

Abstract
Major depressive disorder (MDD) is a prevalent neuropsychiatric disease that causes profound social and economic burdens. The impact of MDD is compounded by the limited therapeutic efficacy and delay of weeks to months of currently available medications. These issues highlight the need for more efficacious and faster-acting treatments to alleviate the burdens of MDD. Recent breakthroughs demonstrate that certain drugs, including ketamine and scopolamine, produce rapid and long-lasting antidepressant effects in MDD patients. Moreover, preclinical work has shown that the antidepressant actions of ketamine and scopolamine in rodent models are caused by an increase of extracellular glutamate, elevated BDNF, activation of the mammalian target of rapamycin complex 1 (mTORC1) cascade, and increased number and function of spine synapses in the prefrontal cortex (PFC). Here we review studies showing that both ketamine and scopolamine elicit rapid antidepressant effects through converging molecular and cellular mechanisms in the PFC. In addition, we discuss evidence that selective antagonists of NMDA and muscarinic acetylcholine (mACh) receptor subtypes (i.e., NR2B and M1-AChR) in the PFC produce comparable antidepressant responses. Furthermore, we discuss evidence that ketamine and scopolamine antagonize inhibitory interneurons in the PFC leading to disinhibition of pyramidal neurons and increased extracellular glutamate that promotes the rapid antidepressant responses to these agents. Collectively, these studies indicate that specific NMDA and mACh receptor subtypes on GABAergic interneurons are promising targets for novel rapid-acting antidepressant therapies.
Keywords: Acetylcholine, antidepressant, BDNF, depression, GABA, glutamate, ketamine, mTORC1, scopolamine, synapse
Introduction
Despite advances in the fields of psychiatry and neuropharmacology over the years, major depressive disorder (MDD) continues to be a leading cause of disability in the world with a lifetime risk of 7-12% in men and 20-25% risk in women [1-3]. In addition, MDD patients often have comorbid psychiatric illnesses, such as anxiety and substance abuse, along with an increased risk of other systemic chronic disorders, including cardiovascular disease, cancer, diabetes and obesity [4]. These debilitating characteristics contribute to the significant social and economic burdens caused by MDD [5]. The impact of this illness is compounded by the limited therapeutic efficacy and time-lag of weeks to months of currently available medications. Thus, these issues highlight the need for more efficacious and faster-acting treatments to alleviate the problems of MDD.
In order to facilitate the development of more effective MDD treatments a thorough understanding of the patho-physiology of this disease is needed. The debilitating effects of MDD have been hypothesized to result from the dysfunction of an interconnected network of brain regions that collectively regulate mood [6]. Clinical and preclinical data indicate that depressive behaviors stem from dysfunction in several integrated brain regions, including the prefrontal cortex (PFC) and hippocampus (HPC). For instance, recent evidence shows that depressive behavior is linked to decreased synapse-related genes and loss of synapses in the PFC [7]. These neurobiological effects are precipitated by chronic exposure to stress, which promotes development of depressive behaviors in humans and rodent models [8-10]. In this context, it is important to develop treatments that can attenuate the stress-induced synaptic deficits and neuronal atrophy that underlies altered mood and depression.
Recent clinical studies demonstrate that ketamine produces long-lasting mood-enhancing effects, even in MDD patients that have failed to respond to two or more typical agents and are considered treatment-resistant [11-13]. Though these results are promising, ketamine also produces dissociative and psychomimetic side effects [14] and has abuse potential, especially at higher doses [15, 16]. Separate clinical studies also demonstrate that scopolamine produces rapid and sustained antidepressant responses in treatment-resistant patients [17-20]. Compared to ketamine, scopolamine has limited adverse effects particularly at concentrations used for antidepressant therapy, however, it can elicit cognitive deficits [21, 22]. In order to harness the antidepressant potential of ketamine and scopolamine, it is imperative to understand how they function in the brain and to use that knowledge to develop new therapies that are safer and can be widely prescribed. A key aspect of this endeavor is to characterize the brain areas and cell types that mediate the antidepressant actions of ketamine and scopolamine and how regulation of these cells produces a behavioral response. Preclinical studies show that the rapid antidepressant responses to ketamine, as well as scopolamine, are caused by increased glutamate neurotransmission and activation of the mechanistic target of rapamycin complex 1 (mTORC1) signaling in the PFC [23, 24]. The mTORC1 signaling pathway is an activity/energy/growth sensor that has been implicated in protein synthesis-dependent synaptic plasticity [25]. In fact, following ketamine and scopolamine treatment pyramidal neurons in the PFC show enhanced number and function of dendritic spines [23, 24]. Importantly, increased synapse number and function in the PFC in response to ketamine or scopolamine reverses the pathophysiology associated with stress and depression [26, 27]. The converging mechanisms by which ketamine and scopolamine produce an antidepressant effect suggest that there are common neurophysiological pathways underlying the actions of these rapid-acting antidepressants.
An important component that appears to initiate the molecular signaling required for rapid antidepressant responses is the fast (10-60 minutes) elevation or burst of glutamate in the PFC following ketamine and scopolamine administration [23, 28]. In fact, the neurocircuitry in the PFC is thought to provide a framework to enhance glutamate neurotransmission following ketamine and scopolamine administration. The purpose of this review is to summarize the molecular and cellular pathways that underlie the action of rapid antidepressant drugs and how they can lead to structural changes in the PFC that reverse synaptic deficits associated with MDD. Furthermore, we discuss how preferential antagonists of specific N-methyl-D-aspartate (NMDA) and muscarinic acetylcholine (mACh) receptor subtypes can promote glutamate transmission in the PFC and provide evidence that inhibitory interneurons may be a key mediator of these neurophysiological responses.
Behavioral, Molecular, and Structural Consequences of Stress and Depression
Recent studies have built on an extensive literature demonstrating the myriad ways that depression affects brain structure and activity. Human neuroimaging studies illustrate the long-term effects of stress on cortical, sub-cortical, and limbic structures implicated in depression [29]. One area of particular interest is the prefrontal cortex (PFC), which regulates and stabilizes the activity of much of the network [30]. In humans, the PFC is thought to be involved in self-evaluation and other self-referential activities, including the regulation of emotion and mood [31]. The prominent role of the PFC in depression is evident from studies demonstrating that deep brain stimulation of this brain region relieves depressive symptoms [32, 33]. Neuroimaging reports demonstrate that the volume and connectivity of the subgenual PFC and cingulate cortex is decreased in depressed individuals [34, 35]. Corresponding with these findings human postmortem studies have found a reduction in cell body size [36] as well as a loss of synapses in the dorsolateral PFC [7]. Region-specific changes of neuronal morphology, especially loss of synapses, contributes to the loss of functional connectivity found in human imaging studies. Gene expression analyses of depressed patients compared to healthy controls have found dysregulation of genes encoding proteins involved in synapse formation and glutamate signaling [37]. These pathological features are observed in models of chronic stress that promote development of depressive-like behavior. For instance, studies in rodents showed that long-term stress caused a decrease in the number and size of dendrites in layer 2/3 of the PFC [38, 39]. These findings are consistent with other rodent models that show reduced length and branching of the apical dendrites of layer V pyramidal neurons in the PFC in response to chronic stress. Coinciding with changes in dendritic branching, chronic stress also reduces spine density in the PFC [40, 41].
Another brain region of interest in MDD is the hippocampus (HPC), which plays a key role in cognitive functions such as spatial and declarative memory and anxiety, as well as in regulation of the hypothalamic-pituitary-adrenal (HPA) axis. HPC volume is reduced in depressed individuals and antidepressant therapy has been shown to reverse this stress-induced atrophy [42, 43]. Furthermore, work by McEwen and colleagues using rodent models of stress and depression report decreased number and length of apical dendrites in CA3 pyramidal neurons of the HPC and reduced synaptic connectivity in response to chronic stress paradigms [44]. The amygdala, a subcortical structure involved in emotional memory formation and retrieval, has primarily been studied for its role in fear conditioning and memory. In addition, the amygdala exhibits a dysfunctional activity profile in individuals suffering from depression and anxiety [45, 46]. Additional brain regions that are altered in animal and human studies of depression include the hypothalamus and nucleus accumbens [47]. While depression should be considered a system-wide disorder, the PFC is a key target for study as it receives and sends substantial projections throughout the brain to both cortical and subcortical regions that have been implicated in depression. In this context, it is important to work towards a thorough understanding of the pathophysiology of depression in order to develop better therapeutic agents that prevent or reverse these stress-induced cellular and molecular deficits in the PFC, as well as other brain regions.
