

ABSTRACT
As an exclusively human pathogen, Streptococcus pyogenes (the group A streptococcus [GAS]) has specifically adapted to evade host innate immunity and survive in multiple tissue niches, including blood. GAS can overcome the metabolic constraints of the blood environment and expresses various immunomodulatory factors necessary for survival and immune cell resistance. Here we present our investigation of one such factor, the predicted LysR family transcriptional regulator CpsY. The encoding gene, cpsY, was initially identified as being required for GAS survival in a transposon-site hybridization (TraSH) screen in whole human blood. CpsY is homologous with transcriptional regulators of Streptococcus mutans (MetR), Streptococcus iniae (CpsY), and Streptococcus agalactiae (MtaR) that regulate methionine transport, amino acid metabolism, resistance to neutrophil-mediated killing, and survival in vivo. Our investigation indicated that CpsY is involved in GAS resistance to innate immune cells of its human host. However, GAS CpsY does not manifest the in vitro phenotypes of its homologs in other streptococcal species. GAS CpsY appears to regulate a small set of genes that is markedly different from the regulons of its homologs. The differential expression of these genes depends on the growth medium, and CpsY modestly influences their expression. The GAS CpsY regulon includes known virulence factors (mntE, speB, spd, nga [spn], prtS [SpyCEP], and sse) and cell surface-associated factors of GAS (emm1, mur1.2, sibA [cdhA], and M5005_Spy0500). Intriguingly, the loss of CpsY in GAS does not result in virulence defects in murine models of infection, suggesting that CpsY function in immune evasion is specific to the human host.
KEYWORDS: CpsY, LysR family regulators, human-specific virulence, immune evasion, transcriptional regulation
INTRODUCTION
As an exclusively human pathogen, Streptococcus pyogenes, or group A streptococcus (GAS), has specifically adapted to evade host innate immunity and survive in multiple tissue niches. GAS primarily colonizes the epithelia of the nasopharynx and skin, but a number of clinically important serotypes are capable of invading deep tissue and the bloodstream, resulting in pathologies with a high mortality rate (1, 2). The invasive phenotype in some of these strains has been linked to mutation of global transcriptional regulators, such as the CovRS/CsrRS two-component system (TCS), which regulates important virulence factors such as SpeB protease, streptokinase, streptodornase, and the hyaluronic acid capsule (3–5). Transcriptional regulators of GAS are crucial to immune evasion, as observed for both stand-alone regulators like Mga (6, 7) and TCSs like Ihk/Irr (8, 9) and CovRS/CsrRS (10). In the case of the latter TCS pathways, adaptation of these regulators following exposure of GAS to macrophages enhanced survival of GAS during reinfection of immune cells (9). Thus, control of virulence factor expression through transcriptional regulator modulation has significant consequences for GAS-associated disease development.
Pathogenic streptococci exposed to host blood employ a variety of strategies to survive and propagate in this environment. The GAS cell surface specifically interacts with a high number of blood plasma proteins, and differences have been observed between the interaction profiles of invasive and noninvasive GAS strains (11). The host coagulation cascade functions during bacterial infection of the bloodstream to entrap bacteria within a fibrin clot for killing. However, GAS can circumvent this mechanism by sequestering host plasminogen on the bacterial cell surface, converting it into active plasmin via streptokinase, and using it to degrade the fibrin network, thus liberating GAS from clots (reviewed in reference 12). Investigation of isogenic invasive and noninvasive GAS isolates found that expression of immunomodulatory factors (e.g., C5a peptidase, Gls24, and M protein) increased in response to growth in whole blood, particularly in invasive strains, and were necessary for survival in whole blood and neutrophil resistance (13).
GAS is also able to overcome the metabolic constraints imposed on it by the blood environment. It possesses a metal transport operon (mtsABC) (14) as well as several dedicated hemoprotein and iron acquisition systems (siaABC, shr, htsABC) (15–17) that are controlled by multiple regulators (mtsR and perR; reviewed in reference 18) and are necessary for survival in blood (19). Regulation of amino acid metabolism is required for both growth and immune evasion. For example, arginine catabolism through CodY-mediated control of the arcABC operon (20) contributes to resistance to inducible nitric oxide synthase (iNOS)-mediated production of nitric oxide (21). Access to carbon sources directly impacts immune evasion, as recently demonstrated by loss of the fruB-encoded 1-phosphopfrucokinase adversely affecting GAS survival in human blood and resistance to phagocytes (22). Thus, it is important to understand how GAS adapts to specific host niches, metabolically and through immune evasion, to effectively address the rising incidence of invasive GAS strains (reviewed in reference 23).
