

Abstract
The lectin-like, oxidized low-density lipoprotein (ox-LDL) receptor-1 (LOX-1)/ox-LDL system contributes to atherosclerosis and may be involved in cartilage degeneration. The purpose of this study was to determine whether the LOX-1/ox-LDL system contributes to age-related osteoarthritis (OA) in vivo, using LOX-1 knockout (LOX-1 KO) mice. Knee cartilage from 6, 12, and 18-month old (n = 10/group) C57Bl/6 wild-type (WT) and LOX-1 KO mice was evaluated by determining the Osteoarthritis Research Society International (OARSI) score of Safranin-O stained samples. The prevalence of knee OA in both mouse strains was also investigated. Expression levels of LOX-1, ox-LDL, runt-related transcription factor-2 (Runx2), type-X collagen (COL X), and matrix metalloproteinase-13 (MMP-13) in the articular chondrocytes were analyzed immunohistologically. No significant difference was observed in the mean scores of WT (2.00±0.61) and LOX-1 KO mice (2.00±0.49) at 6 months of age (P=1.00, n=10). At 12 and 18 months of age, the mean scores of LOX-1 KO mice (3.75±0.93 and 5.50±0.78) were significantly lower than those of WT mice (5.25±1.14 and 9.00±1.01; P<0.001 in both cases; n=10). The prevalence of OA in LOX-1 KO mice was lower than that in WT mice at 12 and 18 months of age (40 vs 70%, 70 vs 90%, respectively; n=10). The expression levels of Runx2, COL X, and MMP-13 in articular chondrocytes significantly decreased in LOX-1 KO, mice compared with those in WT mice. The study indicated that the LOX-1/ox-LDL system in chondrocytes plays a role in the pathogenesis of age-related knee OA, which is potentially a target for preventing OA progression.
Key words: Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), oxidized low-density lipoprotein (ox-LDL), mouse, knee osteoarthritis, aging
Introduction
Oxidized low-density lipoprotein (ox-LDL) is produced by LDL oxidation at sites of oxidative stress and inflammation and plays an important role in the pathogenesis of atherosclerosis.1 Lectin-like ox-LDL receptor-1 (LOX-1) is an important receptor for ox-LDL, was cloned from cultured bovine aortic endothelial cells,2 and is expressed in various cell types, including endothelial cells,3,4 vascular smooth muscle cells,5 macrophages,6 and chondrocytes.7,8
LOX-1 plays roles in the degeneration of articular cartilage in vivo and in vitro. Nakagawa et al. demonstrated LOX-1 expression and the presence of ox-LDL in articular chondrocytes in zymosan-induced arthritis (ZIA).7 Further, they showed that rat articular chondrocytes under basal culture conditions expressed LOX-1 and that ox-LDL binding to LOX-1 reduced the viability of cultured chondrocytes.9 Ox-LDL in human synovial fluid10 and co-expression of LOX-1 and ox-LDL in human osteoarthritis (OA) cartilage were reported.8,11 Previously, we demonstrated in vitro that ox-LDL binding to LOX-1 increased the production of intracellular reactive oxygen species (ROS) in bovine articular chondrocytes (BACs),12 induced premature senescence in articular chondrocytes by suppressing telomerase activity,13 and promoted a hypertrophic chondrocyte-like phenotype through oxidative stress in cultured BACs.14 A point of interest is whether the LOX-1/ox-LDL system can contribute to development of OA induced by mechanical stress. Recently, we addressed this question and demonstrated that LOX-1 KO mice show resistance, using a murine knee OA model, by destabilizing the medial meniscus.15
Genetic predisposition, obesity, female gender, greater bone density, and joint laxity are risk factors of primary OA.16,17 Although these factors may increase the risk for developing OA in select populations, and mechanical loading that exceeds the ability to repair is another universal risk factor, the most important risk factor among all populations is aging.18, 9 Regarding age-related OA, recent evidence indicates that mitosis and DNA synthesis in chondrocytes decline with age, and they become less responsive to anabolic cytokines and mechanical stimuli. Further, chondrocyte cell senescence in age-related OA was suggested because telomere erosion and expression of senescence markers may be attributable to oxidative damage.20,21
The aim of the current study was the histological observation of OA development in the articular cartilages of wild-type (WT) and LOX-1 knockout (KO) mice, maintained for up to 18 months of age. We hypothesized that the LOX-1/ox-LDL system is involved in the age-related OA in vivo and demonstrated the involvement of the LOX-1/ox-LDL system in the development and progression of age-related knee OA, using LOX-1 KO mice.
Materials and Methods
Mice
LOX-1+/+ C57BL/6 Jcl mice (WT) were provided by Nihon CLEA (Tokyo, Japan). LOX-1-/- C57BL/6 Jcl mice (KO) were originally generated by Sawamura et al.22 and were provided by the National Cerebral and Cardiovascular Center (Osaka, Japan). Pups were genotyped by Southern blotting using an external probe, as previously described.22 Mice were housed in cages with access to food and water ad libitum in a temperature-controlled room with a 12-h dark/12-h light cycle. All animal experiments were performed in accordance with protocols approved by the Animal Care and Use Committee of our hospital.
Body weight measurements
We measured mouse body weights at 6, 12, and 18 months of age.
Age-related OA model
To monitor age-related OA, WT and LOX-1 KO mice were examined at 6, 12, and 18 months of age.
