

ABSTRACT
This study explored the origin of age-related granules in the apolipoprotein E gene knockout (ApoE−/−) B6 background mice brains following chronic gingival infection with Porphyromonas gingivalis for 24 weeks. Intracerebral localization of P. gingivalis was detected by fluorescence in situ hybridization (FISH) and its protease by immunohistochemistry. The age-related granules were observed by periodic acid–Schiff (PAS), silver impregnation, and immunostaining. FISH showed intracerebral dissemination of P. gingivalis cells (p = 0.001). PAS and silver impregnation demonstrated the presence of larger inclusions restricted to the CA1, CA2, and dentate gyrus sectors of the hippocampus. A specific monoclonal antibody to bacterial peptidoglycan detected clusters of granules with variable sizes in mice brains infected with P. gingivalis (p = 0.004), and also highlighted areas of diffuse punctate staining equating to physical tissue damage. Mouse immunoglobulin G was observed in the capillaries of the cerebral parenchyma of all P. gingivalis–infected brains (p = 0.001), and on pyramidal neurons in some severely affected mice, compared with the sham-infected mice. Gingipains was also observed in microvessels of the hippocampus in the infected mice. This study supports the possibility of early appearance of age-related granules in ApoE−/− mice following inflammation-mediated tissue injury, accompanied by loss of cerebral blood-brain barrier integrity.
KEYWORDS: Age-related granules, blood–brain barrier damage, infection, inflammation, injury
The origins of murine age-related granules, first observed in the brains of senescence-accelerated mouse (SAM) models [1], continue to intrigue scientists. These inclusions appear around 7–8 months of age in SAM mice and have an intimate association with astrocytes [2]. The age-related granules stain positively with periodic acid–Schiff (PAS) and display argyrophilia following silver impregnation. Their location is largely restricted to the stratum radiatum of the hippocampus [1,2]. It was initially thought that the high inbreeding of SAM mice may have conferred an inheritable genetic susceptibility locus in these mice that enabled the emergence of age-related granules, as they were absent in mice without the B6 genetic background [2]. Further reports of the same inclusions being observed in the brains of SAMP8 (B6) mice strengthened this genetic inheritance theory [2,3], but conclusive evidence is still lacking.
Greater densities of these inclusions are seen in regions of the brain that bear a higher burden of neuropathological hallmarks (amyloid-beta Aβ, and tau tangles) of Alzheimer’s disease (AD), suggesting a potential role of these granules in learning or memory deficits [4,5]. Many antigenic protein components are reportedly involved in the etiology of age-related granules. Among them, heparan sulfate proteoglycan (HSPG) is considered to be significant [3], as HSPG is seen as a plausible substrate onto which extracellular Aβ plaques may become deposited in the suitable host [6–9].
Numerous studies have highlighted a commonality between the aging human brain and apolipoprotein E gene knockout (ApoE−/−) mice in that they both demonstrate ongoing damage to the blood–brain barrier (BBB) [10–12] albeit in different anatomical areas. How cerebral BBB damage initiates in humans is poorly understood, but in ApoE−/− mice brains, compromised BBB integrity appears to be secondary to experimentally induced intracerebral tissue injury [13]. ApoE−/− mice also demonstrate altered innate immune responses, especially after bacterial infections and in response to the lipopolysaccharide (LPS) endotoxin [14,15]. During bacterial infections, these mice activate acute phase innate immune responses in which the immediate immunologic response (acute phase) is elicited by tissue injury from free radicals, which are released by macrophages and ongoing non-specific microbe killing, as well as via the activated complement cascade [16].
A plausible association between periodontal disease and AD was previously demonstrated by detecting LPS from the oral bacterium Porphyromonas gingivalis in 4/10 AD brain specimens [17]. Subsequently, the intravascular dissemination of P. gingivalis from the same gingival tissue into the brain tissue of ApoE− / − mice was demonstrated [16,18]. Furthermore, in the same study, it was demonstrated that the hippocampal CA neurons were opsonized with complement activation fragments iC3b/C3b/C3d as a consequence of local P. gingivalis entry [16]. It was concluded that the innate immune activation serves as a critical risk factor for the development of AD inflammatory pathology, with secondary tissue damage in the ApoE−/− phenotype following periodontal bacterial infection.
A reduction in the number of age-related granules following antioxidant therapy in ApoE−/− mice was reported [19], potentiating the impact of inflammation to inflict injury on the cerebral parenchyma. The antioxidant therapy data [19] and the common feature of a defective BBB in humans and ApoE−/− mice [10,11,13] together served as the rationale behind the initiation of this study to explore the role of periodontal infection in augmenting earlier occurrence of age-related granules. This study tested the hypothesis of whether chronic periodontal infection accelerated the appearance of age-related granules in ApoE−/− mice brains.
Materials and methods
In vivo infection mouse model
Male ApoE−/− mice (strain B6.129P2-Apoetm1Unc/J) were obtained from Jackson Laboratories (Bar Harbor, MA). At 10 weeks of age, the mice were randomly assigned to sham-infected or infected groups via the oral route with P. gingivalis FDC 381 (5 × 109 bacteria per mL), as described previously [18]. The University of Florida Animal Care Services has an assurance with the Office of Laboratory Animal Welfare (OLAW) and follows US Public Health Services (PHS) policy, the Animal Welfare Act and Animal Welfare Regulations, and the Guide for the Care and Use of Laboratory Animals. The University of Florida is also accredited with the Association for Assessment and Accreditation of Laboratory Animal Care. All experimental procedures with the mice were approved by the Institutional Animal Care and Use Committee at the University of Florida (Gainesville, FL; Protocol # 201304539) and the Animal Welfare and Ethics Review Board at the University of Central Lancashire (Preston, UK; reference no: RE/12/04) before the study began.