In a transposon site hybridization (TraSH) forward genetic screen of M1T1 GAS 5448, cpsY was found to be required for survival in whole human blood (19). GAS CpsY displays 83% identity with MetR, a LysR family transcriptional regulator of Streptococcus mutans shown to activate transcription of genes involved in methionine biosynthesis (metE) and uptake (atmB) in a homocysteine-dependent manner (24). Homologs of CpsY found in Streptococcus iniae and Streptococcus agalactiae (MtaR) have been characterized as virulence determinants that regulate methionine transport (25), amino acid metabolism (26), and cell wall modifications necessary for resistance to neutrophil-mediated killing and survival in vivo (27–29). Thus, CpsY appears to play a crucial role as a transcriptional regulator in the development of streptococcal invasive disease through the promotion of immune cell resistance and host niche adaptation.
Here, we present evidence that the CpsY transcriptional regulator of GAS acts as a regulator of resistance to innate immune cells and survival in human blood in a manner distinct from those of other streptococcal pathogens. Loss of CpsY in GAS does not produce the methionine-dependent in vitro growth defects or the in vivo survival phenotypes observed with other streptococcal species in mouse models of infection. However, disruption of CpsY expression significantly impacts GAS resistance to human phagocytic cells, indicating that CpsY function in immune evasion is specific to the human host. Transcriptome analysis revealed that CpsY influences the expression of a small number of genes, both known and hypothetical, under our experimental conditions. The regulon includes genes encoding cell wall-modifying proteins and surface proteins, suggesting that CpsY-mediated resistance to host innate immunity may require alterations to the GAS cell envelope.
RESULTS
CpsY is required for GAS survival in whole human blood.
The cpsY gene was originally identified as necessary for GAS survival in heparinized whole human blood in a transposon mutagenesis (TraSH) screen (19). To further characterize the role of CpsY in GAS virulence, we generated a stable insertional inactivation mutant, the (ι)cpsY mutant, and a strain rescued for the mutation (the cpsYR strain) in the M1T1 5448 background initially used in the blood screen (see Materials and Methods). The survival in heparinized whole human blood of the mutant and its rescue strain was tested using the Lancefield blood bactericidal assay (19, 30). The (ι)cpsY mutant had a significantly lower multiplication factor (MF) in heparinized whole blood than did wild-type 5448 and the cpsYR strain (Fig. 1A), confirming the requirement of cpsY for GAS survival in blood. To assess whether the observed phenotype resulted from decreased resistance to host immune cells found in whole blood or to the metabolic environment of blood, growth of 5448, the (ι)cpsY mutant, and the cpsYR rescue strain in the presence of human plasma was also examined. In plasma isolated from heparinized whole donor blood equilibrated 1:1 with tissue culture medium (RPMI 1640, 2.05 mM l-glutamine), no appreciable defects in growth were observed in the (ι)cpsY or the cpsYR strain relative to the 5448 strain (Fig. 1B).
FIG 1.
