

Abstract
Secreted protein acidic and rich in cysteine (SPARC/osteonectin/BM40) is one of the most abundant non-collagenous protein expressed in mineralized tissues. This review will focus on elucidating functional roles of SPARC in bone formation building upon results from non-mineralized cells and tissues, the phenotype of SPARC-null bones, and recent discoveries of human diseases with either dysregulated expression of SPARC or mutations in the gene encoding SPARC that give rise to bone pathologies. The capacity of SPARC to influence pathways involved in extracellular matrix assembly such as procollagen processing and collagen fibril formation as well as the capacity to influence osteoblast differentiation and osteoclast activity will be addressed. In addition, the potential for SPARC to regulate cross-linking of extracellular matrix proteins by members of the transglutaminase family of enzymes is explored. Elucidating defined biological functions of SPARC in terms of bone formation and turnover are critical. Further insight into specific cellular mechanisms involved in the formation and homeostasis of mineralized tissues will lead to a better understanding of disease progression.
Introduction
SPARC, also referred to as osteonectin or basement membrane protein 40 (BM-40), is a 32 kDa calcium-binding matricellular protein [1, 2]. At one time SPARC was thought to be expressed specifically in mineralized tissues (thus the name osteonectin) but was subsequently demonstrated to have a much wider expression pattern in both mineralized and non-mineralized tissues [3]. Typically, expression of SPARC is closely aligned with that of fibrillar collagens such as collagen type I. The protein component of the pre-mineralized bone matrix (osteoid) is composed mainly of collagen type I (94%) and a number of noncollagenous proteins. The mechanical properties of bone are largely attributed to the mineral composition of the osteoid in the form of hydroxyapatite (HA) - a complex of calcium and phosphate - that mineralizes the collagenous matrix. Generally, calcified bone is comprised of ∼ 70% HA, 25% matrix, and 5% water (IOF). SPARC contains both a collagen-binding domain and an HA binding region [4]. In the osteoid, SPARC has been proposed to bind collagen and HA crystals and release calcium ions perhaps enhancing mineralization of the collagen matrix in bones [3]. The function of SPARC in mineralized tissues both during homeostasis and disease has yet to be fully defined, however a significant amount of insight into SPARC activity in mineralized tissues has emanated from analysis of SPARC-null mice and identified mutations in the gene encoding SPARC in human disease. Herein we present a review of relevant SPARC activities that have the potential to influence mineralized tissue based on: 1) phenotypic changes detected in SPARC-null bones; 2) recently discovered mutations in the gene encoding SPARC in patients with idiopathic osteoporosis and osteogenesis imperfecta (OI); 3) regulation of transglutaminase (TG) activity by SPARC in the periodontal ligament, and 4) Factor XIII/TG/SPARC expression in new bone formation. We also examine the evidence that SPARC regulates the activity of osteoblasts and osteoclasts: the two primary cell types regulating bone homeostasis and repair.
Structural Features and Expression
SPARC is encoded by a single gene that generates a secreted, monomeric, glycosylated polypeptide. SPARC has four defined domains: 1) an N-terminal low-affinity, high capacity, calcium-binding domain that contains the mineral binding region, 2) a Cysteine-rich domain, 3) a hydrophilic region, and 4) an extracellular Ca2+ (EC) domain with an E-F hand motif at the C-terminus that encompasses the collagen binding domain. The helix-loop-helix structure (EF hand motif) is a domain characteristic of some calcium-binding proteins and is composed of two alpha helices and one short loop region [5]. Discovery of the SPARC E-F hand was unusual in that other proteins with an E-F hand motif were intracellular, thus the structure of the SPARC E-F hand was the first described for an extracellular protein [6]. The human gene that encodes SPARC is 26.5kb, located on chromosome 5q31-q33 and contains 10 exons and 9 introns [7]. SPARC is a glycoprotein shown to undergo differential glycosylation dependent upon tissue-specific expression. Importantly, the glycosylated form of SPARC expressed in bone binds collagen with higher affinity than the form found in platelets [8].
SPARC is secreted by osteoblasts in bone during bone formation [9]. The collagen-binding domain of SPARC, at the C-terminus is separated from the hydroxyapatite-binding site at the N-terminal region. The separation of these two interacting domains has been predicted to facilitate mineralization of collagen during bone formation [3]. Similar to patterns of SPARC expression in other tissues, levels of SPARC are high in immature bone and are associated with mineralization of collagen whereas a decrease in SPARC expression is observed in mature bone homeostasis [10]. Notably, SPARC is expressed at high levels in osseous tissue with high turnover such as in active osteoblasts and bone marrow progenitor cells, odontoblasts (dentin-forming cells), periodontal ligament fibroblasts, hypertrophic chondrocytes, and osteoid [4, 9-12]. In addition to osteoblasts, SPARC is also synthesized by other cell types present in mineralized tissues including endothelial cells and fibroblasts [13]. During injury, recruited macrophages express SPARC and SPARC is also found in platelet granules [14,15].
In vitro, SPARC activities are implicated in the regulation of cell attachment and cell spreading. Increased levels of SPARC have been shown to inhibit cell spreading and cell attachment in cultured endothelial cells, disrupt focal adhesions in fibroblasts, and increase the production and activity of matrix metalloproteinases leading to matrix degradation in some cell types [13]. SPARC also influences a number of growth factors, some of which are bound directly by SPARC, to regulate cell proliferation. Generally, SPARC is characterized as an inducer of cell rounding and as an inhibitor of cell proliferation in cultured cells [13].