Mechanisms of Rapid-Acting Antidepressants
Preclinical studies have found that the behavioral and synaptic deficits linked to stress and depression can be reversed following chronic treatment with typical antidepressants. Chronic administration of the selective serotonin-reuptake inhibitor (SSRI) fluoxetine is able to increase spine density [48] and block the stress-induced atrophy of dendrites and spines following chronic stressors [49, 50]. Release of a major neurotrophic factor, brain-derived neurotrophic factor (BDNF), is implicated in antidepressant actions as it has been shown that mice with the BDNF Val66Met polymorphism, which decreases activity-dependent BDNF release, are unresponsive to chronic fluoxetine treatment [51, 52]. Phar- macological blockade of BDNF receptor signaling also attenuates the antidepressant behavioral actions of fluoxetine [53]. Similar studies reveal a comparable role of fibroblast growth factor (FGF)-2 in the behavioral effects of SSRIs [54]. It is important to note that these synaptic, as well as behavioral actions of SSRIs require chronic administration (2-3 weeks). This is consistent with clinical reports that current antidepressant treatments require at least two weeks for symptom improvement and six to eight weeks of chronic administration before a full therapeutic response is observed [55, 56]. Complicating the use of these antidepressant treatments is that up to two-thirds of patients fail to respond to the first antidepressant prescribed [57].
In contrast, a seminal study by Berman and colleagues found that a single dose of ketamine, an NMDA receptor antagonist, produces rapid antidepressant effects in treatment-resistant patients within hours of administration and that these effects are sustained for 7-10 days [14]. Several preclinical studies set out to investigate the underlying cellular and molecular mechanisms of ketamine and other rapid-acting antidepressants using rodent models. Behavioral and molecular studies illustrate the rapid actions of these agents, correlating behavior with alterations in signaling pathways and neuroplasticity. A single treatment with ketamine reverses CUS-induced anhedonia, anxiety and diminished spine density, and these effects are dependent on mTORC1 signaling [24, 58]. The mTORC1 pathway has been identified as an important mediator of protein- and activity-dependent synaptogenesis [25]. In addition to ketamine, scopolamine and mGluR2/3 antagonists, have all demonstrated promise as rapid-acting antidepressants through stimulation of mTORC1 signaling. Prior administration with the selective mTORC1 inhibitor rapamycin blocks the behavioral and synaptogenic actions of these agents, further supporting a role for the mTORC1 pathway [24, 58-60]. While it is clear that mTORC1 signaling is an integral molecular mechanism in the behavioral effects of these drugs several cell types are required to facilitate this molecular cascade.
Glutamate burst in the PFC as a trigger for rapid antidepressants
An early, key event that precedes mTORC1 activation is increased extracellular glutamate in the PFC. For example, acute NMDA or mACh receptor antagonism produced a rapid glutamate burst in the PFC within 10 to 60 minutes [23, 28, 61] (Fig. 1). It is important to note that glutamate elevation occurs with very low levels of scopolamine (25 ug/kg) and sub-anesthetic doses of ketamine (10 mg/kg) [28, 62]. The increased glutamate in the PFC is considered integral for the effects of ketamine and scopolamine as pretreatment with an AMPA receptor antagonist blocks the cellular and behavioral responses to these agents [23, 24, 63]. This is consistent with findings that show AMPA receptor potentiators increase BDNF levels and produce antidepressant-like effects [64]. Thus, increased glutamate release in the PFC following ketamine or scopolamine activates AMPA receptors to promote the rapid antidepressant effects observed, including increased release of BDNF and binding to TrkB receptors which leads to stimulation of mTORC1 signaling (Fig. 1) [24, 65]. In fact, a recent study has shown that antibody-mediated sequestration of BDNF in the PFC blocks the antidepressant effects of ketamine [66]. In support of these findings the synaptic and behavioral actions of ketamine are blocked in mice homozygous for the BDNF Val66Met polymorphism (Met/Met)[67], which leads to diminished activity-dependent release of BDNF and decreased PFC pyramidal neuron function [68, 69]. It is important to note that while these molecular signaling cascades occur quickly (within minutes) following ketamine and scopolamine treatment it likely takes several hours for enhanced synaptic responses to be observed, consistent with the time course for the therapeutic responses to these agents. In all, these studies demonstrate that an initial burst of glutamate in the PFC promotes downstream molecular signaling that mediates the rapid antidepressant actions of ketamine and scopolamine.
Fig. (1).
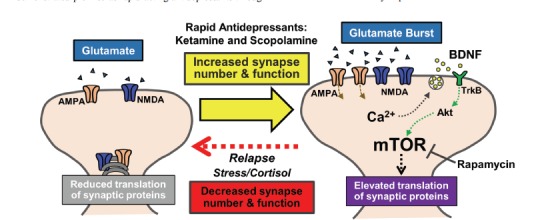
Molecular mechanisms mediating the synaptic plasticity caused by rapid-acting antidepressants. Treatment with ketamine or scopolamine causes glutamate influx in the PFC promoting AMPA receptor activation which initiates BDNF release. Subsequent binding of BDNF to TrkB receptors results in activation of mTORC1 signaling that contributes to increased synaptic plasticity underlying rapid antidepressant responses.
Rapid-acting antidepressants may engage specific neurotransmitter receptor subtypes to elicit behavioral responses
Recent studies suggest that selective NMDA and mACh receptor subtypes can be targeted to produce behavioral responses comparable to non-selective antagonists, providing insight to the receptor subtypes underlying the actions of ketamine or scopolamine [70]. This is supported by a clinical study demonstrating a rapid antidepressant response to a selective NR2B receptor antagonist, ifenprodil also known as CP-101, 606 (0.5 mg/kg/hour i.v. over 1.5 hours) [71]. Evidence from preclinical studies also demonstrate ketamine-like effects of NR2B selective antagonists. Administration of the selective NR2B antagonist Ro 25-6981 (10 mg/kg i.p.) increases mTORC1 signaling in PFC, leading to rapid anti- depressant responses in rodent models [24, 58]. Consistent with these findings, a recent study has found that genetic knockout of NR2B in CaMKII+ neurons results in blockade of antidepressant effects following ketamine. However, it is worth noting that mice lacking NR2B in CaMKII+ neurons showed hyperlocomotion, which confounds interpretation of these findings as an antidepressant response in the forced swim test is mimicked by increased locomotor activity [72]. Similar studies revealed preferential blockade of mACh receptor subtypes recapitulate the molecular and behavioral effects of scopolamine. For example, telenzepine, which preferentially antagonizes M1-ACh receptors, produces antidepressant responses similar to scopolamine [23]. Reinforcing these findings, Witkin, et al. have recently shown that mice lacking M1- or M2-ACh receptor do not show antidepressant responses following scopolamine treatment [73]. Taken together, these studies suggest that specific receptor subtypes mediate, in part, the molecular and behavioral responses of ketamine and scopolamine (Fig. 2).
Fig. (2).
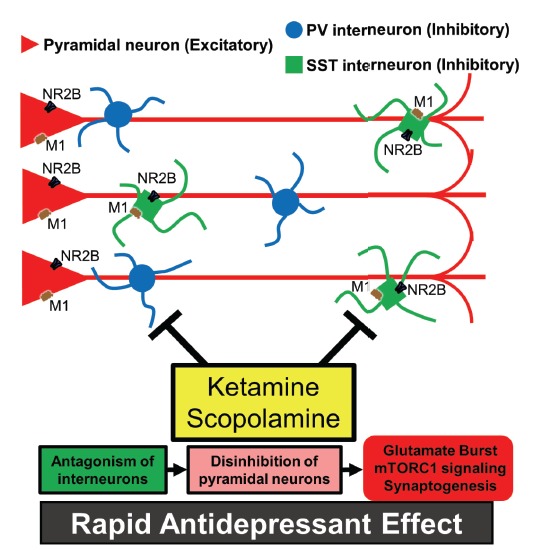
Neurocircuitry in the PFC may facilitate glutamate influx through antagonism of interneurons leading to disinhibition of pyramidal neurons. Evidence suggests that interneurons tonically regulate pyramidal neuron activation, thus ketamine or scopolamine may target specific NMDA or mACh receptors on particular subsets of interneurons leading to glutamate influx in the PFC and antidepressant behavioral effects.
