | Female | Caucasian | Cardiac arrhythmia |
| DS 6 | 14 | 19.9 | Male | Indian | Cardiopulmonary arrest: congenital heart disease |
| DS-AD 1 | 5.3 | 57 | Female | Unknown | Seizure disorder |
| DS-AD 2 | 3 | 63 | Female | Unknown | Respiratory |
| DS-AD 3 | 6 | 63 | Female | Unknown | Unknown |
| DS-AD 4 | 4.5 | 55 | Male | Unknown | Pneumonia |
| DS-AD 5 | 10.5 | 61 | Male | Unknown | Unknown |
| DS-AD 6 | 3 | 57 | Female | Unknown | Pneumonia |
| Control O 1 | 5 | 47.3 | Female | Caucasian | Pneumonia |
| Control O 2 | 8 | 64 | Female | Unknown | Myocardial infarction |
| Control O 3 | 17 | 56.8 | Male | Caucasian | HACVD |
| Control O 4 | 16 | 55.3 | Male | Caucasian | Arteriosclerotic cardiovascular disease |
| Control O 5 | 4.5 | 65 | Male | Unknown | Cardiac arrest |
| Control O 6 | 2.7 | 67 | Male | Unknown | Cardiomyopathy |
AD, Alzheimer's disease; DS, Down syndrome; HACVD, hypertensive arteriosclerotic cardiovascular disease; HCVD, hypertensive cardiovascular disease; PMI, postmortem interval.
Specificity of ubiquitin enrichment kit: isolation of endogenously ubiquitinylated protein complexes
The aim of this study was to analyze the profile of polyubiquitinated proteins in brain homogenates from DS patients before and after development of AD. To achieve this aim, we selected a specific Ub enrichment kit, the only one available for efficient isolation of endogenously polyubiquitinated protein complexes, not mono-, and suitable for mass spectrometry (MS)-based proteomics. To test the specificity of the kit employed for this study, we performed a number of experiments. The workflow for isolating endogenously ubiquitinylated protein complexes for proteomic analysis is shown in Figure 1. First, immunoprecipitation (IP) using anti-Ub antibody (provided by the kit) was performed to pull down ubiquitinylated proteins (Fig. 1A). The IP fraction (bound fraction, BF) and the supernatant (nonbound fraction, NBF) were blotted with anti-Ub antibody (Fig. 1, A1 and A2). Subsequently, the NBF—containing all the proteins lacking Ub modification—was mixed with the polyubiquitin affinity resin for evaluating nonspecific protein binding (false-positive hits). Figure 1B shows the NBF eluted from the affinity resin (total protein stain) and analyzed using anti-Ub antibody. The null signal observed excludes nonspecific binding of nonubiquitinated proteins by the resin. In addition, the BF was passed through the polyubiquitin affinity resin (Fig. 1, B1 and B2). The resin binds polymers of Ub containing four or more Ub subunits. Therefore, monoubiquitinated proteins and short-chain polymers are recovered in the flow-through (FT), while the polyubiquitinated (longer than four Ub subunits) proteins were collected in the eluate.
FIG. 1.

Isolation of endogenously polyubiquitinated protein complexes by use of the ubiquitin enrichment kit. A workflow for isolating endogenously ubiquitinylated protein complexes for proteomic analysis is shown. (A) Ubiquitinylated proteins were isolated from brain homogenates by IP. IP fraction (BF) and the supernatant (NBF) were loaded on an SDS-PAGE gel and stained for total protein expression (A1) and subsequently blotted on nitrocellulose membrane and stained with a polyclonal anti-Ub antibody (anti-Ub Ab) to determine enrichment efficiency (A2). (B) The IP fractions (BF and NBF) were further processed with the enrichment kit (Thermo) containing the polyubiquitin affinity resin (Poly-Ub AR). BF, NBF, and FT were loaded in SDS page (B1) and blotted with antiubiquitin antibody (B2). (C) Heat denaturation of high-affinity resin using high temperature (60°) was used to destroy the bond between resin and poly-Ub antibody. Poly-Ub was detected by performing a Western blot (agarose-only control; C1). BF, bound fraction; FT, flow-through; IP, immunoprecipitation; NBF, nonbound fraction; Poly-Ub, polymeric Ub; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; Ub, ubiquitin.
Furthermore, the specificity of the ligand to recognize polyubiquitinated proteins in human samples has been demonstrated by breaking the interaction between ligand and substrate. Specifically, samples were passed through heat-denatured resin and processed by Western blot (agarose resin-only negative control) as shown in Figure 1C.
Polyubiquitinylation levels
Figure 2A shows an increase of poly-Ub-conjugated proteins in both DS and DS/AD subjects compared with their age-matched controls (fold increase for DS vs. Ctr Y is 1.4 and DS/AD vs. Ctr O is 2.5). Moreover, the total polyubiquitinylation levels increased significantly between DS and DS/AD (1.6-fold).
FIG. 2.

Poly-Ub levels. (A) Total levels of poly-Ub-bound proteins in DS (n = 6/group) and DS/AD (n = 6/group) cases compared with their age-matched controls (n = 6/group). (B, C) Increased poly-Ub of two types of ubiquitin chains [K63 (B) and K48 (C)] in both DS (n = 6/group) and DS/AD (n = 6/group) cases compared with their age-matched controls (n = 6/group). Representative bands are shown and protein levels (A−C, upper bands) were normalized per total protein load, named total load (A−C, lower bands). Densitometric values are shown as percentage of Ctr Y set as 100%. Mean ± SEM (DS vs. Ctr y *p < 0.05; DS/AD vs. DS **p < 0.01; DS/AD vs. Ctr O ***p < 0.001; DS vs. Ctr Y and DS/AD vs. DS [for K48] *p < 0.05; DS vs. Ctr Y, DS/AD vs. Ctr O, and DS/AD vs. DS [for K63] **p < 0.01; Ctr O vs. Ctr Y #p < 0.05 one-way ANOVA). AD, Alzheimer's disease; ANOVA, analysis of variance; DS, Down syndrome; DS/AD, Down syndrome with Alzheimer's disease; SEM, standard error of the mean.
Based on these results, we examined the two most prominent types of poly-Ub chains: K63 and K48. Our results showed the same trend for K63 and K48 ubiquitin chains (Fig. 2B, C): the poly-Ub K63 and K48 displayed, respectively, an increase of 1.6 and 2.1 in DS compared with Ctr Y and 2 and 3.5 in DS/AD compared with Ctr O age-matched controls. A significant increase for both K63 and K48 was also evident by comparing DS/AD versus DS (Fig. 2B, C).
Proteomic profile of polyubiquitinated proteins
We further investigated the polyubiquitinated proteome in the four groups to identify specific proteins showing different levels of polyubiquitinylation. Brain (frontal cortex) homogenates (n = 6 per group) were processed using a specific kit for the isolation of intracellular poly-Ub-conjugated proteins. Through the use of a high-binding affinity resin, polyubiquitinated proteins were isolated from brain samples, and the bound proteins were eluted from the affinity resin and separated on two-dimensional (2D) gels. In Figure 2, a representative 2D map of polyubiquitinated proteins (Fig. 3B) compared with a 2D expression gel (Fig. 3A) is shown. The second 2D expression gel was run in parallel to normalize variation of polyubiquitinylation levels with respect to specific protein levels.
FIG. 3.
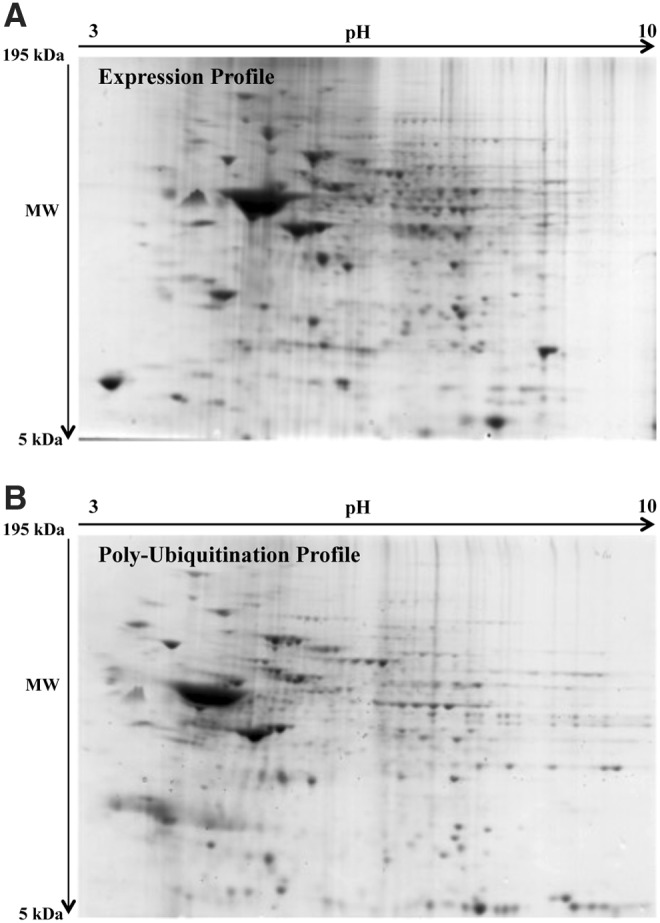
Proteomic profile of representative 2D blots. (A) Representative SYPRO Ruby-stained expression gel from a DS case. (B) SYPRO Ruby-stained polyubiquitinated gel in a DS case. SYPRO Ruby-stained gel images were obtained using a Chemidoc MP System at different times of exposure for the expression of a poly-Ub profile. 2D, two-dimensional.