Histological evaluation of OA
Knee joints were fixed in 4% paraformaldehyde at 4°C for 12 h. After washing with phosphate-buffered saline (PBS), samples were decalcified in 10% ethylenediaminetetraacetic acid solution at 4°C for 2 weeks and then embedded in paraffin. Coronary sections (4-µm) were cut and mounted onto slides. Each sample was stained with Safranin O (SFO) for the histological evaluation of cartilage degeneration during OA progression.2 The sections were deparaffinized in xylene and dehydrated using an ethanol gradient, and then immersed in SFO solution (Sigma-Aldrich, St. Louis, MO, USA) for 10 min. Sections were washed in PBS for 5 min, and then immersed in 1% Fast Green solution (Wako, Osaka, Japan) for 5 min. The OARSI scoring system24 was used to evaluate the extent of cartilage degeneration. Each score is defined as follows: 0, normal cartilage; 0.5, proteoglycan loss with an intact surface; 1, superficial fibrillation without cartilage loss; 2, vertical clefts and loss of surface lamina; 3, vertical clefts/erosion of the calcified layer lesion for 1-25% of the quadrant width; 4, lesion reaches the calcified cartilage for 25-50% of the quadrant width; 5, lesion reaches the calcified cartilage for 50-75% of the quadrant width; and 6, lesion reaches the calcified cartilage for >75% of the quadrant width. Evaluations of the articular cartilage were performed for all 4 quadrants in each sample using a light microscope (BIOREVO BZ-9000; Keyence, Osaka, Japan) at 100× magnification. The summed score for the knee joint (determined by summing the scores for the femoral condyle and tibial plateau) was used to measure the extent of cartilage destruction. Evaluations were made by 2 independent investigators (YO, FN), who were blinded to the experimental groups, in order to avoid observer bias.
Prevalence of knee OA
We examined the prevalence of OA divided by the OARSI score in both WT and LOX-1 KO mice at each time point. The medial tibia plateau score was used. The OA score was divided into 3 groups: ≤1, between 1 and 3, and ≥3.
Immunohistochemistry
To visualize LOX-1 and ox-LDL expression and to investigate the involvement of cartilage cell hypertrophy in OA progression, we performed immunohistochemistry against LOX-1, ox-LDL, runt-related transcription factor-2 (Runx2), and type-X collagen (COL X). Immunohistochemistry was also used to detect matrix metalloproteinases 13 (MMP-13), which is a cartilage matrix-degrading enzyme. Tissue sections adjacent to those used for evaluating cartilage degeneration were deparaffinized in xylene and dehydrated through an ethanol gradient. Endogenous peroxidase activity was blocked by immersion in 3% H2O2 in methanol for 5 min. The sections were then incubated in blocking solution (3% bovine serum in PBS) at room temperature for 15 min, followed by incubation in a diluted primary antibody for 2 h at room temperature. The primary antibodies used included a rabbit anti-mouse LOX-1 polyclonal antibody [Abcam, Cambridge, UK; 1:100 dilution in 0.1% Triton-X 100-Tris-buffered saline (TBS) containing 10% Block Ace; DS Pharma Biomedical, Osaka, Japan], a rabbit anti-mouse ox-LDL polyclonal antibody (LSL, Tokyo, Japan; 1:100 dilution in 0.1% Triton-TBS containing 10% Block Ace), a rabbit anti-mouse Runx2 polyclonal antibody (LSL; 1:1,000 dilution in 0.1% Triton-TBS containing 10% Block Ace), a rabbit anti-mouse COL X polyclonal antibody (LSL; 1:500 dilution in 0.1% Triton-TBS containing 10% Block Ace), and a rabbit anti-mouse MMP-13 polyclonal antibody (Abcam; 1:200 dilution in 0.1% Triton-TBS containing 10% Block Ace). After incubation with the primary antibody, the LOX-1 and ox-LDL-labeled sections were washed in PBS and incubated with a horseradish peroxidase-conjugated donkey anti-rabbit IgG secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:1,000 dilution) for 30 min at room temperature. The sections were then washed with PBS, immersed in a diaminobenzidine solution (Agilent Technologies, Santa Clara, CA, USA) for 10 min at room temperature to visualize the immunoreactivity, and counterstained with hematoxylin. The Runx2- and COL X-labeled sections were immersed in PBS, incubated with a fluorescein isothiocyanate-conjugated chicken anti-rabbit IgG secondary antibody (Santa Cruz; CA, USA; 1:1,000) for 30 min at room temperature, and then imaged by fluorescence microscopy. As a negative control, tissue sections were incubated with a non-immunized rabbit IgG as a substitute for the primary antibody. As a positive control, endothelial cell immunoreactivity was tested in LOX-1 and ox-LDL-labeled sections, and growth plate staining was tested in Runx2 and COL X-labeled sections. The positive cell rate for LOX-1, ox-LDL, Runx2, COL X, and MMP-13 expression was measured as previously described.15 Cells positive for LOX-1, ox-LDL, and MMP-13 staining were counted at 200-fold magnification using an optical microscope. Those positive for Runx2 and COL X staining were counted using a fluorescence microscope (BIOREVO BZ-9000; Keyence). To observe and count positive cells in the entire medial tibial plateau, 2 images at 200-fold magnification were combined and analyzed as a single image. The percentage of positive cells was calculated as the number of stained cells relative to the number of total chondrocytes in the medial tibial plateau.
