

Abstract
Purpose of Review
Craniofacial disorders are among the most common human birth defects and present an enormous health care and social burden. The development of animal models has been instrumental to investigate fundamental questions in craniofacial biology and this knowledge is critical to understand the etiology and pathogenesis of these disorders.
Recent findings
The vast majority of craniofacial disorders arise from abnormal development of the neural crest, a multipotent and migratory cell population. Therefore, defining the pathogenesis of these conditions starts with a deep understanding of the mechanisms that preside over neural crest formation and its role in craniofacial development.
Summary
This review discusses several studies using Xenopus embryos to model human craniofacial conditions, and emphasizes the strength of this system to inform important biological processes as they relate to human craniofacial development and disease.
Keywords: Xenopus, Neural crest, Pharyngeal arches, Craniofacial disorders
Introduction
Craniofacial disorders are among the most common birth defects, second only to congenital heart diseases. They represent a very diverse and complex group of anomalies, with their prevalence varying greatly across geographic areas and ethnic groups (Table 1, [1]). The most common disorders are orofacial clefts, which occur when the lip and/or the roof of the mouth do not form properly, and craniosynostosis, which corresponds to a premature fusion of the skull bones. Craniofacial anomalies represent an enormous medical and social burden. Children with craniofacial defects face a greater risk for physical, learning, emotional and developmental challenges, and their integration in the society is often difficult. Therefore, there is a tremendous need to develop appropriate experimental model systems to gain insights into the cellular pathogenesis of these devastating conditions. This fundamental knowledge is ultimately critical to develop assays for early detection of these craniofacial anomalies, and devise new therapeutic strategies to minimize defects at birth.
Table 1. Most common craniofacial disorders and their incidence.
From the Report of WHO Meetings on International Collaborative Research on Craniofacial Anomalies (Modified from [1]).
| Craniofacial disorders | Prevalance per 10,000 live births |
|---|---|
| Cleft lip +/- cleft palate | |
| Caucasian | 10 |
| Japanese | 20 |
| Native American | 36 |
| African American | 3 |
| Cleft palate only | 5 |
| Craniosynostosis | 3 |
| Otomandibular dysplasias | 1.2 |
| Mandibulofacial dysostosis | 0.2 |
| CHARGE syndrome | 1 |
| Holoprosencephaly | 1.2 |
| Stickler syndrome | 1 |
| Fetal alcohol syndrome | 2 |
For several decades, the basic understanding of most human craniofacial anomalies was primarily rooted in patient studies that were largely descriptive. Recent advances in next-generation sequencing technologies have transformed the current understanding of the genetic basis of these conditions. This knowledge has been also greatly expanded at the cellular and molecular levels by studies using animal models, including Xenopus. The South African clawed-frog Xenopus laevis has long been used to investigate fundamental questions in developmental biology. It is an especially powerful system to explore the molecular mechanisms underlying gene function. The large size of the Xenopus embryo and its development outside of the body of the female makes it an ideal system to alter gene expression by microinjection of mRNA or morpholino antisense oligonucleotides. The first cleavage divides the Xenopus embryo along the left/right axis, therefore injection of one cell at the 2-cell stage, Nieuwkoop and Faber (NF) stage 2 [2], can affect gene function in only one side of the embryo, while the other side serves as an internal control. Moreover, the embryo has an extremely reproducible pattern of cleavage, allowing for the manipulation of gene expression in restricted lineages of the developing embryo by injection at the 16-cell stage (NF stage 5) to 32-cell stage (NF stage 6) [3], [4]. The Xenopus animal cap explant assay has especially proven to be a remarkably versatile system to assess the ability of exogenously expressed transcription factors or signaling molecules to influence patterns of gene expression. Moreover, TALEN and CRISPR/Cas9 technologies have been successfully used to introduce targeted modifications in Xenopus genome, specifically in Xenopus tropicalis, which is genetically a more tractable system and is complementary to Xenopus laevis [5], [6]. Xenopus species as model organisms also benefit from many well-developed resources (collections of biological, expression and genomic data) available via “Xenbase” (wwww.xenbase.org, RRID: SCR 003280). It is also a very powerful alternative to the more costly and less tractable mouse model, and the evolutionarily more distant zebrafish.
Taking advantage of all these attributes, Xenopus has proven to be an especially effective model system to study craniofacial development and morphogenesis. In this review, we describe the basic principles underlying craniofacial development and the role of the neural crest in this process, and summarize the studies that have used Xenopus to model human craniofacial disorders.
A conserved blueprint of craniofacial development
The craniofacial skeleton has been an intriguing area of research for centuries, from the earliest comparative drawings of animals to the current comprehensive studies on the anatomical and molecular pathways involved in head development. The evolutionary changes in craniofacial structures seen in vertebrates mirror their adaptive functional changes, and it is the unique features of the head that distinguish vertebrates from other chordates [7]. Comparative analyses in multiple animal models indicate that the basic principles underlying craniofacial development are well conserved [8], [9]. Central to vertebrate craniofacial morphogenesis is a unique population of progenitor cells known as the neural crest (NC), which has been one of the driving forces of vertebrate evolution [10]. In this section, we present an overview of the mechanisms underlying NC formation and its contribution to craniofacial structures.
Neural crest development
The NC is a multipotent and migratory cell population, with the extraordinary ability to give rise to a broad array of cell types including connective tissue, bones, smooth muscles, neurons and pigment cells, thus contributing to multiple organ systems. Amphibian species were among the first vertebrates examined to analyze the contribution of the NC to the development of the skull [11]. Subsequent studies in a number of organisms including birds, mouse and fish demonstrated that the general pattern of skull development was very well conserved across species, with only a few variations in the sequence of bone formation and ossification [12], [13].
Formation of the NC is a multi-step process regulated by multiple signaling events that are facilitated by molecules of bone morphogenetic proteins (Bmp), Wnt and fibroblast growth factors (Fgf) families. During gastrulation, NC progenitors are induced in the embryonic ectoderm at the border of the neural plate (future central nervous system) and the non-neural ectoderm (future epidermis), in a region known as the neural plate border (NPB). During neurulation, NC progenitors end up at the most dorsal aspect of the neural tube, and upon neural tube closure NC cells (NCCs) separate from neighboring cells and migrate into the periphery (Fig 1A). As they assume their fate, NCCs start to express a unique repertoire of genes, which define their new molecular identity, distinguishing them from adjacent cell populations (reviewed in [14], [15]).
Figure 1. Neural crest development and its craniofacial derivatives.
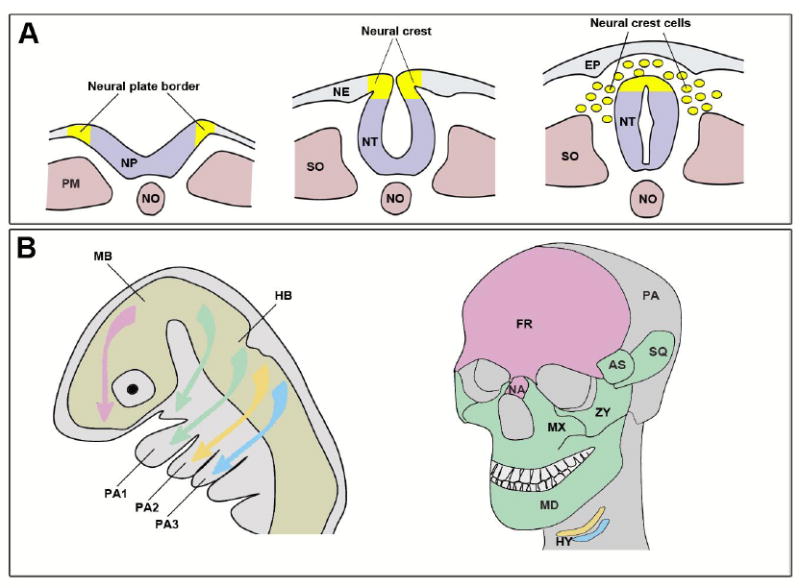
(A) At the beginning of neurulation, the precursors of the NC are located at the NPB, the boundary between the neural plate (NP) and the non-neural ectoderm (NE). As the neural plate folds into a tube, the NC forming region ends up at the dorsal aspect of the neural tube (NT). NCCs delamination and migration is initiated upon neural tube closure. EP, epidermis; PM, paraxial mesoderm; NO, notochord; SO, somite. (B) Colonization of the head and pharyngeal arches by NCCs. Each population of NCCs is color-coded based on its origin in the embryo (left) and the derivatives it produces in the adult head (right). After [23]. AS, alisphenoid bone; PA1–PA3, pharyngeal arches 1-3; FR, frontal bone; HB, hindbrain; HY, hyoid bone; MB, midbrain; MD, mandible; MX, maxillary bone; NA, nasal bone; PA, parietal bone; SQ, squamosal bone; ZY, zygomatic bone. The middle ear ossicles derived from the NC in PA1 (maleus and incus) and PA2 (stapes) are not shown.
In response to signaling events mediated by Bmp, Wnt and Fgf, distinct sets of transcriptions factors are sequentially activated at the NPB. First a set of genes, known as NPB specifiers, is broadly activated in this territory, and their expression domain typically encompasses the NC progenitor domain and other cell types arising from this region. This set of transcription factors includes several members of the Pax, Msx, Tfap2, Dlx5 and Zic families. These factors then activate a second set of genes more restricted to the NC territory, known as NC specifiers, which include genes of the Snail, Fox and Sox family of transcriptional regulators, among others. These factors are essential for the activation of NC effector genes whose function is to regulate NCCs migration and their differentiation a long distinct lineages. The correct temporal and spatial expression of these genes is central to the development of the NC. This regulatory cascade driving NC formation is well conserved among vertebrates, and only minor variations in this blueprint have been reported [16], [17], [18].
