

Abstract
Aiming at development of potent antitubulin agents targeting colchicine-binding site, a series of novel 5-indolyl-7-arylimidazo[1,2-a]pyridine-8-carbonitrilederivatives (5a–5v and 7a–7h) were designed based on bioisosterism and hybridization strategies. All these compounds were concisely synthesized via a three-step process and examined against five human cancer cell lines (HT-29, A549, MKN-45, MDA-MB-231 and SMMC-7721) along with a normal human cell (L02) in vitro. A structure-activity relationships (SARs) study was carried out and optimization towards this series of compounds in cellular assay resulted in the discovery of 5k, which displayed similar or better antitumor potency against the tested cancer cells with IC50 value ranging from 0.02 to 1.22 μM superior to CA-4 and Crolibulin. Significantly, a cell cycle study disclosed the ability of 5k to arrest cell cycle at the G2/M phase, and immunofluorescence assay as well as a colchicine competition assay revealed that tubulin polymerization was disturbed by 5k by binding to the colchicine site. Moreover, the molecular modeling mode showed the posture of 5k and Crolibulin was similar in the colchcine-binding pocket of tubulin as identified with the SARs and pharmacological results. Together, all these results rationalized 5k might serve as a promising lead for a novel class of antitubulin agents for cancer treatments.
Microtubules, which formed by the self-assembly of α- and β-tubulin heterodimers, are an essential eukaryotic protein that plays critical roles in cell division, intracellular trafficking, cell migration and angiogenesis1,2,3,4. Therefore, anticancer therapy based on microtubule targeting agents (MTAs) is receiving growing attentions in drug discovery5. MTAs (Fig. 1) are known to interact with tubulin via at least four binding sites: the laulimalide, taxane (stabilisers of microtubules), vinca alkaloid and colchicine sites (destabilisers of microtubules)6,7. The taxanes and vinca alkaloids have been approved in clinical practice as antitumor agents8,9. Despite numerous efforts, none of the colchicine-binding site inhibitors (CBSIs) reached the market10, thus the discovery of antitubulin CBSIs is still of substantial efforts, which have led to the identification of several classes of inhibitors.
Figure 1. The representative antitubulin agents and target compounds (5a–5v and 7a–7h).

Combretastatin A-4 (CA-4), a natural cis-stilbene derivative isolated from the bark of South Africa tree Combretum caffrum, is a highly effective natural antitubulin agent strongly effecting microtubules dynamics by binding to the colchicine site which exhibited excellent antiproliferative activity against a wide panel of cell lines11,12. However, two major obstacles of CA-4 remain unsolved: the poor solubility and acquired multidrug resistance13, thus stimulating significant interest in diverse CA-4 analogs. Despite initial SARs study with CA-4 analogs indicated that the cis-olefin configuration at linking bridge was essential for optimal activity14, deficiency of the cis-olefin prone to isomerize to chemically more stable trans-configuration resulted in a dramatic loss in antitumor potency. Therefore, a variety of heteroaromatic-linkaged CA-4 derivatives, such as imidazole15, isoxazole16, thiophene17, pyrazole18 and indole group19 as link-bridge, were disclosed to retain the appropriate configuration of two adjacent aromatic A-ring and B-ring required for bioactivity. For example, the oxazoline derivative a (Fig. 1) with oxazoline motif and indolyl scaffold instead of vinyl linkage and B-ring in CA-4 respectively, displayed potent cytotoxic activity. Also, structurally related CA-4 analogue b that incorporated an indolyl moiety was reported as potent tubulin polymerization inhibitor19.
Crolibulin, a CBSIs under phase II clinical trials20,21, is active against various experimental tumors and exhibit potent inhibition of mitosis at the G2/M stage22, while its undue toxicity, for example, cardiovascular toxicity and neurotoxicity, impinged its clinical development23,24. The SARs study revealed that the cyano group and aromatic A-ring of Crolibulin are essential elements for exerting biological activity25,26,27. Thus, in an attempt to design novel antitubulin agents targeting colchicines-binding site, we were stimulated to optimize the chromene moiety by applying bioisosterism strategy.
Recently, the concept of “privileged medicinal scaffolds” has emerged as one of the guiding principles of drug discovery28,29,30. These frameworks commonly consist of a rigid heterocyclic ring system that assigns well-defined orientation of appended functionalities for target recognition. In this regard, imidazo[1,2-a]pyridines (IMPs) skeleton acted as the privileged medicinal scaffold are of particular utility serving for the generation of small-molecule ligands with pronounced antitumor, antiviral or anti-inflammatory efficacy31,32,33, such as GSK923295A34 and Alpidem35. Herein, our efforts were towards to optimize the 4H-chromenes-3-carbonitrile scaffold in Crolibulin by replacement with imidazo[1,2-a]pyridine-8-carbonitrile fragment based on bioisosterism strategy.
In the continuation of SARs study of restricted CA-4 analogs, we envisioned that imidazo[1,2-a]pyridine-8-carbonitrile fragment could serve as link-bridge instead of the cis-olefin in CA-4, while the favorable indolyl group was introduced into the B-ring by employing hybridization principle. Therefore, a series of novel 7-phenylimidazo[1,2-a]pyridine-8-carbonitrile derivatives conjugated with indolyl moiety (5a–5v) as antitubulin agents were designed and synthesized. Eventually, diverse substituents were introduced to the terminal phenyl fragment and indole portion attempting to investigate the influence on antitumor efficacy by regulating the electronic and steric effects. To further optimize properties of this series of compounds, various hydrophilic N-aliphatic aminomethyl groups were imported into IMPs moiety at C-2 position to afford derivatives 7a–7h.
In this study, a series of novel 7-arylimidazo[1,2-a]pyridine-8-carbonitrile derivatives bearing indolyl moiety were firstly designed and optimized as colchicines-binding-site targeted antitubulin agents. All the compounds were prepared via a concise three-step process involving acylation reaction, one-pot coupling reaction and cyclization transformation, and their structures were determined by MS, 1H-NMR, 13C-NMR and element analysis. Finally, antiproliferative activities of the target compounds were evaluated against five human cancer cell lines and one normal human cell in cellular assay. Also, the primary mechanism of the lead compound 5k on the inhibition of tubulin assembly was examined in vitro by cell cycle arrest, immunofluorescence assay and colchicine competition assay, which indicating a G2/M phase arrest and cellular microtubule depolymerization by binding to the colchicine site, as was in accordance with the docking mode study.
