

Abstract
We report a non-destructive biochemical technique, termed “Glyco-seek”, for analysis of O-GlcNAcylated proteins. Glyco-seek combines chemoenzymatic labeling, proximity ligation, and quantitative polymerase chain reaction to detect O-GlcNAcylated proteins with ultrahigh sensitivity. Our glycan-specific assay can be paired with traditional proximity ligation assays to simultaneously determine the change in total protein levels. We show that Glyco-seek detects attomoles of glycoproteins of interest from cell lysates, with sensitivity several orders of magnitude higher than that of current techniques. We used the method to directly assay the O-GlcNAcylation status of a low-abundance transcription factor from cell lysates without need for isolation or enrichment.
Protein O-GlcNAcylation is an intracellular post-translational modification thought to regulate many cellular processes. O-GlcNAc is installed by a single enzyme, O-GlcNAc transferase (OGT), and removed by O-GlcNAc hydrolase (OGA).1 The modification is widespread—more than 3000 O-GlcNAc sites have been discovered in eukaryotic proteomes—and is thought to be a signaling modification more similar to phosphorylation than to canonical forms of glycosylation.2
Many human pathologies are marked by aberrant O-GlcNAcylation. Dysregulated O-GlcNAcylation of phospho-fructokinase, glycogen synthase kinase 3β, and CaMKII has been associated with breast cancer,3 diabetes,4 and cardiovascular diseases,5 respectively. Hypo-glycosylation of the tau protein leads to an Alzheimer’s disease-like state in a mouse model.6 O-GlcNAc also regulates pluripotency and reprogramming in stem cells through the modification of numerous transcription factors.7 Specifically, the O-GlcNAcylation of master pluripotency regulator OCT4 was shown to increase the ability of hESCs to maintain pluripotency.8 Accordingly, there is much interest in profiling O-GlcNAcylation states of proteins and their dynamics.
Immunoblotting and mass spectrometry (MS) are the primary methods used to interrogate protein O-GlcNAcylation. To detect modification of a specific protein, Western blots are typically paired with a prior precipitation step with either target-specific antibodies or anti-O-GlcNAc affinity reagents. This enrichment process can be labor-intensive and lead to sample loss due to the relatively low affinity of lectins and the notoriously poor affinity and specificity of anti-O-GlcNAc antibodies.9
The elegant and generalizable “Click-it” method, first reported by Hsieh-Wilson and co-workers, has emerged as an alternative to O-GlcNAc antibodies and lectins, and has achieved widespread use.10 The two-step “Click-it” method involves treatment with a permissive galactosyltransferase that appends an azide-functionalized monosaccharide (N-azidoacetylgalactosamine, GalNAz) onto the O-GlcNAc residue (Figure 1a).
Figure 1.

Glyco-seek enables the ultrasensitive detection of O-GlcNAcylation with polymerase chain reaction (PCR). (a) O-GlcNAc residues on proteins are tagged via chemoenzymatic transfer of GalNAz and subsequent ligation with alkynyl biotin via click chemistry. (b) Biotinylated glycoproteins are incubated with antibody–DNA conjugates targeted to either biotin or the protein of interest. Hybridization with a short strand of DNA that is complementary to both single-stranded DNAs creates a double-stranded segment which is joined with DNA ligase. The resultant duplex DNA is then detected by standard qPCR methods.
The azide is then reacted with an alkyne–biotin probe, enabling enrichment with streptavidin or detection on a Western blot. This commercially available technology is now widely used and has been employed in several important studies.11 However, as all O-GlcNAcylated proteins in a cell lysate are simultaneously biotinylated using this method, the detection of the O-GlcNAcylation state of a particular protein requires its further purification for subsequent analysis—a time-consuming endeavor that also results in sample loss.
In typical MS experiments, the glycosidic linkage between O-GlcNAc and its underlying Ser or Thr residue is vulnerable to fragmention and easily lost.12 Although softer ionization techniques, such as electron-transfer dissociation, can partially mitigate cleavage of glycosidic linkages,13 in practice the technique is limited to a handful of laboratories with the appropriate expertise and instrumentation. Also, MS methods inherently detect the most abundant species that are present, while suppressing signals from low-abundance species such as transcription factors, many of which are thought to be regulated by O-GlcNAc.7 Accordingly, the generation of high-quality MS data requires a large amount of sample, followed by rigorous and labor-intensive enrichment to ensure adequate representation of low-abundance glycoproteins. These requirements frustrate the analysis of biologically important samples that may be too precious (e.g., primary cell lines, clinical samples) or challenging and expensive (e.g., stem cells) to culture on large scale. Thus, the development of an operationally simple, sensitive, and quantitative platform for determining the O-GlcNAcylation states of proteins of interest remains an unmet need.