SPARC in Procollagen Processing and Fibril Assembly
Fibrillar collagen is the primary structural component of interstitial extracellular matrix (ECM) and maintains mechanical stability of tissues. The ECM is composed of both collagenous and non-collagenous networks, proteoglycans, and glycosaminoglycans (GAGs) [16]. Collagens are defined as trimeric molecules comprised of repeating glycine-proline-hydroxyproline triplets that form a right-handed triple-helical structure composed of either homotrimeric or heterotrimeric combinations of alpha subunits depending on the collagen type. Currently 27 different collagen types have been identified. Primary components of the ECM include collagens type I, II, III, IV, V, X, XI, and XII [16]. Collagen I, the main structural protein in connective tissues and bone, is the most abundant protein in mammalian species composing up to 35% of whole-body protein content. Collagen II and XI are dominant in cartilage fibrils. Collagen X is expressed by hypertrophic chondrocytes during endochondral ossification and is used as a marker of new bone formation [17]. Collagen III is associated with tooth dentin, alveolar bone, lens, bone, cartilage, and tendon. Collagen IV binds the heparan sulfate proteoglycan perlecan and is primarily localized in sheet-like structure as a component of the basal lamina associated with endothelial and epithelial cells in mineralized tissue [18]. Evidence that SPARC binds fibrillar collagens I, III, and V in addition to basement membrane collagen IV, implicate SPARC in the assembly of connective tissue ECM as well as basement membranes [16].
Collagen I is initially secreted from cells as trimeric procollagen composed of two alpha 1 chains and one alpha 2 chain. Procollagen I undergoes a number of post-translational modifications in the endoplasmic reticulum and the Golgi apparatus. Nucleation of the triple helical procollagen molecule initiates within the C-propeptide recognition sequences and brings together appropriate alpha subunits to form each type of collagen [16]. The rod-like, triple-helical procollagen with globular N- and C-terminal propeptides is then secreted into the extracellular space. The N- and C-terminal propeptides are cleaved off by N and C-terminal propeptide proteinases, an event that can occur at intracellular as well as extracellular sites, and results in monomeric triple helical collagen that is incorporated into insoluble collagen fibrils [19]. Covalent crosslinks within and between triple-helical collagen molecules stabilize mature fibrils [20].
A lack of SPARC expression has been shown to diminish mature collagen accumulation in the ECM of many connective tissues [2, 21]. For example, SPARC-null mice exhibit decreased total collagen I content in skin, bone, and periodontal ligament. Likewise, there is a diminished deposition of fibrotic collagen in tissues in the absence of SPARC. Studies in various tissues (cardiac, dermal, neoplastic, periodontal) have shown a correlation between increased levels of SPARC expression and stimulated increases in collagen deposition and assembly into the ECM [22]. Over-expression of exogenous SPARC led to increases in collagen-rich ECM in the trabecular meshwork surrounding the eye [23]. Increases in soluble collagen in SPARC-null mice also suggest that incorporation of collagen into insoluble collagen fibrils is not efficient in the absence of SPARC [24]. Similar differences in procollagen processing by SPARC-null versus WT cells are observed in dermal and cardiac fibroblast cultures [25, 26]. Studies performed in fibroblasts have suggested that SPARC acts to facilitate procollagen processing by limiting procollagen association with cell surface receptors [25, 26]. The SPARC binding site on collagen overlaps with that of the collagen receptor Discoidin Domain Receptors (DDR) 1 and 2 [27]. Hence, SPARC bound to collagen I might limit binding to cell surface DDR2 and therefore decrease signaling initiated via DDR2 as well as possibly allowing for more efficient incorporation of collagen into insoluble ECM. Speculatively, increased interaction of soluble collagen with DDR2 in the absence of SPARC might tether procollagen at cell surfaces leading to increased turn over of collagen at the cell surface at the expense of collagen deposition into the ECM.
What insight might be gained from activities attributed to SPARC in procollagen processing events by soft tissue fibroblasts that might apply to mineralized tissues? Clearly, differences in procollagen processing, assembly, and collagen fibril formation might have significant impacts on bone formation. As discussed below, decreases in collagen content and maturity have been detected in SPARC-null bones. To date, procollagen processing by SPARC-null versus WT osteoblasts has not been addressed. Whether similar SPARC-dependent mechanisms used by fibroblasts control procollagen processing and assembly used by osteoblasts should prove enlightening for future studies. Another extracellular mechanism by which SPARC was recently shown to affect collagen fibril diameter and content, relevant to mineralized tissues, was the regulation of transglutaminase enzyme activity on collagen [28].
Transglutaminase
Transglutaminases (TGs) are a family of proteins with extracellular crosslinking activity [29]. TGs catalyze the formation of covalent isopeptide bonds and have been shown to cross-link ECM proteins such as fibronectin and fibrillar collagens. Studies carried out in cartilage revealed that SPARC could also serve as a substrate for TG [30]. Tissue transglutaminase type 2 (TG2) is the most ubiquitously expressed of the TG family and, along with Factor XIII, is implicated in bone biology [31, 32]. The catalytic function of TG2 and Factor XIII is defined as a protein transamidation reaction that occurs between glutamine and lysine residues resulting in the formation of a y-glutamyl-ε-lysyl covalent bond. The crosslinking of ECM proteins by transglutaminase is predicted to promote assembly and/or enhance stabilization of collagenous and noncollagenous ECMs [29].
Cui et al. demonstrated in osteoblast cultures that Factor XIII catalyzed the cross-linking of fibronectin from serum (plasma fibronectin; pFn), as opposed to the EDA-splice variant of fibronectin endogenously expressed by osteoblasts (cellular Fn; cFn) [33]. Formation of a cross-linked fibronectin matrix derived from pFn was required for subsequent deposition and assembly of a collagen network. Inhibition of Factor XIII activity, but not that of TG2, decreased collagen deposition and mineralization in osteoblast cultures [33]. Hence, cross-linking of pFn by Factor XIII was predicted to be an essential phase of bone formation that precedes collagen deposition.
Another study revealed enhanced osteoblast cell adhesion properties of two matricellular proteins mediated by TG2 crosslinks: osteopontin (OPN) and bone sialoprotein (BSP). It was confirmed that both OPN and BSP serve as substrates for TG2 and that their modification by TG2 enhanced the formation of cellular extensions in osteoblast cell cultures. Similar to Factor XIII, TG2 is found to localize to osteoid, osteoblasts, and osteocytes and it is hypothesized that these two enzymes work synergistically to oligomerize substrates [34].