Result and Discussion
Chemistry
series of 5-indolyl-7-arylimidazo[1,2-a]pyridine-8-carbonitrile derivatives (5a-5v) and their intermediates were synthesized according to the pathways described in Fig. 2. Acylation reaction of 1H-indole or 5-bromo-1H-indole with acetyl chloride in the presence of stannic chloride in dichloromethane and nitromethane co-solvent for 3.5 h afforded compounds 2a–2b. Then, 2-amino-4,6-diarylnicotinonitrile derivatives 4a–4v could be concisely assembled in a one-pot coupling reaction of four components, namely 2a or 2b, aromatic formaldehyde, malononitrile and ammonium acetate. In this section, the one-pot coupling protocol as an efficient method for preparation of pyridines, has been developed allowing reaction conditions to be accessed in organic synthesis36. Subsequently, 4a–4v were subjected to further cyclization transformation with 2-chloroacetaldehyde in refluxing ethanol on exposure to sodium bicarbonate gave rise to the desired target compounds 5a–5v in good yields (Supplementary Information).
Figure 2. Reagents and conditions.

(i) stannic chloride, acetyl chloride, dichloromethane, nitromethane, 0 °C to r.t., 3.5h; (ii) ammonium acetate, toluene, reflux; (iii) 2-chloroacetaldehyde, sodium bicarbonate, ethanol, reflux.
The target compounds 7a–7h were prepared according to the method as depicted in Fig. 3. The synthesized 4k reacted with 1,3-dichloropropan-2-one in ethanol refluxed for 8 h to generate 7-(2-chloro-4-fluorophenyl)-2-chloromethyl-5-(1H-indol-3-yl)imidazo[1,2-a]pyridine-8-carbonitrile (6). Eventually, nucleophilic substitution of intermediate 6 with a variety of N-aliphatic amines afforded the corresponding compounds 7a–7h in satisfied yield (Supplementary Information).
Figure 3. Reagents and conditions.

(i) 1,3-dichloropropan-2-one, ethanol, reflux; (ii) N-aliphatic amines, ethanol, reflux.
Bioactivity
In vitro antiproliferative activity and SARs study
To evaluate the antiproliferative activity, all synthesized derivatives (5a–5v and 7a–7h) were investigated for their activity in vitro against a panel of cancer cell lines including HT-29 (human colon cancer), H460 (human lung cancer), A549 (human lung carcinoma), MKN-45 (human gastric cancer) and SMMC-7721 (human liver cancer) by MTT assay. CA-4 and Crolibulin were served as positive control, and the results expressed as half-maximal inhibitory concentration (IC50). IC50 values are the concentrations that cause 50% inhibition of cancer cell growth (μM). Data represent the mean values ± standard deviation (SD) of independent experiments performed in triplicate.
As shown in Table 1, all the evaluated compounds (5a–5v) bearing imidazo[1,2-a]pyridines (IMPs) scaffold showed moderate to significant cytotoxic activity against HT-29, H460, A549 and MKN45 cell lines with potency in a dozen nanomolar range, but displayed at least 2-fold less sensitivity on SMMC-7721 cell as indicated by the IC50 values. Moreover, most compounds exhibited excellent cytotoxicity against HT-29, reflecting good selectivity for colon cancer. Significantly, five compounds (5e, 5f, 5i, 5j and 5k) demonstrated prominent cytotoxicity with IC50 values ranging from 0.02 to 26.15 μM, which was comparable or superior to Crolibulin and CA-4. Notably, the candidate 5k displayed pronounced activity with IC50 values of 0.02 μM, 0.05 μM, 0.57 μM, 0.14 μM and 1.22 μM against HT-29, H460, A549, MKN-45 and SMMC-7721 cell lines respectively, which was about 0.60- to 26-fold more potent than Crolibulin, and almost twice over the average of CA-4.
Table 1. Structures and cytotoxicity of compounds 5a–5v.
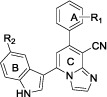 | |||||||
|---|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | IC50 (μM) ±SDa |
||||
| HT29 | H460 | A549 | MKN45 | SMMC-7721 | |||
| 5a | H | H | 0.30 ± 0.04 | 2.79 ± 0.52 | 86.42 ± 7.33 | 79.67 ± 8.10 | 89.43 ± 2.60 |
| 5b | 2-Me | H | 0.16 ± 0.04 | 46.28 ± 5.26 | 0.23 ± 0.04 | 1.52 ± 0.15 | 0.82 ± 0.06 |
| 5c | 3-OMe | H | 0.24 ± 0.01 | 17.50 ± 1.53 | 12.23 ± 1.89 | 1.24 ± 0.08 | 76.15 ± 8.98 |
| 5d | 4-OMe | H | 0.43 ± 0.03 | 9.61 ± 0.76 | 15.47 ± 2.13 | 1.64 ± 0.07 | 71.53 ± 9.43 |
| 5e | 2-F | H | 0.09 ± 0.01 | 1.16 ± 0.12 | 5.67 ± 0.23 | 0.96 ± 0.06 | 26.15 ± 2.82 |
| 5f | 2-Cl | H | 0.07 ± 0.004 | 0.18 ± 0.08 | 0.95 ± 0.07 | 0.38 ± 0.06 | 13.83 ± 2.23 |
| 5g | 3-Br | H | >100 | 6.89 ± 0.67 | >100 | 64.0 ± 7.14 | 79.20 ± 6.87 |
| 5h | 4-Br | H | 0.12 ± 0.02 | 0.95 ± 0.03 | 8.54 ± 0.93 | 2.76 ± 0.47 | 53.45 ± 7.76 |
| 5i | 2,4-di-F | H | 0.08 ± 0.001 | 0.04 ± 0.001 | 0.12 ± 0.03 | 0.03 ± 0.006 | 13.56 ± 2.38 |
| 5j | 3,4-di-F | H | 0.03 ± 0.009 | 0.076 ± 0.01 | 0. 46 ± 0.06 | 1.43 ± 0.12 | 18.54 ± 2.76 |
| 5k | 2-Cl, 4-F | H | 0.02 ± 0.001 | 0.05 ± 0.008 | 0.57 ± 0.08 | 0.14 ± 0.03 | 1.22 ± 0.20 |
| 5l | 3-Br, 4-OH | H | 0.92 ± 0.08 | 1.07 ± 0.12 | >100 | 10.72 ± 1.85 | 80.15 ± 7.54 |
| 5m | 3,5-di-Br, 4-OH | H | 2.13 ± 0.12 | 1.15 ± 0.13 | 2.45 ± 0.37 | 18.32 ± 2.13 | 20.47 ± 3.53 |
| 5n | 2-Br, 4-OH, 5-OMe | H | 1.16 ± 0.13 | 9.61 ± 0.74 | 80.16 ± 9.68 | 14.54 ± 2.43 | 97.16 ± 6.54 |
| 5o | 3,5-di-isopropyl, 4-OH | H | 92.63 ± 5.38 | 42.56 ± 5.17 | 96.16 ± 7.62 | >100 | 77.64 ± 6.43 |
| 5p | 2,3,4-tri-OH | H | 1.48 ± 0.27 | 0.25 ± 0.031 | 1.18 ± 0.21 | 9.67 ± 0.73 | 10.44 ± 1.64 |
| 5q | 2,3,5-tri-OMe | H | 0.46 ± 0.03 | 0. 64 ± 0.04 | 1.75 ± 0.13 | 1.74 ± 0.15 | 27.36 ± 2.32 |
| 5r | H | Br | 1.74 ± 0.14 | 11.43 ± 1.68 | 67.32 ± 7.43 | 7.38 ± 0.63 | >100 |
| 5s | 2-F | Br | 2.86 ± 0.38 | 17.25 ± 1.34 | 67.32 ± 4.59 | 31.25 ± 5.79 | >100 |
| 5t | 2-Cl | Br | 0.58 ± 0.06 | 80.71 ± 6.98 | >100 | 56.82 ± 4.29 | >100 |
| 5u | 4-Br | Br | 0.73 ± 0.09 | 1.93 ± 0.33 | 21.39 ± 2.58 | 38.14 ± 4.42 | 79.32 ± 5.14 |
| 5v | 2-Cl, 4-F | Br | 0.26 ± 0.03 | 2.13 ± 0.24 | 3.57 ± 0.23 | 7.35 ± 0.54 | 56.34 ± 4.87 |
| CA-4 | 0.05 ± 0.01 | 0.08 ± 0.01 | 0.43 ± 0.053 | 0.11 ± 0.02 | 1.92 ± 0.11 | ||
| Crolibulin | 0.52 ± 0.02 | 0.03 ± 0.007 | ND | 0.17 ± 0.03 | ND | ||
aSD: standard deviation.