To this end, we sought to create a proximity ligation assay (PLA) for the detection of protein O-GlcNAcylation (Figure 1b). PLA leverages the amplification power of the polymerase chain reaction (PCR) by linking the presence of the target analytes to the production of a PCR amplicon which can be detected with extreme sensitivity.14 Briefly, a PCR amplicon is split into two halves of ssDNA which are then covalently attached to two antibodies, one binding to the protein of interest, the other to biotinylated O-GlcNAc. Next, a short (20 bp) bridge oligo hybridizes with the two DNA halves, enabling DNA ligase to join them into a complete PCR amplicon. Next, the quantity of the amplicon can be detected by qPCR. Several groups have artfully employed PLA methods to image proteins and their modifications,15 establishing precedent for our adaptation to a chemoenzymatic platform with qPCR readout.
To establish proof-of-principle, we first implemented Glyco-seek using purified alpha-Crystallin, a well-known O-GlcNAcylated protein that is commercially available.16 We first biotinylated O-GlcNAc residues on alpha-Crystallin with the commercial Click-It kit. We then synthesized two antibody–DNA conjugates using sulfo-SMCC cross-linking to thiolated oligonucleotides.17 The single-stranded DNA (ssDNA) sequences (~50 bp) used were designed to minimize secondary structure and self-dimerization, as previously reported18 (Set 4, Table S1). The anti-biotin antibody was linked to the 5′ end of the PCR amplicon, and the anti-alpha-Crystallin antibody was conjugated to 3′ end. This latter ssDNA strand was also 5′-phosphorylated to facilitate ligation. The conjugation stoichiometry for both constructs was 2.5 DNA/antibody, as determined by UV–vis analysis in comparison to a standard curve. Conjugation was also confirmed by observing a shift in the apparent molecular weights of both proteins by SDS-PAGE (Figure S1).
We incubated the two antibody–DNA conjugates at low concentrations (500 pM) with 2 μL of alpha-Crystallin solution in PBS in a dilution series. In one sample, we omitted the galactosyltransferase from the Click-It step. This sample should lack biotinylation and therefore produce no significant signal in the Glyco-seek assay. The samples were diluted in ligation buffer and treated with the bridge oligo and DNA ligase, and the ligated oligonucleotides were then pre-amplified by 13 rounds of PCR.18 The sample was then analyzed by qPCR. We observed a strong dose-dependent signal, suggesting that the presence of O-GlcNAcylated alpha-Crystallin was driving ligation. Importantly, the signal was not observed with the sample lacking biotinylation (Figure S2a).
To confirm that the qPCR signal was dependent on O-GlcNAcylation of alpha-Crystallin, we pretreated the protein with two different doses of OGA to remove O-GlcNAc residues. A control sample was treated with heat-killed OGA. The samples were analyzed as above. Samples treated with a high dose of OGA produced a significantly lower signal than those treated with lower amounts OGA or with heat-killed OGA, confirming that the qPCR readout reflects O-GlcNAcylation status (Figure S2b).
Many studies of protein O-GlcNAcylation focus on changes in the extent of glycosylation during dynamic cellular processes. We sought to develop a Glyco-seek method for simultaneously probing a protein’s abundance as well as its extent of O-GlcNAcylation, both of which can change in response to cell stimuli. We accomplished this by performing two PLA assays in parallel: one that reports on O-GlcNAcylation as above and another that reports on protein abundance. We prepared two sets of alpha-Crystallin samples. One was treated with heat-killed OGA and the other with the active enzyme to remove O-GlcNAc residues. We then subjected both samples to two different proximity ligation analyses. First, we used anti-biotin and anti-protein proximity probes. The signal generated from this set reports on the abundance of the glycosylated protein. Next, we used the standard PLA method for protein detection to quantitate the absolute level of alpha-Crystallin in the sample.14 Briefly, we split an anti-alpha-Crystallin polyclonal antibody into two pools that were conjugated to the two halves of the PCR amplicon (Set 4, Table S1). The combined conjugates provide antibodies that can bind simultaneously to distinct epitopes on the protein surface in a glycosylation-independent manner. We confirmed this by comparing the signals derived from OGA-treated and heat-killed OGA-treated alpha-Crystallin, which were indistinguishable (Figure S2c).