Role of inhibitory interneurons in the pathophysiology of depression and rapid antidepressant responses
While it is intriguing that specific receptor subtypes may mediate some of the rapid antidepressant responses of ketamine and scopolamine, it remains unclear if these receptors are expressed by distinct neuronal subsets and how the microcircuitry of the PFC can promote increased glutamate neurotransmission. The rapid and transient glutamate burst following ketamine or scopolamine administration appears to contradict their role in neurotransmission, because they act to antagonize receptors that usually increase neuronal activation. Based on these findings it has been postulated that interneurons releasing the inhibitory neurotransmitter γ-aminobutyric acid (GABA) play an important role in mediating glutamate release from pyramidal cells. In particular, one current hypothesis is that ketamine and scopolamine antagonize specific receptor subtypes on inhibitory interneurons that lead to disinhibition of glutamatergic pyramidal neurons causing a subsequent glutamate burst in the PFC. Understanding the modulation of pyramidal neurons in the PFC by interneurons may lend insight into the cellular mediators underlying the effects of rapid-acting antidepressants. There is also evidence from postmortem studies of depressed subjects for alterations in the number of GABA interneuron subtypes that further elucidate the potential role of these important cells in the actions of ketamine and scopolamine. The next sections discuss evidence for altered GABA function in depression and role of these interneurons in the response to rapid-acting antidepressants.
PFC interneuron deficits in stress and depression
In the PFC approximately 70-80% of neocortical neurons are excitatory pyramidal neurons and the other 20-30% are inhibitory interneurons. To a greater extent than pyramidal neurons, inhibitory interneurons in the PFC have a variety of morphologies and dynamic anatomical connections with other neurons that often determine cortical function [74, 75]. For example, different subtypes of interneurons express an array of peptides (e.g., somatostatin (SST), neuropeptide Y (NPY), vasoactive intestinal peptide (VIP)) and calcium-binding proteins (e.g., parvalbumin (PV), calbindin (CB)) [76]. Two distinct subtypes of interneurons are of interest based on their location and projection/connection pattern in the PFC. The first type, the PV-expressing interneurons, are primarily basket or chandelier cells, both expressed throughout cortical layers II-VI. While basket cells target the cell body and proximal dendrites, chandelier cells target the axon segment. In general, PV interneurons are fast-spiking interneurons [77]. The second type, the SST-expressing interneurons, the Martinotti interneurons, typically inhibit the dendritic tufts of pyramidal cells located in cortical layer I, where their axons project horizontally enabling one interneuron to inhibit the activity of many pyramidal cells [78-80]. Compared to the fast-spiking PV interneuron, the SST interneurons are more slowly activated and have more sustained effects on pyramidal cells [81, 82].
Dysfunction of homeostatic glutamate-GABA signaling is thought to underlie many neurological and psychiatric diseases. These alterations in glutamate-GABA signaling may be related to changes in the function of interneurons. Interestingly, histological analyses reveal that subsets of interneurons are reduced in the PFC of depressed patients, suggesting a key role for interneuron function in the pathogenesis of depression [83]. Other findings indicate that subsets of interneurons may be altered as down-regulation of SST levels has been reported in mood disorders [84]. These findings are supported by recent postmortem reports showing that depressed subjects have reductions in SST mRNA levels in the PFC in the absence of changes in other GABA-related genes (i.e., PV, GAD67) [85]. Further, transgenic models implicate SST dysfunction in depression as inhibition of SST interneurons leads to increased anxiety- and depressive-like behaviors [86]. Collectively, these data suggest that there are deficits in interneuron function that may contribute to the decreased mood and emotionality observed in MDD [87].
A potential role for GABAergic inter- neurons in the neurophysiological and behavioral responses to rapid-acting antidepressants
Though preclinical studies indicate that mTORC1 signaling and synaptic plasticity in PFC pyramidal neurons underlie the rapid antidepressant responses in rodents, it remains unclear which specific cellular targets mediate the initial actions of ketamine or scopolamine. The glutamate burst in the PFC is likely derived from stimulation of pyramidal neurons in this region [88]. Thus, ketamine or scopolamine either directly activate pyramidal neurons or do so indirectly by inhibition of GABAergic interneuron firing, thereby reducing inhibition of pyramidal neurons and stimulating glutamate release (Fig. 2).
Ketamine
Direct activation of pyramidal neurons seems paradoxical since NMDA receptor antagonists block neuronal activation, however, clinical and preclinical studies show that NMDA receptor antagonists increase pyramidal neuron activity in the PFC [89-92]. These data suggest that in vivo ketamine and scopolamine, at the low doses used for clinical and preclinical studies, may act via GABA interneurons to disinhibit pyramidal neuron firing. In support of this possibility, studies in awake rats show that NMDA receptor antagonism with MK801 reduce interneuron firing prior to increasing pyramidal neuron firing [88]. These findings suggest that during basal, resting state conditions, pyramidal cell firing is inhibited by presynaptic GABA-releasing inter- neurons [93, 94]. Therefore, it is plausible that low-dose ketamine can block NMDA receptors on subsets of inter- neurons resulting in disinhibition of pyramidal cells causing a subsequent glutamate burst within the PFC.
The role of NMDA receptors in specific subsets of interneurons has not been elucidated fully, however, some neurophysiological studies lend insight. Based on spiking signatures PFC interneurons appear to express variable levels of NMDA receptors. For instance, fast-spiking interneurons (indicative of PV subtypes) have limited NMDA currents during patch clamp studies in adult mouse cortical slices. In contrast, regular-spiking and low threshold-spiking interneurons (characteristic of SST) maintained regular NMDA currents in slices from adult animals. Furthermore, studies using ifenprodil, a selective NR1/NR2B receptor antagonist, reveal that approximately 50% of NMDA receptors on interneurons contain NR2B [95]. These results show that NMDA receptors are enriched in putative SST interneurons and roughly half are NR2B-containing NMDA receptors suggesting that these interneurons may be critical to the antidepressant effects of ketamine and other NR2B selective compounds.
While these studies implicate interneurons in the PFC in the effects of ketamine, other reports indicate that ketamine causes direct neurophysiological effects on pyramidal neurons. For instance, incubation of brain slices with high concentrations of ketamine reduces miniature excitatory post-synaptic potentials in pyramidal neurons. Following this short-term decrement in NMDA receptor activity, field potentials of pyramidal neurons in the CA1 region of the hippocampus are enhanced (i.e., 1 hour after ketamine) [65, 96, 97]. These findings suggest that ketamine directly influences pyramidal neurons in the CA1 of the HPC, however, it is important to consider the region-specific neurocircuitry underlying these responses [98]. For instance, interneurons in the PFC and HPC provide persistent inhibitory tone that is integral to control the excitability of pyramidal neurons. In fact, interneurons exhibit more frequent spiking activity that promote a depolarized state, removing Mg2+ and leaving more NMDA receptor channels in the open state. These properties may make NMDA receptors on interneurons more sensitive to NMDA receptor antagonism particularly at the low ketamine concentrations used to produce antidepressant responses [99].
Scopolamine
Separate findings suggest that comparable cellular mechanisms and neurophysiological dynamics are engaged following scopolamine. Despite the role of acetylcholine as a critical neuromodulator recent studies show that mACh receptors in the PFC may not have a prominent role in the direct excitation of cortical pyramidal neurons [100]. Prior studies have shown using single-cell mRNA analyses that pyramidal neurons and interneurons in the PFC express varied combinations of mACh receptors, including M1-ACh receptor [101]. Expression of M1-ACh receptors on pyramidal cells and interneurons in the cortex has been confirmed using immunohistology and in situ hybridization. The neurophysiological role of mACh receptors on neurons remains to be fully elucidated, but evidence indicates that M1-ACh receptors modulate both cortical pyramidal and interneuron activity [102, 103]. These findings are supported by a previous study that infusion of scopolamine increases extracellular glutamate in the striatum [104]. On the other hand, a recent study analyzing layer V pyramidal neurons suggested that less than 10% of the inward current caused by acetylcholine can be attributed to mACh receptors [105]. In all, these findings suggest that scopolamine may not act directly through pyramidal neurons to increase glutamate in the PFC. It will be important to further define the interneuron subsets that express M1-ACh receptors and determine how these interneurons modulate pyramidal neurons following scopolamine treatment.
As indicated above the distribution and projections of interneurons likely play a fundamental role in the neurophysiological response following ketamine or scopolamine and further analyses of these properties will undoubtedly reveal novel drug mechanisms. The influence of specific interneuron subtypes in the PFC can be explored through genetic or viral-mediated molecular approaches to target them selectively. For instance, transgenic mice expressing Cre-recombinase in PV or SST interneurons can be targeted with optogenetic, designer drug receptors, or receptor knockdown constructs to uncover their functions in the neurophysiological effects of ketamine and scopolamine. Moreover, it remains to be explored whether or not rapid-acting antidepressants influence the synaptic plasticity of interneurons or attenuate their functional deficits in models of depression. Other important considerations are fluctuations in expression levels of specific NMDA and mACh receptors on interneurons or pyramidal neurons in the PFC or HPC throughout adolescence and adulthood and how this might impact its effectiveness as an antidepressant [96, 106, 107]. These fundamental questions will provide important insight into the mediators of rapid-acting antidepressants.