NCCs migration is a complex process, as cells adopt highly stereotyped migration routes and appear to be directed by local signaling cues rather than being guided by their intrinsic cellular makeup [19]. Grafting experiments and single-cell lineage tracing studies have contributed immensely to our understanding of NCCs migration and much of what we know on these processes comes from experiments performed in birds and frogs [20]. Typically, NCCs delaminate from the neuroepithelium at the end of neurulation and undergo an epithelial-to-mesenchymal transition (EMT), coordinated by changes in the expression of cell adhesion molecules. Once they leave the neuroepithelium, the cells quickly form streams and migrate as continuous rostrocaudal waves using both positive and negative guidance cues from the surrounding tissues [20], and follow a fairly similar pattern of migration in all vertebrates. In the most anterior domain of the NC, known as the cranial NC, the NCCs that contribute to the cranial ganglia migrate dorsally where as those contributing to face and neck cartilages migrate further ventrally to populate the branchial/pharyngeal arches [20]. Upon reaching their designated sites, NCCs become developmentally restricted and activate the expression of a unique set of genes characteristic for their fate.
Development of the craniofacial structures
Craniofacial structures arise as a result of complex interactions between the NC and the head mesenchyme to give rise to very specialized structures, such as the sensory ganglia, a bony skull that protects the brain, and a muscularized jaw that allows for feeding [21], [10]. This intricate process is controlled by a combination of genetic and environmental factors. Vertebrates exhibit a relatively conserved pattern of craniofacial morphogenesis, with some variations in the timing of progenitor cells formation and in the position of tissue boundaries. While the overall morphology of the skull can differ significantly between species, the molecular and developmental pathways that underlie the formation of the skull remain relatively conserved [13]. Given these similarities, several animal models are used in the study of cranial morphogenesis, each with its unique advantages and limitations.
Head development involves a series of well-orchestrated morphogenetic events that result in the formation of uniquely patterned cartilages, bones, muscles and nerves in the head. During this process, the NC provides the main source of craniofacial mesenchyme. Cranial NCCs originate from progenitors in the posterior midbrain, and segmental units of the hindbrain known as rhombomeres (R). The most rostral NCCs migrate into the frontonasal prominence to give rise to the frontal and nasal bones (Fig 1B). NCCs from R2, R4 and R6 migrate as three individual streams that populate the 1st, 2nd and 3rd pharyngeal arches respectively, which are also known as the hyoid, mandibular and branchial arches (Fig 1B). The branchial arch further splits into two streams: the anterior branchial and the posterior branchial arches. R3 and R5 typically produce fewer cells, which join adjacent streams. The pharyngeal arches are transient embryonic structures that form on either sides of the developing pharynx. They give rise to most of the differentiated tissues of the head and neck. The pharyngeal arches are externally lined by the ectoderm that produces the epidermis and forms the neurogenic placodes, which make the sensory neurons of the arch-associated ganglia. Internally, the endoderm forms the epithelium lining the pharynx and the pharyngeal glands (thymus, thyroid and parathyroid). Each arch has a core of NCCs surrounding a group of mesoderm-derived cells. The mesoderm forms the musculature of the head and the endothelial cells of the arch arteries, while NCCs develop into craniofacial skeletal elements specific for each pharyngeal arch (Fig 1B, [22], [23], [24], [9], [25]). The first pharyngeal arch is composed of two prominences: the maxillary prominence in which the NC forms the maxillary, zygomatic and temporal bones; and the mandibular prominence in which the NC gives rise to the mandible and two of the middle ear bones (malleus and incus). In the second pharyngeal arch, the NC forms the third middle ear ossicle (stapes), styloid process and lesser horn of the hyoid, while the greater horn of the hyoid is derived from the third pharyngeal arch NC. The NC in the most caudal pharyngeal arches develops into the laryngeal cartilages (Fig 1B).
Craniofacial disorders modeled in Xenopus
There is abundant literature describing the mechanisms regulating NC development in Xenopus at the molecular level [14], [15]. In fact, Xenopus and chicken are probably two of the most widely used systems to study NC formation and migration, and most of our understanding of these processes was first described in these species. Xenopus NC progenitors can be easily identified at different stages of development, pre- and post-migration, and visualized by the expression of a broad range of genes (Fig 2A, B). The NC is also very accessible at these early stages and explants can be easily dissected and cultured in vitro to analyze the developmental potential of these cells and/or their migratory properties [26], [27], [28], [29]. Studies using cell labeling and grafting experiments have established very detailed fate maps of cranial NCCs migration, and identified its contribution to the craniofacial structures of the tadpoles, pre-metamorphic froglets, as well as its long-term contributions to the persistent cartilages in the post-metamorphic skulls (Fig 2C, [30], [31], [32], [33]. Xenopus has also proven to be a very suitable system to study cranial ossification and suture patterning [13]. It takes approximately four days from fertilization to reach the tadpole stage, at which stage the mouth, craniofacial cartilages, and others head features can be clearly identified by gross morphology or with the appropriate staining (Fig 2D, F). Finally, by microinjection of mRNA or antisense oligonucleotides (morpholino) at the 2-cell stage (NF stage 2) or later we can manipulate gene function in one half of the embryo and use the other half as an internal control in each experiment (Fig 2E, F).
Figure 2. Xenopus: a model to study craniofacial development.
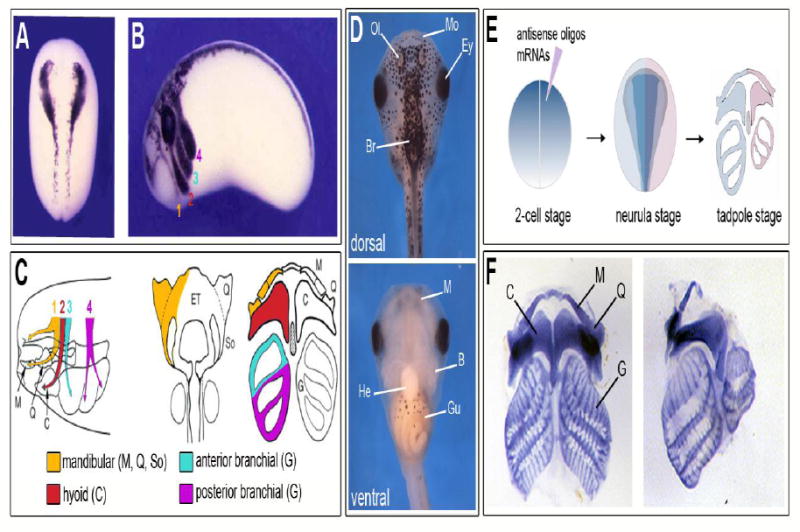
(A) Expression of the transcription factor sox10 in premigratory NC progenitors visualized by in situ hybridization at the neurula stage (NF stage 15). Dorsal view, anterior to top. (B) At the tailbud stage (NF stage 28) sox10 expression is detected in the four streams of NCCs migrating towards the pharyngeal arches, the mandibular (1; orange), hyoid (2; red), anterior branchial (3; green) and posterior branchial (4; purple) streams. Lateral view, anterior to left, dorsal to top. (A, B) Modified from [110]. (C) Diagram illustrating the migration pattern and contribution of the NC to cranial skeletal elements. The mandibular neural crest segment (1; orange) contributes to Meckel’s (M), palatoquadrate (Q), and subocular (So) cartilages; the hyoid neural crest segment (2; red) contributes to the ceratohyal (C) cartilages; the anterior (3; green) and posterior (4; purple) branchial NC segments contribute to the cartilages of the gills (G). Modified from [30] and [111]. (D) Dorsal (upper panel) and ventral (lower panel) views of stage 45 tadpole. Anterior to top. Br, brain; Ey, eye; Gu, gut; He, heart; Mo, mouth; Ol, olfactory pit. (E) Diagram illustrating the approach to manipulate gene expression in Xenopus embryos and analyze the consequences on craniofacial structures. (F) Examples of flat-mounts alcian blue-stained craniofacial skeletal elements from a control (left side) and a Sox8-depleted (right side) stage 45 tadpoles. Modified from [112].
All these features make Xenopus a system of choice to analyze the developmental mechanisms underlying craniofacial abnormalities. In the following sections, we summarize studies that have used Xenopus to model several human craniofacial disorders that have either a genetic component (gene mutation), or are the result of exposure to harmful environmental factors (teratogens).
Fetal alcohol syndrome
Fetal Alcohol Syndrome (FAS) is a collection of developmental, behavioral and physiological conditions resulting from prenatal exposure to ethanol, typically from alcohol use by pregnant mothers. A 2002 report from the Centers for Disease Control and Prevention (CDC) estimated that there are “0.2 to 1.5 infants with FAS for every 1,000 live births in certain areas of the United States” (cdc.gov/ncbddd/fasd/). Though there are descriptions of similar disorders as early as Greek and Roman mythology, the first clinical description of this disease was published in 1973 [34]. In this report, the physical abnormalities were classified in terms of delays in physical growth, craniofacial abnormalities, limb defects, and other anomalies. Over the years, the definition of FAS has been refined and broadened to include cognitive and behavioral issues, however craniofacial anomalies are still considered one of the hallmarks of FAS [35]. In the initial reports, the most common defects included microcephaly and short palpebral fissures of the eyes in a majority of the cases. Other manifestations such as epicanthal folds, maxillary hypoplasia, cleft palate, median facial clefts (in rare and severe cases), micrognathia (undersized jaw), pre and post-natal growth retardation, and central nervous system deficiencies were observed at a lower frequency [34], [36]. For a comprehensive description of craniofacial and dental anomalies in FAS patients see [37]. It is clear that the range of defects is directly dependent upon the age of the embryo at the time of exposure, as well as the dose and duration of exposure.