A significant challenge for other analytical techniques is suppressing the background signal derived from protein-rich samples such as serum or cell lysate.19 The dual recognition mechanism of proximity ligation assays should yield superior signal-to-noise, as a single off-target binding event is not sufficient to generate a full amplicon.14 To test this, we serially diluted alpha-Crystallin into serum or Jurkat cell lysate. The alpha-Crystallin was pretreated as before, with either active or heat-killed OGA. A third sample was diluted into lysate from Jurkat cells whose cell surface glycoproteins had been globally biotinylated via metabolic labeling with N-azidoacetylmannosamine and copper-free click chemistry with a commonly used reactive cyclooctyne conjugate, DBCO–biotin.20 This sample was designed to emulate an environment where there are many off-target biotinylated glycoproteins. The samples were analyzed with both biotin/alpha-Crystallin proximity probes to detect protein glycosylation (Figure 2, bottom panels) and alpha-Crystallin/alpha-Crystallin probes to detect protein levels (Figure 2, top panels). We observed an O-GlcNAc-dependent signal in the biotin/alpha-Crystallin assays and an O-GlcNAc-independent signal in the protein-detecting assay, with little to no interference from various matrices (Figure 2a–c). Notably, even lysates with a large excess of biotinylated irrelevant glycoproteins caused only modest interference at high dilution factors when detecting O-GlcNAcylated alpha-Crystallin, a protein with a low-occupancy glycosite.16 These results demonstrate that Glyco-seek detects both glycosylated and total protein levels in a variety of complex environments and in a highly specific manner.
Figure 2.

Glyco-seek simultaneously detects protein abundance and glycosylation state in complex environments. Alpha-Crystallin was chemoenzymatically biotinylated and then diluted into either serum (a), Jurkat cell lysate (b), or non-specifically biotinylated Jurkat cell lysate (c). These samples were then analyzed for total protein level (row 1) or protein glycosylation levels (row 2). Samples were pretreated with either active OGA or heat-killed enzyme as a negative control. The X-axis displays the concentration of the protein in the sample, while the Y-axis is the Ct value in comparison to a blank. All data points are from technical triplicates, and the error bars represent the standard deviation.
Since many O-GlcNAcylated proteins are present at low abundance and are often sub-stoichiometrically modified, sensitivity of analytical techniques is of great importance.19 As a PCR-based technique, Glyco-seek exploits exponential amplification of nucleic acids to achieve a high level of sensitivity. We performed a head-to-head comparison between Glyco-seek and a Western blot probed with the same anti-biotin antibody. Glyco-seek’s sensitivity exceeded that of the Western blot by at least 3 orders of magnitude (Figures 3 and S3). Glyco-seek analysis of samples with as low as 0.5 amol of glycosylated alpha-Crystallin still generated signal above the blank, highlighting this technique’s great analytical sensitivity.
Figure 3.
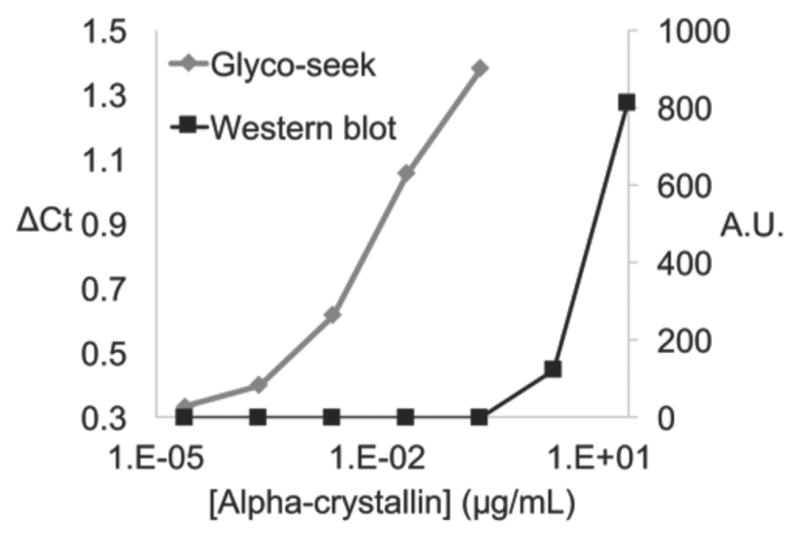
Glyco-seek is several orders of magnitude more sensitive than a comparable Western blot. Alpha-Crystallin with biotinylated O-GlcNAc residues was diluted in PBS and then analyzed by either Western blot or Glyco-seek. Densitometry was used to quantify the bands observed from the Western blot. The right Y-axis displays arbitrary intensity units.