Future Directions
Despite the efforts to better understand the molecular and cellular mechanisms underlying rapid-acting antidepressants there is significant work yet to be performed. There is strong evidence showing that ketamine and scopolamine cause a glutamate burst in the PFC that stimulates molecular signaling pathways to promote synapse formation and function. The behavioral responses are dependent on this synaptic plasticity as these effects reverse the synaptic deficits observed following stress and depression. Increased synaptic plasticity by ketamine and scopolamine occurs following antagonism of NMDA or mACh receptors suggesting that transient inhibition of tonic glutamate or acetylcholine neurotransmission in the PFC is required [108]. Based on these findings it is important to determine the neurons that modulate the release of glutamate in the PFC and develop tools to control these processes. Additional factors such as alterations in dopamine or acetylcholine levels in the PFC during stress or depression and in the presence of rapid antidepressants need to be examined [28, 61, 109]. In all, further research into the neurocircuitry of the PFC in the presence of rapid-acting antidepressants may highlight novel therapeutic targets for pharmacological treatment of MDD.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Kessler R.C., Chiu W.T., Demler O., Merikangas K.R., Walters E.E. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry. 2005;62(6):617–627. doi: 10.1001/archpsyc.62.6.617. [http://dx.doi.org/10.1001/archpsyc.62.6.617]. [PMID: 15939839]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kessler R.C. Epidemiology of women and depression. J. Affect. Disord. 2003;74(1):5–13. doi: 10.1016/s0165-0327(02)00426-3. [http://dx.doi.org/10.1016/S0165-0327(02)00426-3]. [PMID: 12646294]. [DOI] [PubMed] [Google Scholar]
- 3.Murray C.J., Atkinson C., Bhalla K., Birbeck G., Burstein R., Chou D., Dellavalle R., Danaei G., Ezzati M., Fahimi A., Flaxman D. Foreman; Gabriel, S.; Gakidou, E.; Kassebaum, N.; Khatibzadeh, S.; Lim, S.; Lipshultz, S.E.; London, S.; Lopez; MacIntyre, M.F.; Mokdad, A.H.; Moran, A.; Moran, A.E.; Mozaffarian, D.; Murphy, T.; Naghavi, M.; Pope, C.; Roberts, T.; Salomon, J.; Schwebel, D.C.; Shahraz, S.; Sleet, D.A.; Murray; Abraham, J.; Ali, M.K.; Atkinson, C.; Bartels, D.H.; Bhalla, K.; Birbeck, G.; Burstein, R.; Chen, H.; Criqui, M.H.; Dahodwala; Jarlais; Ding, E.L.; Dorsey, E.R.; Ebel, B.E.; Ezzati, M.; Fahami; Flaxman, S.; Flaxman, A.D.; Gonzalez-Medina, D.; Grant, B.; Hagan, H.; Hoffman, H.; Kassebaum, N.; Khatibzadeh, S.; Leasher, J.L.; Lin, J.; Lipshultz, S.E.; Lozano, R.; Lu, Y.; Mallinger, L.; McDermott, M.M.; Micha, R.; Miller, T.R.; Mokdad, A.A.; Mokdad, A.H.; Mozaffarian, D.; Naghavi, M.; Narayan, K.M.; Omer, S.B.; Pelizzari, P.M.; Phillips, D.; Ranganathan, D.; Rivara, F.P.; Roberts, T.; Sampson, U.; Sanman, E.; Sapkota, A.; Schwebel, D.C.; Sharaz, S.; Shivakoti, R.; Singh, G.M.; Singh, D.; Tavakkoli, M.; Towbin, J.A.; Wilkinson, J.D.; Zabetian, A.; Murray; Abraham, J.; Ali, M.K.; Alvardo, M.; Atkinson, C.; Baddour, L.M.; Benjamin, E.J.; Bhalla, K.; Birbeck, G.; Bolliger, I.; Burstein, R.; Carnahan, E.; Chou, D.; Chugh, S.S.; Cohen, A.; Colson, K.E.; Cooper, L.T.; Couser, W.; Criqui, M.H.; Dabhadkar, K.C.; Dellavalle, R.P.; Jarlais; Dicker, D.; Dorsey, E.R.; Duber, H.; Ebel, B.E.; Engell, R.E.; Ezzati, M.; Felson, D.T.; Finucane, M.M.; Flaxman, S.; Flaxman, A.D.; Fleming, T.; Foreman; Forouzanfar, M.H.; Freedman, G.; Freeman, M.K.; Gakidou, E.; Gillum, R.F.; Gonzalez-Medina, D.; Gosselin, R.; Gutierrez, H.R.; Hagan, H.; Havmoeller, R.; Hoffman, H.; Jacobsen, K.H.; James, S.L.; Jasrasaria, R.; Jayarman, S.; Johns, N.; Kassebaum, N.; Khatibzadeh, S.; Lan, Q.; Leasher, J.L.; Lim, S.; Lipshultz, S.E.; London, S.; Lopez; Lozano, R.; Lu, Y.; Mallinger, L.; Meltzer, M.; Mensah, G.A.; Michaud, C.; Miller, T.R.; Mock, C.; Moffitt, T.E.; Mokdad, A.A.; Mokdad, A.H.; Moran, A.; Naghavi, M.; Narayan, K.M.; Nelson, R.G.; Olives, C.; Omer, S.B.; Ortblad, K.; Ostro, B.; Pelizzari, P.M.; Phillips, D.; Raju, M.; Razavi, H.; Ritz, B.; Roberts, T.; Sacco, R.L.; Salomon, J.; Sampson, U.; Schwebel, D.C.; Shahraz, S.; Shibuya, K.; Silberberg, D.; Singh, J.A.; Steenland, K.; Taylor, J.A.; Thurston, G.D.; Vavilala, M.S.; Vos, T.; Wagner, G.R.; Weinstock, M.A.; Weisskopf, M.G.; Wulf, S.; Murray, The state of US health, 19902010: burden of diseases, injuries, and risk factors. JAMA. 2013;310(6):591–608. doi: 10.1001/jama.2013.13805. [http://dx.doi.org/10.1001/jama.2013.13805]. [PMID: 23842577]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman D.P., Perry G.S., Strine T.W. The vital link between chronic disease and depressive disorders. Prev. Chronic Dis. 2005;2(1):A14. [PMID: 15670467]. [PMC free article] [PubMed] [Google Scholar]
- 5.Wang P.S., Simon G., Kessler R.C. The economic burden of depression and the cost-effectiveness of treatment. Int. J. Methods Psychiatr. Res. 2003;12(1):22–33. doi: 10.1002/mpr.139. [http://dx.doi.org/10.1002/ mpr.139]. [PMID: 12830307]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russo S.J., Nestler E.J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 2013;14(9):609–625. doi: 10.1038/nrn3381. [http://dx.doi. org/10.1038/nrn3381]. [PMID: 23942470]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang H.J., Voleti B., Hajszan T., Rajkowska G., Stockmeier C.A., Licznerski P., Lepack A., Majik M.S., Jeong L.S., Banasr M., Son H., Duman R.S. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 2012;18(9):1413–1417. doi: 10.1038/nm.2886. [http://dx.doi.org/10.1038/nm.2886]. [PMID: 22885997]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ota K.T., Liu R.J., Voleti B., Maldonado-Aviles J.G., Duric V., Iwata M., Dutheil S., Duman C., Boikess S., Lewis D.A., Stockmeier C.A., DiLeone R.J., Rex C., Aghajanian G.K., Duman R.S. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat. Med. 2014;20(5):531–535. doi: 10.1038/nm.3513. [http:// dx.doi.org/10.1038/nm.3513]. [PMID: 24728411]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ota K.T., Andres W., Lewis D.A., Stockmeier C.A., Duman R.S. BICC1 expression is elevated in depressed subjects and contributes to depressive behavior in rodents. Neuropsychopharmacology. 2015;40(3):711–718. doi: 10.1038/npp.2014.227. [http://dx.doi.org/10.1038/ npp.2014.227]. [PMID: 25178406]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Licznerski P., Duman R.S. Remodeling of axo-spinous synapses in the pathophysiology and treatment of depression. Neuroscience. 2013;251:33–50. doi: 10.1016/j.neuroscience.2012.09.057. [http://dx.doi.org/10.1016/j.neuroscience.2012. 09.057]. [PMID: 23036622]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zarate C., Duman R.S., Liu G., Sartori S., Quiroz J., Murck H. New paradigms for treatment-resistant depression. Ann. N. Y. Acad. Sci. 2013;1292(1):21–31. doi: 10.1111/nyas.12223. [http://dx.doi.org/10.1111/nyas.12223]. [PMID: 23876043]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aan Het Rot M., Zarate C.A., Jr, Charney D.S., Mathew S.J. Ketamine for depression: where do we go from here? Biol. Psychiatry. 