Localization of P. gingivalis in brain tissue by fluorescence in situ hybridization
Formalin-fixed, paraffin-embedded tissue blocks (temporal lobe inclusive of the hippocampus) were sectioned (5 µm thick). Fluorescence in situ hybridization (FISH) was performed using probes specific to bacterial ribosomal 16S rRNA gene sequences on rehydrated cortical (frontotemporal lobe) tissue sections from sham-infected and P. gingivalis–infected mice, as described previously [18]. Briefly, tissue sections were probed with 5 mg/mL of P. gingivalis 16S rRNA-specific oligonucleotide POGI [20] 5ʹ-CAA TAC TCG TAT CGC CCG TTA TTC-3ʹ labeled with Alexa Fluor 568 dye (Invitrogen, Carlsbad, CA) in hybridization solution. Following hybridization with the probe, slides were counterstained with DRAQ5 (Thermo Fisher Scientific, Asheville, NC) and mounted in Mowiol 4–88 (Sigma–Aldrich, St. Louis, MO). Data were acquired with a 63× objective on a Leica DMIRB microscope equipped with a Photometrics cascade-cooled EMCCD camera, controlled by the open-source software package μManager (http://www.micromanager.org). Images were processed using ImageJ (NCBI). Blocking buffer, hybridization buffer, and wash buffer all contained protectRNA (Sigma–Aldrich) to protect bacterial RNA from degradation [19].
Detection of P. gingivalis gingipains in the brain
Snap-frozen unfixed infected mouse brain tissue sections from the hippocampus were cut, as previously described [16]. The sections were stabilized in cold acetone for 15 min and washed in phosphate-buffered saline (PBS) before being incubated for 30 min at room temperature in block buffer (PBS containing 0.01% Tween 20 and 0.01% normal goat serum) followed by overnight incubation in mouse anti-gingipains (clone 1A1, from Prof. MA Curtis, London, UK) diluted 1/200 in block buffer. The secondary detection was performed with biotin-labelled anti-mouse Ig antibody (Vectastain ABC kit PK-4002), as per the manufacturer’s instructions. Sections were treated with DAB/hydrogen peroxide/nickel enhancement (Vectastain substrate kit SK-4100), again according to the manufacturer’s instructions. Following a light nuclear counterstain, all sections were examined using the Nikon Eclipse E200 microscope. Images of representative tissue sites were recorded using the Nikon DS-L2 v.441 software.
Detection of age-related granules by neutral methods
PAS reaction
Rehydrated paraffin wax sections from the cortex, inclusive of the hippocampus, from sham-infected ApoE−/− and P. gingivalis–infected mice were treated in commercial PAS histochemical reagent (Sigma 395–2™), according to the manufacturer’s instructions. The sections were dehydrated, cleared in xylene, and mounted under a glass coverslip in DPX mounting medium (Thermo Fisher Scientific).
Silver impregnation
Rehydrated semi-serial sections from the sham-infected and P. gingivalis–infected mice brains were impregnated with silver, according to a published method used on acrylic resin sections [21]. The resin-based method was adapted to rehydrated, fixed tissue sections.
Immunohistochemistry: pan detection of age-related granules
Rehydrated, fixed brain tissue sections were pretreated in 10% aqueous sodium dodecyl sulfate (SDS) and stained, as described by Miklossy et al. [22], with an overnight incubation at 4°C with the primary antibody to bacterial-specific peptidoglycan (Clone MAB995) diluted 1/200. The secondary detection/examination and imaging were performed as described above.
Demonstration of mouse immunoglobulin G in cerebral microvasculature
Anti-mouse immunoglobulin G (IgG) immunohistochemistry was performed using the published method of Fullerton et al. [23] with the peroxidase mouse-IgG ABC kit PK-4002 (Vector Labs, Peterborough, UK). Sections were developed in DAB/hydrogen peroxide/nickel and subsequently examined and imaged as described above.
Statistical analysis
Data are presented as mean ± standard deviation (n ≥ 3 replicates per treatment). The non-parametric Mann–Whitney U-test was performed to compare each group of infected mice with the sham-infected mice. Differences were considered significant at p ≤ 0.05.
Results
Demonstration of bacteria in tissue sections from the frontotemporal lobe
The sham-infected FISH sections did not show any bacterial hybridization product (Figure 1(a)). The P. gingivalis–infected brain sections (6/6; p = 0.001) showed localization of the hybridization product with the rRNA probe to bacterial aggregates that were located at the perinuclear regions of brain cells (marker antibody identity of cells was not performed; Figure 1(b), arrowheads) and extracellularly in the frontotemporal lobe of the cerebral cortex tissue sections as green dots (Figure 1(b), boxed areas) in the 24-week-infected mice.
Figure 1.