Interestingly, the expression of SPARC was recently shown to influence TG activity in periodontal ligament (PDL). As mentioned previously, an absence of SPARC expression results in decreased levels of collagen I in many connective tissues in mice. In addition to reduced collagen content, collagen fibrils assembled in the absence of SPARC are smaller and more uniform in diameter versus those in WT tissues. PDL, the connective tissue lying between the alveolar bone and the tooth, is one example. Accordingly, SPARC-null PDL was shown to have decreased mechanical strength in comparison to WT [28]. SPARC-null PDL also demonstrated increased TG activity. Specifically, TG activity on collagen I was shown to be enhanced with abrogated SPARC expression. Administration of TG inhibitors to SPARC-null PDL resulted in increased collagen content and the generation of larger collagen fibers. The increase in collagen content and the rescued morphology of the collagen fibers was associated with enhanced mechanical strength of the PDL [28]. This study raises the possibility that SPARC bound to collagen reduces accessibility of TGs to certain glutamine and/or lysine residues on collagen. Over-modification by TG on collagen might lock collagen fibrils into a smaller diameter preventing further fiber expansion and reducing overall collagen content and mechanical strength of soft tissues. TGs might also be an essential factor in cross-linking of collagen fibers in bone formation and/or SPARC might facilitate a reduction in TG binding to collagen in bones. Additional studies examining the mechanisms of TG regulation by SPARC are necessary. Future experiments to identify TG family members that participate in collagen cross-linking and the role of these enzymes in regulating bone matrix formation in vivo will be enlightening.
SPARC-null Bone Phenotype
Overt phenotypic differences in SPARC-null versus WT mice are represented in both mineralized and non-mineralized tissues. As SPARC is one of the most abundant non-collagenous ECM protein found in bone, the expression of SPARC would be predicted to influence bone biology. Initial characterization of SPARC-null mice indicated no overt phenotypic differences in skeletal development in the absence of SPARC [35]. However, subsequent studies have revealed significant differences in the bone phenotype in SPARC-null versus WT mice. Relevant to this review, SPARC-null mice have a curly tail, demonstrate decreases in bone mineral density, develop low-turnover osteopenia at an early age, and exhibit slight decreases in the length of femurs [36, 37]. Despite these phenotypic abnormalities, the absence of SPARC expression does not significantly influence lifespan, reproduction, or overall body weight. Similar to connective tissue, skeletal defects in SPARC-null mice were most pronounced under conditions of active bone remodeling. For example, SPARC-null mice develop accelerated intervertebral disc degeneration [38]. In addition, whereas WT mice exhibit significant increases in bone mineral density in response to bone-anabolic parathyroid hormone treatment, SPARC-null mice do not [39].
The first reported skeletal phenotype in SPARC-null mice was early onset osteopenia [36]. Osteopenia is characterized by decreased bone mineral density and is considered by some clinicians to be a precursor to osteoporosis. The osteopenia indicative of SPARC-null mice became progressively worse with age. At 11 weeks, radiographic differences in trabecular bone density of femurs were detected in SPARC-null versus WT mice. By 17 weeks of age, SPARC-null femurs demonstrated reduced biomechanical properties versus WT bones as measured by a 3-point bending test. Whereas WT bones gained strength from 11 to 17 weeks, the bones of SPARC-null mice did not as evidenced by lower measured maximum load (N), maximum displacement (mm), and stiffness (N/mm) [36]. SPARC-null mice have decreased numbers of osteoblasts and osteoclasts and an ∼50% reduction in bone-formation rates [36]. SPARC-null tibia at 17 weeks had 50% less trabecular bone volume compared to WT tibia as indicated by decreases in trabecular number and increases in trabecular spacing quantitated by histomorphometric measurements. Trabecular bone volume of SPARC-null tibia decreased further with age (36 weeks) to 70% less than WT trabecular bone volume [36]. Frequently, decreased bone formation in both the trabecular and cortical bone compartments is characteristic of osteopenia. Interestingly, in SPARC-null mice, osteopenia was primarily observed in trabecular bone where bone resorption was greater than bone formation leading to a decrease in bone mass that was compounded over time. Trabecular bone is frequently more affected in deficiencies characterized by reduced bone mineral density and SPARC expression was more readily detected in extracts from trabecular versus cortical bone [3].
Two identified haplotypes in the osteonectin 3′ UTR were found to associate with bone mineral density (BMD). Haplotype A containing a single nucleotide polymorphism at SNP1599 (SNP1599G) was found at a higher frequency in severe osteoporotic patients with decreased BMD, whereas haplotype B (SNP1599C) was more commonly associated with healthy controls and higher BMD levels [40]. To discern whether SNP1599C versus SNP1599G influenced the expression of SPARC in vivo, mice carrying either SNP1599C or SNP1599G were genetically engineered [41]. Mice carrying the SNP1599G variant produced lower amounts of SPARC, deposited less mineralized matrix, demonstrated decreased trabecular bone volume with age, and decreased overall bone formation rates in comparison to mice carrying SNP1599C variant and to control mice. Increased cortical bone area in response to intermittent PTH treatment was less in mice with SNPS1599G than mice carrying the SNP1599C variant. Importantly, SNP1599G introduced a novel miR-433 binding site in the 3′UTR of SPARC [40]. SPARC expression levels were decreased by the inclusion of SNP1599G, presumably through inhibition mediated by miR-433 [41]. The 3′ UTR of many mRNAs is known to have regulatory effects on gene expression through modulation of mRNA stability, targeting, and/or translation. Accordingly, SNPs within the SPARC 3′UTR might influence synthesis of SPARC and contribute to variations in expression patterns among individuals. During osteoblastic differentiation, the expression of miR-433 decreases and inhibitors of miR-433 promote osteoblast differentiation [41]. Hence miR-433 is predicted to act as an inhibitor of osteoblast differentiation.