The SARs based on IC50 values (Table 1) showed that different substitution patterns of the aromatic A-ring and B-ring attached on IMPs skeleton had a vital impact on the cytotoxicity. It is worth noting that introduction of bromine group at position C-5 of B-ring resulted in a 1.5- to 13.8-fold decreased in potency as compared with the relative hydrogenous compounds (5a vs. 5r, 5e vs. 5s, 5f vs. 5t, and 5h vs. 5u), in good agreement with the low expand ability of the B-ring pocket at the colchicine site. Obviously, embedment of substituents into A-ring (5b–5n) led to a dramatic increase in cytotoxicity against MKN-45 and A549 cells relative to non-substituted phenyl analogue 5a. Interestingly, the halogenated phenyl analogue (5h, IC50 = 0.12 μM) was about 2.5-fold more potent than 5a and almost 5.8-fold more active than Crolibulin (IC50 = 0.52 μM) on HT-29 cell, indicating the presence of electron-withdrawing halogen group on A-ring would impart enhanced activity. By contrast, the electron-donating groups, with the exception of compound 5b with methyl group, weakened the biological activity slightly, as might be the reason of compounds 5e and 5f with 2-F or 2-Cl groups exhibited at least 1.7-fold increased potency over 5a (H), 5c (3-OMe) and 5d (4-OMe). Generally, in contrast with the mono-electron-withdrawing functions on phenyl portion, the introduction of double-electron-withdrawing groups showed evidently improved cytotoxicity against H460 and A549 cell lines as well as a slight increase for other three cancer cells. In particular, shifting the mono-halogen group (e.g. 5e) to a double halogen substitutions at both C-2 and C-4 positions (e.g. 5k) turned out to enhance the biological activity, speculating the latter might form stronger interactions with target protein through hydrogen bonds or polar contact concluded from docking study. For instance, the 2,4-difluorophenyl analog 5i (IC50 = 0.04 μM) was about 29-fold more active in H460 than the 3-bromophenyl analog 5h (IC50 = 1.16 μM). While, a dramatic loss in cytotoxic potency was observed for derivatives 5m–5q with tri-substituents on phenyl ring against five cell lines. In addition, compounds bearing small substituents, such as 5p with 2,3,4-tri-OH (HT-29, IC50 = 1.48 μM) and 5q with 2,3,5-tri-OMe (HT-29, IC50 = 0.46 μM), displayed more potent cytotoxicity than 5o possessing a bulky 3,5-di-isopropyl-4-OH group (HT-29, IC50 = 92.63 μM), suggesting the substituted group at this region was fairly sensitive to a substituent’s size.
To further optimize structural skeleton and explore the extending SARs, we prepared a series of derivatives 7a–7h by introducing several N-aliphatic amino groups into the position C-2 on the IMP skeleton of the most active candidate 5k. The pharmacological data of 7a–7h was illustrated in Table 2. Though inferior to the relative lead 5k, the in vitro cellular assay revealed that these compounds exhibited promising anticancer activity with IC50 values ranging from 0.03 to 42.57 μM against tested five cancer cells (Table 2). Especially, it is noteworthy to mention that 7b with ethylamino moiety (HT-29, IC50 = 0.03 μM) displayed the optimal antiproliferative activity which was about 17-fold and 1.7-fold more active than the reference Crolibulin (HT-29, IC50 = 0.52 μM) and CA-4 (HT-29, IC50 = 0.05 μM), respectively. However, an increase in the steric hindrance or a replacement with tertiary amino group (e.g. dimethylamino, 4-methylpiperidinyl and morpholinyl, et al.) strongly diminished the efficiency as observed in compounds 7d, 7f and 7h. The negative effect on cytotoxic activity of a bulk amino group might be attributed to prevent the test molecule from reaching the colchicine-binding-site of tubulin and thus result in a low activity. To our delight, compounds 7a–7h exhibited pronounced activity against HT-29 cell superior to on other four cells with a 3- to 1000-fold increase in IC50 values, indicating the prominent selectivity on HT-29. Additional SARs and further optimization are in progress and will be reported in a due course.
Table 2. Structures and cytotoxicity of compounds 7a–7 h.