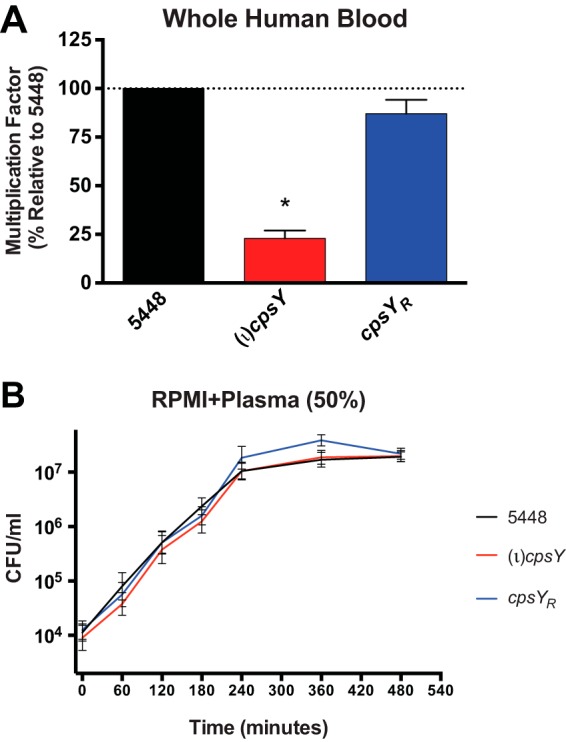
CpsY is required for GAS survival in whole human blood. Growth of wild-type GAS 5448, an isogenic (ι)cpsY mutant, and its rescue (cpsYR) strain were assayed in the human blood environment. (A) A Lancefield bactericidal assay was performed by monitoring growth in heparinized whole human blood. Wild-type GAS 5448 had a mean multiplication factor (MF) of 75.2 ± 6.9 in the donor blood assayed. Data are presented as percent growth in blood, corresponding to the MF of the mutant and rescue divided by the MF of the wild type × 100. Data represent means ± SEMs from four independent experiments. *, P < 0.05. (B) Growth of GAS was measured in RPMI 1640 supplemented with plasma (50%) obtained from heparinized whole blood from human donors by plating aliquots at the indicated time points to quantify CFU. Results of a representative experiment out of three biological replicates are shown.
Loss of CpsY does not adversely affect GAS growth in vitro.
CpsY homologs in other streptococcal species have been shown to play a role in amino acid metabolism, impacting growth in various lab culture media and in human plasma. Finding no phenotype for growth in plasma, we examined whether the (ι)cpsY strain exhibited any growth defects in commonly used media. Growth of (ι)cpsY and its rescue strain (cpsYR) was comparable to that of the 5448 parent in rich medium (Todd-Hewitt medium supplemented with 0.2% yeast extract [THY]) (Fig. 2A). Likewise, in chemically defined medium supplemented with 0.5% glucose (CDM) and in low-glucose, high-peptide C medium, the GAS (ι)cpsY strain and its wild-type parent showed no differences in growth rates or yields (Fig. 2B and C). This was in contrast to the case with other streptococci, for which disruption of cpsY led to significant growth phenotypes in these media (25, 28). In S. agalactiae (GBS), growth defects for mutants of the CpsY homolog MtaR were rescued by supplementing culture media with methionine. However, for GAS, addition of methionine to CDM had no effect on growth of either 5448 or the (ι)cpsY mutant (see Fig. S1A in the supplemental material).
FIG 2.

CpsY is not required for growth of GAS in vitro. Growth curves for the (ι)cpsY mutant and its rescue (cpsYR) strain in enriched THY medium (A), chemically defined medium (CDM) (B), and C medium (C) were measured by absorbance using a Klett-Summerson colorimeter and expressed in Klett units compared to the value for wild-type 5448. Data are the results from three biological replicates. (D) Limited carbohydrate utilization profiles of 5448, the (ι)cpsY mutant and its rescue (cpsYR), assayed using the API 50 CH assay. Results after incubation at 37°C for 24 h and 48 h are shown. Listed are selected carbon sources from a panel of 50 total carbohydrate sources, with complete utilization (white boxes), partial utilization (gray boxes), and no utilization (black boxes) based on the colorimetric indicator dye shown for each.
To further explore if the metabolic environment of blood contributed to the reduced survival phenotype observed for the (ι)cpsY strain, we tested whether loss of CpsY impacted carbohydrate metabolism using a bioMérieux API 50 CH assay strip that tests utilization of a subset of 49 carbohydrates (Fig. 2D). No significant changes in the utilization of different carbon sources were observed between 5448 and the (ι)cpsY and cpsyR strains after either 24 h or 48 h of incubation. Thus, it appears that the reduced survival of the (ι)cpsY mutant in whole human blood is not due to metabolic defects in carbon utilization.
Loss of CpsY reduces GAS resistance to opsonophagocytic killing.