Statistical analysis
All data were expressed as the mean ± standard deviation. The scores of each group were compared using Student’s t-test. P-values of less than 0.05 were considered significant. All data were analyzed with Stat Mate software for Windows, version 4.01.
Results
Body weight
The mean body weights of WT and LOX-1 KO mice measured at 6, 12, and 18 months are summarized in Table 1. The mean body weights were not significantly different between WT and LOX-1 KO mice at any time point tested.
Table 1.
Body weights at each experimental time.
| Strain | Age (months) | ||
|---|---|---|---|
| 6 | 12 | 18 | |
| WT (g) (n=10) | 32.87±3.27 | 37.74±3.45 | 38.44±4.66 |
| KO (g) (n=10) | 32.13±2.29 | 37.01 ±3.60 | 37.39±2.40 |
| P-value | 0.57 | 0.65 | 0.53 |
WT, wild-type mice; KO, knockout mice. Data are presented as mean ± SD.
Osteoarthritis development
First, we confirmed by SFO staining that knee OA did not develop by 9 weeks of age and that no significant difference in cartilage degeneration occurred between WT and LOX-1 KO mice (data not shown). At 6 months of age, staining was reduced in the articular cartilage of WT and LOX-1 KO mice (Figure 1A). The summed arthritic scores of WT mice were 1.00 ± 0.74 for the medial compartment (medial tibia plateau and femur) and 1.00±0.44 for the lateral compartment (lateral tibia plateau and femur) at 6 months of age (Figure 1B). The summed arthritic scores for LOX-1 KO mice were 1.00±0.58 for the medial compartment and 1.00±0.39 for the lateral compartment at 6 months (Figure 1B). There were no significant differences between the summed arthritic scores for WT and LOX-1 KO mice at 6 months of age (Figure 1B). At 12 and 18 months, cartilage degeneration had progressed in both WT and LOX-1 KO mice, and prominent differences in SFO staining were observed (Figure 1A). In WT mice, moderate to severe cartilage degeneration was observed, whereas only mild changes were observed in LOX-1 KO mice at 12 and 18 months (Figure 1A). WT mice had summed arthritic scores of 2.25±1.02 for the medial compartment and 3.00±1.09 for the lateral compartment at 12 months (Figure 1B). LOX-1 KO mice had summed arthritic scores of 1.50±0.69 for the medial compartment and 2.25±1.02 for the lateral compartment at 12 months, which were significantly lower than those for WT mice (P<0.01, P=0.031, n=10; Figure 1B). At 18 months of age, WT mice had summed arthritic scores of 4.00±1.09 for the medial compartment and 5.00±0.64 for the lateral compartment (Figure 1B). The summed scores of LOX-1 KO mice were significantly lower at 2.50±0.73 for the medial compartment and 3.00±0.69 for the lateral compartment at 18 months (P<0.001, P<0.001, n=10; Figure 1B).
Figure 1.
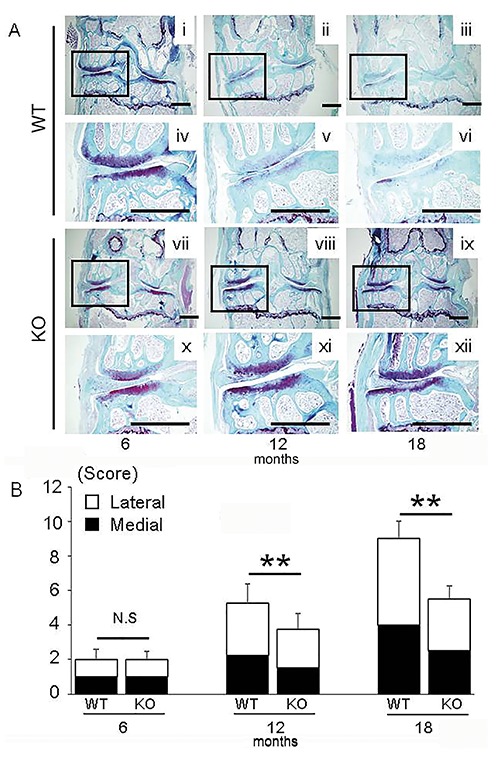
A SFO staining of knee joints in WT mice (i-iii) and LOX-1 KO mice (vii-ix) at 6, 12, and 18 months. Insets show higher-magnification views of the medial compartment from WT (iv-vi) and LOX-1 KO mice (x-xii). B) The summed OARSI scores of tibias and femurs in WT and LOX-1 KO mice at 6, 12, and 18 months. The white and black coloring represents the scores of lateral and medial compartments. Values shown represent the mean ± SD. **P<0.001. Scale bar: 500 µm.
Prevalence of OA
We examined the prevalence of OA divided by arthritic score of medial tibia plateau in both WT mice and LOX-1 KO mice at each time point (Figure 2). At 6 months of age, 8 WT and 8 LOX1- KO mice had a score of ≤1. Two WT and 2 LOX1-KO mice had scores between 1 and 3. No difference was observed in the prevalence of OA divided by the score between WT and LOX-1 KO mice at 6 months. During the time course, the overall OA grade increased in both WT and LOX-1 KO mice. At 12 months of age, the number of WT mice with scores of ≤1, between 1 and 3, and ≥3 was 3, 3, and 4, respectively, whereas these prevalences were 4, 6, and 0, respectively, in LOX-1 KO mice. At 18 months of age, 1, 2, and 7 WT mice had scores of ≤1, between 1 and 3, and ≥3, respectively, while 3, 3, and 4 LOX-1 KO mice had scores of ≤1, between 1 and 3, and ≥3, respectively. At 12 and 18 months of age, fewer LOX-1 KO mice had high OA grades than did WT mice.