The intensities of polyubiquitinated protein spots showed a different profile in both DS/AD and DS compared with their respective matched controls (Fig. 4). The poly-Ub analyses identified 16 polyubiquitinated spots (normalized to specific protein levels) in the frontal cortex of DS and DS/AD (Fig. 4 and Table 2). These identified proteins are discussed in the following text.
FIG. 4.

Proteomic profile of representative 2D gels with proteins differentially polyubiquitinated in Ctr Y and Ctr O cases versus DS and DS/AD cases. The comparison (A) is shown for DS versus Ctr Y; (B) between DS/AD and DS; (C) between DS/AD and Ctr O; and (D) between Ctr O and Ctr Y. The spot numbers are reported in Table 2.
Table 2.
List of Identified Polyubiquitinated Proteins by Mass Spectrometry
| Spot | SSP | Uni Prot N° | N° peptides | MW | Protein | DS vs. Ctr Ya | DS/AD vs. Ctr Oa | DS/AD vs. DSa | Ctr O vs. Ctr Ya |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3408 | P12277 | 3 | 42.64 | Creatine kinase B-type | 2.11 | 2.28 | ||
| Q99961/2 | 2 | 41.49 | Endophilin-A2 and -A1 | 2.11 | 2.28 | ||||
| 2 | 7511 | P25705 | 3 | 59.75 | ATP synthase subunit alpha, mitochondrial | 1.67 | |||
| 3 | 3202 | P32119 | 6 | 21.88 | Peroxiredoxin-2 | 2.59 | 3.45 | 2.04 | |
| 4 | 3203 | D6R956 | 2 | 26.84 | Ubiquitin carboxyl-terminal hydrolase L1 | 2.75 | 3.8 | 2.05 | |
| 5 | 6303 | P04083 | 2 | 38.71 | Annexin A1 | 2.96 | |||
| 6 | 4613 | Q16555 | 3 | 62.29 | Dihydropyrimidinase-related protein 2 | 27.34 | 4.5 | ||
| P25705 | 3 | 59.75 | ATP synthase subunit alpha | 27.34 | 4.5 | ||||
| P21281 | 2 | 56.5 | V-type proton ATPase subunit B, brain isoform | 27.34 | 4.5 | ||||
| 7 | 3605 | P10809 | 7 | 61.05 | 60 kDa heat shock protein | 7.22 | |||
| 8 | 4811 | Q16891 | 15 | 82.62 | Mitochondrial inner membrane protein | 2 | |||
| P06396 | 8 | 85.69 | Gelsolin | 2 | |||||
| P46459 | 9 | 82.59 | Vesicle-fusing ATPase | 2 | |||||
| A2A274 | 7 | 87.82 | Aconitate hydratase | 2 | |||||
| 9 | 2402 | P60709/P63261 | 2 | 41.73 | Actin cytoplasmic 1 and 2 | 4.5 | |||
| 10 | 5202 | P30041 | 2 | 25.03 | Peroxiredoxin-6 | 25.03 | |||
| 11 | 2603 | Q13509 | 3 | 50.43 | Tubulin beta-3 chain and -6 chain | 0.32 | |||
| P04350 | 2 | 49.58 | Tubulin beta-4A | 0.32 | |||||
| P30153 | 3 | 65.3 | Serine/threonine protein phosphatase 2A | 0.32 | |||||
| Q9UMX0 | 2 | 62.51 | Ubiquilin-1 | 0.32 | |||||
| 12 | 3409 | Q13885 | 3 | 49.9 | Tubulin beta-2A | 0.17 | 0.22 | ||
| P31930 | 3 | 52.64 | Cytochrome b-c1 complex subunit 1 | 0.17 | 0.22 | ||||
| Q16352 | 4 | 55.39 | Alpha-internexin | 0.17 | 0.22 | ||||
| Q9Y2T3 | 2 | 52.83 | Guanine deaminase | 0.17 | 0.22 | ||||
| F5H5D3 | 3 | 57.73 | Tubulin alpha-1B chain | 0.17 | 0.22 | ||||
| 13 | 4505 | P30101 | 4 | 56.78 | Protein disulfide isomerase A3 | 5.12 | |||
| P21281 | 3 | 56.5 | V-type proton ATPase subunit B, brain isoform | 5.12 | |||||
| 14 | 5608 | P55809 | 2 | 56.16 | Succinyl-CoA:3- transferase 1 | 0.48 | |||
| O43175 | 3 | 56.65 | D-3-phosphoglycerate dehydrogenase | 0.48 | |||||
| 15 | 1207 | B7Z596 | 4 | 31.75 | Tropomyosin alpha-1 chain | 2.13 | |||
| P06753 | 2 | 28.95 | Tropomyosin alpha-3 chain | 2.13 | |||||
| P61981 | 5 | 28.3 | 14-3-3 protein gamma | 2.13 | |||||
| 16 | 6602 | P04075 | 14 | 45.26 | Fructose bisphosphate aldolase A | 3.06 | |||
| P09972 | 11 | 39.45 | Fructose bisphosphate aldolase C | 3.06 | |||||
| P24752 | 6 | 45.20 | Acetyl-CoA acetyltransferase, mitochondrial | 3.06 | |||||
| P62333 | 7 | 44.17 | 26S protease regulatory subunit 10B | 3.06 |
The fold of increase/decrease of polyubiquitylation of a specific protein, in the groups of analysis, is indicated in the last four columns.
ATP, adenosine triphosphate; Ctr O, control old; Ctr Y, control young; DS/AD, Down syndrome with Alzheimer's disease; MW, molecular weight; SSP, standard spot.
Down syndrome versus Ctr Y
Comparing DS with Ctr Y cases, proteins with greater polyubiquitinylation were identified as creatine kinase B-type (CKB) and endophilin (SH3) A2 and A1 with 2.11-fold increase; adenosine triphosphate (ATP) synthase α (FoF1 complex) with 1.67-fold increase; peroxiredoxin-2 (PRDX2) with 2.59-fold increase; ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCH-L1) with 2.75-fold increase; Annexin A1 (ANXA1) with 2.96-fold increase; dihidropyrimidinase-related protein 2 (DRP-2, also known as collapsin response-mediated protein-2, CRMP-2), ATP synthase α (FoF1 complex), V-type proton ATPase (V0 ATPase) B and 60 kDa heat shock protein (HSP60) with 7.22-fold increase; and mitochondrial inner membrane protein, gelsolin, vesicle-fusing ATPase, and aconitate hydratase with twofold increase.
Down syndrome with Alzheimer's disease versus Ctr O
In this second comparison of DS/AD with Ctr O, there were three spots in common with the previous comparison (DS vs. Ctr Y): CKB and SH3 A2 and A1 with 2.28-fold increased polyubiquitinylation; PRDX2 with 3.45-fold increase, and UCH-L1 with 3.8-fold increased polyubiquitinylation. The proteins found to be polyubiquitinated exclusively in this comparison were tropomyosin alpha-1 and 3 chain, 14-3-3 protein gamma with a 2.13-fold increase; fructose-bisphosphate aldolase A (FBA A), acetyl-CoA acetyltransferase, and mitochondrial 26S protease regulatory subunit 10B with 3.06-fold increase. On the other hand, a group of proteins showed decreased levels of Poly-Ub: tubulin beta-2, cytochrome b-c1 complex subunit 1 (UQCRC 1), alpha-internexin, guanine deaminase, and tubulin alpha-1B chain with 0.17-fold decrease.
Down syndrome with Alzheimer's disease versus DS
We next compared DS without AD pathology (DS) with the DS with AD pathology groups (DS/AD). This comparison allowed us to detect proteins modified as a function of AD neuropathology in DS. Four spots were increasingly polyubiquitinated in DS/AD: PRDX2 with 2.04-fold increase; UCH-L1 with 2.05-fold increase; and protein disulfide isomerase A3 and V0 ATPase B with 5.12-fold increase. The first two proteins are in common with DS versus Ctr Y. Some proteins showed decreased polyubiquitinylation: tubulin beta-3 chain and -6 chain, tubulin beta-4, serine/threonine protein phosphatase 2A, and ubiquilin-1, 2, and 4 with 0.32-fold increase. In addition, another group of spots showed decreased polyubiquitinylation that is in common with DS/AD and Ctr O: tubulin beta-2, cytochrome b-c1 complex subunit 1 (UQCRC 1), alpha-internexin, guanine deaminase, and tubulin alpha-1B chain with 0.22-fold decreased polyubiquitinylation.
Ctr O versus Ctr Y
The comparison between the Ctr Y versus Ctr O groups was used to distinguish age effects from AD neuropathology-dependent differences in polyubiquitinylation observed in DS. The following proteins showed increased polyubiquitinylation: DRP-2, ATP synthase α (FoF1 complex), V0 ATPase B, adenylyl cyclase-associated proteins 1–2, and PRDX6 with 4.5-fold increase; tubulin beta-3 and -6 chain with 25-fold increase.