2012;72(7):537–547. doi: 10.1016/j.biopsych.2012.05.003. [http://dx.doi.org/10.1016/j. biopsych.2012.05.003]. [PMID: 22705040]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diazgranados N., Ibrahim L., Brutsche N.E., Newberg A., Kronstein P., Khalife S., Kammerer W.A., Quezado Z., Luckenbaugh D.A., Salvadore G., Machado-Vieira R., Manji H.K., Zarate C.A. Jr A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry. 2010;67(8):793–802. doi: 10.1001/archgenpsychiatry.2010.90. [http://dx.doi.org/ 10.1001/archgenpsychiatry.2010.90]. [PMID: 20679587]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berman R.M., Cappiello A., Anand A., Oren D.A., Heninger G.R., Charney D.S., Krystal J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry. 2000;47(4):351–354. doi: 10.1016/s0006-3223(99)00230-9. [http://dx.doi.org/10.1016/S0006-3223(99)00230-9]. [PMID: 10686270]. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y., Xu Z., Zhang S., Desrosiers A., Schottenfeld R.S., Chawarski M.C. Profiles of psychiatric symptoms among amphetamine type stimulant and ketamine using inpatients in Wuhan, China. J. Psychiatr. Res. 2014;53:99–102. doi: 10.1016/j.jpsychires.2014.02.010. [http://dx.doi. org/10.1016/j.jpsychires.2014.02.010]. [PMID: 24613031]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun L., Li Q., Li Q., Zhang Y., Liu D., Jiang H., Pan F., Yew D.T. Chronic ketamine exposure induces permanent impairment of brain functions in adolescent cynomolgus monkeys. Addict. Biol. 2014;19(2):185–194. doi: 10.1111/adb.12004. [http://dx.doi.org/10.1111/adb.12004]. [PMID: 23145560]. [DOI] [PubMed] [Google Scholar]
- 17.Drevets W.C., Zarate C.A., Jr, Furey M.L. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol. Psychiatry. 2013;73(12):1156–1163. doi: 10.1016/j.biopsych.2012.09.031. [http://dx.doi. org/10.1016/j.biopsych.2012.09.031]. [PMID: 23200525]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furey M.L., Nugent A.C., Speer A.M., Luckenbaugh D.A., Hoffman E.M., Frankel E., Drevets W.C., Zarate C.A., Jr Baseline mood-state measures as predictors of antidepressant response to scopolamine. Psychiatry Res. 2012;196(1):62–67. doi: 10.1016/j.psychres.2012.01.003. [http:// dx.doi.org/10.1016/j.psychres.2012.01.003]. [PMID: 22349648]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furey M.L., Khanna A., Hoffman E.M., Drevets W.C. Scopolamine produces larger antidepressant and antianxiety effects in women than in men. Neuropsychopharmacology. 2010;35(12):2479–2488. doi: 10.1038/npp.2010.131. [http://dx.doi.org/10.1038/npp.2010.131]. [PMID: 20736989]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furey M.L., Drevets W.C. Antidepressant efficacy of the anti- muscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch. Gen. Psychiatry. 2006;63(10):1121–1129. doi: 10.1001/archpsyc.63.10.1121. [http://dx.doi.org/10.1001/archpsyc.63.10.1121]. [PMID: 17015814]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunderland T., Tariot P.N., Newhouse P.A. Differential responsivity of mood, behavior, and cognition to cholinergic agents in elderly neuropsychiatric populations. Brain Res. 1988;472(4):371–389. doi: 10.1016/0006-8993(88)91227-9. [http://dx.doi.org/10.1016/0006-8993(88)91227-9]. [PMID: 3066441]. [DOI] [PubMed] [Google Scholar]
- 22.Newhouse P.A., Sunderland T., Tariot P.N., Weingartner H., Thompson K., Mellow A.M., Cohen R.M., Murphy D.L. The effects of acute scopolamine in geriatric depression. Arch. Gen. Psychiatry. 1988;45(10):906–912. doi: 10.1001/archpsyc.1988.01800340028004. [http://dx.doi.org/10.1001/ archpsyc.1988.01800340028004]. [PMID: 3048225]. [DOI] [PubMed] [Google Scholar]
- 23.Voleti B., Navarria A., Liu R.J., Banasr M., Li N., Terwilliger R., Sanacora G., Eid T., Aghajanian G., Duman R.S. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol. Psychiatry. 2013;74(10):742–749. doi: 10.1016/j.biopsych.2013.04.025. [http://dx. doi.org/10.1016/j.biopsych.2013.04.025]. [PMID: 23751205]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li N., Lee B., Liu R.J., Banasr M., Dwyer J.M., Iwata M., Li X.Y., Aghajanian G., Duman R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. doi: 10.1126/science.1190287. [http://dx.doi.org/ 10.1126/science.1190287]. [PMID: 20724638]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoeffer C.A., Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33(2):67–75. doi: 10.1016/j.tins.2009.11.003. [http://dx.doi.org/10.1016/j.tins.2009.11.003]. [PMID: 19963289]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duman R.S., Li N. A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. 2012. [DOI] [PMC free article] [PubMed]
- 27.Duman R.S., Aghajanian G.K. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338(6103):68–72. doi: 10.1126/science.1222939. [http://dx.doi.org/10.1126/science.1222939]. [PMID: 23042884]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moghaddam B., Adams B., Verma A., Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997;17(8):2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [PMID: 9092613]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucassen P.J., Pruessner J., Sousa N., Almeida O.F., Van Dam A.M., Rajkowska G., Swaab D.F., Czéh B. Neuropathology of stress. Acta Neuropathol. 2014;127(1):109–135. doi: 10.1007/s00401-013-1223-5. [http://dx.doi. org/10.1007/s00401-013-1223-5]. [PMID: 24318124]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myers-Schulz B., Koenigs M. Functional anatomy of ventromedial prefrontal cortex: implications for mood and anxiety disorders. Mol. Psychiatry. 2012;17(2):132–141. doi: 10.1038/mp.2011.88. [http://dx.doi.org/ 10.1038/mp.2011.88]. [PMID: 21788943]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beer J.S., Lombardo M.V., Bhanji J.P. Roles of medial prefrontal cortex and orbitofrontal cortex in self-evaluation. J. Cogn. Neurosci. 2010;22(9):2108–2119. doi: 10.1162/jocn.2009.21359. [http://dx.doi.org/10.1162/jocn. 2009.21359]. [PMID: 19925187]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holtzheimer P.E., Mayberg H.S. Deep brain stimulation for psychiatric disorders. Annu. Rev. Neurosci. 2011;34:289–307. doi: 10.1146/annurev-neuro-061010-113638. [http://dx.doi.org/10.1146/annurev-neuro-061010-113638]. [PMID: 21692660]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayberg H.S., Lozano A.M., Voon V., McNeely H.E., Seminowicz D., Hamani C., Schwalb J.M., Kennedy S.H. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651–660. doi: 10.1016/j.neuron.2005.02.014. [http://dx.doi.org/10.1016/j.neuron.2005.02.014]. [PMID: 15748841]. [DOI] [PubMed] [Google Scholar]
- 34.Price J.L., Drevets W.C. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn. Sci. 2012;16(1):61–71. doi: 10.1016/j.tics.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 35.Drevets W.C., Price J.L., Furey M.L. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct. Funct. 2008;213(1-2):93–118. doi: 10.1007/s00429-008-0189-x. [http://dx.doi.org/10.1007/s00429-008-0189-x]. [PMID: 18704495]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajkowska G., Miguel-Hidalgo J.J., Wei J., Dilley G., Pittman S.D., Meltzer H.Y., Overholser J.C., Roth B.L., Stockmeier C.A. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry. 1999;45(9):1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [http://dx.doi.org/10.1016/S0006-3223(99)00041-4]. [PMID: 10331101]. [DOI] [PubMed] [Google Scholar]
- 37.Duric V., Banasr M., Stockmeier C.A., Simen A.A., Newton S.S., Overholser J.C., Jurjus G.J., Dieter L., Duman R.S. Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. Int. J. Neuropsychopharmacol. 2013;16(1):69–82. doi: 10.1017/S1461145712000016. [http://dx.doi.org/10.1017/ S1461145712000016]. [PMID: 22339950]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radley J.J., Rocher A.B., Miller M., Janssen W.G., Liston C., Hof P.R., McEwen B.S., Morrison J.H. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb. Cortex. 2006;16(3):313–320. doi: 10.1093/cercor/bhi104. [http://dx.doi.org/10.1093/cercor/ bhi104]. [PMID: 15901656]. [DOI] [PubMed] [Google Scholar]