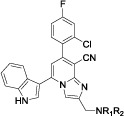 | ||||||
|---|---|---|---|---|---|---|
| Compd. | -NR1R2 | IC50 (μM) ± SDa | ||||
| HT29 | H460 | A549 | MKN45 | SMMC-772 | ||
| 7a |  |
0.04 ± 0.001 | 1.29 ± 0.21 | 5.34 ± 0.67 | 12.19 ± 1.89 | 26.98 ± 3.06 |
| 7b |  |
0.03 ± 0.003 | 0.96 ± 0.06 | 3.54 ± 0.41 | 20.47 ± 2.32 | 21.56 ± 3.16 |
| 7c | 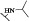 |
0.33 ± 0.04 | 0.96 ± 0.07 | 6.18 ± 0.71 | 22.35 ± 2.51 | 36.67 ± 4.92 |
| 7d | 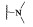 |
0.19 ± 0.02 | 0.71 ± 0.05 | 7.53 ± 0.63 | 16.56 ± 1.13 | 22.13 ± 3.41 |
| 7e | 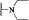 |
0.21 ± 0.03 | 0.67 ± 0.05 | 4.87 ± 0.38 | 13.75 ± 1.79 | 24.61 ± 2.73 |
| 7f | 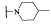 |
0.56 ± 0.07 | 0.85 ± 0.06 | 20.28 ± 2.32 | 1.63 ± 0.28 | 11.35 ± 2.15 |
| 7g |  |
0.19 ± 0.02 | 1.32 ± 0.12 | 8.79 ± 0.62 | 19.17 ± 1.35 | 42.57 ± 4.12 |
| 7h |  |
0.92 ± 0.14 | 1.27 ± 0.14 | 16.86 ± 1.98 | 36.31 ± 5.15 | 28.68 ± 1.79 |
| CA-4 | 0.05 ± 0.01 | 0.08 ± 0.01 | 0.43 ± 0.053 | 0.11 ± 0.02 | 1.92 ± 0.11 | |
| Crolibulin | 0.52 ± 0.02 | 0.03 ± 0.007 | ND | 0.17 ± 0.03 | ND | |
aSD: standard deviation.
The drug candidate potency depends not only on its cytotoxicity in malignant cells, but also on its relative lack of toxicity toward normal cells. Therefore, we evaluated the effects of compound 5b, 5f and 5k on human fetal hepatocyte line L02 (non-cancer cell line) by MTT assay. As shown in Fig. 2B, three compounds almost had negligible effects on the normal cell, indicating a particularly effectivity and hypotoxicity of this series of derivatives.
5k induces G2/M cell cycle arrest
The structural similarities between the target compounds (5a–5v and 7a–7h) with CA-4 and Crolibulin, as well as the promising cytotoxic activity led us to hypothesize that these compounds exert their antiproliferative properties through inhibition of tubulin dynamics, thus inducing mitotic arrest in cancer cells. To test the hypothesis, the lead compound 5k was therefore subjected to human colon cancer HT-29 cell to examine for its effect on cell cycle progression and further to identify whether cell apoptosis was involved in the inhibition of HT-29 cell growth. HT-29 cells were incubated for 24 h with increasing concentrations of 5k at concentration ranging from 20 nM to 1000 nM, and the percent of cells in each phase of the cell cycle was determined by flow cytometry (Fig. 4). Controls were treated with drug vehicle DMSO. Cell cycle analysis revealed that the cells accumulated in G2/M phase of the cell cycle in a concentration-dependent manner, whereas the control cells were mainly in the G1 phase. Results presented in Fig. 2A and C showed that treatment with 5k (0 nM, 20 nM, 200 nM and 1000 nM) resulted in the percentage of cells in the G2/M phase increased from 8.89% to 64.07% significantly, indicating a pronounced cell cycle arrest in G2/M phase. And this effect is characteristic of antimitotic agents disrupting microtubule assembly.
Figure 4. Effects of 5k on cell cycle progression in HT-29 cells.

(A,C): HT-29 cells were incubated with vehicle (DMSO, as CTL), 0, 20, 200 and 1000 nM of 5k for 24 h and collected for cell cycle analysis. (B): the effect of compounds 5b, 5f and 5k on human fetal hepatocyte line L02. Data are expressed as the mean ± SEM.
Effect of 5k on microtubule depolymerization in immunofluorescence studies
In order to further ascertain the characteristic cellular morphological changes that maybe relevant to the antitubulin activity of compound 5k, we evaluated its effect on the microtubule network via tubulin immunostaining. The immunofluorescence analysis using the specific antibodies to α-tubulin revealed that the microtubule network of the cells in negative control group displayed intact organization and arrangement. 5k (32.5 nM) and CA-4 (20 nM) treated cells exhibited cellular microtubule depolymerization with scattered short microtubule fragments in the cytoplasm of HT-29 cells (Fig. 5).
Figure 5. Effect of 5k on microtubules and nuclear condensation.
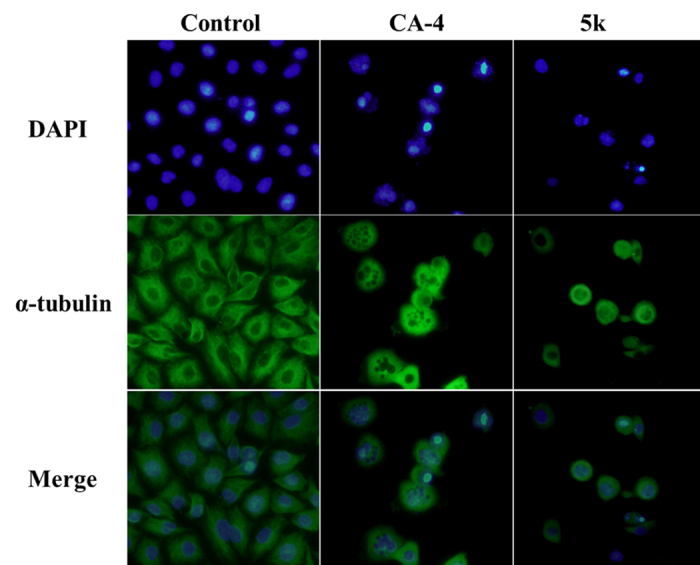
5k induces microtubules depolymerization in HT-29 cells. HT-29 cells were treated with CA-4 (20 nM) or 5k (32.5 nM) for 12 h. After incubation, cells were fixed, reacted with monoclonal anti-tubulin antibody at 4 °C overnight and then reacted with fluorescein isothiocyanate (FITC) conjugated secondary antibody, DAPI was used for nuclear counter-staining, the cellular microtubules were observed under fluorescence microscope (scale bar = 20 μm).
5k binds to tubulin at the colchicine site
To further elucidate the mechanism of 5-indolyl-7-arylimidazo[1,2-a]pyridine-8-carbonitrile derivatives interfering with microtubule formation, a competition binding assay was employed to examine whether the selected 5k directly binds to the colchicine site. As reported, competition between the test compound and colchicine for the binding site will decrease the intrinsic fluorescence of colchicine-tubulin complex by reducing the amount of colchicine bound, which could be used as an index for 5k competition with colchicine in tubulin binding37. As depicted in Fig. 6, 5k decreased the intrinsic colchicine fluorescence in a dose-dependent manner while Taxol exerts scarcely effect on the complex fluorescence for the reason that it binds at a different site on tubulin. Therefore, the results showed that compound 5k strongly bound to the colchicine binding domain of tubulin compared with the known microtubule polymerization inhibitor CA-4. Together, all these findings showed that in both molecular and cellular levels, tubulin polymerization was disturbed by 5k involving in the colchicine-binding-site, denoting the potential tubulin-targeting activity possessed by this compound.