To better understand how developmental pathways are disrupted upon ethanol exposure, FAS has been modeled in several organisms including frogs. In the first attempt to model FAS in Xenopus, blastula (NF stage 6) embryos were exposed to 1-2% ethanol and allowed to develop until the early tailbud stage (NF stage 23-24), when organogenesis begins [38]. In this study, the embryos exhibited defects such as reduced brain and body size, and facial malformations around the mouth. These defects were attributed to failure of mesodermal cells to migrate during gastrulation, resulting in the formation of a smaller neural plate. Subsequent studies have shown that Xenopus embryos were most sensitive to ethanol exposure between late blastula and early/mid-gastrula stages [39]. Ethanol exposure during this period caused changes in gene expression in the dorsal mesoderm, delayed gastrulation movements, and reduced levels of retinoic acid (RA; a posteriorizing factor), which resulted in tadpoles with shorter antero-posterior axis. Ocular anomalies and microcephaly (small head) were also observed and correlated with reduced expression of several transcription factors important for head and eye development, such as otx2, pax6, en2 and tbx3 [40].
A more recent study has focused on the impact of ethanol exposure on NC formation. While 1% ethanol exposure did not affect NPB (msx1, pax3 and zic1) and NC (snai2) specifier genes, it more specifically impaired the expression of genes involved in NCCs migration at a later stage, which has been proposed to be the cause of reduced craniofacial cartilages [41]. Because alcohol consumption reduces folic acid uptake, the same group examined a possible link between the two pathways. Microinjection of 5-mehtyltetrahydrofolate (5-MTHF), the most bioactive form of folic acid, prior to ethanol exposure was able to partially rescue the expression of genes associated with NCCs migration in these embryos [41], suggesting that 5-MTHF could be beneficial for treating FAS. However, the authors did not directly evaluate whether the treatment could also rescue craniofacial structures in these tadpoles. In another study, cranial NCCs explants were exposed to low concentrations of ethanol. This treatment resulted in an initial delay in the number of NCCs emigrating from these explants, associated with a reduction in the overall distance covered by individual cells [42], again consistent with a role of ethanol in disrupting NCCs migration.
Altogether, these studies indicate that exposure to ethanol adversely affected NCCs migration in vivo and in vitro, thereby providing a potential developmental mechanism for FAS associated craniofacial anomalies. Further studies are needed to identify the molecular targets specifically affected by ethanol exposure and how they impact NCCs migration in the head.
Orofacial clefts
Orofacial clefts (OFCs) are among the most common congenital abnormalities affecting the lip, the palate or both (Table 1). Over ten different categories are described in OMIM (Online Mendelian Inheritance in Man; http://www.omim.org). Cleft lip and/or cleft palate occurs about 1 in 750 live births in the United States, with frequencies varying with race, gender and geographic areas [43]. The etiology of OFCs is complex and involves both genetic and environmental factors. OFCs can be syndromic, when occurring with other anomalies, or non-syndromic when they are not associated with other defects. The most recent estimate from patient studies indicates that 70% of OFCs are non-syndromic [44]. OFCs are classified into four broad categories: non-syndromic cleft lip with or without cleft palate, non-syndromic cleft palate alone, syndromic cleft lip with or without cleft palate, and syndromic cleft palate alone, with further subdivisions based upon whether the primary or the secondary palate is affected. For a comprehensive review on OFCs, see [44]. These numerous categories of OFCs suggest that there is a complex regulatory network of genes that orchestrates the processes involved in facial formation, with OFCs being the primary outcome when any of these processes are disrupted. Animal models have informed much of the current understanding of OFCs, despite the obvious and stark differences in facial morphology of the vertebrates, because of the conserved underlying mechanisms. In that respect, Xenopus is an extremely useful experimental model as the orofacial region is readily visible and can be easily dissected and transplanted [45].
The first report using Xenopus to model OFCs investigated the role of RA signaling, a morphogen regulating multiple developmental processes. RA receptor gamma (Rarγ) and retinaldehyde dehydrogenase 2 (Raldh2), the enzyme involved in the synthesis of RA from its precursor retinal, are expressed in the orofacial region of Xenopus tadpoles in a complementary pattern, and depletion of Raldh2 using morpholino-mediated knockdown resulted in dorsal median clefts in about 40% of the cases, a phenotype that could be rescued by treatment with all-trans RA [46]. To rule out the possibility that these defects were not due to an early effect of the morpholino on surrounding tissues, this finding was confirmed in “face transplant” experiments [47]. In this assay, a piece of embryonic tissue containing the presumptive primary mouth from a morpholino-injected donor embryo was transplanted into the same region of an uninjected recipient embryo. This transplantation again resulted in mouth clefting in 80% of the embryos. Additionally, blocking RA signaling by treatment of tadpoles with a pharmacological inhibitor of Rarγ (BMS-453) resulted in median clefts. Altogether these results confirmed a direct role for RA signaling in orofacial development. Furthermore, the authors were able to demonstrate that RA signaling regulated the expression of two transcription factors, lhx8 and msx2, which in concert can direct the tissue growth and differentiation of the upper lip and primary palate formation [46],
Xenopus was also used to investigate the role of folate metabolism in orofacial development. Folic acid supplementation during pregnancy has been known to reduce the risk of neural tube defects and OFCs in infants [48]. Folate is converted into tetrahydrofolate, which is the precursor for synthesis of purines and pyrimidines, through a pathway involving the enzyme dihydrofolate reductase (DHFR) [49]. DHFR is expressed in the orofacial region and mutations in DHFR have been linked to non-syndromic OFCs [50]. Both folate and RA signaling are known to functionally overlap during mouth formation. To understand the role of folate metabolism in OFCs, the impact of folate reduction on Xenopus orofacial morphogenesis was examined alone, as well as in context of RA signaling. Using morpholino and pharmacological inhibitors targeting Dhfr, and face transplants the studies demonstrated that loss of folate metabolism can lead to alterations in the shape of the mouth and a narrower, underdeveloped face due to loss of jaw muscles and cartilages [51], a set of defects rescued by treatment with folinic acid.
These findings indicate that folate plays a distinct role from RA in facial morphogenesis, and suggest that folate deficiencies could significantly contribute to multifactorial OFCs. Therefore, Xenopus offers many tools that can be used to elucidate the intersection of various signaling pathways in the etiology of OFCs.
Campomelic dysplasia
Campomelic dysplasia (CD; OMIM # 114290) is a rare autosomal dominant skeletal dysmorphology syndrome characterized by congenital bowing of the long bones, associated with ambiguous genitalia or male to female sex reversal in 75% of XY CD patients. CD was first described in 1971 but was first formally reported in 1994 as an autosomal dominant skeletal disorder [52]. Affected newborns typically die in the first year of life due to respiratory distress. Craniofacial manifestations of CD include macrocephaly (enlarged head), depressed nasal bridge, hypertelorism (increased distance between the eyes), macroglossia (enlarged tongue) and micrognathia with or without cleft palate [53]. Adults who survive this condition often display respiratory problems, sensorineural deafness, developmental delay, short stature and orthopedic problems in addition to facial anomalies [54], [55]. The underlying cause of CD is haploinsufficiency of the transcription factor Sox9. Nonsense, missense and frameshift mutations in the SOX9 coding region accounts for the majority of cases of CD [56], [52], [57], reviewed in [58].
Sox9 belongs to the SoxE group, which has two additional members Sox8 and Sox10, and all three SoxE proteins are critical for the development of NC progenitors [59]. Sox9 expression has been described in NC progenitors and pharyngeal arch mesenchyme in several species including Xenopus, suggesting a conserved function during craniofacial development. In Xenopus embryos, Sox9 knockdown by injection of morpholino antisense oligonucleotides resulted in failure to form neural folds and also disrupted the expression of multiple NC specifiers, including snai2 and foxd3, in a dose-dependent fashion [60]. At later stages, these embryos lacked the ability to form pharyngeal arches and at the tadpole stage, exhibited loss of Meckel’s cartilage (precursor of the lower jaw), with milder reductions of the branchial and cerathoyal cartilages [60], which are cartilage elements derived from the mandibular, hyoid and branchial NC (Fig 2C, [30]). Thus, Sox9 was found to be critical for craniofacial morphogenesis due to its requirement in NC progenitors [60], [61]. In another study, Sox9 depletion in Xenopus embryos was shown to affect otic placode formation, which later gives rise to the inner ear the organ responsible for hearing and balance [62]. Reduction in Sox9 expression early in development resulted in loss of genes typically associated with otic placode development, and in the most extreme cases these animals failed to form a morphologically discernible otic vesicle, indicating that Sox9 is a critical regulator of inner ear formation, consistent with the observation that some CD patients are affected by sensorineural deafness.
These studies have provided a mechanistic basis for the craniofacial anomalies associated with CD, tracing back the origin of these defects to a deficiency in NC progenitors formation and otic plocode development, and as such advancing our understanding of the etiology of this disease and the nature of the developmental processes altered by sSOX9 mutations.
DiGeorge syndrome
DiGeorge sydrome (DGS; OMIM # 188400) is an autosomal dominant disorder caused by a 1.5 to 3.0 Mb hemizygous deletion of chromosome 22q11.2 [63], a region that contains between 35 and 60 genes. It is characterized by a spectrum of defects including craniofacial anomalies such as cleft palate, micrognathia, elongated faces and dental abnormalities. Other features include short stature, hypoplasia of thymus, resulting in cellular immunodeficiency, and cardiac defects. DGS occurs in 1:3000 live births [64]. Haploinsufficiency of the transcription factor Tbx1 was identified as a cause of DGS. Tbx1 belongs to the T-box family of transcription factors, which have been implicated in multiple developmental processes, including limb and heart development. The functions of the T-box gene family are well conserved in vertebrate development [65]. Tbx1 is required for the normal development of pharyngeal arches in a dose-dependent manner, as heterozygous deletion causes disruptions in the fourth pharyngeal arch, while homozygous deletion results in severe disruptions in the entire pharyngeal arch apparatus [66].