Validation experiments
To confirm that the enrichment method employed indeed isolated polyubiquitinated proteins, we performed two further analyses. First, we employed mUbiSiDa—a comprehensive database of ubiquitinylation sites in mammals (11, 60). This online database collects only ubiquitinylation sites with experimental evidence and can be a valuable resource for the study of ubiquitinylation in mammals, especially in humans. All the identified proteins were searched on mUbiSiDa and the list of ubiquitinylation sites for each protein is reported in Table 3.
Table 3.
MUbiSiDa Search
| Protein | Ub sitesa |
|---|---|
| Creatine kinase B-type | K304; K307; K223; K156; K313; K298; K11; K265; K242 |
| Endophilin-A2 and -A1 | K239 |
| ATP synthase subunit alpha, mitochondrial | K305; K175; K531; K261; K126; K230; K161; K434; K316; K498; K539; K194 |
| Peroxiredoxin-2 | K119; K16; K92; K26; K135 |
| Ubiquitin carboxyl-terminal hydrolase L1 | K4; K71; K115; K157; K83; K78; K135 |
| Annexin A1 | K71; K98; K97; K312; K128; K287; K166; K257; K185; K250; K81; K53; K58; K214; K26; K29; K90 |
| Dihydropyrimidinase-related protein 2 | K472 |
| ATP synthase subunit alpha | K305; K175; K531; K261; K126; K230; K161; K434; K316; K498; K539; K194 |
| V-type proton ATPase subunit B, brain isoform | K64; K108 |
| 60 kDa heat shock protein | K96; K233; K469; K160; K72; K133; K75; K359; K352; K250; K292; K191; K218; K417; K58; K125; K481 |
| Mitochondrial inner membrane protein | K516; K101; K581; K506; K110; K451; K640 |
| Gelsolin | K533; K322; K677; K317; K373; K728; K368; K584 |
| Vesicle-fusing ATPase | K529 |
| Aconitate hydratase | K395 |
| Actin cytoplasmic 1 and 2 | K326; K68; K50; K84; K291; K191; K113; K213; K315; K215; K61; K328 |
| Peroxiredoxin-6 | K125; K199; K63; K182; K209 |
| Tubulin beta-3 chain and -6 chain | K252; K324; K216; K336; K58; K297; K350; K362; K379 |
| Tubulin beta-4A | K252; K324; K216; K336; K297; K350; K362 |
| Serine/threonine protein phosphatase 2A | K305; K307; K272; K546; K475; K472; K202; K163; K280; K133; K107; K467; K194; K255; K188; K561; K266; K485; K416; K542 |
| Ubiquilin-1 | K70; K83; K47; K45 |
| Tubulin beta-2A | K103; K252; K324; K216; K336; K58; K297; K350; K362; K379 |
| Cytochrome b-c1 complex subunit 1 | K225 |
| Alpha-internexin | K95; K438; K216; K288; K290 |
| Guanine deaminase | K259; K53; K189; K267 |
| Tubulin alpha-1B chain | K234; K233; K130; K182; K166; K194 |
| Protein disulfide isomerase A3 | K305; K425; K296; K335; K94; K104; K362 |
| V-type proton ATPase subunit B, brain isoform | K64; K108 |
| Succinyl-CoA:3- transferase 1 | K176; K286; K293; K296; K511b |
| D-3-phosphoglycerate dehydrogenase | K69; K289; K394; K33; K21; K129; K351; K364; K58; K380; K146; K384 |
| Tropomyosin alpha-1 chain | K82 |
| Tropomyosin alpha-3 chain | K169; K223; K82; K215; K228; K13; K181 |
| 14-3-3 protein gamma | K152; K50; K69; K142; K127; K162; K10; K125 |
| Fructose bisphosphate aldolase A | K101; K99; K140; K200; K42; K312; K111; K230; K108; K14; K318; K153; K322; K294; K147; K208; K330; K28 |
| Fructose bisphosphate aldolase C | K115; K234; K295; K417; K198; K227; K287 |
| Acetyl-CoA acetyltransferase, mitochondrial | K174; K181 |
| 26S protease regulatory subunit 10B | K34; K274; K48; K168; K206; K322; K20; K7; K197; K298; K383; K180 |
Second, three proteins were selected for validation of the proteomics results. These proteins were immunoprecipitated and then probed by specific antibodies for the two Ub chains: K63 and K48.
Ubiquitin carboxyl-terminal hydrolase isozyme L1
UCH-L1 is involved in the processing of Ub precursors and ubiquitinylated proteins. The proteomic results for UCH-L1 indicated an increased polyubiquitinylation in DS and DS/AD compared with their respective controls and also when comparing DS/AD with DS. We previously reported that UCH-L1 was increasingly carbonylated and also HNE modified in DS and DS/AD (17, 19) brains and in brains from AD cases (7). Data from the validation analysis on UCH-L1 confirmed the elevated poly-Ub K63 in DS and DS/AD compared with their age-matched controls (2- and 1.6-fold, Fig. 5A). Similarly, an increase of poly-Ub K48 between DS and DS/AD (1.5-fold, Fig. 5B) was also observed, while no significant differences were found between the groups on total UCH-L1 protein levels (data not shown).
FIG. 5.

UCH-L1. (A, B) All samples (n = 6/group) were immunoprecipitated with anti-UCH-L1. Immunoprecipitated proteins were separated on SDS-PAGE, transferred on nitrocellulose membranes, and probed with anti-poly-UbK63 (A, upper bands) and anti-poly-UbK48 (B, upper bands). (A) Shows increased levels of poly-UbK63 bound to UCH-L1 in DS and DS/AD compared with their matched controls. (B) Shows an increase in poly-UbK48 bound to UCH-L1 in DS/AD compared with DS. All the IP experiments were normalized on the total amount of UCH-L1 (indicated as Expr.; A, B, lower bands). Representative bands are shown. Densitometric values are shown as percentage of Ctr Y set as 100%. Mean ± SEM (DS vs. Ctr Y [for K63], DS/AD vs. DS [for K48] *p < 0.05; DS/AD vs. Ctr O [for K63] **p < 0.01 one-way ANOVA). UCH-L1, ubiquitin carboxyl-terminal hydrolase isozyme L1.
Gelsolin
Gelsolin is a structural protein and a key regulator of actin filament formation and degradation, depending on its phosphorylation state. A recent study showed that gelsolin exerts protective effects in AD transgenic mice because it has the ability to bind Aβ, thus inhibiting its aggregation into fibrils (36). Our results confirmed an ∼60% increased level of poly-Ub K63 gelsolin in DS compared with Ctr Y (Fig. 6A). In contrast, poly-Ub K48 gelsolin was decreased ∼30% in DS compared with the Ctr Y, which is opposite to poly-UbK63 (Fig. 6B). No differences in total gelsolin protein levels were observed (data not shown). A recent phosphoproteomic study from our laboratory of brains from cases with preclinical AD, amnestic mild cognitive impairment (MCI), and late-state AD identified decreased phosphorylation of gelsolin associated with decreased activity of cytoskeletal remodeling properties of this protein in all stages of AD (70).
FIG. 6.

Gelsolin. (A, B) All samples (n = 6/group) were immunoprecipitated with anti-gelsolin. Immunoprecipitated proteins were separated on SDS-PAGE, transferred on nitrocellulose membranes, and probed with anti-poly-UbK63 (A, upper bands) and anti-poly-UbK48 (B, upper bands). (A) Shows increased level of poly-UbK63 bound to gelsolin in DS compared with Ctr Y, an opposite trend for poly-UbK48 bound to gelsolin in DS compared with Ctr Y is shown (B). All the IP experiments were normalized on the total amount of gelsolin (indicated as Expr.; A, B, lower bands). Representative bands are shown. Densitometric values are shown as percentage of Ctr Y set as 100%. Mean ± SEM (DS vs. Ctr Y [for K63] **p < 0.01; DS vs. Ctr Y [for K48] *p < 0.05 one-way ANOVA).
Ubiquilin-1
Ubiquilin-1 mediates the proteasomal targeting of misfolded or accumulated proteins for degradation by binding to poly-Ub chains and by interacting with proteasome. We found significantly reduced polyubiquitinylation on ubiquilin-1 in DS/AD compared with DS. Data from the validation analysis confirmed a decrease in poly-Ub K63 by ∼50% in DS/AD compared with DS (Fig. 7B). In contrast, poly-UbK48 ubiquilin-1 displayed an opposite trend to poly-Ub K63, that is, an increase (∼30%) in DS/AD and Ctr O. In addition, the poly-Ub K48 modification was increased in DS/AD compared with DS (Fig. 7C). Furthermore, total ubiquilin-1 protein levels were significantly decreased by ∼50% in DS and DS/AD compared with their age-matched controls (Fig. 7A).
FIG. 7.