- 39.Bloss E.B., Puri R., Yuk F., Punsoni M., Hara Y., Janssen W.G., McEwen B.S., Morrison J.H. Morphological and molecular changes in aging rat prelimbic prefrontal cortical synapses. Neurobiol. Aging. 2013;34(1):200–210. doi: 10.1016/j.neurobiolaging.2012.05.014. [http://dx.doi.org/10.1016/ j.neurobiolaging.2012.05.014]. [PMID: 22727942]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu R.J., Aghajanian G.K. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc. Natl. Acad. Sci. USA. 2008;105(1):359–364. doi: 10.1073/pnas.0706679105. [http://dx.doi.org/10.1073/pnas.0706679105]. [PMID: 18172209]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cook S.C., Wellman C.L. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J. Neurobiol. 2004;60(2):236–248. doi: 10.1002/neu.20025. [http://dx.doi.org/10.1002/neu.20025]. [PMID: 15266654]. [DOI] [PubMed] [Google Scholar]
- 42.Sheline Y.I., Gado M.H., Kraemer H.C. Untreated depression and hippocampal volume loss. Am. J. Psychiatry. 2003;160(8):1516–1518. doi: 10.1176/appi.ajp.160.8.1516. [http://dx.doi.org/10.1176/appi.ajp.160.8.1516]. [PMID: 12900317]. [DOI] [PubMed] [Google Scholar]
- 43.MacQueen G., Frodl T. The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol. Psychiatry. 2011;16(3):252–264. doi: 10.1038/mp.2010.80. [http://dx.doi.org/10.1038/mp.2010.80]. [PMID: 20661246]. [DOI] [PubMed] [Google Scholar]
- 44.Vyas A., Mitra R., Shankaranarayana Rao B.S., Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci. 2002;22(15):6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [PMID: 12151561]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drevets W.C. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr. Opin. Neurobiol. 2001;11(2):240–249. doi: 10.1016/s0959-4388(00)00203-8. [http:// dx.doi.org/10.1016/S0959-4388(00)00203-8]. [PMID: 11301246]. [DOI] [PubMed] [Google Scholar]
- 46.Liotti M., Mayberg H.S. The role of functional neuroimaging in the neuropsychology of depression. J. Clin. Exp. Neuropsychol. 2001;23(1):121–136. doi: 10.1076/jcen.23.1.121.1223. [http://dx.doi.org/10.1076/jcen.23.1.121.1223]. [PMID: 11320448]. [DOI] [PubMed] [Google Scholar]
- 47.Christoffel D.J., Golden S.A., Russo S.J. Structural and synaptic plasticity in stress-related disorders. Rev. Neurosci. 2011;22(5):535–549. doi: 10.1515/RNS.2011.044. [http://dx.doi.org/10.1515/RNS.2011.044]. [PMID: 21967517]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ampuero E., Rubio F.J., Falcon R., Sandoval M., Diaz-Veliz G., Gonzalez R.E., Earle N., Dagnino-Subiabre A., Aboitiz F., Orrego F., Wyneken U. Chronic fluoxetine treatment induces structural plasticity and selective changes in glutamate receptor subunits in the rat cerebral cortex. Neuroscience. 2010;169(1):98–108. doi: 10.1016/j.neuroscience.2010.04.035. [http://dx.doi.org/10.1016/j.neuroscience.2010.04.035]. [PMID: 20417256]. [DOI] [PubMed] [Google Scholar]
- 49.Bessa J.M. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol. Psychiatry. 2009;14(8):764–773. doi: 10.1038/mp.2008.119. [http://dx.doi.org/10.1038/ mp.2008.119]. [DOI] [PubMed] [Google Scholar]
- 50.Radley J.J., Sisti H.M., Hao J., Rocher A.B., McCall T., Hof P.R., McEwen B.S., Morrison J.H. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125(1):1–6. doi: 10.1016/j.neuroscience.2004.01.006. [http:// dx.doi.org/10.1016/j.neuroscience.2004.01.006]. [PMID: 15051139]. [DOI] [PubMed] [Google Scholar]
- 51.Chen Z.Y., Jing D., Bath K.G., Ieraci A., Khan T., Siao C.J., Herrera D.G., Toth M., Yang C., McEwen B.S., Hempstead B.L., Lee F.S. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314(5796):140–143. doi: 10.1126/science.1129663. [http://dx.doi.org/10.1126/science.1129663]. [PMID: 17023662]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duman R.S., Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 2012;35(1):47–56. doi: 10.1016/j.tins.2011.11.004. [http://dx.doi.org/10.1016/j.tins.2011.11.004]. [PMID: 22217452]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duman R.S., Monteggia L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry. 2006;59(12):1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [http://dx.doi.org/10.1016/j.biopsych.2006.02.013]. [PMID: 16631126]. [DOI] [PubMed] [Google Scholar]
- 54.Elsayed M., Banasr M., Duric V., Fournier N.M., Licznerski P., Duman R.S. Antidepressant effects of fibroblast growth factor-2 in behavioral and cellular models of depression. Biol. Psychiatry. 2012;72(4):258–265. doi: 10.1016/j.biopsych.2012.03.003. [http://dx.doi.org/10.1016/j.biopsych.2012. 03.003]. [PMID: 22513055]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katz M.M., Houston J.P., Brannan S., Bowden C.L., Berman N., Swann A.C., Frazer A. A multivantaged behavioural method for measuring onset and sequence of the clinical actions of antidepressants. Int. J. Neuropsychopharmacol. 2004;7(4):471–479. doi: 10.1017/S1461145704004407. [http://dx.doi.org/10.1017/S1461145704004407]. [PMID: 15154974]. [DOI] [PubMed] [Google Scholar]
- 56.Katz M.M., Tekell J.L., Bowden C.L., Brannan S., Houston J.P., Berman N., Frazer A. Onset and early behavioral effects of pharmacologically different antidepressants and placebo in depression. Neuropsychopharmacology. 2004;29(3):566–579. doi: 10.1038/sj.npp.1300341. [http://dx.doi.org/10.1038/sj.npp.1300341]. [PMID: 14627997]. [DOI] [PubMed] [Google Scholar]
- 57.Gaynes B.N., Warden D., Trivedi M.H., Wisniewski S.R., Fava M., Rush A.J. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr. Serv. 2009;60(11):1439–1445. doi: 10.1176/ps.2009.60.11.1439. [http://dx.doi.org/10.1176/ps.2009. 60.11.1439]. [PMID: 19880458]. [DOI] [PubMed] [Google Scholar]
- 58.Li N., Liu R.J., Dwyer J.M., Banasr M., Lee B., Son H., Li X.Y., Aghajanian G., Duman R.S. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry. 2011;69(8):754–761. doi: 10.1016/j.biopsych.2010.12.015. [http://dx.doi.org/10.1016/j. biopsych.2010.12.015]. [PMID: 21292242]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu R.J., Fuchikami M., Dwyer J.M., Lepack A.E., Duman R.S., Aghajanian G.K. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38(11):2268–2277. doi: 10.1038/npp.2013.128. [http://dx.doi.org/10.1038/npp.2013.128]. [PMID: 23680942]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dwyer J.M., Lepack A.E., Duman R.S. mGluR2/3 blockade produces rapid and long-lasting reversal of anhedonia caused by chronic stress exposure. J. Mol. Psychiatry. 2013;1(1):15. doi: 10.1186/2049-9256-1-15. [http://dx.doi.org/10.1186/2049-9256-1-15]. [PMID: 25408908]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lorrain D.S., Baccei C.S., Bristow L.J., Anderson J.J., Varney M.A. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117(3):697–706. doi: 10.1016/s0306-4522(02)00652-8. [http://dx.doi. org/10.1016/S0306-4522(02)00652-8]. [PMID: 12617973]. [DOI] [PubMed] [Google Scholar]
- 62.Chowdhury G.M., Behar K.L., Cho W., Thomas M.A., Rothman D.L., Sanacora G. 1H-[13C]-nuclear magnetic resonance spectroscopy measures of ketamines effect on amino acid neurotransmitter metabolism. Biol. Psychiatry. 2012;71(11):1022–1025. doi: 10.1016/j.biopsych.2011.11.006. [http://dx.doi.org/10.1016/j.biopsych.2011.11.006]. [PMID: 22169441]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koike H., Chaki S. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav. Brain Res. 2014;271:111–115. doi: 10.1016/j.bbr.2014.05.065. [http://dx.doi.org/10.1016/j.bbr.2014.05.065]. [PMID: 24909673]. [DOI] [PubMed] [Google Scholar]