The reduced fitness of the M1T1 (ι)cpsY mutant in whole human blood, combined with the absence of growth defects in vitro, suggests that CpsY is involved in the evasion of cellular components of host innate immunity (e.g., neutrophils and macrophages). To examine this possibility, we assessed the viability of both 5448 and the (ι)cpsY mutant in an opsonophagocytic killing assay (OKA). Bacteria were opsonized in plasma obtained from heparinized whole donor blood and presented to either cultured neutrophil-like differentiated HL60 cells or freshly isolated neutrophils or monocytes. The percentages of viable GAS following incubation in the presence or absence of immune cells were calculated by quantifying CFU recovered from wells with immune-cell-challenged bacteria versus bacteria in wells with OKA medium (i.e., RPMI plus 20% plasma) alone.
OKA data showed a reduction in the number of viable (ι)cpsY mutant bacteria in the presence of polymorphonuclear leukocytes (PMNs) (Fig. 3A and B) and monocytes and differentiated HL60 cells (Fig. 3C), suggesting that the loss of CpsY resulted in reduced resistance of GAS to opsonophagocytic killing. Inhibition of phagocytosis by cytochalasin D treatment and heat inactivation of plasma to inactivate opsonins produced similar levels of viable bacteria for 5448, the (ι)cpsY mutant, and the cpsYR rescue strains (Fig. 3B and C, CytoD+HI Plasma). The decreased survival of the (ι)cpsY mutant in the presence of neutrophils likely stems from increased phagocytosis and/or reduced tolerance to intracellular antimicrobial mechanisms of the phagocytes. Additionally, reduced survival of the (ι)cpsY mutant in the neutrophil killing assay was not related to growth of the mutant in the assay medium, as multiplication factors were comparable for all three strains in RPMI 1640–2.05 mM l-glutamine plus plasma (Fig. S1B).
FIG 3.

CpsY is required for GAS resistance to opsonophagocytic killing by human innate immune cells. Recovery of viable wild-type 5448 (black) GAS following immune cell challenge was compared to those of the (ι)cpsY mutant (red) and its cpsYR rescue (blue), with GAS opsonized in either plasma or heat-inactivated plasma (HI Plasma) prior to exposure to untreated or cytochalasin D-treated (CytoD) immune cells. (A) Viable bacteria recovered from challenge by human neutrophils (PMNs). GAS (1 × 105 CFU/well) and neutrophils (1 × 106 CFU/well) were incubated for 2 h at 37°C. Neutrophils were lysed at 30 min, 60 min, and 2 h, and the number of viable GAS was enumerated as CFU recovered. Results of a representative experiment out of a minimum of three biological replicates are shown. (B) Viable bacteria recovered from challenge by human PMNs. GAS (1 × 105 CFU/well) and neutrophils (1 × 106 CFU/well) were incubated for 2 h at 37°C. Neutrophils were then lysed, and the percentage of viable GAS was determined and shown as survival relative to that of wild-type 5448. (C) Viable bacteria recovered from challenge by monocytes and neutrophil-like HL60 cells. GAS (1 × 105 CFU/well) and either monocytes or differentiated HL60 cells (1 × 106 CFU/well) were incubated for 2 h at 37°C. Cells were lysed, and the percentage of viable GAS was determined relative to the survival of 5448. Data shown in panels B and C represent means ± SEMs of the results from at least three biological replicates performed in triplicate, with P less than 0.001 (overall ANOVA). Individual groups were compared using Dunnett's multiple-comparison test (***, P < 0.001; **, P < 0.01).
Absence of CpsY does not impact GAS virulence in mouse models of infection.
Given the reduced resistance to innate immune cells, we examined whether the loss of CpsY impacted GAS virulence in vivo using mouse models of soft tissue (subcutaneous [s.c.]) and systemic (intraperitoneal [i.p.]) GAS infections. The (ι)cpsY mutant showed no significant differences with respect to both 5448 and the cpsYR rescue strain in its ability to cause disease in either model of infection (Fig. 4A and B). Though the lesion size for the (ι)cpsY mutant in the subcutaneous model of infection trended smaller than for 5448 or the cpsYR rescue strain (data not shown), the differences were not statistically significant. This indicates that CpsY does not significantly affect virulence in GAS mouse models of infection. Interestingly, a modified Lancefield blood bactericidal assay employing murine blood found that the (ι)cpsY mutant did not exhibit the same growth defect (Fig. 4C) as observed in whole human blood (Fig. 1A). These data suggest that the role of CpsY is specific to the evasion of human innate immune cells by GAS, which is distinct from that observed for CpsY homologs in other streptococcal species.