Figure 6. Colchicine competition assay.
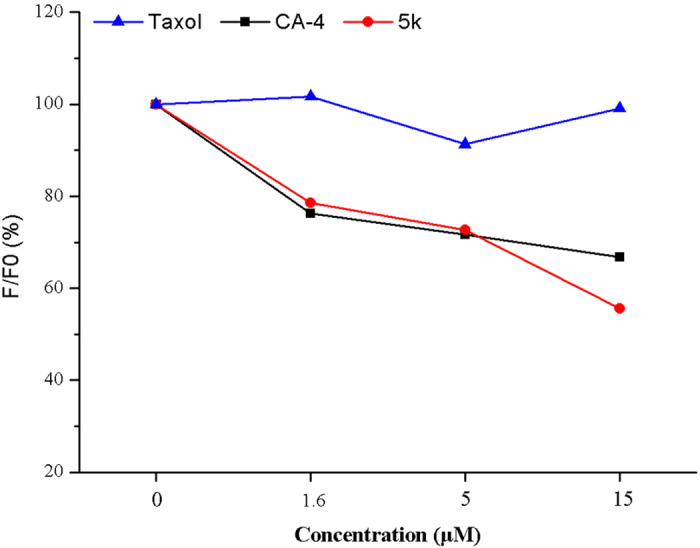
Tubulin was co-incubated with indicated concentrations of Taxol, CA-4 or 5k for 1h, then 5 μM colchicine was added. The fluorescence was measured by Hitachi F-2500 spectrofluorometer.
Molecular modeling of 5k in the colchicine-binding site of tubulin
To make sense of the cytotoxic data observed and the most probable orientation of compound 5k in the colchicine-binding site, molecular docking simulation of 5k to tubulin was carried out using Discovery Studio Program 3.0. The X-ray crystal structure of the DAMA-colchicines-tubulin complex (PDB code: 4O2B)38 was used as the tubulin protein template. The 3D structure of 5k was built using the Sybyl sketch module followed by energy minimization using the Triposforce field.
As expected, docking studies showed that 5k occupies the colchicine binding site shown in the Fig. 7B,C. The main binding driving force was thought to be shape matching with hydrophobic interaction between 5k and tubulin (Fig. 7A). Also, 5k formed two hydrogen bonds with Val181 of α-tubulin and Asp251 of β-tubulin shown in Fig. 7B. Concretely, the imidazo[1,2-a]pyridine (IMP) skeleton could link up to Met259, Ala316, Lys352 and Leu255 by hydrophobic interaction (Fig. 7A), suggesting that introduction of IMP moiety instead of chromenes scaffold retains tubulin-binding potency. The cyano group on the IMP framwork is engaged in a hydrogen bond with the amino group of Val181 of a-tubulin (Fig. 7B). And also, the indole group was involved in the hydrogen bonding with the Asp251 and hydrophobic interactions with the Leu248, Cys241, Leu255 and Ala250, which revealed that indole moiety existing in the structure of 5k could foster potent binding affinity leading to significant antiproliferative activities (Fig. 7A). Moreover, the substituted phenyl moiety also can form hydrophobic interactions with the hydrophobic pocket made up by Ala180, Thr179, Ser178, Asn101, Lys254 and Leu248. Notably, the halogen at both C-2 and C-4 positions of the phenyl ring forms hydrophobic interactions well with the hydrophobic P1 pocket, as may be the reason that introducing halogen groups could enhance the antitubulin activity. Thus, the results from the docking study were in accordance with the SARs analysis (e.g. 5f, 5j and 5k).To further understand the detailed binding mode of the candidate 5k, we thus superimposed Crolibulin and 5k in a 3D model (Fig. 7C). The result disclosed 5k was oriented in the same direction with Crolibulin at the colchicine site. Besides, the structural features of 5k were also in accordance with the common pharmacophores of ligands of the colchicine-binding site reported in literature39. Therefore, the binding mode revealed the posture of 5k is similar to Crolibulin binding at the colchicine site of tubulin.
Figure 7. The binding mode between the active conformation of 5k and tubulin.

(A) 2D diagram of the interaction between 5k and the colchicines binding site. (B) 3D diagram of the interaction between 5k and the colchicine binding site. For clarity, only interacting residues are displayed. The H-bond (yellow arrows) is displayed as dotted arrows. (C) Predicted modes for 5k (green) and Crolibulin (pink) binding at the colchicine-binding site of tubulin (PDB code: 4O2B), and overlapping with each other.
Conclusion
In this paper, in an attempt to develop promising tubulin polymerization inhibitors targeting colchicine-binding site, a series of 5-indolyl-7-arylimidazo[1,2-a]pyridine-8-carbonitrile derivatives (5a–5v and 7a–7h) were discovered according to bioisosterism and hybridization principles. This novel class of compounds was efficiently synthesized via a concise three-step reaction involving acylation reaction, one-pot coupling reaction and cyclization transformation, and the resultant thirty title compounds were determined by MS, 1H-NMR, 13C-NMR and element analysis. Evaluation of target compounds against a panel of cancer cell lines (HT-29, A549, MKN-45, MDA-MB-231 and SMMC-7721) resulted in the discovery of five promising compounds (5e, 5f, 5i, 5j and 5k) with the IC50 value in a dozen nanomolar range, indicating a great potency as antitubulin agents. Exploration of SARs based on IC50 values led to the identification of 5k as a prominent lead, which possessed remarkable antitumor potency against all the tested cancer cells with IC50 values ranging from 0.02 μM to 1.22 μM superior to references CA-4 and Crolibulin. In contrast, its cytotoxic effect on normal human fetal hepatocyte cell line (L02) was minimal. Importantly, the cell cycle analysis, immunofluorescence assay and the colchicine competition assay confirmed that candidate 5k could inhibit cellular tubulin polymerization by binding to the colchicine site, interfere with the mitosis, and at the end lead to G2/M cell cycle arrest in vitro. Moreover, a molecular docking mode revealed that 5k could not only form critical hydrogen bonding interactions and hydrophobic interactions with tubullin but also overlap well with Crolibulin in 3D model, which is consistent with the observed SARs for this class of compounds.
In summary, this is the first report of a novel series of 5-indolyl-7-arylimidazo[1,2-a]pyridine-8-carbonitrile derivatives as potent tubulin-targeting agents and thus, describe the SARs-guided discovery process that lead to a candidate 5k as a potential antitubulin agent targeting colchicines-binding-site. All of these finding suggest the molecular basis of 5k to serve as a probe in the characterization of a new class of tubulin polymerization inhibitors.