- 64.Alt A., Nisenbaum E.S., Bleakman D., Witkin J.M. A role for AMPA receptors in mood disorders. Biochem. Pharmacol. 2006;71(9):1273–1288. doi: 10.1016/j.bcp.2005.12.022. [http://dx.doi.org/10.1016/j.bcp.2005.12.022]. [PMID: 16442080]. [DOI] [PubMed] [Google Scholar]
- 65.Autry A.E., Adachi M., Nosyreva E., Na E.S., Los M.F., Cheng P.F., Kavalali E.T., Monteggia L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475(7354):91–95. doi: 10.1038/nature10130. [http://dx.doi.org/10.1038/nature10130]. [PMID: 21677641]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lepack A.E., Fuchikami M., Dwyer J.M., Banasr M., Duman R.S. BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 2014;18(1):pyu033. doi: 10.1093/ijnp/pyu033. [http://dx.doi.org/10.1093/ijnp/pyu033]. [PMID: 25539510]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu R.J., Lee F.S., Li X.Y., Bambico F., Duman R.S., Aghajanian G.K. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol. Psychiatry. 2012;71(11):996–1005. doi: 10.1016/j.biopsych.2011.09.030. [http:// dx.doi.org/10.1016/j.biopsych.2011.09.030]. [PMID: 22036038]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Egan M.F., Kojima M., Callicott J.H., Goldberg T.E., Kolachana B.S., Bertolino A., Zaitsev E., Gold B., Goldman D., Dean M., Lu B., Weinberger D.R. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112(2):257–269. doi: 10.1016/s0092-8674(03)00035-7. [http://dx.doi.org/10.1016/S0092-8674(03)00035-7]. [PMID: 12553913]. [DOI] [PubMed] [Google Scholar]
- 69.Pattwell S.S., Bath K.G., Perez-Castro R., Lee F.S., Chao M.V., Ninan I. The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J. Neurosci. 2012;32(7):2410–2421. doi: 10.1523/JNEUROSCI.5205-11.2012. [http://dx.doi.org/ 10.1523/JNEUROSCI.5205-11.2012]. [PMID: 22396415]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pochwat B., Pałucha-Poniewiera A., Szewczyk B., Pilc A., Nowak G. NMDA antagonists under investigation for the treatment of major depressive disorder. Expert Opin. Investig. Drugs. 2014;23(9):1181–1192. doi: 10.1517/13543784.2014.918951. [http://dx.doi.org/10.1517/ 13543784.2014.918951]. [PMID: 24818801]. [DOI] [PubMed] [Google Scholar]
- 71.Preskorn S.H., Baker B., Kolluri S., Menniti F.S., Krams M., Landen J.W. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J. Clin. Psychopharmacol. 2008;28(6):631–637. doi: 10.1097/JCP.0b013e31818a6cea. [http://dx.doi.org/10.1097/JCP.0b013 e31818a6cea]. [PMID: 19011431]. [DOI] [PubMed] [Google Scholar]
- 72.Miller O.H., Yang L., Wang C.C., Hargroder E.A., Zhang Y., Delpire E., Hall B.J. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife. 2014;3:e03581. doi: 10.7554/eLife.03581. [http://dx.doi.org/10.7554/eLife.03581]. [PMID: 25340958]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Witkin J.M., Overshiner C., Li X., Catlow J.T., Wishart G.N., Schober D.A., Heinz B.A., Nikolayev A., Tolstikov V.V., Anderson W.H., Higgs R.E., Kuo M.S., Felder C.C. M1 and m2 muscarinic receptor subtypes regulate antidepressant-like effects of the rapidly acting antidepressant scopolamine. J. Pharmacol. Exp. Ther. 2014;351(2):448–456. doi: 10.1124/jpet.114.216804. [http://dx.doi.org/10.1124/jpet.114. 216804]. [PMID: 25187432]. [DOI] [PubMed] [Google Scholar]
- 74.Gupta A., Wang Y., Markram H. Organizing principles for a diversity of GABAergic interneurons and synapses in the neocortex. Science. 2000;287(5451):273–278. doi: 10.1126/science.287.5451.273. [http://dx.doi.org/ 10.1126/science.287.5451.273]. [PMID: 10634775]. [DOI] [PubMed] [Google Scholar]
- 75.DeFelipe J., López-Cruz P.L., Benavides-Piccione R., Bielza C., Larrañaga P., Anderson S., Burkhalter A., Cauli B., Fairén A., Feldmeyer D., Fishell G., Fitzpatrick D., Freund T.F., González-Burgos G., Hestrin S., Hill S., Hof P.R., Huang J., Jones E.G., Kawaguchi Y., Kisvárday Z., Kubota Y., Lewis D.A., Marín O., Markram H., McBain C.J., Meyer H.S., Monyer H., Nelson S.B., Rockland K., Rossier J., Rubenstein J.L., Rudy B., Scanziani M., Shepherd G.M., Sherwood C.C., Staiger J.F., Tamás G., Thomson A., Wang Y., Yuste R., Ascoli G.A. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat. Rev. Neurosci. 2013;14(3):202–216. doi: 10.1038/nrn3444. [http://dx.doi.org/10.1038/nrn3444]. [PMID: 23385869]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Markram H., Toledo-Rodriguez M., Wang Y., Gupta A., Silberberg G., Wu C. Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 2004;5(10):793–807. doi: 10.1038/nrn1519. [http://dx.doi. org/10.1038/nrn1519]. [PMID: 15378039]. [DOI] [PubMed] [Google Scholar]
- 77.Hu H., Gan J., Jonas P. Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science. 2014;345(6196):1255263. doi: 10.1126/science.1255263. [http://dx.doi.org/ 10.1126/science.1255263]. [PMID: 25082707]. [DOI] [PubMed] [Google Scholar]
- 78.McGarry L.M., Packer A.M., Fino E., Nikolenko V., Sippy T., Yuste R. Quantitative classification of somatostatin-positive neocortical interneurons identifies three interneuron subtypes. Front. Neural Circuits. 2010;4:12. doi: 10.3389/fncir.2010.00012. [PMID: 20617186]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Y., Toledo-Rodriguez M., Gupta A., Wu C., Silberberg G., Luo J., Markram H. Anatomical, physiological and molecular properties of Martinotti cells in the somatosensory cortex of the juvenile rat. J. Physiol. 2004;561(Pt 1):65–90. doi: 10.1113/jphysiol.2004.073353. [http://dx.doi.org/ 10.1113/jphysiol.2004.073353]. [PMID: 15331670]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Silberberg G., Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53(5):735–746. doi: 10.1016/j.neuron.2007.02.012. [http://dx.doi.org/10.1016/j.neuron.2007. 02.012]. [PMID: 17329212]. [DOI] [PubMed] [Google Scholar]
- 81.Neske G.T., Patrick S.L., Connors B.W. Contributions of diverse excitatory and inhibitory neurons to recurrent network activity in cerebral cortex. J. Neurosci. 2015;35(3):1089–1105. doi: 10.1523/JNEUROSCI.2279-14.2015. [http://dx. doi.org/10.1523/JNEUROSCI.2279-14.2015]. [PMID: 25609625]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tan Z., Hu H., Huang Z.J., Agmon A. Robust but delayed thalamocortical activation of dendritic-targeting inhibitory interneurons. Proc. Natl. Acad. Sci. USA. 2008;105(6):2187–2192. doi: 10.1073/pnas.0710628105. [http://dx.doi.org/10.1073/pnas.0710628105]. [PMID: 18245383]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rajkowska G. ODwyer, G.; Teleki, Z.; Stockmeier, C.A.; Miguel-Hidalgo, J.J. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32(2):471–482. doi: 10.1038/sj.npp.1301234. [http://dx.doi.org/10.1038/sj.npp.1301234]. [PMID: 17063153]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rubinow D.R., Gold P.W., Post R.M., Ballenger J.C., Cowdry R., Bollinger J., Reichlin S. CSF somatostatin in affective illness. Arch. Gen. Psychiatry. 1983;40(4):409–412. doi: 10.1001/archpsyc.1983.01790040063009. [http://dx.doi.org/ 10.1001/archpsyc.1983.01790040063009]. [PMID: 6132592]. [DOI] [PubMed] [Google Scholar]