FIG 4.

CpsY does not impact GAS virulence in mouse models of infection. Virulence of the (ι)cpsY mutant (red) and the cpsYR rescue strain (blue) was compared to that of wild-type 5448 (black) in the following mouse models. (A) Survival in a murine intraperitoneal (i.p.) model of systemic infection. GAS (∼2 × 108 CFU/mouse) bacteria were injected i.p. into mice (n = 10/strain) and monitored for morbidity over the course of 72 h. Significance was determined using Kaplan-Meier survival analysis and the log rank test. (B) Survival in a murine subcutaneous (s.c.) model of soft tissue infection. GAS (∼2.8 × 108 CFU/mouse) were injected s.c. into mice (n = 10/strain) and monitored for morbidity over the course of 7 days. Significance was determined using Kaplan-Meier survival analysis and the log rank test. (C) Survival in a modified Lancefield bactericidal assay using heparinized whole mouse blood. The data are presented as the multiplication factor (MF) of wild-type 5448 and the (ι)cpsY mutant in whole mouse blood. Data represent GAS survival in blood obtained from multiple mice (n = 4 to 6) and are means ± SEMs of the results of independent experiments.
GAS CpsY regulates a markedly different set of genes to other streptococcal species.
Since CpsY activity in GAS appears to phenotypically differ from that of homologs in other streptococci, we hypothesized that it possessed a regulon different from that in S. mutans, S. iniae, and S. agalactiae. RNA sequencing (RNA-seq) analysis was performed on RNA isolated from either 5448 or (ι)cpsY mutant cultures that were grown to late logarithmic phase in either rich medium (THY) or RPMI 1640 plus 20% human donor plasma for 2 h to simulate OKA conditions. We identified distinct, medium-specific sets of transcripts, consisting of only 25 and 16 genes, respectively, that were differentially expressed in the (ι)cpsY mutant relative to the 5448 parent (Table 1). Notably absent from the list of identified transcripts were genes regulated by CpsY homologs in other streptococci, such as the methionine transport operon atmBDE (metNPQ), the arginine transport operon artPQ, and the peptidoglycan modification-associated gene murA (Table 2). We confirmed these results by quantitative real-time PCR (qRT-PCR) analysis of artP, atmB (metN), and murA transcripts quantified in total RNA isolated at various phases of growth from 5448 and (ι)cpsY strains growing in enriched medium (THY). No changes in the relative expression of these genes were observed between the wild type and the (ι)cpsY mutant at any of the time points examined, with one exception (Fig. 5). Expression of artP showed a 2-fold increase during the late logarithmic phase of growth in the absence of CpsY (Fig. 5B).
TABLE 1.
Genes differentially expressed in the GAS (ι)cpsY mutanta

Data for downregulated genes are shaded.
bRNA-seq analysis results (P < 0.05).
TABLE 2.
Expression of GAS genes CpsY regulated in other streptococcal species
| Spy designation | Annotation | Gene name | Log2 fold changea |
|---|---|---|---|
| Methionine transport/metabolism | |||
| M5005_Spy0146 | Cystathionine beta-lyase | metB (cysD) | −0.421 |
| M5005_Spy0271 | ABC transporter substrate-binding protein | atmB (metQ) | 0.269 |
| M5005_Spy0272 | ABC transporter ATP-binding protein | atmD (metN) | 0.174 |
| M5005_Spy0273 | ABC transporter permease | atmE (metP) | 0.361 |
| Arginine transport | |||
| M5005_Spy1237 | Arginine transport ATP-binding protein | artP (gtr) | 0.765 |
| M5005_Spy1238 | Arginine transporter permease | artQ | 0.785 |
| Cell wall modification | |||
| M5005_Spy0038 | Acyltransferaseb | −0.222 | |
| M5005_Spy0584 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | murA | −0.086 |
RNA-seq analysis results.