Figure 2.
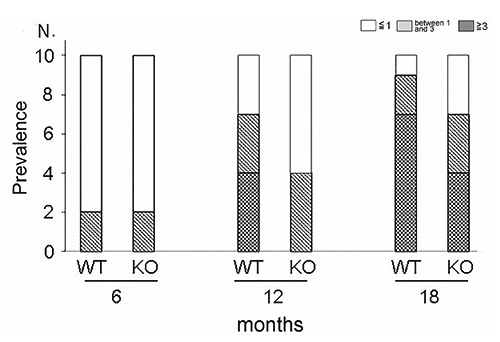
The graph shows the prevalence of knee OA divided by OARSI grade, in both WT and LOX-1 KO mice.
Time course of LOX-1, ox-LDL, Runx2, COL X, and MMP-13 expression in cartilage
LOX-1 and ox-LDL staining was performed with articular cartilage from WT mice, and the staining intensity increased at 12 and 18 months of age, compared to 6 months (Figure 3 G-I, M-O). No LOX-1 or ox-LDL staining was observed in tissue sections of LOX-1 KO mice (Figure 3 J-L, PR). Runx2, COL X, and MMP-13 immunohistochemical staining in WT and LOX-1 KO mice showed results similar to those of LOX-1 and ox-LDL (Figure 4). In the articular cartilage of WT mice, we observed Runx2, COL X, and MMP-13 staining intensities at 12 and 18 months of age, compared to those at 6 months (Figure 4 A-C, G-I, M-O). Runx2, COL X and MMP-13 staining intensity also increased at 12 and 18 months of age, compared to that at 6 months in LOX-1 KO mice, but the intensity was lower than that of WT mice (Figure 4 D-F, J-L, P-R). The following percentages of LOX-1- and ox-LDL-positive cells were measured in WT mice: 6 months, 26.4±0.96 and 22.3±7.03, respectively; 12 months, 40.2±8.16 and 49.8±10.5, respectively; and 18 months, 61.6±7.91 and 59.9±8.49, respectively (Figure 5 A,B). In WT mice, the following percentages of cells positive for Runx2, COL X, and MMP-13 expression were: 6 months, 15.7±5.95, 15.1±5.48, and 7.96±4.41 respectively; 12 months, 35.6±5.60, 38.9±7.50, and 24.0±6.66, respectively; and 18 months: 60.6±7.25, 59.7±5.89, and 42.1±6.88, respectively (Figure 5 C-E). In LOX-1 KO mice, only a few cells were positive for Runx2, COL X, and MMP-13. In LOX-1 KO mice, the Runx2-, COL X-, and MMP-13-positive cell rates were as follows: 6 months, 10.8±3.76, 12.1±4.01, and 7.65±4.17, respectively; 12 months, 13.8±4.31, 16.3±7.50, and 13.0±4.93, respectively; 18 months, 19.2±3.71, 21.7±5.28, and 16.4±6.16, respectively (Figure 5 C-E). At 12 and 18 months, the Runx2-, COL X-, and MMP-13-positive cell rate was significantly lower in LOX-1 KO mice than in WT mice (Runx2, P<0.001, P<0.001; COL X, P<0.001, P<0.001; MMP-13, P=0.014, P<0.001; n=10), as shown in Figure 5 C-E.
Figure 3.
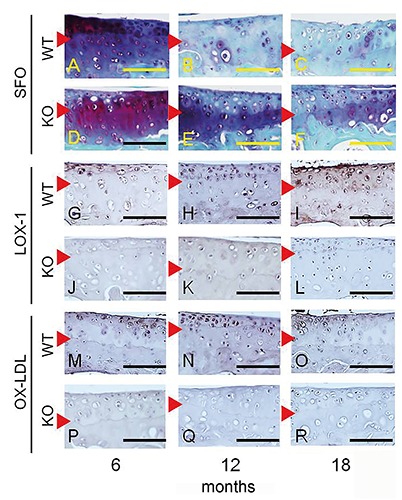
Immunostaining for LOX-1 and ox-LDL expression in the articular cartilage of the medial tibia. Corresponding SFO-stained images for each section (A-F). Positive staining for LOX-1 (G) and ox-LDL (M) in chondrocytes from 6-month-old mice. LOX-1 and ox-LDL staining in chondrocytes increased at 12 months (H, N) and 18 months (I, O) of age. LOX-1 and ox-LDL expression was negative in chondrocytes from LOX-1 KO mice (J-L and P-R). Red arrowhead: tide mark. Scale bar: 100 µm.
Figure 4.