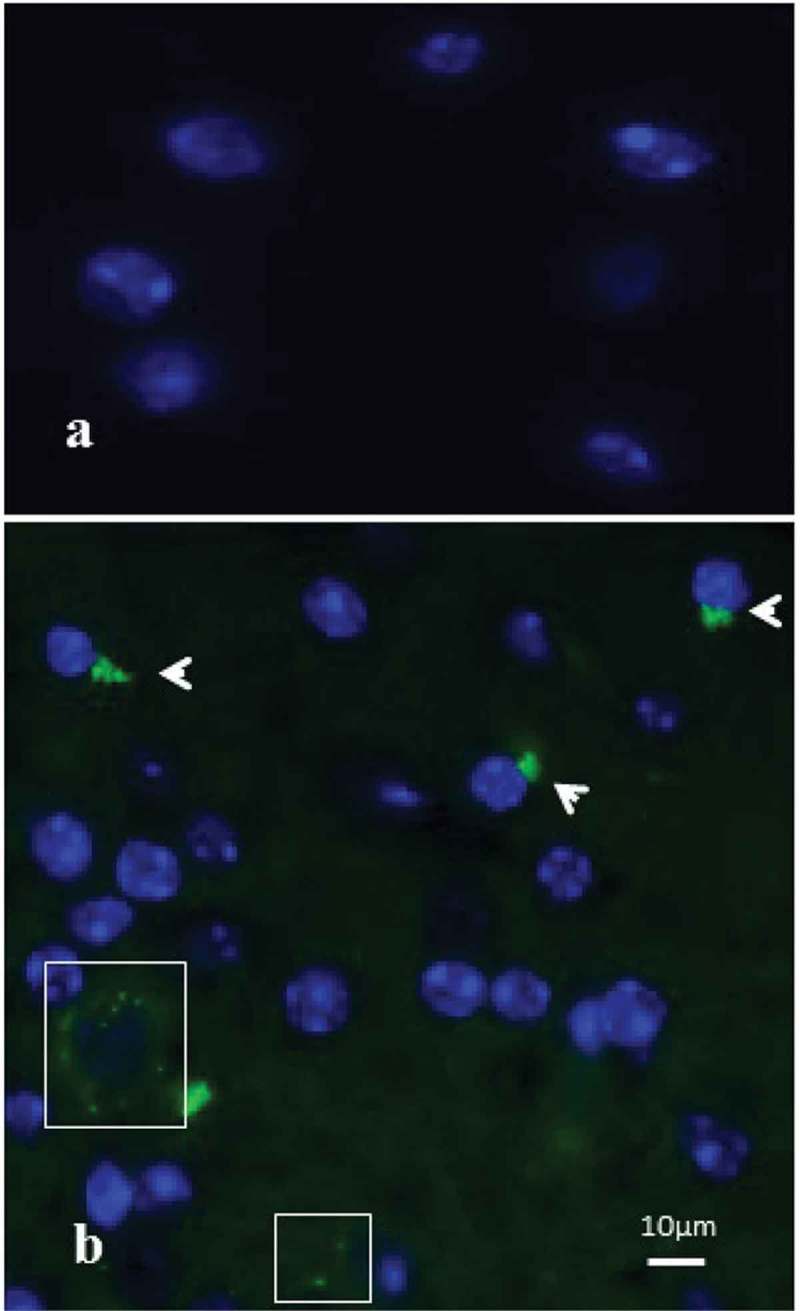
Fluorescent in-situ hybridization (FISH) demonstrating intracellular localization of Porphyromonas gingivalis in temporal cortex tissue sections from apolipoprotein E gene knockout (ApoE−/−) mice. (a) Sham-infected mouse brain, devoid of any bacterial hybridization product. (b) ApoE−/− mouse brain tissue infected with P. gingivalis (24 weeks) revealing the presence of intracellular (arrowheads) P. gingivalis green dots (boxes).
Demonstration of gingipains in the cerebral microvasculature
Following immunohistochemistry with anti-gingipains, little staining was observed in all sections whereby the primary antibody was omitted (Figure 2(a and b)). The anatomical area known to be free of the BBB, namely the choroid plexus (acting as a positive control), from direct brain infection by P. gingivalis, demonstrated anti-gingipains staining (Figure 2(c)). In addition, the anti-gingipains staining was also observed in major and microvessels of the hippocampus from infected mice (Figure 2(d-i)).
Figure 2.
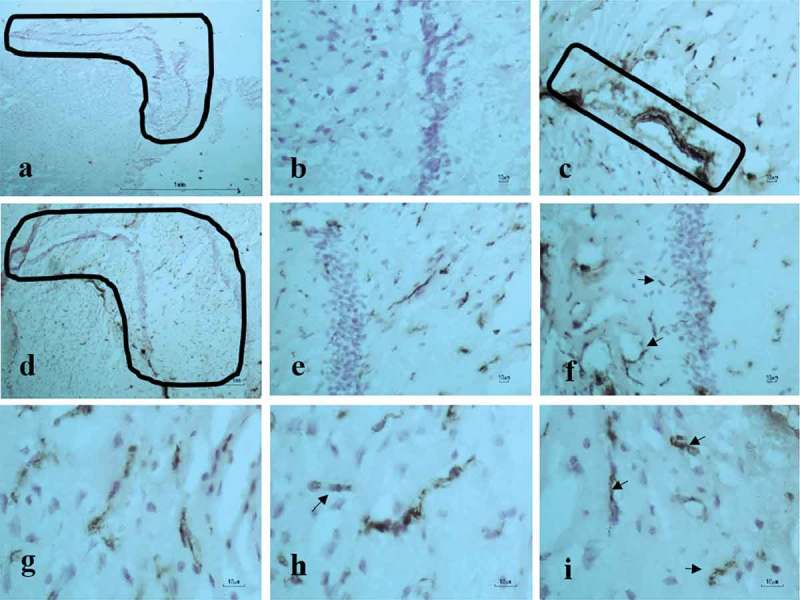
P. gingivalis–infected ApoE−/− mouse brain. Immunohistochemical detection of gingipains in frozen brain tissue sections. (a-b) Negative controls in hippocampus demarcated by an L-shaped box, and (b) higher magnification of the hippocampus adjacent to pyramidal neurons, which are free of staining. (c) Choroid plexus as a positive control for the blood–brain barrier (BBB) free site in the brain. (d) Anti-gingipains staining. (e-i) Specific anti-gingipains antibody staining in the microvessels (arrows) of the hippocampus at higher magnification (see micron bar).
Detection of age-related granules by neutral methods
Infected mice brain sections demonstrated age-related, PAS-positive granules that were generally larger or mature and extracellular in appearance (Figure 3(a), boxes). The two boxed areas with stars show the same granules (Figure 3(b and c)). The granules within the hippocampus of ApoE−/− mice brain sections also demonstrated a degree of argyrophilia, which was difficult to appreciate at low magnification but imperative for showing their location in the CA1 area of the hippocampus (Figure 3(d), box). The same boxed area is shown again in Figure 3(e), demarcated by two boxes and a black star. The inclusions within the smaller rectangular box with broken lines in Figure 3(e) are enlarged in Figure 3(f) (box), and the site flagged by a black star in Figure 3(e) is enlarged to show three clusters of granules close together in Figure 3(g) (star).
Figure 3.