Closer examination of SPARC-null bones has revealed specific alterations in collagen structure, mineral composition, and apatite crystallite morphology in comparison to WT bones [42]. In line with a low bone turnover rate in the absence of SPARC, higher mineral contents and increases in crystal size were found in SPARC-null bones. In addition, infrared microspectroscopic measurements of SPARC-null cortical bone collected from three dimensions of tibia revealed decreased rates of bone formation despite the absence of differences in overall cortical thickness versus WT bones [42]. Hence, SPARC-dependent defects in trabecular bone formation were also observed in cortical bone. Likely, elevated mineral content and highly cross linked matrix generated by slow turnover in the absence of SPARC maintained cortical bone thickness whereas overt decreases in trabecular bone density were more easily detected. Nonetheless, mechanical properties of cortical bone were weakened in SPARC-null mice demonstrated by increased bone fragility despite normal cortical area [36]. Synthesis of mRNA encoding collagen I subunits along with other ECM proteins associated with bone formation were not significantly altered in SPARC-null versus WT mice at 17 weeks of age [36]. Hence, although basic multicellular units (BMUs), the functional unit comprised of osteoblasts and osteoclasts, were reduced in the absence of SPARC, decreases in collagen synthesis did not appear to significantly contribute to reductions in bone matrix remodeling.
SPARC might also influence bone formation through its capacity to bind mineral. In a recent study, a peptide (ON29) derived from the mineral-binding region of SPARC induced changes to the disordered phase and apatite crystallite morphology in an in vitro assay of matrix mineralization [43]. Apatite embedded with ON29 revealed significant decreases in the amount of water molecules in the disordered phase of crystal mineralization and reductions in phosphate content, however calcium content remain unchanged [43]. Decreases in phosphate levels altered the disordered phase of apatite crystal growth by inducing needle-like crystal morphology in culture as opposed to plate-like morphology. Decreased hydroxyl transfer to the disordered phase was observed with peptide treatment, but overall mineral transfer was unaffected [43]. Hence, some aspects of the altered phenotype of SPARC-null bones might arise from deficiencies in crystal mineralization, specifically in the disordered interface, that in turn might influence turnover rates of bone.
Osteogenesis Imperfecta
Osteogenesis imperfect (OI) is a heritable bone fragility disorder most frequently caused by mutations in genes encoding collagen 1α(I) or collagen 2α(I) subunits [44]. The severity of OI demonstrates a broad range among afflicted individuals. There are eight types of osteogenesis imperfecta (I-VIII). The majority of cases of OI (90%) are type I, II, III, and IV and are attributed to genetic mutations in genes coding for type I collagen: COL1A1 or COL1A2 genes [44]. These mutations can lead to <50% decreases in levels of matrix collagen in some cases as well as impaired incorporation of alpha chains into the collagen heterotrimers. Since collagen type I is the main structural protein component of bone matrix, abnormal collagen synthesis diminishes bone mass and increases susceptibility to fracture. Mutations in genes other than COL1 have also been uncovered that are associated with different types of OI. For example, mutations in CRTAP, a cartilage associated protein, and P3H1, prolyl 3-hydroxylase 1, are associated with rare forms of OI [45]. CRTAP and P3H1 proteins are considered chaperone-type proteins that reside in the endoplasmic reticulum and participate in post-translational modification of collagen required for secretion. Additional novel mutations in other genes encoding proteins also thought to act as collagen chaperones have also been recently linked to OI diagnoses [46].
Osteoblasts from patients with the most common forms of OI (Type I and Type IV) have been shown to exhibit reduced rates of growth relative to cells from age-matched individuals [47]. Osteoblasts from patients with OI have also been reported to display decreased amounts of total collagen, SPARC, and proteoglycans as well as defects in metabolism [48, 49]. Whether the decreased levels of SPARC are a result of abnormal Ol bone matrix or whether decreased levels of SPARC contribute to this disease phenotype is not resolved. Hydroxyproline incorporation reflecting collagen deposition into insoluble matrix by osteoblasts increases during initial periods of deposition and then level off and subsequently decline with time [48]. In contrast, osteoblasts from OI cultures exhibit reduced amounts of insoluble collagen deposition and increased synthesis of the glycosaminoglycan, hyaluronan, that lead to higher matrix levels of hyaluronan that are maintained over time in comparison to ECM deposited by osteoblasts from control individuals. In bone, hyaluronan is thought to function to create extracellular space that facilitates matrix deposition. The elevated levels of hyaluronan in cultured osteoblasts from patients with OI likely reflect defects in collagen secretion or deposition that might contribute to decreases in rates of proliferation by these cells.
Recently, mutations in the gene encoding SPARC were identified in two individuals with recessive OI using whole-exome sequencing [50]. Both of the effected individuals demonstrated severe bone fragility and early-onset scoliosis. The mutations in the gene encoding SPARC in individuals with OI were found to be glycine to arginine missense mutations in exon 7 from one individual and in exon 9 from the second individual [50]. The sequence of the collagen-binding site in SPARC is characterized as a “phenylalanine (Phe) pocket”. Argl66 and Glu263 are conserved amino acids in the extracellular Ca2+ (EC) binding domain of SPARC. These amino acids form a salt bridge necessary for collagen binding in the “Phe pocket”. Mutations in Arg 166 or Glu 263 disrupt the pocket and significantly reduce SPARC affinity for collagen I. Hence, the collagen-binding function of SPARC is hypothesized to play a critical role in collagen deposition in bone. Dermal fibroblasts isolated from individuals with SPARC mutations associated with OI exhibited kinetic differences in collagen I triple helical assembly including delayed procollagen I secretion [50]. Despite phenotypic similarities, the recent study distinguished OI caused by mutations in the gene encoding SPARC from previously characterized OI type VI. SPARC was thus identified as one of the known non-collagenous proteins in which mutations can cause a severe form of OI.