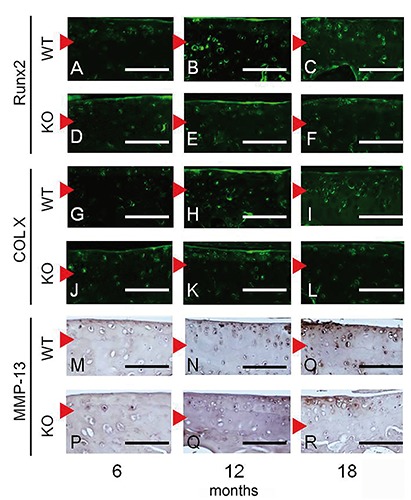
Immunostaining for Runx2, COL X, and MMP-13 expression in articular cartilage of the medial tibia. Runx2- and COL X-positive chondrocytes were observed in WT (A and G) and LOX-1 KO mice (D and J) at 6 months of age. Fluorescence staining for Runx2 and COL X increased in WT (B, C, H, and I) and LOX-1 KO mice (E, F, K, and L) at 12 and 18 months. Weak MMP-13 staining was observed in chondrocytes at 6 months in WT (M) and LOX-1 KO mice (P). MMP-13 expression increased in chondrocytes from WT (N and O) and LOX-1 KO mice (Q and R) at 12 and 18 months. Red arrowhead: tide mark. Scale bar: 100 µm.
Figure 5.
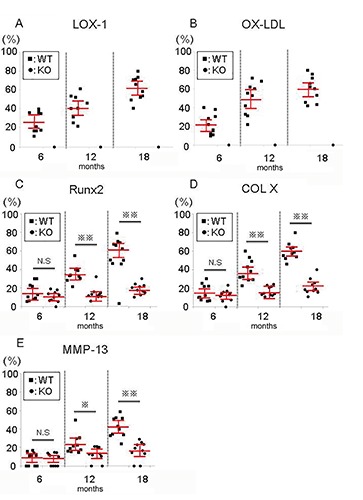
The positive-cell rate for LOX-1 (A), ox-LDL (B), Runx2 (C), COL X (D), or MMP-13 (E) from WT and LOX-1 KO mice at 6, 12, and 18 months of age. The positive-cell rate was calculated as the number of positive cells divided by the total number of chondrocytes in the articular cartilage of the medial tibia. All values are shown with black dots. The red bars show the average and 95% confidence interval. *P<0.05, **P<0.001.
Discussion
Previously, we showed that the LOX-1/ox-LDL system might be involved in age-related cartilage degeneration in vitro.13 However, this has not yet been determined in vivo. To address this, we observed age-related OA in LOX-1 KO mice. The loss of LOX-1 reduced the progression of age-related cartilage degeneration in the murine knee. Furthermore, LOX-1/ox-LDL expression increased with OA progression in WT mice. These findings suggested that LOX-1 is important for cartilage degeneration during age-related OA progression in vivo.
It is also possible that subtle differences can serve as risk factors for OA development during a long observational period. For example, small differences of body weight, atherosclerosis,25 or fat metabolism26 can influence OA development in KO mice, even if LOX-1 signaling may cause those differences. In the current study, the mean body weights of LOX-1 KO mice were slightly lower than those of WT mice although the differences were not statistically significant. However, this small difference in body weight may attenuate OA development in LOX-1 KO mice, considering that body weight has been implicated as a leading factor in knee OA.27,28
Chondrocyte senescence plays a major role during the development of age-related OA.29,30 Interestingly, telomere shortening was also detected in chondrocytes isolated from the articular cartilage of older adults.3 We previously reported that ox-LDL binding to LOX-1 promotes stress-induced premature senescence in chondrocytes, resulting in suppressed telomerase activity.13 Furthermore, oxidative changes are important for chondrocyte senescence in cartilage degeneration.32,33 The ox-LDL–LOX-1 interaction induces ROS production in BACs.12 In this study, we demonstrated that cartilage degeneration was significantly inhibited in LOX-1 KO mice and that the prevalence of severe OA was lower in LOX-1 KO mice at 12 and 18 months of age. Although LOX-1 and ox-LDL expression increased during the time course in chondrocytes from WT mice, their expression levels did not increase in chondrocytes of LOX-1 KO mice. These findings indicated that the LOX-1/ox-LDL system may be involved in chondrocyte senescence in vivo, which may contribute to the suppression of OA development; here, we provide evidence that LOX-1 deficiency inhibits age-related cartilage degeneration.
Recently, some studies have reported that endochondral ossification signals, which cause hypertrophy and apoptosis in chondrocytes, are involved in age-related OA development.34,35 Moreover, while hypertrophic chondrocyte differentiation was observed during endochondral bone formation in growth plates,36-38 a hypertrophic phenotype was also observed in an age-related OA mouse model and human OA chondrocytes.39,40 We previously reported that ox-LDL-LOX-1 binding induces ROS production12 and ROS was recently shown to induce chondrocyte hypertrophy.41 We previously reported in vitro that the LOX-1/ox-LDL system induces chondrocyte hypertrophy via Runx2 expression and that LOX-1 knockdown reduces Runx2 expression.14 In the current study, the expression of Runx2 (a transcription factor associated with chondrocyte hypertrophy) and COL X (a marker of hypertrophic chondrocytes) increased in articular chondrocytes from WT mice during OA development. However, in LOX-1 KO mice, Runx2 and COL X expression was reduced and OA progression was inhibited. These findings led us to hypothesize that the LOX-1/ox-LDL system is involved in age-related cartilage degeneration via chondrocyte hypertrophy in vivo. It is well established that cartilage degeneration involves various enzymes, including MMPs.42,43 MMP-13 is a major cartilage degradation enzyme that contributes to OA progression.44,45 In age-related OA, MMP-13 plays a crucial role in cartilage degeneration.46,47 In this study, MMP-13 was significantly up- regulated in the OA cartilage of WT mice compared with that in LOX-1 KO mice. These findings indicated that LOX-1/ox-LDL-induced Runx2 and COL X expression in hypertrophic chondrocytes may induce MMP-13 expression. In addition, MMP-13 expression increased later than Runx2 and COL X expression. Previous reports have suggested that Runx2 induces MMP-13 secretion.48 Although previous reports have demonstrated a direct activation of MMP-13 transcription by Runx2,49,50 a time lag between Runx2 and MMP-13 expression was observed in this study. Runx2 induced by the LOX-1/ox-LDL system may directly induce COL X expression and hypertrophic chondrocytes may induce MMP-13 expression during the progression of aging-related OA. These results indicate that the LOX-1/ox-LDL system may be involved in cartilage degeneration via MMP-13 expression in vivo.