We next sought to detect O-GlcNAc on endogenous proteins in cell lysates using Glyco-seek. As a proof-of-principle, we created a Glyco-seek assay to detect the glycosylation status of c-Rel protein. C-Rel is a member of the nuclear factor κB (NFκB) transcription factor family.20 Altered O-GlcNAcylation of c-Rel has been shown to modify its DNA binding and transactivation properties.20 Increased c-Rel glycosylation can be induced by addition of the OGA inhibitors into the culture media.20 We treated Jurkat cells with the OGA inhibitor thiamet G (TMG) and labeled lysates produced from these cells with the Click-It kit. Increased glycosylation of c-Rel was confirmed by immunoprecipitation followed by Western blot analysis (Figure S4). The labeled lysate was subjected to two sets of analysis. First, to detect the abundance of the protein, a pair of anti-c-Rel antibodies (Set 2, Table S1) was used as in a traditional PLA. Second, anti-biotin and anti-c-Rel proximity probes were used to detect the O-GlcNAcylation status of c-Rel. These separate analyses revealed a significant increase of O-GlcNAcylation on c-Rel while the c-Rel protein level remained unpertubed by TMG treatment, as expected20 (Figure 4). Together, these results imply that TMG treatment increases the O-GlcNAc modification stoichiometry on c-Rel rather than elevating c-Rel protein expression20 and that Glyco-seek faithfully recapitulates these changes while using minute amounts of samples.
Figure 4.

Glyco-seek analysis of endogenous O-GlcNAcylated c-Rel. Jurkat cells were treated with the OGA inhibitor thiamet G (TMG) to increase the amount of O-GlcNAcylation, or vehicle (DMSO) as a control. Another sample was immunodepleted of c-Rel. After chemoenzymatic biotinylation of O-GlcNAc residues, all samples were analyzed for protein and glycosylation levels.
A final concern we sought to address is off-target specificity. Poorly characterized antibodies can bind unrelated proteins and lead to false changes in signal.21 For example, TMG treatment globally elevates O-GlcNAcylation. An increase in signal from Glyco-seek could be the result of off-target ligation to an unrelated glycoprotein. To rule out this possibility, we removed c-Rel from Jurkat lysates via immunodepletion. In this experiment, we added anti-c-Rel polyclonal antibody to lysates from cells cultured with TMG. The sample was then incubated with protein G resin to remove c-Rel. The c-Rel-depleted supernatant was subjected to chemoenzymatic biotinylation and analysis with Glyco-seek, which showed a marked decrease in signal (Figure 4). We also performed immunodepletion with isotype control antibody to verify that this decrease of signal indeed results from c-Rel depletion (Figure S5). These experiments provide strong evidence that the O-GlcNAc-dependent signal is specific to c-Rel and does not result from off-target proteins that may also have undergone elevation in O-GlcNAcylation.
In summary, we developed a technique for sensitive and specific detection of protein O-GlcNAcylation in complex samples including serum and whole-cell lysates. By converting the glycosylation status of a protein into a detectable PCR amplicon, we attained sensitivity several orders of magnitude beyond that of more conventional Western blot methods.
The non-destructive nature of this assay stands in contrast to MS assays that rely on protein digestion and fragmentation. The ultrasensitivity and low sample consumption (2 μL) facilitate the study of precious samples and low-abundance glycoproteins. The widespread availability of qPCR empowers laboratories to monitor protein O-GlcNAcylation in their biological systems of interest.
We envision Glyco-seek as complementary to other methods that are better suited for de novo discovery of previously unknown glycoproteins, such as MS-driven glycoproteomics platforms.22 Glyco-seek does not lend itself particularly well to de novo discovery of previously unknown glycoproteins, as MS can accomplish. Rather, Glyco-seek is a directed approach to glycoprotein analysis, analogous to other popular large-scale analytical platforms, such as DNA, antibody, and protein microarrays, where libraries of known targets are analyzed for changes in response to biological stimuli. We consider Glyco-seek to be complementary to these approaches, offering a sensitive and convenient analysis of targeted candidate glycoproteins.
Glyco-seek also has the potential to be a highly multiplexed tool via DNA-barcoding. This functionality could benefit from well-established high-throughput methods for constructing libraries of antibody–DNA conjugates.23 Besides, other forms of glycosylation are also amenable to profiling by Glyco-seek. All that is required is a reagent that binds to the target glycan, and which can be conjugated to ssDNA. We envision that oligonucleotide aptamers, lectins, and many emerging antibody variants will find synergy with the Glyco-seek method.