SPARC as a Regulator of Osteoblast and Osteoclast Activity
As mentioned, evidence that SPARC influences osteoblast and osteoclast activity was noted in SPARC-null mice [4]. SPARC is expressed by osteoblasts undergoing active matrix deposition. Interestingly, during in vitro differentiation of osteoblasts in WT cells, SPARC mRNA levels remain relatively constant [51]. However, levels of SPARC protein were shown to increase during the matrix deposition phase of osteoblast culture differentiation. Levels of SPARC protein are highest at times coinciding with initial stages of differentiation and subsequently decrease as cells mature and begin to express mature osteoblastic markers [51]. The differences in expression of SPARC protein versus mRNA suggested a significant role for post-transcriptional regulation of SPARC in osteoblasts. Notably, SPARC mRNA has a significantly long half-life, which would be predicted to affect the efficacy of transcriptional regulation as a primary mediator of expression. Regions in the 3′UTR of SPARC mRNA contain highly conserved sequences that interact with miR29. Kapinas et al., presented strong evidence that post-transcriptional regulation of SPARC mRNA by members of the miR-29 family was a primary mechanism by which levels of SPARC were regulated in osteoblasts. Increased levels of miR-29 during osteoblast differentiation correlates with decreased amounts of SPARC protein levels [52].
Osteoblasts and adipocytes differentiate from a common mesenchymal precursor cell originating in the bone marrow. Delany et al. have reported that osteoblastic precursors were reduced in SPARC-null mice and that induction of osteoblast differentiation from SPARC-null bone marrow derived cells was impaired [51]. Cell populations isolated from SPARC-null bone marrow demonstrated fewer osteoblastic cells when grown under conditions favoring osteoblastogenesis. Concomitantly, a higher number of adipocytes were detected in SPARC-null versus WT cultures in which SPARC-null cells expressed higher levels of adipsin mRNA, a marker of mature adipocytes [51]. Interestingly, diminished cell survival also indicative of SPARC-null bone marrow derived cells was rescued by reintroduction of SPARC expression through transfection of SPARC-expressing vectors but not by incubation with recombinant SPARC protein. Whereas SPARC-null osteoblasts differentiated in vitro showed similar levels of osteoblast differentiation markers osteopontin and bone sialoprotein. Significantly lower levels of osteocalcin mRNA were detected versus WT cells. In addition, attenuated formation of mineralized nodules was observed in SPARC-null cultures that suggested that SPARC-null osteoblast do not undergo a complete process of maturation [51]. Hence, cultures derived from SPARC-null mice displayed a higher propensity for the differentiation of adipocytes at the expense of competent osteoblast differentiation. Interestingly, an increase in adiposity of SPARC-null bone marrow has also been described [39].
Parathyroid hormone (PTH) is a potent stimulator of bone remodeling that supports formation of new bone over those processes that influence bone resorption. Presently, intermittent administration of PTH is a commonly used bone-anabolic therapeutic option for osteoporosis patients. To test whether SPARC might influence PTH efficacy, WT and SPARC-null mice were treated with PTH and assessed for differences in bone formation [39]. As expected, PTH treatment elevated whole body bone mineral density in WT mice however, notably, no increases in bone mineral density were observed in SPARC-null mice. Although the osteoblastic response was similar in WT and SPARC-null mice treated with PTH, SPARC-null mice had a significant increase in the number of osteoclasts and the associated eroded surface of bone in comparison to WT mice treated with PTH [39]. PTH treatment is known to result in the formation of multinucleated osteoclasts through the increase in expression of RANK-ligand accompanied by a decrease in osteoprotegerin. Interestingly, bone marrow cells isolated from SPARC-null mice treated with PTH showed a greater propensity for osteoclast differentiation in comparison to WT cultures and suggested that in the absence of SPARC, PTH has an enhanced effect on osteoclastogenesis over that of WT cells [39]. Observed increases in levels of expression of RANKL in PTH treated SPARC-null mice might contribute to increased osteoclast formation although other cellular mechanisms could also be important as expression of osteoprotegerin (OPG) was also increased in SPARC-null cells rendering the RANKL/OPG ratio relatively unchanged [39].
Osteoclasts and macrophages arise from the same precursor monocyte cell lineage that also derives from bone marrow cell populations. Expression of SPARC by some monocyte/macrophage cell lines and by monocyte/macrophage populations in tissues has been reported [15, 53, 54]. To date, the expression of SPARC by osteoclasts, either in vitro or in vivo, has not been reported. As mentioned above, in PTH-treated mice, SPARC appeared to limit osteoclast formation however the mechanism by which SPARC acts to influence osteoclast activity is not defined and might involve 1) direct interaction of extracellular SPARC with osteoclasts, 2) an influence on other cell types that support osteoclast activity such as immune cells or osteoblasts, or 3) reside in differences in mineralized ECM assembled in the absence of SPARC.
Conclusion
In conclusion, studies from mice and man have revealed that SPARC clearly plays a critical role in regulating bone remodeling and maintaining bone mass and quality. The mechanisms by which SPARC influences bone formation, maintenance, and repair might occur through multiple pathways that include the regulation of procollagen processing and assembly in the bone matrix, cross-linking, mineralization, and/or osteoblast/osteoclast differentiation and activity. As each of these processes are tightly linked to one another, perturbations in one pathway might induce a ripple effect leading to consequences for other mediators of bone biology. In non-mineralized tissues, SPARC has been implicated as a significant contributor to fibrotic diseases such as those in skin, heart, liver, and kidney [20]. Given the close association of SPARC with collagen I expression in both mineralized and non-mineralized tissues and the capacity of SPARC to bind to collagens strongly suggest that SPARC is critical in the production and/or deposition of collagen. The recent identification of mutations in SPARC specifically in the collagen binding pocket region that associate with osteogenesis imperfecta lend credence to the concept that the collagen-binding function of SPARC is essential for proper ECM assembly and architecture.
Figure 1. Schematic of SPARC Activity in Formation of Mineralized Matrix.