There are some limitations to the current study. First, the mechanism connecting aging with the LOX-1/ox-LDL system was not fully elucidated. Although our results suggested that aging may enhance the function of the LOX-1/ox-LDL system, further studies are required to completely elucidate a direct correlation.
Another limitation of this study was the lack of insight concerning the origin of ox-LDL expression. Although the origin of ox-LDL expression was not clear, we considered 3 possible modes of association between ox-LDL and articular chondrocytes. First, ox-LDL in the systemic circulating blood might enter the joint fluid through the synovial vasculature. Second, LDL in the systemic circulating blood might enter the joint fluid and be oxidized in the inflamed joint cavity.51 Third, LDL penetrating the degraded cartilage matrix might become oxidized by ROS produced by chondrocytes. We previously reported that chondrocytes can produce ROS after mechanical stimulation, such as cyclic tensile stress,12 but it was difficult to confirm the timing of oxidation with the current study design. Because ox-LDL is a large protein of approximately 550 kDa,52 it is unlikely that ox-LDL penetrates the extracellular matrix of normal articular cartilage and binds to LOX-1 expressed on chondrocytes. However, degenerative OA cartilage is known to release high-molecular weight molecules such as chondroitin sulfate, keratin sulfate, and hyaluronic acid from the inner layer, suggesting the possibility that ox-LDL permeates into degenerated cartilage. Thus, it is possible that oxidation occurred because of chondrocyte senescence in WT and LOX-1 KO mice. The expression of ox-LDL was confirmed in WT mice, but not in LOX-1 KO mice, probably because ox-LDL was not internalized through the LOX-1 receptor. Further studies are required to determine the location of LDL oxidation and the exact mechanism by which ox-LDL regulates OA pathogenesis. These investigations could include the injection of soluble LOX-1 into the murine knee joint.53 Furthermore, generating additional data showing the effects of blocking LOX-1 signaling using small interfering RNA, antagonists of LOX-1 signaling, or neutralizing antibodies for OA induction would potentially strengthen the model proposed in this study.
Despite these limitations, we have demonstrated that the LOX-1/ox-LDL system is involved in age-related OA progression in vivo and that the pathology of OA is age-related, as in arteriosclerosis. It is well known that various lifestyle-related diseases such as hyperlipidemia, hypertension, and diabetes mellitus involve oxidative stress and that these conditions can cause arteriosclerosis via the LOX-1/ox-LDL system. Therefore, treating or otherwise addressing various lifestyle-related diseases that cause oxidative stress may help to prevent OA. We believe that this study provides valuable information for understanding the pathology of OA as a lifestyle-related disease.
In conclusion, we demonstrated the effects of LOX-1 deficiency on OA development in age-related OA in the murine knee. Our data showed that the LOX-1/ox-LDL system played a role in cartilage degeneration during age-related OA progression. Our data suggest that addressing or treating lifestyle-related diseases may help to prevent OA.
Acknowledgments
We thank Mr. Katsumi Okumoto (Department of Life Science, Kindai University Hospital) for providing technical assistance, which helped in performing the experiments.