Mouse models have provided important information on the role of Tbx1 in the pathogenesis of DGS [67], [68]. While a direct attempt to model DGS in Xenopus has not been reported, some studies have investigated Tbx1 function in this organism. In Xenopus, like in other species, tbx1 is not expressed in the NC, rather it is expressed in the ectoderm, mesoderm and endoderm components of the pharyngeal arches [69]. To analyze the function of Tbx1 in the frog, a repressor form of the protein was generated by replacing its transactivation domain by the repressor domain of Drosophila engrailed protein. This dominant interfering mutant disrupted the development of the head and pharyngeal structures in a dose-dependent manner when injected in the early embryo. Typically, the anterior-posterior axis was shortened, Meckel’s cartilage was reduced or absent, and tadpoles had small and abnormally shaped heads [69]. In another study, loss of Tbx1 function was generated by injection of morpholino antisense oligonucleotides. The corresponding tadpoles displayed hypoplastic craniofacial cartilages (including highly reduced subocular and Meckel’s cartilages), underdeveloped head muscles, and a delay in NCCs migration at early stages [70].
Altogether these studies indicate that Tbx1 has a conserved function during head development, positioning Xenopus as a viable model to study its function in relation to the mechanisms underlying the craniofacial defects associated with DGS. Because Tbx1 is not expressed in the NC, defects in NC development are occurring in a non-cell autonomous fashion in DGS, therefore it is critical to identify the targets that mediate its activity.
CHARGE syndrome
CHARGE syndrome (OMIM # 214800) was reported in 1979 as a collection of congenital malformations with nonrandom occurrence [71], with an estimated incidence of 1 in every 12,000 live births [72]. CHARGE is an acronym derived from the malformations associated with this condition: Coloboma, Heart defect, Atresia choanae, Retarded growth and development, Genital hypoplasia, and Ear anomalies/deafness [73]. Of these, choanal atresia (blockage or narrowing of the nasal passage), coloboma of the eyes (missing tissue in or around the eye), ear and cranial nerve anomalies are considered to be the major criteria for a CHARGE syndrome diagnosis. The craniofacial abnormalities in CHARGE syndrome also include orofacial clefts and facial nerve palsy [74]. CHARGE syndrome results from heterozygous mutations in the gene encoding the chromodomain helicase DNA-binding protein 7 (CHD7), an ATP-dependent chromatin remodeler. These mutations result in the production of truncated proteins [75], and the condition is inherited in an autosomal dominant pattern [76].
Chd7 knockdown in Xenopus, by injection of morpholino antisense oligonucleotides, resulted in failure of NCCs to migrate to the pharyngeal arches, a phenotype rescued by expression of human CHD7 [77]. Further analysis revealed that in Chd7-depleted embryos the expression of NPB specifier genes such as pax3, zic1 and msx1 was largely unperturbed, indicating that competence to form NC progenitors was not affected in these embryos. However, two NC specifier genes (twist1 and snai2), which are involved in EMT, as well as sox9, were downregulated. Interestingly, overexpression of a dominant negative Chd7, by substitution of a conserved lysine residue in its ATPase domain, also disrupted NCCs migration, indicating that the ATPase domain of Chd7 was important to the process. As development progresses, the corresponding tadpoles exhibited a number of defects resembling those seen in CHARGE syndrome patients [77]. Mechanistically, this study demonstrated that in vitro CHD7 interacts with a chromatin-remodeling complex of the SWI/SNF family called PBAF (polybromo- and BRG1-associated factor-containing complex), as part of a large chromatin remodeling complex that occupies the regulatory elements controlling the NC-specific expression of SOX9 and TWIST1 [77]. This complex of CHD7 and PBAF in turn enables NC gene expression and NCCs migration. Consistent with these observations, CHD7 has also been shown to regulate semaphorin-3A expression, another factor involved in NCCs guidance [78].
These studies have significantly improved our understanding of the function of CHD7 and its cofactors in the pathogenesis of CHARGE syndrome, and the pathways that are critically altered in this disease. During NC formation, CHD7 and PBAF control the expression of several important NC specifier genes, which in turn allow these cells to acquire their identity and migratory properties.
Smith-Magenis syndrome
Smith-Magenis Syndrome (SMS; OMIM # 182290) is a complex disorder with multiple congenital defects, including craniofacial and skeletal anomalies, obesity, intellectual disability, and a neurobehavioral component characterized by self-destructive behavior and attention-seeking [79]. The craniofacial defects that are frequently associated with SMS and include cleft lip/palate, midface hypoplasia, flat nasal bridge, brachycephaly (flattened head), and prognathia (misaligned maxilla and mandible). SMS is sporadic, with estimated incidence of 1 in 15,000-25,000 births. While first described in 1982, it is often underdiagnosed due to the broad spectrum of symptoms [80]. SMS results from a haploinsufficiency of the RAI1 (retinoic acid-induced 1) protein caused either by a microdeletion of chromosome 17p11.2 that contains the gene, or mutations in the RAI1 gene alone [81]. Most patients have a common 4 Mb deletion, but smaller deletions are also found, and the minimal deletion region known to occur encompasses around 25 genes [82]. The severity of the phenotype is exacerbated in patients carrying larger deletions [81].
RAI1 encodes a transcription factor whose cellular function is unclear. It was initially identified as a gene induced by RA in mouse embryonal carcinoma cells [83]. The sequence and structure of RAI1 is relatively conserved among vertebrates, with 44% identity between Xenopus and human proteins for example. In Xenopus, Rai1 is also upregulated by RA treatment. It is expressed in the neural plate at early stages of development, and in the pharyngeal arches, the otic vesicle and the developing central nervous system at later stages of development [84]. Morpholino-mediated knockdown of Rai1 resulted in dose-dependent alterations in head and body axis development. Upon further analysis, these embryos exhibited midface hypoplasia, an abnormal rounder mouth and failure of snout outgrowth. Morphometric analyses of these morphant embryos showed that these alterations were consistent with the craniofacial anomalies observed in SMS [84]. The expression of the NC gene tfap2a was also markedly reduced in the pharyngeal arches of these embryos. They appeared to have less tfap2a-positive cells and the NC streams were ill-defined, suggesting that the number of NC progenitors was reduced and/or their migration was affected. Consistent with these observations, almost all the NC-derived cartilages (ethmoid plate, Meckel’s, ceratohyal, infrarostral and branchial) were reduced at the tadpole stage [84]. Finally, Rai1 depletion had also adverse consequences on nervous system development, characterized by increased apoptosis in the forebrain, reduced forebrain ventricle, abnormal patterns of nerve tracts and decrease levels of brain derived neurotrophic factor (BDNF), a factor that supports neuronal survival.
This Xenopus model of SMS not only recapitulates the craniofacial abnormalities seen in SMS patients but also provides some mechanistic insights into the causes of these defects that may reside in the disruption of NC specification and/or migration. It is still unclear how RAI1 functions in the RA signaling pathway to regulate NC development and additional work with this model is needed to address this issue.
Andersen-Tawil syndrome
Andersen-Tawil Syndrome (ATS; OMIM # 170390), also called Andersen-Cardiodysrhythmic Periodic Paralysis, or Long QT syndrome 7, is a rare, autosomal-dominant multisystem disorder characterized by a clinical “triad” of symptoms: ventricular arrhythmias, episodic muscle paralysis, and facial skeletal anomalies [85]. The exact prevalence of ATS is not known but can be roughly surmised from the prevalence of periodic paralysis, as ATS is often diagnosed in patients presenting with primary periodic paralysis. Primary periodic paralysis occurs with an estimated prevalence of 1:100,000, and currently, ATS is diagnosed in less than 10% of the paralysis patients [86]. Facial dysmorphologies occur in 78% of ATS patients and include low-set ears, cleft palate, mandibular hypoplasia, hypertelorism, micrognathia, a broad forehead, and dental anomalies [87], [88]. ATS is classified as a Kir 2.1 channelopathy as it is caused by heterozygous mutations in KCNJ2 gene, which encodes an inwardly rectifying K+ channel (Kir) protein, KIR 2.1 [87]. Kir channels allow for greater flow of K+ into the cell to maintain homeostasis and membrane potential, and most of the 40 mutations identified in KIR 2.1 of ATS patients are found mainly in the conserved regions of the protein [89], [90].
Xenopus oocytes were used to express ten versions of human KCNJ2 gene carrying mutations known to result in ATS, to assess their functional impact on ion channel function. These mutations were shown to have dominant-negative effect on KIR 2.1, completely suppressing its normal channel function in this system, and adversely affecting cardiac and skeletal muscle excitability in the mutation carriers [91]. This study was important as it highlighted the emerging role of ion channels in developmental processes. The specific role of Kir 2.1 during craniofacial development was addressed more recently. In Xenopus, kcnj2 is initially expressed at early neurula stage (NF stage 14) in a broad dorso-anterior domain, which later becomes regionalized anteriorly, consistent with a potential role in craniofacial morphogenesis [92]. Previous studies from this group showed that there is a unique pattern of membrane resting potential (Vmem) in the ectoderm of the embryo that is critical to maintain gene expression domains as development progresses [93]. Injection of either wild-type (WT) Kcnj2 or a variant carrying one of the known ATS mutations led to changes in the normal pattern of Vmem at the neurula stage, and resulted in craniofacial anomalies at the tadpole stage. However, Kcnj2 mutant proteins caused defects at a higher frequency than the WT [92]. The craniofacial anomalies observed in these tadpoles included misshapen eyes, small or bent Meckel’s cartilage, undersized branchial arches and pigmented optic nerve. Interestingly, misexpression of other channels that also alter the Vmem caused similar craniofacial anomalies. This effect was not seen upon overexpression of an electroneutral ion channel, indicating that rather than the specific identity of the channel, it is the maintenance of the bioelectric state of the embryo that is critical for craniofacial morphogenesis [92].