Experimental Procedures
Reagents and instrumentation
All melting points were obtained on a Büchi Melting Point B-540 apparatus (BüchiLabortechnik, Flawil, Switzerland) and were uncorrected. Mass spectra (MS) were taken in ESI mode on Agilent 1100 LC-MS (Agilent, palo Alto, CA, USA). 1H NMR and 13C NMR spectra were performed using Bruker 400 MHz and 100 MHz spectrometers (Bruker Bioscience, respectively, Billerica, MA, USA) with TMS as an internal standard. Column chromatography was run on silica gel (200–300 mesh) from Qingdao Ocean Chemicals (Qingdao, Shandong, China). Unless otherwise noted, all materials were obtained from commercially available sources and were used without further purification.
Chemistry section
General procedure for Preparation of compounds (2a–2b)
A 500 mL oven-dried three-necked flask was deal with septa and drained/backfilled with nitrogen gas (N2) three times before starting the reaction. A solution of anhydrous stannic chloride (62.4 g, 240 mmol) in dichloromethane (150 mL) was added to a solution of 1H-indole or 5-bromo-1H-indole (200 mmol) in dry dichloromethane (200 mL) at −5 °C and the mixture was stirred at 0 °C for 30 min. Then a solution of acetyl chloride (15.6 g, 200 mmol) in dry dichloromethane/nitromethane co-solvent (1:1, 50 mL) was added successively. Subsequently, the resulting mixture was stirred at 0 °C for 30 min and then warmed to room temperature stirring for 3 h. After monitored by TLC, the reaction mixture was quenched with ice-water, the suspension solution was filtered and the filtrate was extracted with ethyl acetate (100 mL × 2). Afterward, the combined organic phase was washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The removal of the solvent yielded a brown residue that was purified by washing-up with ethanol for 1 h to furnish 2a as a white solid.
General procedure for the preparation of compounds (4a–4r)
To a solution of substituted benzaldehydes (100 mmol) in toluene (250 mL) was added 2a or 2b (100 mmol), ammonium acetate (18.48 g, 240 mmol) and malononitrile (6.6 g, 100 mmol) at room temperature. Then the solution was refluxed until the completion of the reaction indicated by TLC (about 8 h). Up on cooling to room temperature, the solvent was removed under vacumm, and then, the residue was triturated with ethanol (100 mL) and filtered to give the corresponding crude product. Afterward, the crude product was transited to anhydrous ethanol (120 mL) and stirred overnight at room temperature. Finally, the suspension liquid was filtered off, dried over MgSO4 to afford 4a-4v.
General procedure for the preparation of compounds (5a - 5 v)
To a solution of 4a-4v (1.6 mmol) and sodium bicarbonate (0.14 g, 1.6 mmol) in absolute ethanol (30 mL) were added to 40% 2-chloroacetaldehyde (3.14 g, 16 mmol). The mixture was refluxed and stirred for 4 h. After the solvent was evaporated, the residue was obtained by filtration and washed with distilled water (30 mL) to give the corresponding pure compounds 5a-5v.
Preparation of 7-(2-chloro-4-fluorophenyl)-2-chloromethyl-5-(1H-indol-3-yl)imidazo[1,2-a] pyridine-8-carbonitrile (6)
To a mixture of 4k (5.0 g, 13.8 mmol) and 1,3-dichloropropan-2-one (18.2 g, 144.1 mmol) in ethanol (50 mL) were added and refluxed for 10 h. After the completion of thereaction indicated by TLC, the solvent was evaporated and then the residue was washed with ethyl ether (50 mL) for 2 h to give the corresponding crude product in 64.7% yield.
General procedure for the preparation of compounds (7a–7h)
A stirring mixture of 6 (0.5 g, 1.2 mmol) and an appropriate N-aliphatic amines (1.8 mmol) in ethanol (15 mL) was stirred at reflux for 5 h. After cooling to room temperature, the solvent was evaporated in vacuum to afford yellow residue. The formed precipitate was dissolved in DMF and purified on a silica gel column (petroleum ether/ethyl acetate, 3:1) to give pure target product 7a–7h.
(Thirty target compounds were determined by MS, 1H-NMR, 13C-NMR and element analysis, and the detailed information is in Supplementary Information).
Biological section
Cell lines and culture conditions.The human colon carcinoma cell line HT-29, the human pulmonary carcinoma cell line A549, the human gastric carcinoma cell line MKN-45, the human breast cancer cell line MDA-MB-231 and human heptocarcinoma cellline SMMC-7721were cultured in RPMI-1640 mediumcontaining 10% FBS, 100 U/mL streptomycin and 100 U/mL penicillinat 37 °C in humidified atmosphere with 5% CO2. All of the cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA).
MTT assay
The antiproliferative activities in vitro of Crolibulin and all the target compounds were determined by an MTT assay40. Briefly, cells were seeded into 96-well plates at a density of 1–3 × 104/well (depends on the cell growth rate). 24 h later, triplicate wells were treated with media and the agents. After 72 h ofincubation at 37 °C in 5% CO2, the drug containing medium wasremoved and replaced by 100 mL of fresh medium with 5 mg/mLMTT solution. After 4 h of incubation, the medium with MTT was removed, and 100 mL of DMSO was added to each well. The plates were gently agitated until the purple formazan crystals were dissolved, and the OD490 was determinedusing a microplate reader (MK3, Thermo, Germany). The data were calculated and plotted as the percent viability compared to the control. The 50% inhibitory concentration (IC50) was defined as theconcentration that reduced the absorbance of the untreated wells by 50% of the vehicle in the MTT assay.
Cell cycle phase distribution
For flow cytometric analysis of DNA content, cells inexponential growth were treated with compound for 24 h41. The cells treated with compoundwere collected, washed twice with PBS, and then fixed with 75% alcohol overnight. Then, the cells were washed with PBS and resuspended in 100 μL of PBS, 200 mg/mL RNase wasadded for 30 min to eliminate the interference of RNA, and 20 mL/L propidium iodide (PI; Sigma) was added for 30 min.Then, the cells were washed, and the DNA content wasdetected by flow cytometer (BD Accuri C6).
Immunofluorescence staining assay
Immunostaining was carried out to detect microtubule associatedtubulin protein after exposure to CA-4 and 5k42. The HT-29 cells were seeded at 1 × 104 per well on a 24 wellplate and grown for 24 h. Cells were treated with CA-4 or 5k for 12 h. Cells in the control group were treated with culture medium. The control and treated cells were fixed with 4% formaldehydein PBS for 30 min at −20 °C, then washed twice with PBS and permeabilized with 0.1% (v/v) Triton X-100 in PBS for 5 min. Then, the cells were blocked with 3% bovine serum albumin(BSA) in PBS for 30 min. The primary α-tubulin antibody wasdiluted (1:100) with 2% BSA in PBS and incubated overnight at 4 °C. The cells were washed with PBS to remove unbound primaryantibody and then cells were incubated with FITC-conjugated antimousesecondary antibody, diluted (1:100) with 2% BSA in PBS, for 2 h at 37 °C. The cells were washed with PBS to remove unbound secondary antibody, nucleus was stained with 4,6-diamino-2-phenolindoldihydrochloride (DAPI) and then, immunofluorescencewas detected using a fluorescence microscope (Olympus,Tokyo, Japan).