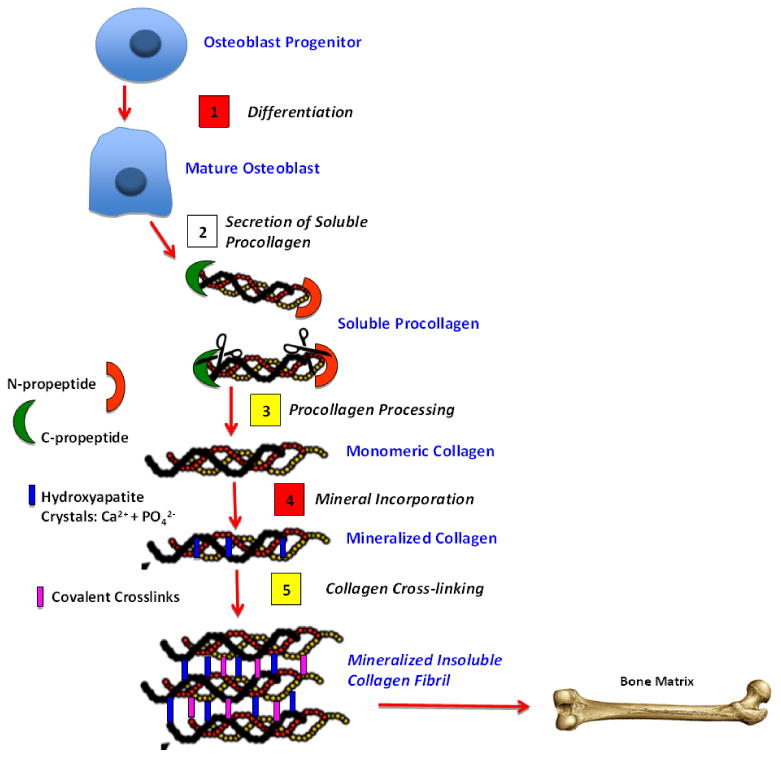
Critical steps in bone formation include: 1) osteoblast differentiation 2) procollagen secretion 3) procollagen processing 4) mineral incorporation, and 5) collagen cross-linking. Steps shown with yellow boxes are processes regulated by SPARC that potentially influence bone formation based on results from cells and/or non-mineralized tissues. Steps shown with red boxes are processes shown to be altered in SPARC-null bones.
Table 1. A list of activities attributed to SPARC in mineralized and non-mineralized cells and tissues.
| In Vitro | In Vivo | |
|---|---|---|
|
| ||
| Collagen Production | SPARC-null fibroblasts (fb)): No change in levels of mRNA for collagen I (22) |
SPARC-null tissues: No change in levels of mRNA for collagen I, (50) ↑ soluble collagen (24) ↓ total collagen content (22) ↓fibrotic collagen (22) |
|
| ||
| Collagen Assembly | SPARC-null fb: Altered procollagen processing ↑cell surface-associated collagen (25, 26) |
SPARC-null PDL: ↓ collagen content, smaller collagen fibers (11) ↑TG activity on collagen I (28) ↑TG collagen crosslinks (28) |
| SPARC-null bones: ↑collagen content (36) ↑collagen maturity (36, 42) Larger, less uniform fibril diameters | ||
|
| ||
| Mineralization | SPARC Peptide ON29: ↓apatite crystal H2O content (43) ↓phosphate content (43) |
SPARC-null bones: ↑mineral content in young mice (42) ↓ mineral content in older mice (42) |
|
| ||
| Bone Properties | N/A | SPARC-null bones: ↓biomechanical properties: 3-point bending of femur (36) ↓bone turnover rate (36) ↓long bone length (37) ↑ early-onset trabecular osteopenia (36) Normal cortical area, ↓ cortical fragility (37) ↑ adiposity of bone marrow (37) ↑ intervertebral disc degeneration (38) |
| Missense (G→R) mutation in gene encoding SPARC disrupts collagen binding pocket → severe OI (50) | ||
| SNP1599 of 3′UTR SPARC → idiopathic osteoporosis - ↓SPARC, ↓deposited matrix, ↓cortical bone area, ↓ trabecular bone with age (40, 41) | ||
|
| ||
| Osteoblasts | SPARC expressed by differentiating osteoblasts, Regulated by miR29 (52) |
SPARC-null bones: ↓basic multicellular units (BMUs) (36) |
| SPARC-null bone-marrow derived cells: ↓OB precursor cells (51) ↑adipocytes differentiated in vitro (51) ↓mineralized OB nodules (51) | ||
|
| ||
| Osteoclasts | Do not express SPARC | SPARC-null bones: OC numbers ↑with PTH stimulation (39) |
| SPARC-null cells: ↑PTH effect on osteoclastogenesis (39) | ||
Research Highlights.
The regulation of procollagen processing by SPARC and potential roles in bone matrix assembly
Functional implications of SPARC in the regulation of collagen cross-linking by transglutaminase
Phenotypic differences characteristic of SPARC-null bones, osteoblasts, and osteoclasts
SPARC mutations identified in patients with OI and idiopathic osteoporosis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Bradshaw AD. Diverse biological functions of the SPARC family of proteins. Int J Biochem Cell Biol. 2012;44:480–8. doi: 10.1016/j.biocel.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol. 2014;37:1–14. doi: 10.1016/j.matbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML, Martin GR. Osteonectin, a bone-specific protein linking mineral to collagen. Cell. 1981;26:99–105. doi: 10.1016/0092-8674(81)90037-4. [DOI] [PubMed] [Google Scholar]
- 4.Delany AM, Hankenson KD. Thrombospondin-2 and SPARC/osteonectin are critical regulators of bone remodeling. J Cell Commun Signal. 2009;3:227–38. doi: 10.1007/s12079-009-0076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest. 2001;107:1049–54. doi: 10.1172/JCI12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hohenester E, Maurer P, Hohenadl C, Timpl R, Jansonius JN, Engel J. Structure of a novel extracellular Ca(2+)-binding module in BM-40. Nat Struct Biol. 1996;3:67–73. doi: 10.1038/nsb0196-67. [DOI] [PubMed] [Google Scholar]
- 7.Swaroop A, Kogan BL, Francke U. Molecular analysis of the cDNA for human SPARC/osteonectin/BM-40: sequence, expression, and localization of the gene to chromosome 5q31-q33. Genomics. 1988;2:37–47. doi: 10.1016/0888-7543(88)90107-3. [DOI] [PubMed] [Google Scholar]
- 8.Kelm RJ, Jr, Mann KG. The collagen binding specificity of bone and platelet osteonectin is related to differences in glycosylation. J Biol Chem. 1991;266:9632–9. [PubMed] [Google Scholar]
- 9.Holland PW, Harper SJ, McVey JH, Hogan BL. ln vivo expression of mRNA for the Ca++-binding protein SPARC (osteonectin) revealed by in situ hybridization. J Cell Biol. 1987;105:473–82. doi: 10.1083/jcb.105.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mundlos S, Schwahn B, Reichert T, Zabel B. Distribution of osteonectin mRNA and protein during human embryonic and fetal development. J Histochem Cytochem. 1992;40:283–91. doi: 10.1177/40.2.1552170. [DOI] [PubMed] [Google Scholar]
- 11.Trombetta JM, Bradshaw AD. SPARC/osteonectin functions to maintain homeostasis of the collagenous extracellular matrix in the periodontal ligament. J Histochem Cytochem. 2010;58:871–9. doi: 10.1369/jhc.2010.956144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sage H, Vernon RB, Decker J, Funk S, Iruela-Arispe ML. Distribution of the calcium-binding protein SPARC in tissues of embryonic and adult mice. J Histochem Cytochem. 1989;37:819–29. doi: 10.1177/37.6.2723400. [DOI] [PubMed] [Google Scholar]