This report also demonstrated that alterations in the Vmem affected the expression of NC and cranial placodes specific genes, including six1, pax6, snai2, sox10 and foxe3, further confirming that alteration of the bioelectric state can affect gene expression domains, resulting in mis-patterned embryos. Finally, using optogenetics to spatially and temporally alter the Vmem of the embryo, the work showed that maintaining a normal pattern of Vmem in the ectoderm during early neurula stages is especially critical for normal craniofacial development - altering the Vmem during late neurulation did not impact craniofacial development [92].
Taken together, this study highlights the importance of bioelectric gradients in development, and indicates that Kcnj2 is critical for craniofacial morphogenesis by maintaining the bioelectric state of the ectoderm. Perturbations in this state may underlie the pathogenesis of craniofacial defects seen in ATS patients. This work also emphasizes the versatility of Xenopus with the use of optogenetics to study craniofacial development. Future studies investigating how the mechanotransduction of bioelectric signals can affect gene expression domains will greatly advance our understanding of the role of ion channels in development and diseases.
Hamamy syndrome
Hamamy syndrome (OMIM # 611174) is an extremely rare condition. Only 5 affected individuals have been described in two consanguineous families, a Turkish and a Jordanian family [94], [95]. It is an autosomal recessive craniofacial syndrome characterized by severe hypertelorism with upslanted palpebral fissures, brachycephaly, abnormal ears, enamel hypoplasia and sensorineural deafness. Associated features include osteopenia (low bone density) with chronic bone fractures, congenital heart defects and anemia. A recent study identified 2 homozygous missense mutations in the IRX5 gene that segregated with the disease in these two families [95].
IRX5 belongs to the Iroquois family of transcriptional regulators. These proteins act as pre-patterning factors by imposing specific positional identity in embryonic tissues [96]. Irx5 knockdown by morpholino injection in Xenopus embryos showed reduced twist1 expression in the first pharyngeal arch, indicative of a role for Irx5 in NC formation. This phenotype was efficiently rescued by expression of WT mouse Irx5. To gain mechanistic insights into the role of Irx5, the authors performed a microarray experiment and found that cxcl12, which encodes a chemokine essential for cranial NCCs migration [97], was strongly upregulated in Irx5-depleted embryos. Consistent with this finding, in situ hybridization revealed that cxcl12 was strongly upregulated in the cranial region of Irx5-depleted embryos. As a confirmation that both factors were acting in the same pathway, the authors were able to demonstrate that the branchial arch phenotype of Irx5-depleted embryos (reduced twist1 expression) could be rescued by Cxcl12 knockdown. These results suggest that Irx5 deficiency deregulates Cxcl12 expression in the head thereby interfering with the cues required for proper cranial NCCs migration.
While this study has not directly demonstrated that Irx5-depleted Xenopus tadpoles have craniofacial skeletal defects similar to those seen in Hamamy patients, the work is important as it provides significant information on the potential mechanisms underlying this condition. Here, the defects are not intrinsic to the NC, as seen in other craniofacial diseases discussed in this review, but rather the result of the disruption of signaling cues guiding the NCCs migration to their final location.
Musculocontractural Ehlers-Danlos syndrome
Musculocontractural Ehlers-Danlos syndrome type 1 (EDSMC1; OMIM # 601776) is a clinically and genetically heterogeneous group of disorders that affect connective tissues. They are mainly characterized by skin hyperextensibility, articular hypermobility and tissue fragility. The condition also has a distinctive craniofacial component including micrognathia, high and/or cleft palate, brachycephaly, hypertelorism, downslanting palpebral fissures and low-set ears [98], [99]. It is caused by autosomal recessive loss-of-function mutations in genes encoding dermatan sulfate (DS) biosynthetic enzymes: dermatan 4-O-sulfotransferase 1 (CHST14) or DS epimerase-1 (DSE).
DS is a glycosaminoglycan attached to serine residues of core proteins, typically extracellular matrix proteins [100]. In Xenopus dse is expressed in the epidermis and the NC at the neurula stage (NF stage 17). Dse knockdown led to a reduction of NC-derived mandibular, hyoid and branchial cartilages at the tadpole stage, as well as other NC derivatives, pointing to a requirement of Dse for NC development [101]. Early on in embryogenesis, Dse knockdown did not affect the formation of early NC progenitors at the NPB. However, it disrupted the expression of genes involved in EMT (snai2 and twist1) in the pharyngeal arches at the tailbud stage (NF stage 28), suggesting a potential role of Dse in NCCs migration. To confirm this possibility, the authors used homotypic transplantation of GFP labeled cranial NC explants derived from Dse-depleted donors into WT host embryos, and showed that very few labeled cells were able to populate the pharyngeal arches in the host. Similarly, in cranial NC explants excised from Dse-deficient embryos and cultured on a fibronectin substrate, NCCs failed to spread, migrate and form protrusions [101].
This study demonstrates that impaired NCCs migration in Dse-depleted embryos is the primary cause of reduced craniofacial skeletal structures in these tadpoles. More broadly, these findings suggest that defective NCCs migration may account for the craniofacial anomalies in EDSCM1 patients, providing important new information on the underlying disease mechanisms.
Nager syndrome
Nager syndrome (OMIM # 154400) is the most common form of acrofacial dysostosis (a group of diseases that combine craniofacial and limb defects), it was first described by Nager and De Reynier in 1948 [102]. The craniofacial abnormalities include downslanting palpebral fissures, malar hypoplasia (underdeveloped chick bones), micrognathia, atresia of the external auditory canal as well as other external ear defects, and cleft palate. The affected structures are primarily derived from the first and second pharyngeal arches. These malformations are associated with preaxial limb defects, such as radial and thumb hypoplasia or aplasia, duplication of thumbs or proximal radioulnar synostosis (proximal fusion of the radius and ulna). With less than 100 cases described in the literature, the prevalence of Nager syndrome is not known [103]. Haploinsufficiency of SF3B4 (Splicing factor 3b, subunit 4) has been identified as a major cause of Nager syndrome, accounting for ~60% of the cases [104], [105], [106]. SF3B4 encodes SAP49, one of the core proteins of the pre-mRNA spliceosomal complex, which is the cellular machinery that removes the introns to join exons together and produce mature mRNAs [107].
In order to model Nager syndrome in Xenopus, Sf3b4 was knocked down in the early embryos by injection of two distinct morpholino antisense oligonucleotides. Both antisenses had a similar outcome, and the injected tadpoles exhibited hypoplastic craniofacial cartilages. Meckel’s, ceratohyal and branchial cartilages were all affected ranging from relatively mild size reductions to complete loss of some cartilages [108]. This phenotype is consistent with the craniofacial defects seen in Nager syndrome patients. Around the time of NC induction, morpholino injected embryos showed a dramatic reduction of several NC specifier genes (snai2, sox10 and twist1). Importantly, this phenotype could be rescued by injection of WT human SF3B4 mRNA, but not by mRNAs carrying mutations that have been associated with Nager syndrome [108]. The reduction in NC genes expression in Sf3b4-depleted embryos was associated with an increase in cell death in the ectoderm, indicative of an early loss of NC progenitors in these embryos [108], and suggesting that this mechanism may underlie the pathogenesis of Nager syndrome.
This work points to an important role of Sf3b4 in the formation and survival of NC progenitors. This is reminiscent of another condition, Treacher Collins syndrome (TCS, OMIM # 154500), which has similar craniofacial defects, involving structures derived form the first and second pharyngeal arches. Mouse models of TCS indicate that the condition is caused by a depletion of NC progenitors through a mechanism involving apoptosis [109], suggesting that the craniofacial pathogenesis in both syndromes share the same mechanism. An important challenge now is to identify the molecular targets of SF3B4 to determine whether its role in NC formation depends entirely on its splicing activity, presumably by targeting transcripts essential for NC formation, or an unrelated function of the protein.
Conclusions
In this review, we have summarized studies investigating the pathogenesis of a broad array of human craniofacial disorders using Xenopus as a model system. These studies illustrate that Xenopus is an extremely powerful system to inform important biological processes as they relate to human craniofacial development and disease. Most of these conditions can be traced to defects in the development of the NC, induction of progenitors, survival and/or migration, that affect the NC itself or the environment in which it navigates. Modeling these conditions in Xenopus as well as other model organisms has the tremendous potential to provide novel insights into the etiology and pathogenesis of these diseases, since these studies are impossible to conduct in humans. Ultimately, this information can be used to inform therapeutic strategies to prevent or minimize craniofacial defects at birth.
Acknowledgments
Work in the Saint-Jeannet Lab is supported by the National Institutes of Health (NIH), grant numbers R01DE25806 and R01DE25468.
Footnotes
Conflict of interest
Aditi Dubey and Jean-Pierre Saint-Jeannet declare that they have no conflict of interest.