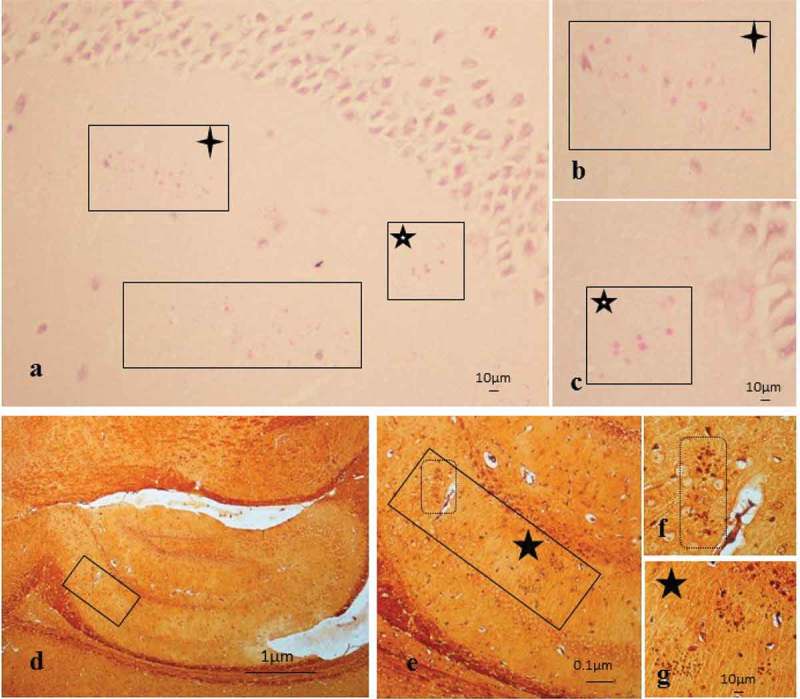
Age-related granules in the brain tissue of infected mice. (a-c) Periodic acid–Schiff reaction in a P. gingivalis tissue section from the hippocampus region showing the mature (large) age-related granules demarcated by boxes. (b-c) Boxes with four-tailed stars in (a and b) and five-tailed star with white core (a and c) are the same regions of the hippocampus, with scale bar shown for clarity. (d) Methenamine silver impregnated tissue section showing an overview of the location of inclusions in the CA1 area of the hippocampus; the same inclusions shown in (e–g) are denoted by boxes and stars.
Immunohistochemistry
Pan-detection of age-related granules
P. gingivalis–infected (24 weeks) ApoE−/− mouse brain tissue sections stained with the bacterial-specific ascites monoclonal anti-peptidoglycan antibody demonstrated 10/12 brains with numerous clusters (p = 0.004) that were of variable size and shape (immature and mature; Figure 4(a-c), arrows and boxes). Their location was observed in close proximity to CA2 pyramidal neurons (Figure 4(a and b), smaller rectangle) and within the CA1 subfield of the hippocampus (Figure 4). The rectangular area in Figure 4(a) is enlarged in Figure 4(b), illustrating the presence of age-related granules in the stratum radiatum (Figure 4(b), arrow and a box with arrows and a broken line circle). The encircled cluster of granules and arrows in box in Figure 4(b) is shown more clearly in Figure 4(c), accompanied by many more clusters in its vicinity. P. gingivalis–infected mice brains demonstrated areas of diffuse punctate staining within the hippocampus (stratum radiatum area of the CA1 and CA2l Figure 4(d)). Only 2/12 sham-infected mice at 24 weeks contained significant numbers of age-related granules observed largely within the stratum radiatum and CA1 (Figure 4(e-g), arrow and boxes) and CA2 regions of the hippocampus. No areas with diffuse punctate staining were evident in the hippocampus of any of the sham-infected mice brains (Figure 4(h)).
Figure 4.
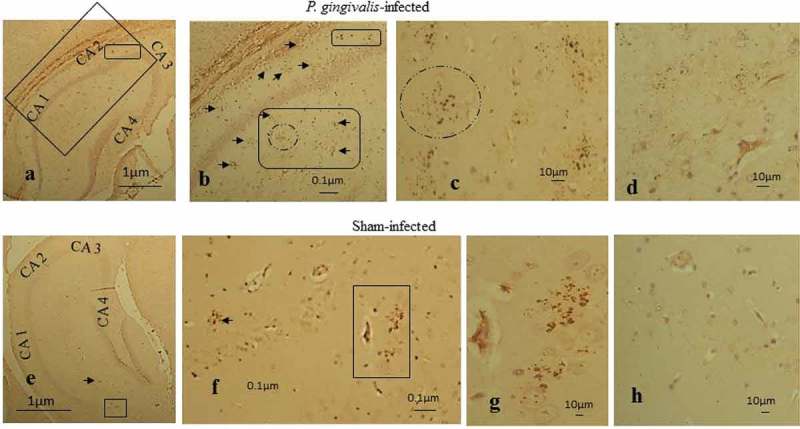
Peptidoglycan immunostaining of infected ApoE−/− mouse brains with hippocampal CA areas labeled (CA1-3). (a-c) P. gingivalis–infected tissue section with numerous clusters located at the boundary of CA2 neurons (smaller box) and (b) in the stratum radiatum CA1 (arrows and larger box with circle). (d) Diffuse immunostaining with anti-peptidoglycan antibody. (e) Fewer age-related granules in the CA1 region of the hippocampus in sham-infected mice indicated by small arrow and box. (f) Arrow pointing to a cluster and two clusters within box and in (g) without the box. (h) Sham-infected ApoE−/− mouse brain devoid of any diffuse staining.
Demonstration of IgG in cerebral microvasculature
At 12 weeks of infection, both ApoE−/− mouse brains infected with P. gingivalis and sham-infected mice presented plasma IgG in the cerebellar cortical tissue vasculature (arteries, arterioles, veins, venules, and capillaries) and the choroid plexus (Figure 5(a-c)) but not in the capillaries of the cerebral parenchymal tissues. The controls in which the primary antibody was omitted consistently remained negative (Figure 5(d-g)).
Figure 5.