References
- 1.Zhang PY, Xu X, Li XC. Cardiovascular diseases: oxidative damage and antioxidant protection. Eur Rev Med Pharmacol Sci 2014;18:3091-6. [PubMed] [Google Scholar]
- 2.Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997;386:73-7. [DOI] [PubMed] [Google Scholar]
- 3.Kume N, Murase T, Moriwaki H, Aoyama T, Sawamura T, Masaki T, et al. Inducible expression of lectin-like oxidized LDL receptor-1 in vascular endothelial cells. Circ Res 1998;83:322-7. [DOI] [PubMed] [Google Scholar]
- 4.Aoyama T, Fujiwara H, Masaki T, Sawamura T. Induction of lectin-like oxidized LDL receptor by oxidized LDL and lysophosphatidylcholine in cultured endothelial cells. J Mol Cell Cardiol 1999;31:2101-14. [DOI] [PubMed] [Google Scholar]
- 5.Hofnagel O, Luechtenborg B, Stolle K, Lorkowski S, Eschert H, Plenz G, et al. Proinflammatory cytokines regulate LOX-1 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2004;24:1789-95. [DOI] [PubMed] [Google Scholar]
- 6.Kume N, Moriwaki H, Kataoka H, Minami M, Murase T, Sawamura T, et al. Inducible expression of LOX-1, a novel receptor for oxidized LDL, in macrophages and vascular smooth muscle cells. Ann N Y Acad Sci 2000;902:323-7. [DOI] [PubMed] [Google Scholar]
- 7.Nakagawa T, Akagi M, Hoshikawa H, Chen M, Yasuda T, Mukai S, et al. Lectin-like oxidized low-density lipoprotein receptor 1 mediates leukocyte infiltration and articular cartilage destruction in rat zymosan-induced arthritis. Arthritis Rheum 2002;46:2486-94. [DOI] [PubMed] [Google Scholar]
- 8.Akagi M, Kanata S, Mori S, Itabe H, Sawamura T, Hamanishi C. Possible involvement of the oxidized low-density lipoprotein/lectin-like oxidized low-density lipoprotein receptor-1 system in pathogenesis and progression of human osteoarthritis. Osteoarthritis Cartilage 2007;15:281-90. [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa T, Yasuda T, Hoshikawa H, Shimizu M, Kakinuma T, Chen M, et al. LOX-1 expressed in cultured rat chondrocytes mediates oxidized LDL-induced cell death-possible role of dephosphorylation of Akt. Biochem Biophys Res Commun 2002;299:91-7. [DOI] [PubMed] [Google Scholar]
- 10.Kakinuma T, Yasuda T, Nakagawa T, Hiramitsu T, Akiyoshi M, Akagi M, et al. Lectin-like oxidized low-density lipoprotein receptor1 mediates matrix metalloproteinase 3 synthesis enhanced by oxidized low-density lipoprotein in rheumatoid arthritis cartilage. Arthritis Rheum 2004;50:3495-503. [DOI] [PubMed] [Google Scholar]
- 11.Simopoulou T, Malizos KN, Tsezou A. Lectin-like oxidized low density lipoprotein receptor 1 (LOX-1) expression in human articular chondrocytes. Clin Exp Rheumatol 2007;25:605-12. [PubMed] [Google Scholar]
- 12.Nishimura S, Akagi M, Yoshida K, Hayakawa S, Sawamura T, Munakata H, et al. Oxidized low-density lipoprotein (ox-LDL) binding to lectin-like ox-LDL receptor 1 (LOX-1) in cultured bovine articular chondrocytes increases production of intracellular reactive oxygen species (ROS) resulting in the activation of NF-κB. Osteoarthritis Cartilage 2004;12:568-76. [DOI] [PubMed] [Google Scholar]
- 13.Zushi S, Akagi M, Kishimoto H, Teramura T, Sawamura T, Hamanishi C. Induction of bovine articular chondrocyte senescence with oxidized low-density lipoprotein through lectin-like oxidized low-density lipoprotein receptor 1. Arthritis Rheum 2009;60:3007-16. [DOI] [PubMed] [Google Scholar]
- 14.Kishimoto H, Akagi M, Zushi S, Teramura T, Onodera Y, Sawamura T, et al. Induction of hypertrophic chondrocyte-like phenotypes by oxidized LDL in cultured bovine articular chondrocytes through increase in oxidative stress. Osteoarthritis Cartilage 2010;18:1284-90. [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto K, Mori S, Oda Y, Nakano A, Sawamura T, Akagi M. Lectin-like oxidized low density lipoprotein receptor 1-deficient mice show resistance to instability-induced osteoarthritis. Scand J Rheumato. 2016;45:412-22. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Jordan JM. Epidemiology of osteoarthritis. Clin Geriatr Med. 2010;26:355-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bierma-Zeinstra SM, Koes BW. Risk factors and prognostic factors of hip and knee osteoarthritis. Nat Clin Pract Rheumatol 2007;3:78-85. [DOI] [PubMed] [Google Scholar]
- 18.Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage 2009;17:971-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shane Anderson A, Loeser RF. Why is osteoarthritis an age-related disease?. Best Pract Res Clin Rheumatol 2010;24:15-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price JS, Waters JG, Darrah C, Pennington C, Edwards DR, Donell ST, et al. The role of chondrocyte senescence in osteoarthritis. Aging Cell 2002;1:57-65. [DOI] [PubMed] [Google Scholar]
- 21.Martin JA, Klingelhutz AJ, Moussavi-Harami F, Buckwalter JA. Effects of oxidative damage and telomerase activity on human articular cartilage chondrocyte senescence. J Gerontol A Biol Sci Med Sci 2004;59:324-37. [DOI] [PubMed] [Google Scholar]
- 22.Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res 2007;100:1634-42. [DOI] [PubMed] [Google Scholar]
- 23.Musumeci G, Castrogiovanni P, Mazzone V, Szychlinska MA, Castorina S, Loreto C. Histochemistry as a unique approach for investigating normal and osteoarthritic cartilage. Eur J Histochem 2014;58:2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 2010;18:S17-23. [DOI] [PubMed] [Google Scholar]