Compliance with Ethics Guidelines
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Bibliography
Recently published papers of particular interest have been highlighted as:
•Of importance
••Of major importance
- 1.Shaw DW. Global Strategies to Reduce the Health Care Burden of Craniofacial Anomalies: Report of WHO Meetings on International Collaborative Research on Craniofacial Anomalies. The Cleft Palate-Craniofacial Journal. 2004;41(3):238–43. doi: 10.1597/03-214.1. [DOI] [PubMed] [Google Scholar]
- 2.Nieuwkoop Pieter Dirk, Faber J. Normal table of Xenopus laevis (Daudin). A systematical and chronological survey of the development from the fertilized egg till the end of metamorphosis. 2. Amsterdam: North-Holland Publishing Company; 1956. Guilders. [Google Scholar]
- 3.Moody SA. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Developmental Biology. 1987;119(2):560–78. doi: 10.1016/0012-1606(87)90059-5. http://dx.doi.org/10.1016/0012-1606(87)90059-5. [DOI] [PubMed] [Google Scholar]
- 4.Moody SA. Fates of the blastomeres of the 32-cell-stage Xenopus embryo. Developmental Biology. 1987;122(2):300–19. doi: 10.1016/0012-1606(87)90296-x. http://dx.doi.org/10.1016/0012-1606(87)90296-X. [DOI] [PubMed] [Google Scholar]
- 5.Blitz IL, Biesinger J, Xie X, Cho KWY. Biallelic genome modification in F0Xenopus tropicalis embryos using the CRISPR/Cas system. genesis. 2013;51(12):827–34. doi: 10.1002/dvg.22719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakayama T, Fish MB, Fisher M, Oomen-Hajagos J, Thomsen GH, Grainger RM. Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. genesis. 2013;51(12):835–43. doi: 10.1002/dvg.22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gans C, Northcutt RG. Neural Crest and the Origin of Vertebrates. A New Head Science. 1983;220(4594):268–73. doi: 10.1126/science.220.4594.268. [DOI] [PubMed] [Google Scholar]
- 8.Helms JA, Cordero D, Tapadia MD. New insights into craniofacial morphogenesis. Development. 2005;132(5):851–61. doi: 10.1242/dev.01705. [DOI] [PubMed] [Google Scholar]
- 9.Cordero DR, Brugmann S, Chu Y, Bajpai R, Jame M, Helms JA. Cranial neural crest cells on the move: Their roles in craniofacial development. American Journal of Medical Genetics Part A. 2011;155(2):270–9. doi: 10.1002/ajmg.a.33702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Douarin NM, Kalcheim C. The neural crest. Cambridge: Cambridge University Press; 1999. [Google Scholar]
- 11.Hörstadius SO. The neural crest : its properties and derivatives in the light of experimental research. London [u.a.]: Oxford University Press; 1950. [Google Scholar]
- 12.Trueb L, Hanken J. Skeletal development in Xenopus laevis (Anura: Pipidae) Journal of Morphology. 1992;214(1):1–41. doi: 10.1002/jmor.1052140102. [DOI] [PubMed] [Google Scholar]
- 13.Slater BJ, Liu KJ, Kwan MD, Quarto N, Longaker MT. Cranial Osteogenesis and Suture Morphology in Xenopus laevis: A Unique Model System for Studying Craniofacial Development. PLoS ONE. 2009;4(1):e3914. doi: 10.1371/journal.pone.0003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bae C-J, Saint-Jeannet J-P. Neural Crest Cells. Boston: Academic Press; 2014. Chapter 2 - Induction and Specification of Neural Crest Cells: Extracellular Signals and Transcriptional Switches A2 - Trainor Paul A; pp. 27–49. [Google Scholar]
- 15.Stuhlmiller TJ, García-Castro MI. Current perspectives of the signaling pathways directing neural crest induction. Cellular and Molecular Life Sciences. 2012;69(22):3715–37. doi: 10.1007/s00018-012-0991-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meulemans D, Bronner-Fraser M. Gene-Regulatory Interactions in Neural Crest Evolution and Development. Developmental Cell. 2004;7(3):291–9. doi: 10.1016/j.devcel.2004.08.007. http://dx.doi.org/10.1016/j.devcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Betancur P, Bronner-Fraser M, Sauka-Spengler T. Assembling Neural Crest Regulatory Circuits into a Gene Regulatory Network. Annual Review of Cell and Developmental Biology. 2010;26(1):581–603. doi: 10.1146/annurev.cellbio.042308.113245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simões-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142(2):242–57. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helms JA, Schneider RA. Cranial skeletal biology. Nature. 2003;423(6937):326–31. doi: 10.1038/nature01656. [DOI] [PubMed] [Google Scholar]
- 20.Theveneau E, Mayor R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Developmental Biology. 2012;366(1):34–54. doi: 10.1016/j.ydbio.2011.12.041. http://dx.doi.org/10.1016/j.ydbio.2011.12.041. [DOI] [PubMed] [Google Scholar]
- 21.Chai Y, Maxson RE. Recent advances in craniofacial morphogenesis. Developmental Dynamics. 2006;235(9):2353–75. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- 22.Trainor PA, Krumlauf R. Patterning the cranial neural crest: Hinbrain segmentation and hox gene plasticity. Nat Rev Neurosci. 2000;1(2):116–24. doi: 10.1038/35039056. [DOI] [PubMed] [Google Scholar]
- 23.Santagati F, Rijli FM. Cranial neural crest and the building of the vertebrate head. Nat Rev Neurosci. 2003;4(10):806–18. doi: 10.1038/nrn1221. [DOI] [PubMed] [Google Scholar]
- 24.Minoux M, Rijli FM. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development. 2010;137(16):2605–21. doi: 10.1242/dev.040048. [DOI] [PubMed] [Google Scholar]
- 25.Cesario JM, Malt AL, Jeong J. Developmental Genetics of the Pharyngeal Arch System. Colloquium Series on Developmental Biology. 2015;2(1):1–108. doi: 10.4199/C00127ED1V01Y201503DEB006. [DOI] [Google Scholar]
- 26.Borchers A, Epperlein H-H, Wedlich D. An assay system to study migratory behavior of cranial neural crest cells in Xenopus. Development Genes and Evolution. 2000;210(4):217–22. doi: 10.1007/s004270050307. [DOI] [PubMed] [Google Scholar]
- 27.Alfandari D, Cousin H, Gaultier A, Hoffstrom BG, DeSimone DW. Integrin α5β supports the migration of Xenopus cranial neural crest on fibronectin. Developmental Biology. 2003;260(2):449–64. doi: 10.1016/s0012-1606(03)00277-x. http://dx.doi.org/10.1016/S0012-1606(03)00277-X. [DOI] [PubMed] [Google Scholar]
- 28.Theveneau E, Mayor R. Beads on the Run: Beads as Alternative Tools for Chemotaxis Assays. In: Wells CM, Parsons M, editors. Cell Migration: Developmental Methods and Protocols. Totowa, NJ: Humana Press; 2011. pp. 449–60. [DOI] [PubMed] [Google Scholar]
- 29.Milet C, Monsoro-Burq AH. Dissection of Xenopus laevis Neural Crest for in vitro. Explant Culture or in vivo Transplantation. 2014;(85):e51118. doi: 10.3791/51118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadaghiani B, Thiébaud CH. Neural crest development in the Xenopus laevis embryo, studied by interspecific transplantation and scanning electron microscopy. Developmental Biology. 1987;124(1):91–110. doi: 10.1016/0012-1606(87)90463-5. http://dx.doi.org/10.1016/0012-1606(87)90463-5. [DOI] [PubMed] [Google Scholar]
- 31.Gross JB, Hanken J. Use of fluorescent dextran conjugates as a long-term marker of osteogenic neural crest in frogs. Developmental Dynamics. 2004;230(1):100–6. doi: 10.1002/dvdy.20036. [DOI] [PubMed] [Google Scholar]
- 32.Gross JB, Hanken J. Cranial neural crest contributes to the bony skull vault in adult Xenopus laevis: Insights from cell labeling studies. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution. 2005;304B(2):169–76. doi: 10.1002/jez.b.21028. [DOI] [PubMed] [Google Scholar]
- 33.Gross JB, Hanken J. Segmentation of the vertebrate skull: neural-crest derivation of adult cartilages in the clawed frog, Xenopus laevis. Integrative and Comparative Biology. 2008;48(5):681–96. doi: 10.1093/icb/icn077. [DOI] [PubMed] [Google Scholar]
- 34.Jones K, Smith D. Recognition of the Fetal Alcohol Syndrome in early infancy. The Lancet. 1973;302(7836):999–1001. doi: 10.1016/s0140-6736(73)91092-1. http://dx.doi.org/10.1016/S0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- 35.Riley EP, Infante MA, Warren KR. Fetal Alcohol Spectrum Disorders: An Overview. Neuropsychology review. 2011;21(2):73–80. doi: 10.1007/s11065-011-9166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson VP, Swayze VW, Sato Y, Andreasen NC. Fetal alcohol syndrome: Craniofacial and central nervous system manifestations. American Journal of Medical Genetics. 1996;61(4):329–39. doi: 10.1002/(SICI)1096-8628(1996020261:4<329::AID-AJMG6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 37.Sant’Anna LB, Tosello DO. Fetal alcohol syndrome and developing craniofacial and dental structures – a review. Orthodontics & Craniofacial Research. 2006;9(4):172–85. doi: 10.1111/j.1601-6343.2006.00377.x. [DOI] [PubMed] [Google Scholar]
- 38.Nakatsuji N. Craniofacial malformation in Xenopus laevis tadpoles caused by the exposure of early embryos to ethanol. Teratology. 1983;28(2):299–305. doi: 10.1002/tera.1420280220. [DOI] [PubMed] [Google Scholar]