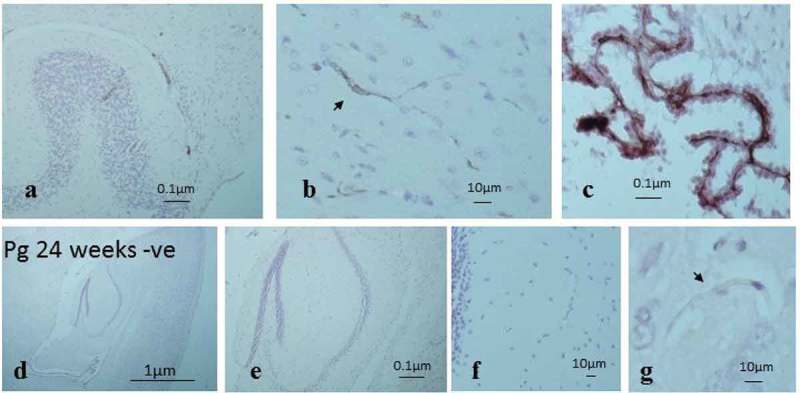
Demonstration of mouse IgG in the cerebellar cortex of sham-infected mouse brain tissue to confirm the impaired BBB. (a) A cerebellar folium showing larger vessels positive for IgG. (b) Capillary (arrow) from the cerebellum labelled with IgG. (c) Choroid plexus as a positive internal tissue control as it lacks the BBB. (d-g) Brain tissue sections from infected ApoE−/− mouse as negative controls (anti mouse-IgG omitted) showing they remained free of any staining.
At 24 weeks of infection, all P. gingivalis–infected brains (n = 12) demonstrated IgG within the expected areas (arteries, arterioles, veins, venules, choroid plexus) and especially in the capillaries of the cerebral and stratum radiatum CA3 regions of the hippocampus (Figure 6(a-f), box). Overall, P. gingivalis–infected mice brains at 24 weeks had greater numbers of age-related granules and appeared with more widespread IgG in cerebral capillaries (p = 0.001), and in some mouse brains, the IgG also bound to pyramidal neurons (Figure 6(c and d)). Capillaries in the stratum radiatum CA3 regions of the hippocampus of some P. gingivalis–infected mice appeared more structurally damaged (Figure 6(e and f) from 6c boxed area) than those from similar regions of the brain in other infected mice (Figure 6(g-i), arrows).
Figure 6.
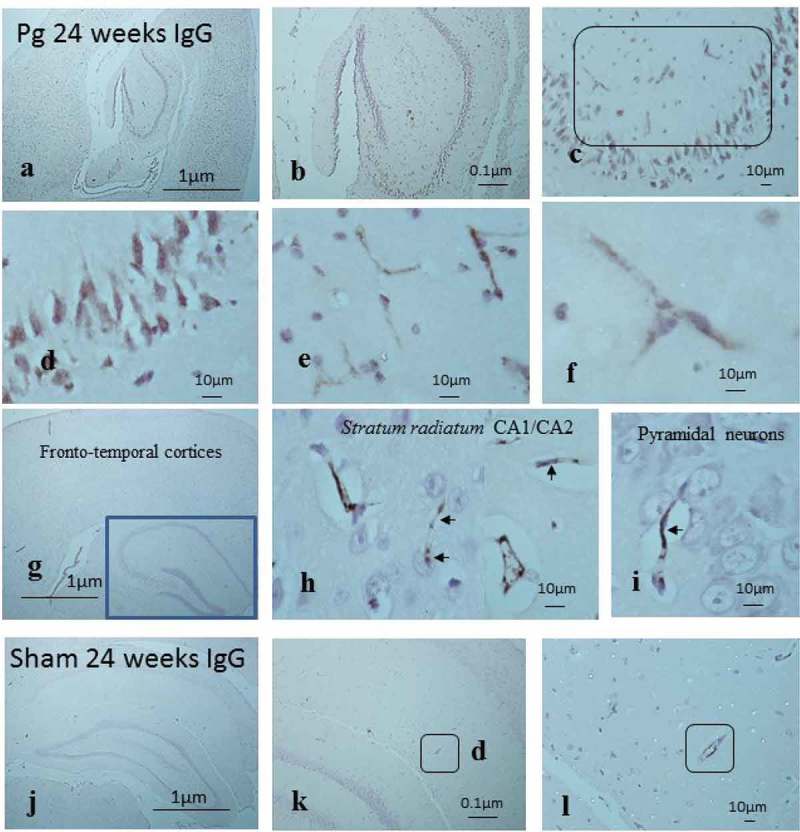
Demonstration of mouse IgG in the cortical brain including the hippocampus to show an impaired BBB due to infection. (a-c) P. gingivalis–infected brain with IgG immunostaining not only on severely damaged capillaries in the stratum radiatum (c, box) of the hippocampus but also on pyramidal neurons. (b-d) Regions from (c) at higher magnification in (e) and (f) (see micron bar). (g-h) Another P. gingivalis–infected brain with IgG immunostaining to illustrate structurally less damaged capillaries in the stratum radiatum (h) of the hippocampus but also (i) not opsonized to pyramidal neurons. (j-l) Sham-infected brains exposed to labeled anti-IgG (k-l) show larger vessels immune-positive for mouse IgG (boxes).
At 24 weeks, the 10/12 sham-infected brains with insignificant numbers of inclusion clusters also demonstrated IgG within the expected areas (larger vessels, choroid plexus; Figure 6(j–l), boxes) but not in the cerebral/hippocampal capillaries. The 2/12 sham-infected mice brains that demonstrated numerous age-related clusters also demonstrated IgG within cerebral/hippocampal capillaries (not shown).