Competitive tubulin-binding assay
Tubulin was co-incubated with various concentrations of Taxol, CA-4 or 5k respectively at 37 °C for 1 h. Then colchicine was added to a final concentration of 5 μM43. Fluorescence was determined using a Hitachi F-2500 spectrofluorometer (Tokyo, Japan) at excitation wavelengths of 365 nm and emission wavelengths of 435 nm. Blank values (buffer alone) as background were subtracted from all samples. Then the inhibition rate (IR) was calculated as follows: IR = (F ÷ F0) × 100% where F0 is the fluorescence of the 5 μM colchicine-tubulin complex, and F is the fluorescence of a given concentration of Taxol, 5k or CA-4 (0 μM, 1.6 μM, 5 μM and 15 μM) competition with the 5 μM colchicine-tubulin complex. Taxol, not binding in the colchicine-site of tubulin, was added as a negative control.
Molecular Docking
The molecular modelling studies were performed with Accelrys Discovery Studio 3.044. The crystal structure of tubulin complexedwith DAMA-colchicine (PDB code: 4O2B) was retrieved from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/). In the docking process, the protein protocol was prepared by several operations, including standardization of atom names and insertion of missingatoms in residues. Then, the receptor model was typed with the CHARMm force field and a binding sphere with radius of 15.0 Å was defined through the original ligand (DAMA-colchicine) as the binding site. The Crolibulin and 5k were drawn with Chemdraw and fully minimized using the CHARMm force field. Finally, they were docked into the binding site using the CDOCKER protocol with the default settings. And figures were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 1.4.1. Schrödinger, LLC).
Statistical analysis
Statistical analysis was performed with Origin Version 8.0 software package. Data were expressed as means ± standard deviation (SD). Comparisons between groups were performed with analysis of non-parametric test.
Additional Information
How to cite this article: Zhai, X. et al. Discovery and Optimization of Novel 5-Indolyl-7-arylimidazo[1,2-a]pyridine-8-carbonitrile Derivatives as Potent Antitubulin Agents Targeting Colchicine-binding Site. Sci. Rep. 7, 43398; doi: 10.1038/srep43398 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The work was supported by National Natural Science Foundation of China (No. 81273357), Program for Liaoning Excellent Talents in University (No. LR2014030) and Development Project of Ministry of Education Innovation Team (No. IRT1073).
Footnotes
The authors declare no competing financial interests.
Author Contributions X.Z. and P.G. designed the research. X.W., J.W. and J.L. performed the chemical experiments. D.Z., X.Y. and T.Z. performed all other experiments, N.J., and T.J. analyzed the data. X.Z. and X.W. wrote the main manuscript, and all authors reviewed and commented on the manuscript.
References
- Jordan M. A. & Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 4, 253–265 (2004). [DOI] [PubMed] [Google Scholar]
- Wang Y. T. et al. Synthesis, biological evaluation, and molecular docking studies of novel 1-benzene acyl-2-(1-methylindol-3-yl)-benzimidazole derivatives as potential tubulin polymerization inhibitors. European Journal of Medicinal Chemistry 99, 125–137 (2015). [DOI] [PubMed] [Google Scholar]
- Kamal A. et al. Design and synthesis of pyrazole-oxindole conjugates targeting tubulin polymerization as new anticancer agents. European Journal of Medicinal Chemistry 92, 501–513 (2015). [DOI] [PubMed] [Google Scholar]
- Liu Y. N. et al. Design, Synthesis, and Biological Evaluation of 1-Methyl-1,4-dihydroindeno[1,2-c]pyrazole Analogues as Potential Anticancer Agents Targeting Tubulin Colchicine Binding Site. Journal of Medicinal Chemistry 59, 5341–5355 (2016). [DOI] [PubMed] [Google Scholar]
- Dumontet C. & Jordan M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nature reviews. Drug discovery 9, 790–803 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alday P. H. & Correia J. J. Macromolecular interaction of halichondrin B analogues eribulin (E7389) and ER-076349 with tubulin by analytical ultracentrifugation. Biochemistry 48, 7927–7938 (2009). [DOI] [PubMed] [Google Scholar]
- Guan Q. et al. Synthesis and biological evaluation of novel 3,4-diaryl-1,2,5-selenadiazol analogues of combretastatin A-4. European Journal of Medicinal Chemistry 87, 1–9 (2014). [DOI] [PubMed] [Google Scholar]
- Ceresoli G. L. & Zucali P. A. Vinca alkaloids in the therapeutic management of malignant pleural mesothelioma. Cancer Treat Revive 41, 853–858 (2015). [DOI] [PubMed] [Google Scholar]
- Perez E. A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Molecular cancer therapeutics 8, 2086–2095 (2009). [DOI] [PubMed] [Google Scholar]
- Roman B. I. et al. Assessment of the antineoplastic potential of chalcones in animal models. Current Medicinal Chemistry 20, 186–221 (2013). [DOI] [PubMed] [Google Scholar]
- Hamel E. Antimitotic natural products and their interactions with tubulin. Medicinal Research Reviews 16, 207–231 (1996). [DOI] [PubMed] [Google Scholar]
- Pettit G. R., et al. Isolation and structure of combretastatin. Canadian Journal of Chemistry 60, 1374–1376 (1982). [Google Scholar]
- Simoni D. et al. Heterocyclic and Phenyl Double-Bond-Locked Combretastatin Analogues Possessing Potent Apoptosis-Inducing Activity in HL60 and in MDR Cell Lines. Journal of Medicinal Chemistry 48, 723–736 (2005). [DOI] [PubMed] [Google Scholar]
- Hsieh H. P., Liou J. P. & Mahindroo N. Pharmaceutical Design of Antimitotic Agents Based on Combretastatins. Current Pharmaceutical Design 11, 1655–1677 (2005). [DOI] [PubMed] [Google Scholar]
- Bonezzi K. et al. Vascular Disrupting Activity of Tubulin-Binding 1,5-Diaryl-1H-imidazoles. Journal of Medicinal Chemistry 52, 7906–7910 (2009). [DOI] [PubMed] [Google Scholar]
- Kaffy J. et al. Isoxazole-type derivatives related to combretastatin A-4, synthesis and biological evaluation. Bioorganic & Medicinal Chemistry 14, 4067–4077 (2006). [DOI] [PubMed] [Google Scholar]
- Theeramunkong S. et al. Regioselective Suzuki Coupling of Dihaloheteroaromatic Compounds as a Rapid Strategy To Synthesize Potent Rigid Combretastatin Analogues. Journal of Medicinal Chemistry 54, 4977–4986 (2011). [DOI] [PubMed] [Google Scholar]