- 13.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19:816–27. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- 14.Breton-Gorius J, Clezardin P, Guichard J, Debili N, Malaval L, Vainchenker W, et al. Localization of platelet osteonectin at the internal face of the alpha-granule membranes in platelets and megakaryocytes. Blood. 1992;79:936–41. [PubMed] [Google Scholar]
- 15.Reed MJ, Puolakkainen P, Lane TF, Dickerson D, Bornstein P, Sage EH. Differential expression of SPARC and thrombospondin 1 in wound repair: immunolocalization and in situ hybridization. J Histochem Cytochem. 1993;41:1467–77. doi: 10.1177/41.10.8245406. [DOI] [PubMed] [Google Scholar]
- 16.Bradshaw AD. The Extracellular Matrix. In: Bradshaw RAaS, P.D., editor. Encyclopedia of Cell Bioloby. 1. Waltham, MA: Academic Press; 2016. pp. 694–703. [Google Scholar]
- 17.Shen G. The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthod Craniofac Res. 2005;8(1):11–7. doi: 10.1111/j.1601-6343.2004.00308.x. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki T, Gohring W, Mann K, Maurer P, Hohenester E, Knauper V, et al. Limited cleavage of extracellular matrix protein BM-40 by matrix metalloproteinases increases its affinity for collagens. J Biol Chem. 1997;272:9237–43. doi: 10.1074/jbc.272.14.9237. [DOI] [PubMed] [Google Scholar]
- 19.Canty EG, Lu Y, Meadows RS, Shaw MK, Holmes DF, Kadler KE. Coalignment of plasma membrane channels and protrusions (fibripositors) specifies the parallelism of tendon. J Cell Biol. 2004;165:553–63. doi: 10.1083/jcb.200312071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eyre DR, Paz MA, Gallop PM. Cross-linking in collagen and elastin. Annual review of biochemistry. 1984;53:717–48. doi: 10.1146/annurev.bi.53.070184.003441. [DOI] [PubMed] [Google Scholar]
- 21.Bradshaw AD. The role of SPARC in extracellular matrix assembly. J Cell Commun Signal. 2009;3:239–46. doi: 10.1007/s12079-009-0062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trombetta-Esilva J, Bradshaw AD. The Function of SPARC as a Mediator of Fibrosis. The open rheumatology journal. 2012;6:146–55. doi: 10.2174/1874312901206010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh DJ, Kang MH, Ooi YH, Choi KR, Sage EH, Rhee DJ. Overexpression of SPARC in human trabecular meshwork increases intraocular pressure and alters extracellular matrix. Invest Ophthalmol Vis Sci. 2013;54:3309–19. doi: 10.1167/iovs.12-11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradshaw AD, Baicu CF, Rentz TJ, Van Laer AO, Boggs J, Lacy JM, et al. Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation. 2009;119:269–80. doi: 10.1161/CIRCULATIONAHA.108.773424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris BS, Zhang Y, Card L, Rivera LB, Brekken RA, Bradshaw AD. SPARC regulates collagen interaction with cardiac fibroblast cell surfaces. Am J Physiol Heart Circ Physiol. 2011;301:H841–7. doi: 10.1152/ajpheart.01247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rentz TJ, Poobalarahi F, Bornstein P, Sage EH, Bradshaw AD. SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. J Biol Chem. 2007;282:22062–71. doi: 10.1074/jbc.M700167200. [DOI] [PubMed] [Google Scholar]
- 27.Carafoli F, Bihan D, Stathopoulos S, Konitsiotis AD, Kvansakul M, Farndale RW, et al. Crystallographic insight into collagen recognition by discoidin domain receptor 2. Structure. 2009;17:1573–81. doi: 10.1016/j.str.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trombetta-eSilva J, Rosset EA, Hepfer RG, Wright GJ, Baicu C, Yao H, et al. Decreased Mechanical Strength and Collagen Content in SPARC-Null Periodontal Ligament is Reversed by Inhibition of Transglutaminase Activity. Journal of bone and mineral research. 2015 doi: 10.1002/jbmr.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collighan RJ, Griffin M. Transglutaminase 2 cross-linking of matrix proteins: biological significance and medical applications. Amino Acids. 2009;36:659–70. doi: 10.1007/s00726-008-0190-y. [DOI] [PubMed] [Google Scholar]
- 30.Aeschlimann D, Kaupp O, Paulsson M. Transglutaminase-catalyzed matrix cross-linking in differentiating cartilage: identification of osteonectin as a major glutaminyl substrate. J Cell Biol. 1995;129:881–92. doi: 10.1083/jcb.129.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nurminskaya M, Kaartinen MT. Transglutaminases in mineralized tissues. Front Biosci. 2006;11:1591–606. doi: 10.2741/1907. [DOI] [PubMed] [Google Scholar]