Discussion
The current study was undertaken to investigate whether chronic periodontal bacterial infection could increase circulating inflammatory mediators and could enable bacteria or their structures/antigenic determinants to disseminate into the brain, thereby providing the necessary inflammatory catalyst to trigger the occurrence of PAS-positive, argyrophilic age-related granules. Previous observations in P. gingivalis–infected ApoE−/− mice revealed that chronic periodontal infection provided a higher burden of inflammatory mediators [18] and demonstrated that P. gingivalis was able to initiate microglial cell activation and promote the synthesis of innate immune inflammatory proteins of the complement cascade [16]. This has direct implications for the survival of neurons, as they lack adequate protection from regulatory proteins on their cell surface membranes [24]. Thus, the presence of a higher inflammatory burden as a result of systemic proliferation of bacteria [18], genetic susceptibility toward recurrent infections [25,26], and direct entry of bacteria (P. gingivalis) into the brain as a result of chronic bacterial infection [16] would provide a catalyst capable of triggering cerebral injury.
Veurink et al. [19] observed that feeding mice with antioxidants resulted in the occurrence of fewer numbers of age-related inclusions in their brains, suggesting an inverse association between inflammatory stress and inclusion appearance in ApoE−/− mouse brains. It was also reported that ApoE−/− mice have a leaky BBB starting at the age of 2 weeks [12], which can be detected in the cerebellar cortex in an age-dependent manner [11,23]. Notably, the cerebral BBB appears to be more resilient to physical injury in a normal host and requires the impact of initial experimental injury to breach it [13]. In vivo observations from various studies [19,23], along with subsequent human data regarding defective hippocampal BBB [10], also support the likelihood of multiple antigenic proteins (infection-related proteins, IgM, and other serum components) associated with age-related granules.
The results from this study are in agreement with a previous study [16] in which the same P. gingivalis (FDC 381) strain was identified as having accessed the cerebral parenchyma of the infected group of ApoE−/− mouse brains. The direct implication of P. gingivalis invasion into the brain is the introduction of its proteinase enzymes known as gingipains, which are intrinsically associated with the outer surface membrane of the bacterium and break down connective tissues. These proteinases represent an alternative source of physical tissue injury to secondary inflammatory mediators, and demonstrate direct, progressive injury to cerebral microvasculature in which endothelial cells and their tight junction proteins become eroded.
The neutral histology techniques confirmed PAS-positive argyrophilic granules in the hippocampus of infected ApoE−/− mice, and their distribution resembled previous observations [1–3,27,28]. The immunohistochemistry results with the anti-peptidoglycan antibody indicated an enhanced occurrence of age-related granules in infected brains, together with diffuse areas of immunoreactivity when compared with sham-infected mice. The diffuse immunostaining can be equated to physical injury resulting from non-specific innate immune responses, such as oxidative stress and complement activation [16,19], and gingipains. The presence of host IgG antibodies within cerebral capillaries and their leakage into tissues of infected mice suggests that the BBB was severely impaired by 24-week infections. These observations further support the concept of the BBB becoming more permeable in the cerebral cortices following infection-mediated injury [13] and erosion of endothelial tight junctional proteins allowing IgG leakage. These data rationalize the observation of fewer age-related granules in the ApoE−/− mice following antioxidant treatment without infection [19].
Numerous studies have reported the ability of bacteria and their structures to access AD brains [17,29–32]. Once within the brain, they are likely to modulate the activation of the complement cascade [16], eventually giving rise to the development of characteristic pathological hallmarks of AD in the appropriate host. Poor memory has also been reported to be associated with multiple infections [33] and high acute phase inflammatory mediators such as interleukin 1 alpha (IL-1α) and IL-1β in the elderly and human clinical AD patients [34]. Thus, the high inflammatory milieu in which IL-1α and P. gingivalis specific IgG antibodies (humoral response) detected in the serum following chronic periodontal disease [18] could potentially contribute to the development of cognitive deficiencies in these mice. However, the scope of this retrospective study was restricted to assessing the occurrence of age-related granules only.
Age-related granules containing cellular debris originate from cells that are highly vulnerable to damage by reactive oxidative processes and higher inflammatory/pyroptosis mediated programmed cell death [35,36]. A likely explanation for the occurrence of these inclusions in the hippocampus over time in ApoE−/− mice could be their higher endogenous vascular inflammation [25,26], and oxidative stress, which results from an already impaired immuno-modulatory function of macrophages [15,37,38]. These processes likely lead to the ‘aging process’, which contributes to slow physical injury surpassing the threshold of protection. Thus, the net effect of the aging process is functional demise of BBB integrity, especially in the cerebral parenchyma, giving rise to increased numbers of age-related granules over time.
The fact that the infected mouse brains overall demonstrated more clusters of age-related granules in the hippocampus compared with the sham-infected mice at 24 weeks suggests that once pyroptotic cell death and physical injury to this anatomical area has occurred, the BBB becomes leaky. In another words, the formation and maturation of age-related granules becomes more accelerated following P. ginivalis infection of the brain than that during normal aging processes in ApoE−/− mice free of infection. The experiments were not conducted on animals maintained in pathogen-free conditions, and the likelihood of the ApoE−/− mice over the course of 24 weeks having harbored an irrelevant infection (both sham and infected mice) and indirectly contributed to peptidoglycan immunostaining is acknowledged but remains to be investigated. In this study, all 12 infected mouse brains demonstrated IgG in the cerebral capillaries, and only 2/12 mice did not show vast numbers of inclusions, which suggests differences in the threshold potential for handling inflammation in each mouse.
Significantly, a human equivalent to murine age-related granules, known as corpora amylacea, is known to occur in aged and demented human brains [39]. Corpora amylacea bear striking similarities with astrocytes [40]. Recent evidence also confirms that elderly and demented brains have a compromised physical BBB through the endothelial cells in capillaries [10,41], suggesting a common infection-related mechanism in the origin of these inclusions in both humans and ApoE−/− mice.
Microbleeds in the immuno-privileged areas of the brain are detrimental to normal functioning of the neurons because of the toxicity associated with some of the serum products (C-reactive protein) and metallic compounds. C-reactive protein has the ability to activate microglia and astrocytes, and entry of metallic compounds (Fe, Al) into the brain may aid in Aβ fibril formation. In addition, there remains a possibility that vascular amyloid proteins (Aβ, amyloidogenic proteins such as serum amyloid A from host and bacterial origins) may transfer to the brain and act as niduses for AD Aβ plaque deposition [42]. Additionally, the brain, in reaction to peripheral infections, may activate the proteases that cleave Aβ from its parent amyloid precursor protein on brain cell membranes and result in senile plaques [30–34,36–44].