Supplementary Material
Acknowledgments
We thank Michelle Chang and Ming Hammond for technical assistance. This work was funded by NIH grant R21 DK108781A to C.R.B.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b03861.
Experimental procedures and characterization data, Table S1, and Figures S1–S5 (PDF)
References
- 1.Love DC, Hanover JA. Sci Signaling. 2005;2005(312):re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Gucek M, Hart GW. Proc Natl Acad Sci USA. 2008;105:13793. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, III, Peters EC, Driggers EM, Hsieh-Wilson LC. Science. 2012;337:975. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parker G, Taylor R, Jones D, McClain D. J Biol Chem. 2004;279:20636. doi: 10.1074/jbc.M312139200. [DOI] [PubMed] [Google Scholar]
- 5.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Nature. 2013;502:372. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Nat Chem Biol. 2012;8:393. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- 7.Özcan S, Andrali SS, Cantrell JE. Biochim Biophys Acta, Gene Regul Mech. 2010;1799:353. doi: 10.1016/j.bbagrm.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang H, Kim TW, Yoon S, Choi SY, Kang TW, Kim SY, Kwon YW, Cho EJ, Youn HD. Cell Stem Cell. 2012;11:62. doi: 10.1016/j.stem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ma J, Hart GW. Clin Prot. 2014;11:1. doi: 10.1186/1559-0275-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Isono T. PLoS One. 2011;6:18959. doi: 10.1371/journal.pone.0018959. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mond JJ, Lees A, Snapper CM. Annu Rev Immunol. 1995;13:655. doi: 10.1146/annurev.iy.13.040195.003255. [DOI] [PubMed] [Google Scholar]
- 10.(a) Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Qasba PK, Hsieh-Wilson LC. J Am Chem Soc. 2003;125:16162. doi: 10.1021/ja038545r. [DOI] [PubMed] [Google Scholar]; (b) Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, Hsieh-Wilson LC. J Am Chem Soc. 2008;130:11576. doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dehennaut V, Slomianny MC, Page A, Vercoutter-Edouart AS, Jessus C, Michalski JC, Vilain JP, Bodart JF, Lefebvre T. Mol Cell Proteomics. 2008;7:2229. doi: 10.1074/mcp.M700494-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Haynes PA, Aebersold R. Anal Chem. 2000;72:5402. doi: 10.1021/ac000512w. [DOI] [PubMed] [Google Scholar]
- 13.Reinhold VN, Reinhold BB, Costello CE. Anal Chem. 1995;67:1772. doi: 10.1021/ac00107a005. [DOI] [PubMed] [Google Scholar]
- 14.Gullberg M, Gústafsdóttir SM, Schallmeiner E, Jarvius J, Bjarnegård M, Betsholtz C, Landegren U, Fredriksson S. Proc Natl Acad Sci USA. 2004;101:8420. doi: 10.1073/pnas.0400552101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Tong QH, Tao T, Xie LQ, Lu H. J Biosens Bioelectron. 2016;80:385. doi: 10.1016/j.bios.2016.02.006. [DOI] [PubMed] [Google Scholar]; (b) Pinto R, Carvalho AS, Conze T, et al. J Cell Mol Med. 2012;16:1474. doi: 10.1111/j.1582-4934.2011.01436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chalkley RJ, Burlingame AL. J Am Soc Mass Spectrom. 2001;12:1106. doi: 10.1016/s1044-0305(01)00295-1. [DOI] [PubMed] [Google Scholar]
- 17.Duval M, Posner MR, Cavacini LA. J Virology. 2008;82:4671. doi: 10.1128/JVI.02499-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai CT, Robinson PV, Spencer CA, Bertozzi CR. ACS Cent Sci. 2016;2:139. doi: 10.1021/acscentsci.5b00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domon B, Aebersold R. Science. 2006;312:212. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 20.Robinson PV, de Almeida-Escobedo G, de Groot AE, McKechnie JL, Bertozzi CR. J Am Chem Soc. 2015;137:10452. doi: 10.1021/jacs.5b04279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bucur O, Pennarun B, Stancu AL, Nadler M, Muraru MS, Bertomeu T, Khosravi-Far R. Apoptosis. 2013;18:1154. doi: 10.1007/s10495-013-0880-0. [DOI] [PubMed] [Google Scholar]
- 22.Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, Bertozzi CR. Nat Methods. 2015;12:561. doi: 10.1038/nmeth.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fredriksson S, Dixon W, Ji H, Koong AC, Mindrinos M, Davis RW. Nat Methods. 2007;4:327. doi: 10.1038/nmeth1020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.