- 32.Deasey S, Grichenko O, Du S, Nurminskaya M. Characterization of the transglutaminase gene family in zebrafish and in vivo analysis of transglutaminase-dependent bone mineralization. Amino Acids. 2011 doi: 10.1007/s00726-011-1021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cui C, Wang S, Myneni VD, Hitomi K, Kaartinen MT. Transglutaminase activity arising from Factor XIIIA is required for stabilization and conversion of plasma fibronectin into matrix in osteoblast cultures. Bone. 2014;59:127–38. doi: 10.1016/j.bone.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Forsprecher J, Wang Z, Goldberg HA, Kaartinen MT. Transglutaminase-mediated oligomerization promotes osteoblast adhesive properties of osteopontin and bone sialoprotein. Cell Adh Migr. 2011;5(1):65–72. doi: 10.4161/cam.5.1.13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilmour DT, Lyon GJ, Carlton MB, Sanes JR, Cunningham JM, Anderson JR, et al. Mice deficient for the secreted glycoprotein SPARC/osteonectin/BM40 develop normally but show severe age-onset cataract formation and disruption of the lens. Embo J. 1998;17:1860–70. doi: 10.1093/emboj/17.7.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delany AM, Amling M, Priemel M, Howe C, Baron R, Canalis E. Osteopenia and decreased bone formation in osteonectin-deficient mice. J Clin Invest. 2000;105:915–23. doi: 10.1172/JCI7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mansergh FC, Wells T, Elford C, et al. Osteopenia in Sparc (osteonectin)-deficient mice: characterization of phenotypic determinants of femoral strength and changes in gene expression. Physiol Genomics. 2007;32(1):64–73. doi: 10.1152/physiolgenomics.00151.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruber HE, Sage EH, Norton HJ, Funk S, Ingram J, Hanley EN., Jr Targeted deletion of the SPARC gene accelerates disc degeneration in the aging mouse. J Histochem Cytochem. 2005;53:1131–8. doi: 10.1369/jhc.5A6687.2005. [DOI] [PubMed] [Google Scholar]
- 39.Machado do Reis L, Kessler CB, Adams DJ, Lorenzo J, Jorgetti V, Delany AM. Accentuated osteoclastic response to parathyroid hormone undermines bone mass acquisition in osteonectin-null mice. Bone. 2008;43:264–273. doi: 10.1016/j.bone.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delany AM, McMahon DJ, Powell JS, Greenberg DA, Kurland ES. Osteonectin/SPARC polymorphisms in Caucasian men with idiopathic osteoporosis. Osteoporos Int. 2008;19:969–78. doi: 10.1007/s00198-007-0523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dole NS, Kapinas K, Kessler CB, Yee SP, Adams DJ, Pereira RC, et al. A single nucleotide polymorphism in osteonectin 3′ untranslated region regulates bone volume and is targeted by miR-433. Journal of Bone Min Res. 2015;30:723–32. doi: 10.1002/jbmr.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boskey AL, Moore DJ, Amling M, Canalis E, Delany AM. Infrared analysis of the mineral and matrix in bones of osteonectin-null mice and their wildtype controls. Journal Bone and Mineral research. 2003;18:1005–11. doi: 10.1359/jbmr.2003.18.6.1005. [DOI] [PubMed] [Google Scholar]
- 43.Iline-Vul T, Matlahov I, Grinblat J, Keinan-Adamsky K, Goobes G. Changes to the Disordered Phase and Apatite Crystallite Morphology during Mineralization by an Acidic Mineral Binding Peptide from Osteonectin. Biomacromolecules. 2015;16:2656–63. doi: 10.1021/acs.biomac.5b00465. [DOI] [PubMed] [Google Scholar]
- 44.Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. American journal of medical genetics Part A. 2014;164a:1470–81. doi: 10.1002/ajmg.a.36545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 46.Marini JC, Reich A, Smith SM. Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Current opinion in pediatrics. 2014;26:500–7. doi: 10.1097/MOP.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fedarko NS, D'Avis P, Frazier CR, Burrill MJ, Fergusson V, Tayback M, et al. Cell proliferation of human fibroblasts and osteoblasts in osteogenesis imperfecta: influence of age. Journal of bone and mineral research. 1995;10:1705–12. doi: 10.1002/jbmr.5650101113. [DOI] [PubMed] [Google Scholar]
- 48.Fedarko NS, Sponseller PD, Shapiro JR. Long-term extracellular matrix metabolism by cultured human osteogenesis imperfecta osteoblasts. Journal of bone and mineral research. 1996;11:800–5. doi: 10.1002/jbmr.5650110611. [DOI] [PubMed] [Google Scholar]
- 49.Fedarko NS, Moerike M, Brenner R, Robey PG, Vetter U. Extracellular matrix formation by osteoblasts from patients with osteogenesis imperfecta. Journal of bone and mineral research. 1992;7:921–30. doi: 10.1002/jbmr.5650070809. [DOI] [PubMed] [Google Scholar]
- 50.Mendoza-Londono R, Fahiminiya S, Majewski J, Tetreault M, Nadaf J, Kannu P, et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet. 2015;96:979–85. doi: 10.1016/j.ajhg.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delany AM, Kalajzic I, Bradshaw AD, Sage EH, Canalis E. Osteonectin-null mutation compromises osteoblast formation, maturation, and survival. Endocrinology. 2003;144:2588–96. doi: 10.1210/en.2002-221044. [DOI] [PubMed] [Google Scholar]
- 52.Kapinas K, Kessler CB, Delany AM. miR-29 suppression of osteonectin in osteoblasts: regulation during differentiation and by canonical Wnt signaling. J Cell Biochem. 2009;108:216–24. doi: 10.1002/jcb.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sangaletti S, Di Carlo E, Gariboldi S, Miotti S, Cappetti B, Parenza M, et al. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008;68:9050–9. doi: 10.1158/0008-5472.CAN-08-1327. [DOI] [PubMed] [Google Scholar]
- 54.Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med. 2009;206:113–23. doi: 10.1084/jem.20081244. [DOI] [PMC free article] [PubMed] [Google Scholar]