This study also demonstrates a lag phase during the first 12 weeks of infection (acute injury phase) for inclusion formation, which supports observations from previous reports [1,2]. A plausible explanation for this could be that the tight endothelial cell junctions in the cerebral parenchyma protect the brain from extrinsic insults, increasing the time it takes for local inflammatory events to inflict a physical tissue injury. Overall, it is likely that physical tissue injury, as well as an impaired cerebral BBB resulting from enhanced systemic and local inflammatory burden following infections, are likely to accelerate aging and contribute to the occurrence of age-related granules in ApoE−/− mice.
Conclusion
This is the first investigation that provides evidence of a periodontal bacterial infection resulting in injury of the hippocampus, thereby increasing BBB permeability to toxic vascular components. Although the exact optimal conditions that could result in breaching the BBB and its threshold are still under intense investigation, this study suggests that at least eight consecutive infections of P. gingivalis over 24 weeks are sufficient to cause significant cerebral tissue injury and BBB impairment in the ApoE−/− mouse model. The diffuse staining in the hippocampus is attributed to physical tissue injury resulting from bacterial lysates and the generally high inflammatory burden. Lack of functional/repair protein (ApoE) and inflammation mediated-physical tissue injury culminate in an impaired BBB. Together, these contribute to astrocytic processing of cellular debris containing serum protein components in the form of age-related granules. Finally, the age-related granule formation could represent a host defense mechanism to mask the immunogenic proteins entering the hippocampus from microbleeds.
Disclosure statement
No potential conflict of interest was reported by the authors.
Biographies
Dr Sim K. Singhrao is a Senior Research Fellow in neurodegenerative diseases.
Dr Sasanka Chukkapalli is a Research Associate with an interest in infection mediated cardiovascular disease animal models.
Dr Sophie Poole is an early career scientist interested in Alzheimer's disease research.
Dr Irina Velsko is an early career scientist with an interest in oral diseases.
St John Crean is Professor of Medicine in Dentistry with an interest in oral diseases and dementia.
Lakshmyya Kesavalu is an Associate Professor specialising in generating animal models of periodontal disease.
Funding Statement
This work was supported by the NIH/National Institute for Dental and Craniofacial Research NIDCR; R01DE020820 (LK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The research work undertaken in the UK was part of a PhD studentship fully funded by the University of Central Lancashire.
References
- Akiyama H, Kameyama M, Akiguchi I. Periodic acid-Schiff (PAS)-positive, granular structures increase in the brain of senescence accelerated mouse (SAM) Acta Neuropathol. 1986;72:124–10. doi: 10.1007/BF00685973. [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC, Schwarb P. Age-related deposition of glia-associated fibrillar material in brains of C57BL/6 mice. Neuroscience. 1994;60:875–889. doi: 10.1016/0306-4522(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Kuo H, Ingram DK, Walker LC. Similarities in the age-related hippocampal deposition of periodic acid-schiff-positive granules in the senescence-accelerated mouse P8 and C57BL/6 mouse strains. Neuroscience. 1996;74:733–740. doi: 10.1016/0306-4522(96)00169-8. [DOI] [PubMed] [Google Scholar]
- Miyamoto M, Kiyota Y, Yamazaki N. Age-related changes in learning and memory in the senescence-accelerated mouse (SAM) Physiol Behav. 1986;38:399–406. doi: 10.1016/0031-9384(86)90112-5. [DOI] [PubMed] [Google Scholar]
- Ma Q, Qiang J, Gu P. Age-related autophagy alterations in the brain of senescence accelerated mouse prone 8 (SAMP8) mice. Exp Gerontol. 2011;46:533–541. doi: 10.1016/j.exger.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Su JH, Cummings BJ, Cotman CW. Localization of heparan sulfate glycosaminoglycan and proteoglycan core protein in aged brain and Alzheimer’s disease. Neuroscience. 1992;51:801–813. doi: 10.1016/0306-4522(92)90521-3. [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC, Kuo H. Age related fibrillary deposits in brains of C57BL/6 mice. A review of localisation, staining characteristics, and strain specificity. Mol Neurobiol. 1994:125–133. doi: 10.1007/BF02816112. [DOI] [PubMed] [Google Scholar]
- Snow AD, Wright TN, Nochlin D. Immunocolocalization of heparin sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann-Straussler syndrome, Creutzfeldt-Jakob disease and scrapie. Lab Invest. 1990;63:601–611. [PubMed] [Google Scholar]
- Snow AD, Sekiguchi RT, Nochlin D. Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer’s disease brain. Am J Pathol. 1994;144:337–347. [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Barnes SR, Sweeney MD. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am J Physiol Cell Physiol. 2007;292:C1256–C1262. doi: 10.1152/ajpcell.00563.2005. [DOI] [PubMed] [Google Scholar]
- Sohet F, Daneman R. Genetic mouse models to study blood-brain barrier development and function. Fluids Barriers CNS. 2013;10:3. doi: 10.1186/2045-8118-10-3. PMID:23305182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methia N, Andre P, Hafezi-Moghadam A. ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med. 2001;7:810–815. [PMC free article] [PubMed] [Google Scholar]
- Laskowitz DT, Lee DM, Schmechel D. Altered immune responses in apolipoprotein E-deficient mice. J Lipid Res. 2000;41:613–620. [PubMed] [Google Scholar]
- Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30:1350–1360. doi: 10.1016/j.neurobiolaging.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole S, Singhrao SK, Chukkapalli S. Active invasion of an oral bacterium and infection-induced complement activation in ApoEnull mice brains. J Alzheimers Dis. 2015;43:67–80. doi: 10.3233/JAD-140315. [DOI] [PubMed] [Google Scholar]
- Poole S, Singhrao SK, Kesavalu L. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013;36:665–677. doi: 10.3233/JAD-121918. [DOI] [PubMed] [Google Scholar]
- Velsko IM, Chukkapalli SS, Rivera MF. Active invasion of oral and aortic tissues by Porphyromonas gingivalis in mice causally links periodontitis and atherosclerosis. PLOS One. 2014;9:e97811. doi: 10.1371/journal.pone.0097811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veurink G, Liu D, Taddei K. Reduction of inclusion body pathology in ApoE-deficient mice fed a combination of antioxidants. Free Radic Biol Med. 2003;34:1070–1077. doi: 10.1016/s0891-5849(03)00042-x. [DOI] [PubMed] [Google Scholar]
- Sunde PT, Olsen I, Gobel UB. Fluoresence in situ hybridization (FISH) for direct visualization of bacteria in periapical lesions of asymptomatic root-filled teeth. Microbiology. 2003;149:1095–1102. doi: 10.1099/mic.0.26077-0. [DOI] [PubMed] [Google Scholar]
- Singhrao S, Cole G, Henderson WJ. White embedding allows a multi-method approach to the analysis of brain tissue from patients with Alzheimer’s disease. J Histochem. 1990;22:257–268. doi: 10.1007/BF01387181. [DOI] [PubMed] [Google Scholar]
- Miklossy J, Darekar P, Gern L. Bacterial peptidoglycan in neuritic plaques in Alzheimer’s disease. Azheimer’s Res. 1996;2:95–100. [Google Scholar]
- Fullerton SM, Shirman GA, Strittmatter WJ. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein E knockout mice. Exp Neurol. 2001;169:13–22. doi: 10.1006/exnr.2001.7631. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Rushmere NK. Spontaneous classical pathway activation and deficiency of membrane regulators render human neurons susceptible to complement lysis. Am J Pathol. 2000;157:905–918. doi: 10.1016/S0002-9440(10)64604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselaar SE, Daugherty A. Apolipoprotein E-deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo . J Lipid Res. 1998;39:1740–1743. [PubMed] [Google Scholar]
- de Bont N, Netea MG, Demacker PN. Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J Lipid Res. 1999;40:680–685. [PubMed] [Google Scholar]
- Jucker M, Walker LC, Martin LJ. Age-associated inclusions in normal and transgenic mouse brain. Science. 1992;255:1443–1445. doi: 10.1126/science.1542796. [DOI] [PubMed] [Google Scholar]
- Cana A, Herder V, Hansmann F. Spitzbarth. Characterization of periodic-acid-Schiff-positive granular deposits in the hippocampus of SJL/J mice. Toxicol Pathol. 2015;43:737–742. doi: 10.1177/0192623314564254. [DOI] [PubMed] [Google Scholar]
- MacDonald AB, Miranda JM. Concurrent neocortical borreliosis and Alzheimer’s disease. Hum Pathol. 1987;18:759–761. doi: 10.1016/s0046-8177(87)80252-6. [DOI] [PubMed] [Google Scholar]
- Miklossy J. Chronic inflammation and amyloidogenesis in Alzheimer’s disease - role of spirochetes. J Alzheimer Dis. 2008;13:381–391. doi: 10.3233/jad-2008-13404. [DOI] [PubMed] [Google Scholar]
- Balin B, Little C, Hammond C. Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease. J Alzheimers Dis. 2008;13:371–380. doi: 10.3233/jad-2008-13403. [DOI] [PubMed] [Google Scholar]
- Riviere GR, Riviere K, Smith K. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol. 2002;17:113–118. doi: 10.1046/j.0902-0055.2001.00100.x. [DOI] [PubMed] [Google Scholar]
- Dunn N, Mullee M, Perry V. Association between dementia and infectious disease: evidence from a case-control study. Alzheimer Dis Assoc Disord. 2005;19:91–94. doi: 10.1097/01.wad.0000165511.52746.1f. [DOI] [PubMed] [Google Scholar]
- Holmes C, El-Okl M, Williams AL. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:788–789. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen I, Yilmaz Ö. Modulation of inflammasome activity by Porphyromonas gingivalis in periodontitis and associated systemic diseases. J Oral Microbiol. 2016;8:30385. doi: 10.3402/jom.v8.30385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen I, Singhrao SK. Inflammasome involvement in Alzheimer’s disease. J Alzheimers Dis. 2016;54:45–53. doi: 10.3233/JAD-160197. [DOI] [PubMed] [Google Scholar]
- Ophir G, Amariglio N, Jacob-Hirsch J. Apolipoprotein E4 enhances brain inflammation by modulation of the NF-κB signaling cascade. Neurobiol Dis. 2005;20:709–718. doi: 10.1016/j.nbd.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Tsoi LM, Wong KY, Liu YM. Ho YY Apoprotein E isoform-dependent expression and secretion of pro-inflammatory cytokines TNF-α and IL-6 in macrophages. Arch Biochem Biophys. 2007;460:33–40. doi: 10.1016/j.abb.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Newman GR. Corpora amylacea could be an indicator of neurodegeneration. Neuropathol Appl Neurobiol. 1993;19:269–276. doi: 10.1111/j.1365-2990.1993.tb00437.x. [DOI] [PubMed] [Google Scholar]
- Ramsey HJ. Ultrastructure of corpora amylacea. J Neuropathol Exp Neurol. 1965;24:29–39. doi: 10.1097/00005072-196501000-00003. [DOI] [PubMed] [Google Scholar]
- Halliday MR, Rege SV, Ma Q. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab. 2015 doi: 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy J. Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front Aging Neurosci. 2015;7:46. doi: 10.3389/fnagi.2015.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar DK, Choi SH, Washicosky KJ. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016 doi: 10.1126/scitranslmed.aaf1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy J. Bacterial amyloid and DNA are important constituents of senile plaques: further evidence of the spirochetal and biofilm nature of senile plaques. J Alzheimer’s Dis. 2016;53:1459–1473. doi: 10.3233/JAD-160451. [DOI] [PMC free article] [PubMed] [Google Scholar]