- 39.Yelin R, Ben-Haroush Schyr R, Kot H, Zins S, Frumkin A, Pillemer G, et al. Ethanol exposure affects gene expression in the embryonic organizer and reduces retinoic acid levels. Developmental Biology. 2005;279(1):193–204. doi: 10.1016/j.ydbio.2004.12.014. http://dx.doi.org/10.1016/j.ydbio.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 40.Yelin R, Kot H, Yelin D, Fainsod A. Early molecular effects of ethanol during vertebrate embryogenesis. Differentiation. 2007;75(5):393–403. doi: 10.1111/j.1432-0436.2006.00147.x. http://dx.doi.org/10.1111/j.1432-0436.2006.00147.x. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Li J, Chen C, Gong M, Chen Y, Liu Y, et al. 5-mehtyltetrahydrofolate rescues alcohol-induced neural crest cell migration abnormalities. Molecular Brain. 2014;7(1):67. doi: 10.1186/s13041-014-0067-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Czarnobaj J, Bagnall KM, Bamforth JS, Milos NC. The different effects on cranial and trunk neural crest cell behaviour following exposure to a low concentration of alcohol in vitro. Archives of Oral Biology. 2014;59(5):500–12. doi: 10.1016/j.archoralbio.2014.02.005. http://dx.doi.org/10.1016/j.archoralbio.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Yu W, Serrano M, Miguel SS, Ruest LB, Svoboda KKH. Cleft lip and palate genetics and application in early embryological development. Indian Journal of Plastic Surgery : Official Publication of the Association of Plastic Surgeons of India. 2009;42(Suppl):S35–S50. doi: 10.4103/0970-0358.57185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schutte BC, Murray JC. The Many Faces and Factors of Orofacial Clefts. Human Molecular Genetics. 1999;8(10):1853–9. doi: 10.1093/hmg/8.10.1853. [DOI] [PubMed] [Google Scholar]
- 45.Dickinson AJG. Using frogs faces to dissect the mechanisms underlying human orofacial defects. Seminars in Cell & Developmental Biology. 2016;51:54–63. doi: 10.1016/j.semcdb.2016.01.016. http://dx.doi.org/10.1016/j.semcdb.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kennedy AE, Dickinson AJG. Median facial clefts in Xenopus laevis: Roles of retinoic acid signaling and homeobox genes. Developmental Biology. 2012;365(1):229–40. doi: 10.1016/j.ydbio.2012.02.033. http://dx.doi.org/10.1016/j.ydbio.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 47.Dickinson AJG, Sive HL. The Wnt antagonists Frzb-1 and Crescent locally regulate basement membrane dissolution in the developing primary mouth. Development. 2009;136(7):1071–81. doi: 10.1242/dev.032912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilcox AJ, Lie RT, Solvoll K, Taylor J, McConnaughey DR, Åbyholm F, et al. Folic acid supplements and risk of facial clefts: national population based case-control study. BMJ. 2007;334(7591):464. doi: 10.1136/bmj.39079.618287.0B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lucock M. Folic Acid: Nutritional Biochemistry, Molecular Biology, and Role in Disease Processes. Molecular Genetics and Metabolism. 2000;71(1–2):121–38. doi: 10.1006/mgme.2000.3027. http://dx.doi.org/10.1006/mgme.2000.3027. [DOI] [PubMed] [Google Scholar]
- 50.Martinelli M, Girardi A, Cura F, Carinci F, Morselli PG, Scapoli L. Evidence of the involvement of the DHFR gene in nonsyndromic cleft lip with or without cleft palate. European Journal of Medical Genetics. 2014;57(1):1–4. doi: 10.1016/j.ejmg.2013.12.002. http://dx.doi.org/10.1016/j.ejmg.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 51.Wahl SE, Kennedy AE, Wyatt BH, Moore AD, Pridgen DE, Cherry AM, et al. The role of folate metabolism in orofacial development and clefting. Developmental biology. 2015;405(1):108–22. doi: 10.1016/j.ydbio.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foster JW, Dominguez-Steglich MA, Guioli S, Kwok C, Weller PA, Stevanovic M, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372(6506):525–30. doi: 10.1038/372525a0. [DOI] [PubMed] [Google Scholar]
- 53.Mansour S, Hall CM, Pembrey ME, Young ID. A clinical and genetic study of campomelic dysplasia. Journal of Medical Genetics. 1995;32(6):415–20. doi: 10.1136/jmg.32.6.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Houston CS, Opitz JM, Spranger JW, Macpherson RI, Reed MH, Gilbert EF, et al. The campomelic syndrome: Review, report of 17 cases, and follow-up on the currently 17-year-old boy first reported by Maroteaux et al in 1971. American Journal of Medical Genetics. 1983;15(1):3–28. doi: 10.1002/ajmg.1320150103. [DOI] [PubMed] [Google Scholar]
- 55.Mansour S, Offiah AC, McDowall S, Sim P, Tolmie J, Hall C. The phenotype of survivors of campomelic dysplasia. Journal of Medical Genetics. 2002;39(8):597–602. doi: 10.1136/jmg.39.8.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79(6):1111–20. doi: 10.1016/0092-8674(94)90041-8. http://dx.doi.org/10.1016/0092-8674(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 57.Meyer J, Südbeck P, Held M, Wagner T, Schmitz ML, Dagna Bricarelli F, et al. Mutational Analysis of the SOX9 Gene in Campomelic Dysplasia and Autosomal Sex Reversal: Lack of Genotype/Phenotype Correlations. Human Molecular Genetics. 1997;6(1):91–8. doi: 10.1093/hmg/6.1.91. [DOI] [PubMed] [Google Scholar]
- 58.Lee Y-H, Saint-Jeannet J-P. Sox9 function in craniofacial development and disease. Genesis (New York, NY: 2000) 2011;49(4):200–8. doi: 10.1002/dvg.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong C-S, Saint-Jeannet J-P. Sox proteins and neural crest development. Seminars in Cell & Developmental Biology. 2005;16(6):694–703. doi: 10.1016/j.semcdb.2005.06.005. http://dx.doi.org/10.1016/j.semcdb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Spokony RF, Aoki Y, Saint-Germain N, Magner-Fink E, Saint-Jeannet J-P. The transcription factor Sox9 is required for cranial neural crest development in Xenopus. Development. 2002;129(2):421–32. doi: 10.1242/dev.129.2.421. [DOI] [PubMed] [Google Scholar]
- 61.Lee Y-H, Aoki Y, Hong C-S, Saint-Germain N, Credidio C, Saint-Jeannet J-P. Early requirement of the transcriptional activator Sox9 for neural crest specification in Xenopus. Developmental Biology. 2004;275(1):93–103. doi: 10.1016/j.ydbio.2004.07.036. http://dx.doi.org/10.1016/j.ydbio.2004.07.036. [DOI] [PubMed] [Google Scholar]
- 62.Saint-Germain N, Lee Y-H, Zhang Y, Sargent TD, Saint-Jeannet J-P. Specification of the otic placode depends on Sox9 function in Xenopus. Development. 2004;131(8):1755–63. doi: 10.1242/dev.01066. [DOI] [PubMed] [Google Scholar]
- 63.Scambler PJ. The 22q11 deletion syndromes. Human Molecular Genetics. 2000;9(16):2421–6. doi: 10.1093/hmg/9.16.2421. [DOI] [PubMed] [Google Scholar]
- 64.Fomin ABF, Pastorino AC, Kim CA, Pereira AC, Carneiro- Sampaio M, Abe Jacob CM. DiGeorge Syndrome: a not so rare disease. Clinics. 2010;65(9):865–9. doi: 10.1590/S1807-59322010000900009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Papaioannou VE. The T-box gene family: emerging roles in development, stem cells and cancer. Development (Cambridge, England) 2014;141(20):3819–33. doi: 10.1242/dev.104471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, et al. Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410(6824):97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 67.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27(3):286–91. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 68.Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Human Molecular Genetics. 2002;11(8):915–22. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- 69.Ataliotis P, Ivins S, Mohun TJ, Scambler PJ. XTbx1 is a transcriptional activator involved in head and pharyngeal arch development in Xenopus laevis. Developmental Dynamics. 2005;232(4):979–91. doi: 10.1002/dvdy.20276. [DOI] [PubMed] [Google Scholar]
- 70.Tazumi S, Yabe S, Uchiyama H. Paraxial T-box genes, Tbx6 and Tbx1, are required for cranial chondrogenesis and myogenesis. Developmental Biology. 2010;346(2):170–80. doi: 10.1016/j.ydbio.2010.07.028. http://dx.doi.org/10.1016/j.ydbio.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 71.Hall BD. Choanal atresia and associated multiple anomalies. The Journal of Pediatrics. 1979;95(3):395–8. doi: 10.1016/s0022-3476(79)80513-2. http://dx.doi.org/10.1016/S0022-3476(79)80513-2. [DOI] [PubMed] [Google Scholar]
- 72.Källén K, Robert E, Mastroiacovo P, Castilla EE, Källén B. CHARGE association in newborns: A registry-based study. Teratology. 1999;60(6):334–43. doi: 10.1002/(SICI)1096-9926(19991260:6<334::AID-TERA5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 73.Blake KD, Prasad C. CHARGE syndrome. Orphanet Journal of Rare Diseases. 2006;1:34-. doi: 10.1186/1750-1172-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hughes SS, Welsh HI, Safina NP, Bejaoui K, Ardinger HH. Family history and clefting as major criteria for CHARGE syndrome. American Journal of Medical Genetics Part A. 2014;164(1):48–53. doi: 10.1002/ajmg.a.36192. [DOI] [PubMed] [Google Scholar]
- 75.Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide A-L, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. Journal of Medical Genetics. 2006;43(3):211–317. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, et al. Spectrum of CHD7 Mutations in 110 Individuals with CHARGE Syndrome and Genotype-Phenotype Correlation. The American Journal of Human Genetics. 2006;78(2):303–14. doi: 10.1086/500273. http://dx.doi.org/10.1086/500273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463 doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schulz Y, Wehner P, Opitz L, Salinas-Riester G, Bongers EMHF, van Ravenswaaij-Arts CMA, et al. CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Human Genetics. 2014;133(8):997–1009. doi: 10.1007/s00439-014-1444-2. [DOI] [PubMed] [Google Scholar]