- Ohsumi K. et al. Syntheses and antitumor activity of cis-restricted combretastatins: 5-Membered heterocyclic analogues. Bioorganic & medicinal chemistry letters 8, 3153–3158 (1998). [DOI] [PubMed] [Google Scholar]
- Medarde M. et al. Synthesis and pharmacological activity of diarylindole derivatives. Cytotoxic agents based on combretastatins. Bioorganic & medicinal chemistry letters 9, 2303–2308 (1999). [DOI] [PubMed] [Google Scholar]
- Field J. J., Kanakkanthara A. & Miller J. H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorganic & medicinal chemistry 22, 5050–5059 (2014). [DOI] [PubMed] [Google Scholar]
- Greene T. F. et al. Synthesis and Biochemical Evaluation of 3-Phenoxy-1,4-diarylazetidin-2-ones as Tubulin-Targeting Antitumor Agents. Journal of Medicinal Chemistry 59, 90–113 (2016). [DOI] [PubMed] [Google Scholar]
- Carlson R. O. New tubulin targeting agents currently in clinical development. Expert Opinion on Investigational Drugs 17, 707–722 (2008). [DOI] [PubMed] [Google Scholar]
- Subbiah I. M., Lenihan D. J. & Tsimberidou A. M. Cardiovascular toxicity profiles of vascular-disrupting agents. The oncologist 16, 1120–1130 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read W. L. et al. Pharmacokinetic and pharmacodynamic results of a 4-hr IV administration phase I study with EPC2407, a novel vascular disrupting agent. ASCO Meeting Abstracts 27, 3569 (2009). [Google Scholar]
- Kemnitzer W. et al. Discovery of 4-aryl-4H-chromenes as a new series of apoptosis inducers using a cell- and caspase-based high-throughput screening assay. 2. Structure-activity relationships of the 7- and 5-, 6-, 8-positions. Bioorganic & medicinal chemistry letters 15, 4745–4751 (2005). [DOI] [PubMed] [Google Scholar]
- Kemnitzer W. et al. Discovery of 4-aryl-2-oxo-2H-chromenes as a new series of apoptosis inducers using a cell- and caspase-based high-throughput screening assay. Bioorganic & medicinal chemistry letters 18, 5571–5575 (2008). [DOI] [PubMed] [Google Scholar]
- Kemnitzer W. et al. Discovery of 4-aryl-4H-chromenes as a new series of apoptosis inducers using a cell- and caspase-based high-throughput screening assay. 3. Structure-activity relationships of fused rings at the 7,8-positions. Journal of Medicinal Chemistry 50, 2858–2864 (2007). [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C. et al. Natural Product-like Combinatorial Libraries Based on Privileged Structures. 2. Construction of a 10 000-Membered Benzopyran Library by Directed Split-and-Pool Chemistry Using NanoKans and Optical Encoding. Journal of the American Chemical Society 122, 9954–9967 (2000). [Google Scholar]
- Welsch M. E. et al. Privileged scaffolds for library design and drug discovery. Current Opinion in Chemical Biology 14, 347–361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L. & Barlocco D. Privileged Structures as Leads in Medicinal Chemistry. Current Medicinal Chemistry 13, 65–85 (2006). [PubMed] [Google Scholar]
- Chen X. et al. Discovery of a Novel Series of Imidazo[1,2-a]pyrimidine Derivatives as Potent and Orally Bioavailable Lipoprotein-Associated Phospholipase A2 Inhibitors. Journal of Medicinal Chemistry 58, 8529–8541 (2015). [DOI] [PubMed] [Google Scholar]
- Cannalire R. et al. A Journey around the Medicinal Chemistry of Hepatitis C Virus Inhibitors Targeting NS4B: From Target to Preclinical Drug Candidates. Journal of Medicinal Chemistry 59, 16–41 (2016). [DOI] [PubMed] [Google Scholar]
- Kaminski J. J. et al. Antiulcer agents. 1. Gastric antisecretory and cytoprotective properties of substituted imidazo[1,2-a]pyridines. Journal of Medicinal Chemistry 28, 876–892 (1985). [DOI] [PubMed] [Google Scholar]
- Qian X. et al. Discovery of the First Potent and Selective Inhibitor of Centromere-Associated Protein E: GSK923295. ACS medicinal chemistry letters 1, 30–34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer S. Z. et al. Selectivity for Omega-Receptor Subtypes as a Strategy for the Development of Anxiolytic Drugs. Pharmacopsychiatry 23, 103–107 (1990). [DOI] [PubMed] [Google Scholar]
- Singh M. S. & Chowdhury S. Recent developments in solvent-free multicomponent reactions: a perfect synergy for eco-compatible organic synthesis. Rsc Advances 2, 4547–4592 (2012). [Google Scholar]
- Kamal A. et al. Design and synthesis of imidazo[2,1-b]thiazole-chalcone conjugates: microtubule-destabilizing agents. ChemMedChem 9, 2766–2780 (2014). [DOI] [PubMed] [Google Scholar]
- Driowya M. et al. Synthesis of triazoloquinazolinone based compounds as tubulin polymerization inhibitors and vascular disrupting agents. European Journal of Medicinal Chemistry 115, 393–405 (2016). [DOI] [PubMed] [Google Scholar]
- Lu Y. et al. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharmaceutical research 29, 2943–2971 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa-Goto K. et al. Antitumor agents. 284. New desmosdumotin B analogues with bicyclic B-ring as cytotoxic and antitubulin agents. Journal of Medicinal Chemistry 54, 1244–1255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad A. et al. Synthesis and antimitotic and tubulin interaction profiles of novel pinacol derivatives of podophyllotoxins. Journal of Medicinal Chemistry 55, 6724–6737 (2012). [DOI] [PubMed] [Google Scholar]
- Reddy M. V. R. et al. (Z)-1-Aryl-3-arylamino-2-propen-1-ones, Highly Active Stimulators of Tubulin Polymerization: Synthesis, Structure–Activity Relationship (SAR), Tubulin Polymerization, and Cell Growth Inhibition Studies. Journal of Medicinal Chemistry 55, 5174–5187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. et al. S9, a Novel Anticancer Agent, Exerts Its Anti-Proliferative Activity by Interfering with Both PI3K-Akt-mTOR Signaling and Microtubule Cytoskeleton. PLOS ONE 4, e4881 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Q. et al. Synthesis and evaluation of benzimidazole carbamates bearing indole moieties for antiproliferative and antitubulin activities. Europine Journal of Medicinal Chemistry 87, 306–315 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.