Ubiquilin-1. (A) Expression levels of ubiquilin-1 are significantly decreased in DS (n = 6/group) and DS/AD (n = 6/group) specimens compared with their age-matched controls (n = 6/group). (B, C) All samples (n = 6/group) were immunoprecipitated with anti-ubiquilin-1. Immunoprecipitated proteins were separated on SDS-PAGE, transferred on nitrocellulose membranes, and probed with anti-poly-UbK63 (B) and anti-poly-UbK48 (C). (B) Shows a decrease in poly-Ub K63 bound to ubiquilin-1 (indicated as Poly-UbK63, first bands) in DS/AD compared with DS, in contrast poly-UbK48 bound to Ubiquilin-1 (indicated as Poly-UbK48, second bands) displays an opposite trend compared with poly-Ub K63 (C). All the IP experiments were normalized on the total amount of ubiquilin-1 (indicated as Expr.; third bands). In the upper side of the figure, representative bands are shown. Densitometric values are shown as percentage of Ctr Y set as 100%. Mean ± SEM (DS vs. Ctr Y and DS/AD vs. Ctr O *p < 0.05; DS/AD vs. Ctr O [K48] *p < 0.05; DS/AD vs. DS [K48] *p < 0.05; DS/AD vs. DS [K63] **p < 0.01 one-way ANOVA).
Proteasome activity
An increase of poly-Ub proteins is associated with decreased proteasomal function (47). To investigate the functionality of the UPS, we measured proteasome activity. In particular, we analyzed chymotrypsin-like, trypsin-like, and caspase-like protease activities. Our results showed that all three activities were significantly reduced in DS and DS/AD compared with their age-matched controls (Fig. 8). In addition, chymotrypsin-like and trypsin-like proteasome activity assays were decreased by ∼20% in DS cases compared with Ctr Y (Fig. 8A, B) as well as caspase-like activities showing ∼30% decreased activity (Fig. 8C). In DS/AD versus Ctr O, a significant decrease in all three proteasome activities was observed (Fig. 8A−C), while no significant differences were detected between DS/AD compared with the DS group.
FIG. 8.

Proteasome activities. Chymotrypsin-like (A), trypsin-like (B), and caspase-like (C) proteasome activities are reduced in DS and DS/AD compared with their age-matched controls. Proteasome function was evaluated by enzymatic assay in Ctr Y and Ctr O cases (n = 6/group), DS with and without AD cases (n = 6/group). Fluorescence intensity (Arbitrary Units, AU) as percentage of Ctr Y set as 100%. Mean ± SEM (DS vs. Ctr y and DS/AD vs. Ctr O *p < 0.05; DS vs. Ctr Y **p < 0.01 one-way ANOVA).
Taken together, these results indicate that the proteasome function is reduced in DS before development of AD neuropathology. Thus, there appears to be an altered proteostasis network early in DS brain that would suggest reduced protein degradation and accumulation of misfolded/polyubiquitinated proteins (3).
Discussion
After middle age, the majority of DS individuals develop neuropathology, with characteristic hallmarks of AD, such as deposition of senile plaques and neurofibrillary tangles. Among the putative mechanisms proposed to be common factors in DS and AD neuropathology, defects in PQC have emerged as a unifying mechanism triggering neurodegeneration (4). Previous studies from our group demonstrate impairments of the proteasome system (17) and the reduction of autophagosome function in DS, before and after the development of AD, which suggests that accumulation of oxidized/misfolded protein is an early hallmark in DS brain (59). We showed that some members of the PQC are oxidatively modified in young DS cases before the development of AD and that oxidation of target proteins possibly leads to impaired activity of clearance systems (19). We hypothesize that reduced protein degradation contributes to the accumulation of polyubiquitinated proteins, which, in turn, may act to disturb other cellular functions.
Recent studies suggest that polyubiquitinylation is required for both proteasomal and endosomal–lysosomal degradation (42). Thus, the regulation of ubiquitinylation and deubiquitinylation machinery may play a crucial role in several pathologies associated with a defective PQC, among which neurodegenerative diseases are major representatives. Moreover, disrupted Ub signaling is likely to have broad consequences for neuronal function, including regulation of degradative pathways, endocytic trafficking, inflammation, and DNA repair. Interestingly, poly-Ub accumulates as a consequence of increased oxidative stress levels, possibly due to increased Ub conjugation and also perturbations in protein degradation (66). Several studies have demonstrated that elevated oxidative stress is already present in fetal DS brain (51, 56, 75) and in the amniotic fluid collected from women carrying a fetus with DS (58).
Consistent with these reports, the current study shows that the accumulation of polyubiquitinated proteins, either K48 or K63 localized, is present in younger cases with DS without significant AD pathology, suggesting an early aging phenotype. Considering that Ub has seven lysines in its structure, positioned at amino acid residues, 6, 11, 27, 29, 33, 48, and 63, poly-Ub chains of different structural topology can be synthetized. All chain types are present in both yeast and mammalian cells, although with varying abundance for each different linkage type and the atypical chains are approximately from 10% to 50% [reviewed in Kulathu and Komander (44)]. While the functional roles of poly-K48 and poly-K63 have been well recognized, little is known about the structure/function of the other different lysine types (48). Recent data obtained both in yeast and mammalian cells demonstrate that Lys11 chains signal proteasomal degradation (74). In addition, recent findings highlight the novel role of Lys11 linkages in cell cycle regulation and Met1 linkages in nuclear factor B activation (34, 71).
Little is known about the specific role of the three residues, Lys27, Lys29, and Lys33, which are the closest in the sequence space and are quite complicated for MS analysis. So far, although Lys27 corresponds to ∼10% of all ubiquitinylation events, the function of Lys27-linked polymers has not been clarified, at least in yeast (33). Several studies have shown the involvement of both Lys29- and Lys33-linked chains in multiple cellular processes, such as the regulation of kinase activity of AMPK protein family. Members of this family show specificity for polyubiquitinylation at Lys29 l and Lys33, which is responsible of inhibiting kinase activity, but not associated with its degradation (33). However, the complexity of these events has not yet been elucidated. As well, the Lys6 modification is currently unclear. MS data show that similar to Lys63 chains, the extent of Lys6 modifications does not follow proteasome inhibition (40), suggesting a nonproteolytic role.
The specific amino acid residue that undergoes polyubiquitinylation thus establishes the structural and biological properties of the corresponding chain. Although the above described chain types have a role in both yeast and mammalian cells, their abundance seems to correlate with the specific cell state and environment (1). Further investigation is needed to unravel the physio/pathological role of atypical ubiquitinylation.
We focused our attention on the currently best-characterized role of Lys48 and Lys63 poly-Ub chains, although keeping in mind that ongoing research in this field may suggest novel physiological mechanisms.
Our finding is in agreement with data obtained from DS fetal fibroblasts, suggesting propensity to a progeroid phenotype (23), and fits with the current view that a genetically defined shift in the global ubiquitinylation pattern of the proteome might initiate age-specific brain pathology and deficits (39). By applying a proteomic approach in brain samples purified by affinity chromatography for poly-Ub binding, we were able to identify brain proteins showing an altered pattern of polyubiquitinylation in DS cases before and after the development of AD neuropathology (Fig. 9). Intriguingly, a large number of polyubiquitinated proteins are also carbonylated and/or HNE modified in DS brain as assessed using redox proteomic approaches (Fig. 10) (19).
FIG. 9.

Pie chart representing polyubiquitinated proteins grouped according to their function. The table shows the list of MS/MS-identified proteins with increased poly-Ub among the different groups of analysis. MS, mass spectrometry.
FIG. 10.

Overlap between oxidative modification and polyubiquitinylation of selected proteins in Down syndrome.
By comparing DS versus age-matched controls (Ctr Y), we found that increased polyubiquitinylation is coupled with increased oxidation for PRDX2, HSP60, UCH-L1, V0-ATPase, ATP synthase, CK, and DRP-2. In addition, UCH-L1 and PRDX2 showed increased polyubiquitinylation in DS/AD versus DS and DS/AD versus Ctr O, suggesting a progressive alteration as a function of both aging and trisomy 21.
PRDX2 is oxidized in DS brain [21] and it also displays increased polyubiquitinylation in DS and DS/AD versus their respective age-matched controls. Previous studies demonstrate that PRDX2 protein levels are decreased in DS fetal brains compared with controls (29, 64). Furthermore, a number of studies show decreased antioxidant enzymes in the brains of DS cases, including glutathione peroxidase (GPX) and thioredoxins (25) and an altered superoxide dismutase 1 (SOD-1)/GPX activity ratio (16). Accordingly, intellectual disability in DS has been associated with increased oxidative stress and increased susceptibility to apoptosis, thus ultimately leading to neuronal death (28, 54, 69). Accordingly, cells from DS or an animal model of DS are vulnerable to oxidative insult and have reduced proliferation ability (23, 69).
Selective recognition of oxidized/misfolded proteins by molecular chaperones (HSPs) is the first step toward their elimination. We previously showed that HSP60 is oxidatively modified and now show that it is increasingly polyubiquitinated, thus suggesting impaired function. Similarly, HSP60 is oxidatively modified in AD brain (6). Furthermore, immunohistochemical and Western blot analyses in AD brain tissue show that expression levels of a number of HSPs were elevated in affected regions (12, 26). Moreover, it has been proposed that chaperones may recognize misfolded proteins and in turn regulate their polyubiquitinylation by directly recruiting an E3 ligase (21).