- 85.Sibille E., Morris H.M., Kota R.S., Lewis D.A. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int. J. Neuropsychopharmacol. 2011;14(6):721–734. doi: 10.1017/S1461145710001616. [http://dx. doi.org/10.1017/S1461145710001616]. [PMID: 21226980]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soumier A., Sibille E. Opposing effects of acute versus chronic blockade of frontal cortex somatostatin-positive inhibitory neurons on behavioral emotionality in mice. Neuropsychopharmacology. 2014;39(9):2252–2262. doi: 10.1038/npp.2014.76. [http://dx.doi.org/10.1038/npp.2014.76]. [PMID: 24690741]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Northoff G., Sibille E. Why are cortical GABA neurons relevant to internal focus in depression? A cross-level model linking cellular, biochemical and neural network findings. Mol. Psychiatry. 2014;19(9):966–977. doi: 10.1038/mp.2014.68. [http://dx.doi.org/10.1038/mp.2014.68]. [PMID: 25048001]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Homayoun H., Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007;27(43):11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [http://dx.doi.org/10.1523/JNEUROSCI.2213-07.2007]. [PMID: 17959792]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jackson M.E., Homayoun H., Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc. Natl. Acad. Sci. USA. 2004;101(22):8467–8472. doi: 10.1073/pnas.0308455101. [http://dx.doi.org/10.1073/ pnas.0308455101]. [PMID: 15159546]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lahti A.C., Koffel B., LaPorte D., Tamminga C.A. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13(1):9–19. doi: 10.1016/0893-133X(94)00131-I. [http://dx.doi.org/10.1016/ 0893-133X(94)00131-I]. [PMID: 8526975]. [DOI] [PubMed] [Google Scholar]
- 91.Breier A., Malhotra A.K., Pinals D.A., Weisenfeld N.I., Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am. J. Psychiatry. 1997;154(6):805–811. doi: 10.1176/ajp.154.6.805. [http://dx.doi.org/10.1176/ajp.154.6.805]. [PMID: 9167508]. [DOI] [PubMed] [Google Scholar]
- 92.Jodo E., Suzuki Y., Katayama T., Hoshino K.Y., Takeuchi S., Niwa S., Kayama Y. Activation of medial prefrontal cortex by phencyclidine is mediated via a hippocampo-prefrontal pathway. Cereb. Cortex. 2005;15(5):663–669. doi: 10.1093/cercor/bhh168. [http://dx.doi.org/10.1093/ cercor/bhh168]. [PMID: 15342431]. [DOI] [PubMed] [Google Scholar]
- 93.Constantinidis C., Goldman-Rakic P.S. Correlated discharges among putative pyramidal neurons and interneurons in the primate prefrontal cortex. J. Neurophysiol. 2002;88(6):3487–3497. doi: 10.1152/jn.00188.2002. [http://dx.doi.org/10.1152/jn.00188.2002]. [PMID: 12466463]. [DOI] [PubMed] [Google Scholar]
- 94.Rao S.G., Williams G.V., Goldman-Rakic P.S. Isodirectional tuning of adjacent interneurons and pyramidal cells during working memory: evidence for microcolumnar organization in PFC. J. Neurophysiol. 1999;81(4):1903–1916. doi: 10.1152/jn.1999.81.4.1903. [PMID: 10200225]. [DOI] [PubMed] [Google Scholar]
- 95.Wang H.X., Gao W.J. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology. 2009;34(8):2028–2040. doi: 10.1038/npp.2009.20. [http://dx.doi. org/10.1038/npp.2009.20]. [PMID: 19242405]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nosyreva E., Autry A.E., Kavalali E.T., Monteggia L.M. Age dependence of the rapid antidepressant and synaptic effects of acute NMDA receptor blockade. Front. Mol. Neurosci. 2014;7:94. doi: 10.3389/fnmol.2014.00094. [http://dx.doi.org/10.3389/fnmol.2014.00094]. [PMID: 25520615]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gideons E.S., Kavalali E.T., Monteggia L.M. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc. Natl. Acad. Sci. USA. 2014;111(23):8649–8654. doi: 10.1073/pnas.1323920111. [http://dx.doi.org/10.1073/pnas.1323920111]. [PMID: 24912158]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kavalali E.T., Monteggia L.M. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am. J. Psychiatry. 2012;169(11):1150–1156. doi: 10.1176/appi.ajp.2012.12040531. [http://dx.doi.org/10.1176/appi.ajp.2012.12040531]. [PMID: 23534055]. [DOI] [PubMed] [Google Scholar]
- 99.Moghaddam B., Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37(1):4–15. doi: 10.1038/npp.2011.181. [http:// dx.doi.org/10.1038/npp.2011.181]. [PMID: 21956446]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Picciotto M.R., Higley M.J., Mineur Y.S. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76(1):116–129. doi: 10.1016/j.neuron.2012.08.036. [http://dx. doi.org/10.1016/j.neuron.2012.08.036]. [PMID: 23040810]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Amar M., Lucas-Meunier E., Baux G., Fossier P. Blockade of different muscarinic receptor subtypes changes the equilibrium between excitation and inhibition in rat visual cortex. Neuroscience. 2010;169(4):1610–1620. doi: 10.1016/j.neuroscience.2010.06.019. [http://dx.doi.org/10.1016/j.neuroscience. 2010.06.019]. [PMID: 20600670]. [DOI] [PubMed] [Google Scholar]
- 102.Disney A.A., Reynolds J.H. Expression of m1-type muscarinic acetylcholine receptors by parvalbumin-immunoreactive neurons in the primary visual cortex: a comparative study of rat, guinea pig, ferret, macaque, and human. J. Comp. Neurol. 2014;522(5):986–1003. doi: 10.1002/cne.23456. [http://dx.doi.org/10.1002/cne.23456]. [PMID: 23983014]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gulledge A.T., Bucci D.J., Zhang S.S., Matsui M., Yeh H.H. M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J. Neurosci. 2009;29(31):9888–9902. doi: 10.1523/JNEUROSCI.1366-09.2009. [http://dx.doi.org/10.1523/JNEUROSCI.1366-09.2009]. [PMID: 19657040]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rawls S.M., Mcginty J.F. Muscarinic receptors regulate extracellular glutamate levels in the rat striatum: an in vivo microdialysis study. J. Pharmacol. Exp. Ther. 1998;286(1):91–98. [PMID: 9655846]. [PubMed] [Google Scholar]
- 105.Tian M.K., Bailey C.D., Lambe E.K. Cholinergic excitation in mouse primary vs. associative cortex: region-specific magnitude and receptor balance. Eur. J. Neurosci. 2014;40(4):2608–2618. doi: 10.1111/ejn.12622. [http://dx.doi.org/10.1111/ejn.12622]. [PMID: 24827827]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roux C., Aligny C., Lesueur C., Girault V., Brunel V., Ramdani Y., Genty D., Driouich A., Laquerrière A., Marret S., Brasse-Lagnel C., Gonzalez B.J., Bekri S. NMDA receptor blockade in the developing cortex induces autophagy-mediated death of immature cortical GABAergic interneurons: An ex vivo and in vivo study in Gad67-GFP mice. Exp. Neurol. 2015;267:177–193. doi: 10.1016/j.expneurol.2015.02.037. [http://dx.doi.org/10.1016/j.expneurol.2015.02.037]. [PMID: 25795167]. [DOI] [PubMed] [Google Scholar]
- 107.Aligny C., Roux C., Dourmap N., Ramdani Y., Do-Rego J.C., Jégou S., Leroux P., Leroux-Nicollet I., Marret S., Gonzalez B.J. Ketamine alters cortical integration of GABAergic interneurons and induces long-term sex-dependent impairments in transgenic Gad67-GFP mice. Cell Death Dis. 2014;5:e1311. doi: 10.1038/cddis.2014.275. [http://dx.doi.org/10.1038/cddis.2014.275]. [PMID: 24991763]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Monteggia L.M., Kavalali E.T. Scopolamine and ketamine: evidence of convergence? Biol. Psychiatry. 2013;74(10):712–713. doi: 10.1016/j.biopsych.2013.08.011. [http://dx.doi.org/10.1016/j.biopsych.2013.08.011]. [PMID: 24144324]. [DOI] [PubMed] [Google Scholar]
- 109.Huang G.B., Zhao T., Muna S.S., Bagalkot T.R., Jin H.M., Chae H.J., Chung Y.C. Effects of chronic social defeat stress on behaviour, endoplasmic reticulum proteins and choline acetyltrans- ferase in adolescent mice. Int. J. Neuropsychopharmacol. 2013;16(7):1635–1647. doi: 10.1017/S1461145713000060. [http://dx.doi.org/10.1017/S1461145713000060]. [PMID: 23442729]. [DOI] [PubMed] [Google Scholar]