- 25.Zhuo Q, Yang W, Chen J, Wang Y. Metabolic syndrome meets osteoarthritis. Nat Rev Rheumatol 2012;8:729-37. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Hunter D, Xu J, Ding C. Metabolic triggered inflammation in osteoarthritis. Osteoarthritis Cartilage 2015;23:22-30. [DOI] [PubMed] [Google Scholar]
- 27.Guilak F. Biomechanical factors in osteoarthritis. Best Pract Res Clin Rheumatol 2011;25:815-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu H, Zhang X, Wang H, Zhang Y, Shi Y, Zhang X. Continuous cyclic mechanical tension increases ank expression in endplate chondrocytes through the TGF-β1 and p38 pathway. Eur J Histochem 2013;57:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hui W, Young DA, Rowan AD, Xu X, Cawston TE, Proctor CJ. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann Rheum Dis 2016;75:449-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shane Anderson A, Loeser RF. Why is osteoarthritis an age-related disease? Best Pract Res Clin Rheumatol 2010;24:15-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin JA, Buckwalter JA. Telomere erosion and senescence in human articular cartilage chondrocytes. J Gerontol A Biol Sci Med Sci 2001;56:B172-9. [DOI] [PubMed] [Google Scholar]
- 32.Hui W, Young DA, Rowan AD, Xu X, Cawston TE, Proctor CJ. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann Rheum Dis 2016;75:449-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta 2016;1862:576-91. [DOI] [PubMed] [Google Scholar]
- 34.Baugé C, Girard N, Lhuissier E, Bazille C, Boumediene K. Regulation and role of TGFβ signaling pathway in aging and osteoarthritis joints. Aging Dis 2013;5:394-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujita N, Matsushita T, Ishida K, Kubo S, Matsumoto T, Takayama K, et al. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J Orthop Res 2011;29:511-5. [DOI] [PubMed] [Google Scholar]
- 36.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 1999;5:623-8. [DOI] [PubMed] [Google Scholar]
- 37.Buckwalter JA, Glimcher MJ, Cooper RR, Recker R. Bone biology. II: formation, form, modeling, remodeling, and regulation of cell function. Instr Course Lect 1996;45:387-99. [PubMed] [Google Scholar]
- 38.Xu HG, Zhang W, Zheng Q, Yu YF, Deng LF, Wang H, et al. Investigating conversion of endplate chondrocytes induced by intermittent cyclic mechanical unconfined compression in three-dimensional cultures. Eur J Histochem 2014;58:2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ailixiding M, Aibibula Z, Iwata M, Piao J, Hara Y, Koga D, et al. Pivotal role of Sirt6 in the crosstalk among ageing, metabolic syndrome and osteoarthritis. Biochem Biophys Res Commun. 2015;466:319-26. [DOI] [PubMed] [Google Scholar]
- 40.Aigner T, Reichenberger E, Bertling W, Kirsch T, Stöss H, von der Mark K. Type X collagen expression in osteoarthritic and rheumatoid articular cartilage. Virchows Arch B Cell Pathol Incl Mol Pathol 1993;63:205-11. [DOI] [PubMed] [Google Scholar]
- 41.Morita K, Miyamoto T, Fujita N, Kubota Y, Ito K, Takubo K, et al. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J Exp Med 2007;204:1613-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cawston TE, Wilson AJ. Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol 2006;20:983-1002. [DOI] [PubMed] [Google Scholar]
- 43.Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci 2006;11:529-43. [DOI] [PubMed] [Google Scholar]
- 44.van den Berg WB. Osteoarthritis year 2010 in review: pathomechanisms. Osteoarthritis Cartilage 2011;19:338-41. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, et al. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest 1996;97:761-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blaney Davidson EN, Remst DF, Vitters EL, van Beuningen HM, Blom AB, Goumans MJ, et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J Immunol 2009;182:7937-45. [DOI] [PubMed] [Google Scholar]
- 47.Weng T, Xie Y, Yi L, Huang J, Luo F, Du X, et al. Loss of Vhl in cartilage accelerated the progression of age-associated and surgically induced murine osteoarthritis. Osteoarthritis Cartilage 2014;22:1197-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen CG, Thuillier D, Chin EN, Alliston T. Chondrocyte-intrinsic Smad3 represses Runx2-inducible MMP-13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum 2012;64:3278-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porte D, Tuckermann J, Becker M, Baumann B, Teurich S, Higgins T, et al. Both AP-1 and Cbfa1-like factors are required for the induction of interstitial collagenase by parathyroid hormone. Oncogene 1999;18:667-78. [DOI] [PubMed] [Google Scholar]
- 50.Jiménez MJ, Balbín M, López JM, Alvarez J, Komori T, López-Otín C. Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol Cell Biol 1999;19:4431-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.James MJ, van Reyk D, Rye KA, Dean RT, Cleland LG, Barter PJ, et al. Low density lipoprotein of synovial fluid in inflammatory joint disease is mildly oxidized. Lipids 1998;33:1115-21. [DOI] [PubMed] [Google Scholar]
- 52.Fong LG, Parthasarathy S, Witztum JL, Steinberg D. Nonenzymatic oxidative cleavage of peptide bonds in apoprotein B-100. J Lipid Res. 1987;28:1466-77. [PubMed] [Google Scholar]
- 53.Ishikawa M, Ito H, Akiyoshi M, Kume N, Yoshitomi H, Mitsuoka H, et al. Lectin-like oxidized low-density lipoprotein receptor 1 signal is a potent biomarker and therapeutic target for human rheumatoid arthritis. Arthritis Rheum 2012;64:1024-34. [DOI] [PubMed] [Google Scholar]