Unfolded/damaged proteins that are not refolded by chaperones are tagged for degradation by the proteasome. UCH-L1, one of the members of the proteasomal system, is itself a major target of oxidative damage and it undergoes aberrant polyubiquitinylation during neurodegeneration. UCH-L1 is quite abundant in the brain and acts to hydrolyze Ub chains from the carboxyl terminus to allow the protein targeted for degradation to gain access to the proteasome, and it also plays a key role in maintaining free Ub levels in neurons (46, 55). Mutations and deficiencies of UCH-L1 in humans have been demonstrated to occur in neurodegenerative diseases such as Parkinson's disease and AD (65). UCH-L1, shown in the current study to have increased polyubiquitinylation, was previously identified to be carbonylated in AD and DS brains associated with loss of activity (4, 5). Consistent with reduced UCH-L1 function and UCH-L1 polyubiquitinylation/carbonylation, we also observed decreased proteasomal activities both in DS and DS/AD compared with their respective age-matched controls.
However, in addition to UPS, cells have an endosomal–lysosomal system to remove damaged proteins, and recent studies show that oxidized proteins undergo not only proteasomal but also endosomal–lysosomal degradation (42, 49). In the current study, we found that V0 ATPase is both oxidized and polyubiquitinated (DS vs. Ctr Y and Ctr O vs. Ctr Y), and we hypothesize that these modifications decrease the V0-ATPase proton pump activity consistent with autophagy dysfunction. Accordingly, the Nixon group has demonstrated reduced acidification of lysosomes in AD (73), and genetic mutation of lysosomal ATPase is among the risk factors recognized to contribute to autophagy-related neurodegenerative diseases (37, 52). Furthermore, disturbance of autophagosome formation coupled with hyperactivation of the mTOR pathway is observed in DS brain, before and after development of AD pathology (59). Our current observation that V0-type ATPase is increasingly polyubiquitinated in DS/AD brain compared with DS brain suggests that this protein accumulates with aging in DS.
The integrity of the PQC relies (to varying extents) on adequate levels of ATP since chaperones, UPS, and autophagy are ATP-dependent processes (4). The DS brain is characterized by defects in mitochondrial function (13) and altered glucose metabolism (45), which ultimately culminates in decreased ATP synthesis. We and other groups showed that these alterations are common features of both DS and AD brain and also other neurodegenerative diseases (4). Several enzymes involved in energy metabolism, including CK, ATP synthase, aconitate hydratase, and FBA, are oxidatively modified and polyubiquitinated, likely exerting impaired activity.
The presence of an aging phenotype in cases with DS is also confirmed by comparing Ctr O versus Ctr Y, which provides information regarding alteration of proteins as a function of normal age. We found that the proteins with increased polyubiquitinylation in young versus old controls are also modified in DS. Putative markers of this accelerated aging phenotype are DRP-2, ATP synthase, and V0-ATPase. Previous results show that the level of DRP-2 is decreased in fetal DS brain (2) and increasingly polyubiquitinated both in DS versus Ctr Y and Ctr O versus Ctr Y in the current study. Furthermore, in adult DS brain, the level of DRP-2 is reduced either at protein l or at mRNA level; we also found this protein to be oxidatively modified (19). Indeed, DS is associated with a group of clinical manifestations of accelerated aging (31) that may ultimately culminate in the development of dementia.
We also noted some interesting alterations in gelsolin as a target of polyubiquitinylation in DS. Gelsolin is a cytoskeletal protein that is present both in the cytoplasm and also secreted in biological fluids. Previous studies show an antiamyloidogenic role of gelsolin in AD (10). We previously reported that gelsolin is increasingly carbonylated in the cerebral spinal fluid from both MCI and AD samples (18) and, as noted above, has decreased phosphorylation throughout all stages of AD (69). These findings suggest that alteration of gelsolin may be a consistent feature of AD, and we suggest that this alteration is already evident in young DS brain before the appearance of AD neuropathology. Accordingly, Ji et al. found a correlation between cleaved gelsolin forms and levels of amyloid β-peptide in the frontal cortex from DS subjects (35, 36).
When comparing cases with DS and AD (DS/AD) with DS without AD, we observe not only increased polyubiquitinylation of PRDX2, UCH-L1, DRP-2, and V0-ATPase, as discussed previously, but also proteins showing decreased levels of poly-Ub chains, including tubulin (different isoforms), alpha-internexin, Ser/Thr PP2A phosphatase, ubiquilin, succinyl CoA transferase, D-phosphoglycerate dehydrogenase, and programmed cell death-interacting protein. This trend may be explained in light of published data showing defective ubiquitinylation of proteins in AD due to lower levels of E1 (50). However, although there is growing evidence that polyubiquitinated proteins accumulate with aging and neurodegeneration, there is less evidence of the role played by ubiquitinylating/deubiquitinylating enzymes that also may contribute to our observations. We also would emphasize that all of the proteins with decreased polyubiquitinylation were also not previously identified as oxidatively modified, in contrast to the proteins with increased polyubiquitinylation. Thus, oxidation may be a modification to proteins that leads to polyubiquitinylation, while those proteins not vulnerable to oxidation appear to be less polyubiquitinated. However, our experimental approach does not differentiate which Lys modifications are most prominent (K48 or K63), meaning the finding of decreased polyubiquitinylation may require further experiments to understand the mechanism.
Ubiquilin, known to play a critical role in protein degradation, showed decreased polyubiquitinylation in the current study. The N-terminal sequence of ubiquilin shows similarity with Ub—the ubiquitin-like domain (UBL)—that is followed by a variable central domain and terminates with a conserved 50 amino acid sequence called ubiquitin-associated domain (UBA) (63). Ubiquilin-1 acts as a shuttle to deliver polyubiquitinated proteins to the proteasome or to autophagosomes, or promotes aggresome formation to avoid accumulation of misfolded proteins. The level of ubiquilin-1 is decreased in both DS and DS/AD compared with their respective age-matched controls and we observed a decreased of K63 poly-Ub levels. Single-nucleotide polymorphisms in the ubiquilin-1 gene are likely a risk factor for late-onset AD (20). Stieren et al. demonstrated that overexpression of ubiquilin-1 reduces the Aβ42/40 ratio and prevents APP-induced cellular toxicity (67). Similarly, in AD patients, ubiquilin-1 protein levels are inversely correlated with Braak stage, suggesting that therapies that restore ubiquilin-1 expression levels in the brain may serve as a therapeutic approach for AD (67).
Finally, we compared DS/AD versus age-matched controls to examine changes in poly-Ub as a marker of AD neuropathology specifically and not age. We identified that proteins involved in energy metabolism, antioxidant responses, the cytoskeleton, and synaptic plasticity have altered ubiquitinylation. Some of these proteins were found to be polyubiquitinated in DS before the development of AD neuropathology, while others show a significant change when AD neuropathology is present.
In summary, the disturbance of polyubiquitinylation may be engaged in DS before the appearance of AD neuropathology. However, the genetic underpinnings to explain these findings have yet to be determined. Studies in people with DS show that the overexpression of Chr21 may affect disomic genes as well, thus making the scenario further complicated (62). In the presence of reduced proteasomal activity and impaired autophagy, oxidized/misfolded proteins tagged by poly-Ub chains may accumulate and contribute to a toxic environment for neurons in DS. Furthermore, considering that oxidation/polyubiquitinylation triggers protein aggregation, our findings also are consistent with the notion that there may be deposition of other protein aggregates that may, or not, colocalize/interact with plaques and tangles. Emerging studies are showing that in addition to Aβ and tau, there are many other active players in the formation of protein aggregates. Understanding the composition and chemical properties of identified polyubiquitinated proteins may provide novel insights into the pathogenesis, and potential treatments, of protein deposition disorders, including DS and AD.
Materials and Methods
Subjects
All the human brain samples were obtained from the University of California-Irvine-ADRC Brain Tissue Repository, the Eunice Kennedy Shriver NICHD Brain and Tissue Bank for Developmental Disorders, and the University of Kentucky ADC. Table 1 shows the characteristics and demographic data of the included subjects in the study. DS cases were divided into two groups, with or without sufficient pathology for a neuropathology diagnosis of AD. The age of DS cases is under 40, while the cases with both DS and AD were over the age of 40 years. Likewise, controls were split into two groups: Controls Young (Ctr Y), which match with DS because they are ≤45 years; Controls Old (Ctr O), which match with DS/AD because they are older than 45 years at death. All the results obtained from human autopsy samples were analyzed by considering the difference in postmortem interval (PMI) among groups. Correlation analysis did not show any association between all the reported measures and PMI (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/ars).
A subset of these autopsy cases was used in previous experiments measuring insoluble Aβ, total oxidation, and autophagy pathway as a function of age in DS (9, 19, 59).
Sample preparation
Brain tissues (frontal cortex, around 20 mg per sample) from DS, DS/AD, and controls (n = 6 per group) were thawed in Media 1 lysis buffer (pH 7.4) containing 320 mM sucrose, 1% of 990 mM Tris-HCl (pH = 8.8), 0.098 mM MgCl2, 0.076 mM EDTA, and the proteinase inhibitors, leupeptin (0.5 mg/ml), aprotinin (0.5 mg/ml), and PMSF (40 μg/ml). Homogenates were centrifuged at 14,000 × g for 10 min to remove debris. The brains were homogenized by 20 passes of a Wheaton tissue homogenizer and sonicated. The resulting homogenate was centrifuged at 14,000 × g for 10 min to remove cellular debris. The supernatant was extracted to determine the total protein concentration by the BCA method (Pierce, Rockford, IL).