- 79.Elsea SH, Williams SR. Smith–Magenis syndrome: haploinsufficiency of RAI1 results in altered gene regulation in neurological and metabolic pathways. Expert Reviews in Molecular Medicine. 2011;13 doi: 10.1017/S1462399411001827. [DOI] [PubMed] [Google Scholar]
- 80.Greenberg F, Guzzetta V, Montes de Oca-Luna R, Magenis RE, Smith AC, Richter SF, et al. Molecular analysis of the Smith-Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2) American Journal of Human Genetics. 1991;49(6):1207–18. [PMC free article] [PubMed] [Google Scholar]
- 81.Elsea SH, Girirajan S. Smith-Magenis syndrome. Eur J Hum Genet. 2008;16(4):412–21. doi: 10.1038/sj.ejhg.5202009. http://www.nature.com/ejhg/journal/v16/n4/suppinfo/5202009s1.html. [DOI] [PubMed] [Google Scholar]
- 82.Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet. 2003;33(4):466–8. doi: 10.1038/ng1126. http://www.nature.com/ng/journal/v33/n4/suppinfo/ng1126_S1.html. [DOI] [PubMed] [Google Scholar]
- 83.Imai Y, Suzuki Y, Matsui T, Tohyama M, Wanaka A, Takagi T. Cloning of a retinoic acid-induced gene, GT1, in the embryonal carcinoma cell line P19: neuron-specific expression in the mouse brain. Molecular Brain Research. 1995;31(1–2):1–9. doi: 10.1016/0169-328x(95)00020-s. http://dx.doi.org/10.1016/0169-328X(95)00020-S. [DOI] [PubMed] [Google Scholar]
- 84.Tahir R, Kennedy A, Elsea SH, Dickinson AJ. Retinoic acid induced-1 (Rai1) regulates craniofacial and brain development in Xenopus. Mechanisms of Development. 2014;133:91–104. doi: 10.1016/j.mod.2014.05.004. http://dx.doi.org/10.1016/j.mod.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 85.Nguyen H-L, Pieper GH, Wilders R. Andersen–Tawil syndrome: Clinical and molecular aspects. International Journal of Cardiology. 2013;170(1):1–16. doi: 10.1016/j.ijcard.2013.10.010. http://dx.doi.org/10.1016/j.ijcard.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 86.Tawil R, Ptacek LJ, Pavlakis SG, DeVivo DC, Penn AS, Özdemir C, et al. Andersen’s syndrome: Potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Annals of Neurology. 1994;35(3):326–30. doi: 10.1002/ana.410350313. [DOI] [PubMed] [Google Scholar]
- 87.Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou Sd, Tsunoda A, et al. Mutations in Kir2.1 Cause the Developmental and Episodic Electrical Phenotypes of Andersen’s Syndrome. Cell. 2001;105(4):511–9. doi: 10.1016/s0092-8674(01)00342-7. http://dx.doi.org/10.1016/S0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 88.Yoon G, Oberoi S, Tristani-Firouzi M, Etheridge SP, Quitania L, Kramer JH, et al. Andersen-Tawil syndrome: Prospective cohort analysis and expansion of the phenotype. American Journal of Medical Genetics Part A. 2006;140A(4):312–21. doi: 10.1002/ajmg.a.31092. [DOI] [PubMed] [Google Scholar]
- 89.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiological Reviews. 2010;90(1):291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 90.Wang H, Ma Y, Huynh J, Yu W, Xi Y, Hu P, et al. Functional characterization of KCNJ2 missense variants identified in patients with Andersen-Tawil Syndrome. Journal of the American College of Cardiology. 2012;59(13s1):E718–E. doi: 10.1016/S0735-1097(12)60719. [DOI] [Google Scholar]
- 91 •.Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) The Journal of Clinical Investigation. 2002;110(3):381–8. doi: 10.1172/JCI15183. This is one of the first studies using optogenetics in Xenopus to manipulate spatially and temporally the electrical state of non-neural cells during embryonic development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Adams DS, Uzel SGM, Akagi J, Wlodkowic D, Andreeva V, Yelick PC, et al. Bioelectric signalling via potassium channels: a mechanism for craniofacial dysmorphogenesis in KCNJ2-associated Andersen–Tawil Syndrome. The Journal of Physiology. 2016;594(12):3245–70. doi: 10.1113/JP271930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tseng A, Levin M. Cracking the bioelectric code: Probing endogenous ionic controls of pattern formation. Communicative & Integrative Biology. 2013;6(1):e22595. doi: 10.4161/cib.22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hamamy HA, Teebi AS, Oudjhane K, Shegem NN, Ajlouni KM. Severe hypertelorism, midface prominence, prominent/simple ears, severe myopia, borderline intelligence, and bone fragility in two brothers: New syndrome?†. American Journal of Medical Genetics Part A. 2007;143A(3):229–34. doi: 10.1002/ajmg.a.31594. [DOI] [PubMed] [Google Scholar]
- 95.Bonnard C, Strobl AC, Shboul M, Lee H, Merriman B, Nelson SF, et al. Mutations in IRX5 impair craniofacial development and germ cell migration via SDF1. Nat Genet. 2012;44(6):709–13. doi: 10.1038/ng.2259. http://www.nature.com/ng/journal/v44/n6/abs/ng.2259.html-supplementary-information. [DOI] [PubMed] [Google Scholar]
- 96.Cavodeassi F, Modolell J, Gómez-Skarmeta JL. The Iroquois family of genes: from body building to neural patterning. Development. 2001;128(15):2847–55. doi: 10.1242/dev.128.15.2847. [DOI] [PubMed] [Google Scholar]
- 97.Theveneau E, Marchant L, Kuriyama S, Gull M, Moepps B, Parsons M, et al. Collective Chemotaxis Requires Contact-Dependent Cell Polarity. Developmental Cell. 2010;19(1):39–53. doi: 10.1016/j.devcel.2010.06.012. http://dx.doi.org/10.1016/j.devcel.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Müller T, Mizumoto S, Suresh I, Komatsu Y, Vodopiutz J, Dundar M, et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers–Danlos syndrome. Human Molecular Genetics. 2013;22(18):3761–72. doi: 10.1093/hmg/ddt227. [DOI] [PubMed] [Google Scholar]
- 99.Syx D, Van Damme T, Symoens S, Maiburg MC, van de Laar I, Morton J, et al. Genetic Heterogeneity and Clinical Variability in Musculocontractural Ehlers–Danlos Syndrome Caused by Impaired Dermatan Sulfate Biosynthesis. Human Mutation. 2015;36(5):535–47. doi: 10.1002/humu.22774. [DOI] [PubMed] [Google Scholar]
- 100.Trowbridge JM, Gallo RL. Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology. 2002;12(9):117R–25R. doi: 10.1093/glycob/cwf066. [DOI] [PubMed] [Google Scholar]
- 101 •.Gouignard N, Maccarana M, Strate I, von Stedingk K, Malmström A, Pera EM. Musculocontractural Ehlers–Danlos syndrome and neurocristopathies: dermatan sulfate is required for Xenopus neural crest cells to migrate and adhere to fibronectin. Disease Models & Mechanisms. 2016;9(6):607–20. doi: 10.1242/dmm.024661. This study used a combination of in vivo and in vitro approaches to establish that in Dse-depleted Xenopus embryos NCCs migration was defective, providing a strong basis for the disease mechanisms underlying craniofacial defects in Musculocontractural Ehlers-Danlos syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nager FR, de Reynier JP. Das Gehörorgan Bei Den Angeborenen Kopfmissbildungen. ORL. 1948;10(suppl 2 Suppl. 2):53–90. [Google Scholar]
- 103.Schlieve T, Almusa M, Miloro M, Kolokythas A. Temporomandibular Joint Replacement for Ankylosis Correction in Nager Syndrome: Case Report and Review of the Literature. Journal of Oral and Maxillofacial Surgery. 2012;70(3):616–25. doi: 10.1016/j.joms.2011.02.053. http://dx.doi.org/10.1016/j.joms.2011.02.053. [DOI] [PubMed] [Google Scholar]
- 104.Bernier Francois P, Caluseriu O, Ng S, Schwartzentruber J, Buckingham Kati J, Innes AM, et al. Haploinsufficiency of SF3B4, a Component of the Pre-mRNA Spliceosomal Complex, Causes Nager Syndrome. The American Journal of Human Genetics. 2012;90(5):925–33. doi: 10.1016/j.ajhg.2012.04.004. http://dx.doi.org/10.1016/j.ajhg.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Petit F, Escande F, Jourdain AS, Porchet N, Amiel J, Doray B, et al. Nager syndrome: confirmation of SF3B4 haploinsufficiency as the major cause. Clinical Genetics. 2014;86(3):246–51. doi: 10.1111/cge.12259. [DOI] [PubMed] [Google Scholar]
- 106.Czeschik JC, Voigt C, Alanay Y, Albrecht B, Avci S, FitzPatrick D, et al. Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Human Genetics. 2013;132(8):885–98. doi: 10.1007/s00439-013-1295-2. [DOI] [PubMed] [Google Scholar]
- 107.Will CL, Lührmann R. Spliceosome Structure and Function. Cold Spring Harbor Perspectives in Biology. 2011;3(7) doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Devotta A, Juraver-Geslin H, Gonzalez JA, Hong C-S, Saint-Jeannet J-P. Sf3b4-depleted Xenopus embryos: A model to study the pathogenesis of craniofacial defects in Nager syndrome. Developmental Biology. 2016;415(2):371–82. doi: 10.1016/j.ydbio.2016.02.010. http://dx.doi.org/10.1016/j.ydbio.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey J-P, et al. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proceedings of the National Academy of Sciences. 2006;103(36):13403–8. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aoki Y, Saint-Germain N, Gyda M, Magner-Fink E, Lee Y-H, Credidio C, et al. Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Developmental Biology. 2003;259(1):19–33. doi: 10.1016/s0012-1606(03)00161-1. http://dx.doi.org/10.1016/S0012-1606(03)00161-1. [DOI] [PubMed] [Google Scholar]
- 111.Baltzinger M, Ori M, Pasqualetti M, Nardi I, Rijli FM. Hoxa2 knockdown in Xenopus results in hyoid to mandibular homeosis. Developmental Dynamics. 2005;234(4):858–67. doi: 10.1002/dvdy.20567. [DOI] [PubMed] [Google Scholar]
- 112.O’Donnell M, Hong C-S, Huang X, Delnicki RJ, Saint-Jeannet J-P. Functional analysis of Sox8 during neural crest development in Xenopus. Development. 2006;133(19):3817–26. doi: 10.1242/dev.02558. [DOI] [PubMed] [Google Scholar]