Polyubiquitin affinity resin
A Thermo Scientific Ubiquitin Enrichment Kit [89899] was used for the isolation and study of intracellular poly-Ub-modified proteins. Through the use of a high-binding affinity resin, polyubiquitinated proteins are isolated from tissue lysates. The specificity of the resin to recognize polyubiquitinated proteins in human samples has been demonstrated (Fig. 1). Specifically, representative DS samples were immunoprecipitated (protocol described in the Immunoprecipitation section) using antiubiquitin antibody (provided by the kit, dilution = 1:100). All proteins ubiquitinated and nonubiquitinated were identified, respectively, in the IP fraction (BF) and the supernatant (NBF). An aliquot of the BF was used to evaluate the specificity of the antiubiquitin antibody in IP by Western blot by using a different antiubiquitin antibody (1:500; Santa Cruz Biotechnology, Dallas, TX). Subsequently, an aliquot of BF and NBF was mixed with the polyubiquitin affinity resin (as described below) for evaluating nonspecific protein binding (false-positive hits). Subfractions (eluate and FT) from BF and NBF passed on the polyubiquitin affinity resin were analyzed by Western blot and the membranes were incubated with antiubiquitin antibody (1:500; Santa Cruz Biotechnology). Furthermore, the specificity of the ligand to recognize polyubiquitinated proteins in human samples was demonstrated by breaking the interaction between ligand and substrate, through heat-denatured resin (60°C), and processed by Western blot (agarose resin-only negative control; Fig. 1D).
Isolation of polyubiquitinated proteins
The polyubiquitin affinity resin binds polymers of ubiquitin containing four or more ubiquitin subunits. Monoubiquitinated proteins and short-chain polymers (<4 ubiquitin monomers) are discarded in the FT and not evaluated by our experimental procedure. This is the first study employing 2D electrophoresis directly on proteins eluted from the affinity resin using an isoelectric focusing (IEF) buffer. Explicitly, fresh homogenated samples were incubated with resin (20 μl) in the spin columns overnight at 4°C on an end-over-end rotator (binding of polyubiquitinated proteins). The second day, the columns with samples were immediately centrifuged 5000 × g for 15 s, then columns were washed three times with wash buffer (0.025 M Tris, 0.15 M NaCl; pH 7.2) at 5000 × g for 15 s. At the end of washes, the columns were incubated with samples in IEF buffer for 2 h at 4°C, then centrifuged at 5000 × g for 30 s. The sample elute contained ubiquitin-enriched fractions ready for IEF.
Two-dimensional electrophoresis
For the first-dimension electrophoresis, ∼200 μl of sample was applied to 110-mm pH 3–10 IPG® ReadyStrip (BIORAD, Hercules, CA). The strips were then actively rehydrated in the protean IEF cell (BIORAD) at 50 V for 18 h. The IEF was performed in increasing voltages as follows: 300 V for 1 h, then linear gradient to 8000 V for 5 h, and finally 20,000 V/h. Strips were then stored at −80°C until the second-dimension electrophoresis was to be performed. For the second dimension, the IPG strips, were thawed and equilibrated for 10 min in 50 mM Tris–HCl (pH 6.8), containing 6 M urea, 1% (w/v) sodium dodecyl sulfate (SDS), 30% (v/v) glycerol, and 0.5% dithiothreitol, and then re-equilibrated for 15 min in the same buffer containing 4.5% iodacetamide instead of dithiothreitol. Linear gradient precast criterion Bis-Tris gels (12%; BIORAD) were used to perform second-dimension electrophoresis. Precision Protein™ Standards (BIORAD) were run along with the samples at 200 V for 50 min.
For the detection of polyubiquitinated proteins, gels run as described above were incubated in fixing solution (10% acetic acid, 40% methanol) for 40 min and stained overnight at room temperature with 50 ml SYPRO Ruby gel stain (BIORAD). The SYPRO Ruby gel stain was then removed and gels stored in deionized water until use.
Image analysis
SYPRO Ruby-stained gel images were obtained using a Chemidoc MP System (BIORAD). All the images were saved in TIFF format. Gel imaging was software aided using PD-Quest (BIORAD) imaging software. Briefly, a master gel was selected, followed by normalization of all gels (Control and AD) according to the total spot density. Gel-to-gel analysis was then initiated in two parts. First, manual matching of common spots that could be visualized among the differential 2D gels was performed. After obtaining a significant number of spots, the automated matching of all spots was then initiated. Automated matching is based on user-defined parameters for spot detection. These parameters are based on the faintest spot, the largest spot, and the largest spot cluster that occur in the master gel and are defined by the user. Based on these parameters, the software defines spot centers for the gel. This process generates a large pool of data, ∼400 spots. Only proteins showing computer-determined significant differential levels between the groups being analyzed were considered for identification.
To determine significant differential levels of proteins, analysis sets were created using the analysis set manger software incorporated into the PD-Quest software. The numbers of pixels that occur in a protein spot were computed by the software corresponding to an increase/decrease in protein level. The gel image analysis was conducted first on SYPRO Ruby-stained polyubiquitinated gels and then on SYPRO Ruby-stained expression gels. The two analyses were compared by software to normalize a polyubiquitinylation value to expression value for each spot matched. A quantitative analysis set was created that recognized matched spots with differences in polyubiquitinylation intensity (normalized to expression intensity) that occur in each spot, and a statistical analysis set was created that used Student's t-test at 95% confidence to identify spots with p-values of p < 0.05. Spots with p < 0.05 were considered significant. A Boolean analysis set was created that identified overlapping spots from the aforementioned quantitative and statistical sets. These spots were selected for subsequent mass spectrometric analysis.
Mass spectrometry
Spots of interest were manually excised from gel and submitted to trypsin proteolysis (18). All peptide mixtures were analyzed by a nanoLC LTQ-Orbitrap mass spectrometer platform. Samples were desalted on stage tips according to the Rappsilber protocol (61), resuspended in 0.1% of formic acid, and loaded from an autosampler onto a 10-cm-long silica capillary (360 μm outer diameter, 75 μm i.d. fused silica with a 8 μm inner diameter (i.d.) tip; New Objective, Woburn, MA), handmade and packed with C18 reverse-phase resin (Magic C18AQ, 5 μm particle, 200 Å pore size), using the Dionex Ultimate 3000 system (Dionex, Amsterdam, The Netherlands). Peptides were eluted according to a 95-min gradient from 5% to 80% acetonitrile in 0.1% of formic acid at a flow rate of 300 nl/min. The liquid chromatography system was connected to a linear ion trap–orbitrap hybrid mass spectrometer (LTQ-Orbitrap Discovery; Thermo Fisher Scientific GmbH, Bremen, Germany) equipped with a nanoelectrospray ion source (Thermo Fisher Scientific), operating in the positive ionization mode with a spray voltage of 1.9 kV. The eluted peptides were detected in a precursor MS scan mode by Orbitrap (300–2000 m/z, 30,000 resolution at m/z 400), followed by sequential data-dependent MS/MS scans, in which the five most abundant ions were fragmented in ion trap collision-induced dissociation and analyzed in the linear trap (minimal signal required 500, isolation window of 2 Th, normalized collision energy 35%, removal of 1+ ions or ions with unassigned charge state, and selection of 2+, 3+, and 4+ ions). Data acquisition was controlled by Xcalibur™ software.
The MS/MS spectra were searched by MaxQuant (version 1.3.0.5, Max Planck Institute of Biochemistry, Martinsried) (14), against human UniProtKB fasta database (November 2014). Searching parameters were established as fixed modification of Cys, as carbamidomethyl cysteine, dynamic modification of oxidized Met, and ubiquitinated Lys, up to two missed cleavages for trypsin digestion, tolerance at 10 ppm, and 0.8 Da for precursor ions and fragment ions, respectively.
Proteasome activity assay
Chymotrypsin-like, trypsin-like, and caspase-like activities were assayed in DS and DS/AD samples and compared with their age-matched Ctr according to Keller et al. (38). Briefly, brain tissues were homogenized in proteolysis activity buffer (Tris–HCl 10 mM, pH 7.4, DTT 0.5 mM, ATP 5 mM, 0.035% SDS, and MgCl2 5 mM) at 4°C. Protein determination was performed with the BCA method (Pierce). Proteins (200 μg) were incubated for 30 min at 37°C with the fluorogenic chymotrypsin-like substrate, succinyl-LLVY-4-methylcoumaryl-7-amide, with the trypsin-like substrate, Z-ARR-4-methylcoumaryl-7-amide, and with the caspase-like substrate, Z-LLE-4-methylcoumaryl-7-amide (Sigma-Aldrich, St Louis, MO), to a final concentration of 50 μM. The reaction was blocked in ice and fluorescence measures were obtained at 380/460 nm excitation and emission, respectively. The background fluorescence was obtained by incubating homogenized samples with MG132 (Sigma-Aldrich), a potent, reversible, and cell-permeable proteasome inhibitor, to a final concentration of 60 μM. The incubation with MG 132 was performed 30 min before addition of the proteasome substrate.
Western blot
For Western blot, 30 μg of proteins (Ctr young, Ctr old, and DS with and without AD) was separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), using new Criterion™ TGX (Tris-Glycine eXtended) Stain-Free™ precast gels. The long shelf life TGX formulation includes unique trihalo compounds that allow rapid fluorescence detection of proteins with ChemiDoc™ MP imaging systems. The resulting gel images correspond to the total load of proteins, which is further used to normalize all the blots. Once detected by Chemidoc, the gels were transferred onto a nitrocellulose membrane (BIORAD). Membranes were blocked with 3% bovine serum albumin in T-TBS and incubated for 1 h and 30 min at room temperature with following primary antibodies: total ubiquitin and polyubiquitin, gelsolin, and ubiquilin-1 (1:500; Santa Cruz Biotechnology); UCHL 1 (1:1000; Thermo-Fisher, Rockford, IL); poly-Ub K63 (1:1000; Enzo Life Science, Farmingdale, NY); and poly-Ub K48 (1:1000; Millipore, Darmstadt, Germany) and for 1 h at room temperature with secondary antibody, horseradish peroxidase-conjugated anti-mouse IgG (1:5000; Sigma-Aldrich). Membrane was developed with the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Waltham, MA), acquired with ChemiDoc MP (BIORAD), and analyzed using Image Lab software (BIORAD).
Immunoprecipitation
Three different sample sets (100 μg of proteins) were incubated overnight with IP buffer (10 mM Tris, pH 7.6; 140 mM NaCl; 0.5% NP40, including protease inhibitors) and the following antibodies: UCH-L1 (1:100), gelsolin (1:200), and ubiquilin-1 (1:100), followed by 2 h of incubation with protein G sepharose beats (Santa Cruz Biotechnology), and then washed three times with RIA buffer (10 mM Tris, pH 7.6; 140 mM NaCl; 1% NP40). Proteins were separated by SDS-PAGE, followed by immunoblotting on a nitrocellulose membrane (BIORAD). Membranes were incubated with the antibodies, anti-poly-UbK63 (1:1000, Enzo Life Science) and poly-UbK48 (1:1000, Millipore), and then detected by the peroxidase-conjugated secondary antibody (1:5000; Sigma-Aldrich) with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). Membranes were then acquired with Chemi-Doc MP (BIORAD) and analyzed using Image Lab software (BIORAD). The IP results were normalized on the total amount of the proteins of interest.
Statistical analyses
Statistical analyses of data obtained by PD-QUEST software were performed using Student's t-test. Significance was accepted if the p-value <0.05. All the data are expressed as mean ± standard error of the mean of six independent samples per group. All statistical analyses were performed using a nonparametric one-way analysis of variance with the post hoc Bonferroni t-test. *p < 0.05 was considered significantly different from control. All statistical analyses were performed using GraphPad Prism 5.0 software.
Supplementary Material
Abbreviations Used
- 2D
two-dimensional
- Aβ
amyloid β (Aβ)-peptide
- AD
Alzheimer's disease
- ATP
adenosine triphosphate
- BF
bound fraction
- Chr21
chromosome 21
- CKB
creatine kinase B-type
- Ctr O
control old
- Ctr Y
control young
- DRP-2
dihidropyrimidinase-related protein 2
- DS
Down syndrome
- DS/AD
Down syndrome with Alzheimer's disease
- E2
ubiquitin-conjugating enzyme
- E3
ubiquitin ligase
- FBA A
fructose bisphosphate aldolase A
- FoF1 complex
ATP synthase α
- FT
flow-through
- GPX
glutathione peroxidase
- HNE
4-hydroxynonenal
- HSP
heat shock protein
- IEF
isoelectric focusing
- IP
immunoprecipitation
- Lys
lysine
- MCI
mild cognitive impairment
- MS
mass spectrometry
- NBF
nonbound fraction
- PMI
postmortem interval
- Poly-Ub
polymeric Ub
- PQC
protein quality control system
- PRDX2
peroxiredoxin 2
- PRDX6
peroxiredoxin 6
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- Ser
serine
- SH3
endophilin
- SOD-1
superoxide dismutase 1
- TGX
Tris-Glycine eXtended
- Thr
threonine
- Ub
ubiquitin
- UBA
ubiquitin-associated domain
- UCH-L1
ubiquitin carboxyl-terminal hydrolase isozyme L1
- UPS
ubiquitin-proteasome system
- UQCRC 1
cytochrome b-c1 complex subunit 1
- V0 ATPase
V-type proton ATPase
Acknowledgments
This work was partially supported by fondi di Ateneo Sapienza to M.P. and F.D.D. Brain tissue was acquired by E.H. under funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institute on Aging (Grant No. NIH 1RO1HD064993-01). Autopsy tissue was obtained from the UCI-ADRC (P50AG16573), from the UK ADC (P30AG28383), and from the NICHD Brain and Tissue Bank for Developmental Disorders of the University of Maryland (Baltimore, MD) contract HHSN275200900011C (N01HD90011). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Alfano C, Faggiano S, and Pastore A. The ball and chain of poly-ubiquitin structures. Trends Biochem Sci 41: 371–385, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Becker L, Mito T, Takashima S, and Onodera K. Growth and development of the brain in Down syndrome. Prog Clin Biol Res 373: 133–152, 1991 [PubMed] [Google Scholar]
- 3.Ben-Zvi A, Miller EA, and Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A 106: 14914–14919, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butterfield DA, Di Domenico F, Swomley AM, Head E, and Perluigi M. Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: Overlaps in Down's syndrome and Alzheimer's disease brain. Biochem J 463: 177–189, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butterfield DA, Perluigi M, Reed T, Muharib T, Hughes CP, Robinson RA, and Sultana R. Redox proteomics in selected neurodegenerative disorders: From its infancy to future applications. Antioxid Redox Signal 17: 1610–1655, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calabrese V, Sultana R, Scapagnini G, Guagliano E, Sapienza M, Bella R, Kanski J, Pennisi G, Mancuso C, Stella AM, and Butterfield DA. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer's disease. Antioxid Redox Signal 8: 1975–1986, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, and Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med 33: 562–571, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Cecarini V, Ding Q, and Keller JN. Oxidative inactivation of the proteasome in Alzheimer's disease. Free Radic Res 41: 673–680, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Cenini G, Dowling AL, Beckett TL, Barone E, Mancuso C, Murphy MP, Levine H, 3rd, Lott IT, Schmitt FA, Butterfield DA, and Head E. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta 1822: 130–138, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chauhan V, Ji L, and Chauhan A. Anti-amyloidogenic, anti-oxidant and anti-apoptotic role of gelsolin in Alzheimer's disease. Biogerontology 9: 381–389, 2008 [DOI] [PubMed] [Google Scholar]
- 11.Chen T, Zhou T, He B, Yu H, Guo X, Song X, and Sha J. mUbiSiDa: A comprehensive database for protein ubiquitination sites in mammals. PLoS One 9: e85744, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cisse S, Perry G, Lacoste-Royal G, Cabana T, and Gauvreau D. Immunochemical identification of ubiquitin and heat-shock proteins in corpora amylacea from normal aged and Alzheimer's disease brains. Acta Neuropathol 85: 233–240, 1993 [DOI] [PubMed] [Google Scholar]
- 13.Coskun PE, Wyrembak J, Derbereva O, Melkonian G, Doran E, Lott IT, Head E, Cotman CW, and Wallace DC. Systemic mitochondrial dysfunction and the etiology of Alzheimer's disease and Down syndrome dementia. J Alzheimers Dis 20 Suppl 2: S293–S310, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, and Mann M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc 4: 698–705, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Dantuma NP. and Bott LC. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front Mol Neurosci 7: 70, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Haan JB, Cristiano F, Iannello RC, and Kola I. Cu/Zn-superoxide dismutase and glutathione peroxidase during aging. Biochem Mol Biol Int 35: 1281–1297, 1995 [PubMed] [Google Scholar]
- 17.Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, Cini C, and Perluigi M. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer's disease neuropathology: redox proteomics analysis of human brain. Biochim Biophys Acta 1832: 1249–1259, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Domenico F, Pupo G, Giraldo E, Badia MC, Monllor P, Lloret A, Eugenia Schinina M, Giorgi A, Cini C, Tramutola A, Butterfield DA, Vina J, and Perluigi M. Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic Biol Med 91: 1–9, 2015 [DOI] [PubMed] [Google Scholar]
- 19.Di Domenico F, Pupo G, Tramutola A, Giorgi A, Schinina ME, Coccia R, Head E, Butterfield DA, and Perluigi M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic Biol Med 71: 270–280, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Ayadi A, Stieren ES, Barral JM, and Boehning D. Ubiquilin-1 and protein quality control in Alzheimer disease. Prion 7: 164–169, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esser C, Alberti S, and Hohfeld J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim Biophys Acta 1695: 171–188, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Fergusson J, Landon M, Lowe J, Dawson SP, Layfield R, Hanger DP, and Mayer RJ. Pathological lesions of Alzheimer's disease and dementia with Lewy bodies brains exhibit immunoreactivity to an ATPase that is a regulatory subunit of the 26S proteasome. Neurosci Lett 219: 167–170, 1996 [DOI] [PubMed] [Google Scholar]
- 23.Gimeno A, Garcia-Gimenez JL, Audi L, Toran N, Andaluz P, Dasi F, Vina J, and Pallardo FV. Decreased cell proliferation and higher oxidative stress in fibroblasts from Down syndrome fetuses. Preliminary study. Biochim Biophys Acta 1842: 116–125, 2014 [DOI] [PubMed] [Google Scholar]
- 24.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature 426: 895–899, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Gulesserian T, Engidawork E, Fountoulakis M, and Lubec G. Antioxidant proteins in fetal brain: Superoxide dismutase-1 (SOD-1) protein is not overexpressed in fetal Down syndrome. J Neural Transm Suppl 71–84, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Hamos JE, Oblas B, Pulaski-Salo D, Welch WJ, Bole DG, and Drachman DA. Expression of heat shock proteins in Alzheimer's disease. Neurology 41: 345–350, 1991 [DOI] [PubMed] [Google Scholar]
- 27.Head E, Lott IT, Wilcock DM, and Lemere CA. Aging in Down syndrome and the development of Alzheimer's disease neuropathology. Curr Alzheimer Res 13: 18–29, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Head E, Silverman W, Patterson D, and Lott IT. Aging and Down syndrome. Curr Gerontol Geriatr Res 2012: 412536, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helguera P, Pelsman A, Pigino G, Wolvetang E, Head E, and Busciglio J. ets-2 Promotes the activation of a mitochondrial death pathway in Down's syndrome neurons. J Neurosci 25: 2295–2303, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hershko A, Ciechanover A, Heller H, Haas AL, and Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A 77: 1783–1786, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters HV, and Franceschi C. Accelerated epigenetic aging in Down syndrome. Aging Cell 14: 491–495, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ihara Y, Morishima-Kawashima M, and Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb Perspect Med 2: 1–27, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikeda F. and Dikic I. Atypical ubiquitin chains: New molecular signals. Protein modifications: Beyond the usual suspects' review series. EMBO Rep 9: 536–542, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwai K, Fujita H, and Sasaki Y. Linear ubiquitin chains: NF-kappaB signalling, cell death and beyond. Nat Rev Mol Cell Biol 15: 503–508, 2014 [DOI] [PubMed] [Google Scholar]
- 35.Ji L, Chauhan V, Mehta P, Wegiel J, Mehta S, and Chauhan A. Relationship between proteolytically cleaved gelsolin and levels of amyloid-beta protein in the brains of Down syndrome subjects. J Alzheimers Dis 22: 609–617, 2010 [DOI] [PubMed] [Google Scholar]
- 36.Ji L, Zhao X, and Hua Z. Potential therapeutic implications of gelsolin in Alzheimer's disease. J Alzheimers Dis 44: 13–25, 2015 [DOI] [PubMed] [Google Scholar]
- 37.Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, and Nixon RA. Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A 107: 1630–1635, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keller JN, Hanni KB, and Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem 75: 436–439, 2000 [DOI] [PubMed] [Google Scholar]
- 39.Kevei E. and Hoppe T. Ubiquitin sets the timer: Impacts on aging and longevity. Nat Struct Mol Biol 21: 290–292, 2014 [DOI] [PubMed] [Google Scholar]
- 40.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, and Gygi SP. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44: 325–340, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komander D. and Rape M. The ubiquitin code. Annu Rev Biochem 81: 203–229, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Korolchuk VI, Menzies FM, and Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584: 1393–1398, 2010 [DOI] [PubMed] [Google Scholar]
- 43.Kudo T, Iqbal K, Ravid R, Swaab DF, and Grundke-Iqbal I. Alzheimer disease: Correlation of cerebro-spinal fluid and brain ubiquitin levels. Brain Res 639: 1–7, 1994 [DOI] [PubMed] [Google Scholar]
- 44.Kulathu Y. and Komander D. Atypical ubiquitylation − The unexplored world of poly-ubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol 13: 508–523, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Labudova O, Cairns N, Kitzmuller E, and Lubec G. Impaired brain glucose metabolism in patients with Down syndrome. J Neural Transm Suppl 57: 247–256, 1999 [DOI] [PubMed] [Google Scholar]
- 46.Larsen CN, Krantz BA, and Wilkinson KD. Substrate specificity of deubiquitinating enzymes: Ubiquitin C-terminal hydrolases. Biochemistry 37: 3358–3368, 1998 [DOI] [PubMed] [Google Scholar]
- 47.Lecker SH, Goldberg AL, and Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17: 1807–1819, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Li W. and Ye Y. Poly-ubiquitin chains: functions, structures, and mechanisms. Cell Mol Life Sci 65: 2397–2406, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lilienbaum A. Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol 4: 1–26, 2013 [PMC free article] [PubMed] [Google Scholar]
- 50.Lopez Salon M, Morelli L, Castano EM, Soto EF, and Pasquini JM. Defective ubiquitination of cerebral proteins in Alzheimer's disease. J Neurosci Res 62: 302–310, 2000 [DOI] [PubMed] [Google Scholar]
- 51.Lott IT, Head E, Doran E, and Busciglio J. Beta-amyloid, oxidative stress and Down syndrome. Curr Alzheimer Res 3: 521–528, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Menzies FM, Fleming A, and Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16: 345–357, 2015 [DOI] [PubMed] [Google Scholar]
- 53.Nixon RA. and Yang DS. Autophagy failure in Alzheimer's disease—Locating the primary defect. Neurobiol Dis 43: 38–45, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S, and Smith MA. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol 59: 1011–1017, 2000 [DOI] [PubMed] [Google Scholar]
- 55.Osaka H, Wang YL, Takada K, Takizawa S, Setsuie R, Li H, Sato Y, Nishikawa K, Sun YJ, Sakurai M, Harada T, Hara Y, Kimura I, Chiba S, Namikawa K, Kiyama H, Noda M, Aoki S, and Wada K. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet 12: 1945–1958, 2003 [DOI] [PubMed] [Google Scholar]
- 56.Pagano G. and Castello G. Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv Exp Med Biol 724: 291–299, 2012 [DOI] [PubMed] [Google Scholar]
- 57.Perluigi M, Di Domenico F, and Butterfield DA. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis 84: 39–49, 2015 [DOI] [PubMed] [Google Scholar]
- 58.Perluigi M, di Domenico F, Fiorini A, Cocciolo A, Giorgi A, Foppoli C, Butterfield DA, Giorlandino M, Giorlandino C, Schinina ME, and Coccia R. Oxidative stress occurs early in Down syndrome pregnancy: A redox proteomics analysis of amniotic fluid. Proteomics Clin Appl 5: 167–178, 2011 [DOI] [PubMed] [Google Scholar]
- 59.Perluigi M, Pupo G, Tramutola A, Cini C, Coccia R, Barone E, Head E, Butterfield DA, and Di Domenico F. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim Biophys Acta 1842: 1144–1153, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, and Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78: 365–380, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rappsilber J, Mann M, and Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Roper RJ. and Reeves RH. Understanding the basis for Down syndrome phenotypes. PLoS Genet 2: e50, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rothenberg C. and Monteiro MJ. Ubiquilin at a crossroads in protein degradation pathways. Autophagy 6: 979–980, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sawa A. Neuronal cell death in Down's syndrome. J Neural Transm Suppl 57: 87–97, 1999 [DOI] [PubMed] [Google Scholar]
- 65.Setsuie R. and Wada K. The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int 51: 105–111, 2007 [DOI] [PubMed] [Google Scholar]
- 66.Silva GM, Finley D, and Vogel C. K63 poly-ubiquitination is a new modulator of the oxidative stress response. Nat Struct Mol Biol 22: 116–123, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stieren ES, El Ayadi A, Xiao Y, Siller E, Landsverk ML, Oberhauser AF, Barral JM, and Boehning D. Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J Biol Chem 286: 35689–35698, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tanaka K. and Matsuda N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim Biophys Acta 1843: 197–204, 2014 [DOI] [PubMed] [Google Scholar]
- 69.Tramutola A, Pupo G, Di Domenico F, Barone E, Arena A, Lanzillotta C, Broekaart D, Blarzino C, Head E, Butterfield D, and Perluigi M. Activation of p53 in Down syndrome and in the Ts65Dn mouse brain is associated with a pro-apoptotic phenotype. J Alzheimers Dis 52: 359–371, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Triplett JC, Swomley AM, Cai J, Klein JB, and Butterfield DA. Quantitative phosphoproteomic analyses of the inferior parietal lobule from three different pathological stages of Alzheimer's disease. J Alzheimers Dis 49: 45–62, 2015 [DOI] [PubMed] [Google Scholar]
- 71.Wickliffe KE, Williamson A, Meyer HJ, Kelly A, and Rape M. K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol 21: 656–663, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wiseman FK, Al-Janabi T, Hardy J, Karmiloff-Smith A, Nizetic D, Tybulewicz VL, Fisher EM, and Strydom A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat Rev Neurosci 16: 564–574, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, and Nixon RA. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur J Neurosci 37: 1949–1961, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, and Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137: 133–145, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zana M, Janka Z, and Kalman J. Oxidative stress: A bridge between Down's syndrome and Alzheimer's disease. Neurobiol Aging 28: 648–676, 2007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.