Homolog of oat product in S. iniae (29).
FIG 5.

CpsY of GAS does not regulate the transcripts controlled by homologous proteins in other streptococcal species. Expression of genes atmD (metN) (A), artP (B), and murA (C) was measured using real-time qPCR. Shown are real-time qPCR analyses of relative transcript levels in the (ι)cpsY mutant strain at early logarithmic, late logarithmic, and stationary phases of growth compared to wild type 5448 grown in enriched medium (THY). (D) Expression of the genes mntE, murA2, mur1.2, emm1, adh2, prsA, speB, phaB, fabK, identified as members of the CpsY regulon, was measured using real-time qPCR. Shown are results from real-time qPCR analyses of relative transcript levels in the (ι)cpsY mutant strain in either THY or OKA medium. Error bars represent the standard errors from a minimum of three biological replicates. Dashed lines indicate 2-fold significance. Significance was determined using relative transcript level comparisons to the gene gyrA.
Transcripts that were differentially expressed in OKA medium included several surface-associated factors of GAS (Table 1): the cell wall-anchored M protein (emm1), cell wall-modifying enzymes (mur1.2, sibA [cdhA], and M5005_Spy0500 [or simply Spy0500]), and a manganese-specific efflux pump (mntE [M5005_Spy0980]). Changes in expression of these transcripts were examined using qRT-PCR analysis of total RNA obtained from bacteria incubated in OKA medium (Fig. 5D). This analysis confirmed that CpsY activity is associated with low-level repression of mntE and Spy0500, as well as induction of emm1 and mur1.2. Further examination of the Spy0500 sequence found 79% identity to the N-acetylmuramoyl-l-alanine amidase encoded by the murA gene characterized as regulated by CpsY in S. iniae (29) and MtaR in S. agalactiae (26). Thus, it appears that the sequence annotated in the MGAS5005 genome as murA (M5005_Spy0548) does not correspond to the homolog of the CpsY-regulated murA characterized in other streptococci. In light of this, we refer to M5005_Spy0500 as murA2. Additional differentially expressed transcripts in OKA medium included pyruvate metabolism proteins (aroA2 and M5005_Spy1509) and a set of short hypothetical proteins. These results indicate that GAS CpsY regulates expression of a set of genes that is markedly different from the ones its homologs in other streptococcal species control under the conditions we tested.
None of the transcripts identified in OKA medium, however, were differentially expressed in the (ι)cpsY mutant at late logarithmic growth in THY medium (Table 1). In contrast, differentially expressed genes identified in THY include those of the fatty acid biosynthetic pathway (accA, fabZFGDK, and phaB), genes for metabolic dehydrogenases (adhA [adhP] and adh2 [adhE]), genes involved in cell stress and protein processing (groEL, clpL, prsA, and serS), and genes for several known virulence factors (nga [spn], sse, spyCEP, speB, and spd). Changes in expression of several of these transcripts were also examined using qRT-PCR analysis. Data from said analysis confirmed that in the absence of CpsY activity, expression of adh2, prsA, and, to a large degree, speB (∼30-fold change) is increased during late log growth in THY (Fig. 5D). Conversely, qRT-PCR analysis failed to detect significant changes in the expression of the phaB, fabK, and emm1 genes in the absence of CpsY activity. Overall, the qRT-PCR results supported the RNA-seq data, with a correlation coefficient (R2) calculated to be 0.82 (n = 9 [Fig. S2]). Thus, it appears that the CpsY regulon of GAS comprises a small number of genes of varied functions and is different from that of related streptococcal species, with the exception of artP and murA2 (M5005_Spy0500). Furthermore, GAS CpsY influences expression of its regulon in a medium-dependent manner and, with the exception of speB, the differential expression of CpsY regulon genes is modest (<4-fold).
Absence of CpsY does not influence GAS resistance to cell wall-targeting antimicrobials.
CpsY regulates modifications of the bacterial cell wall in S. iniae that are necessary for resistance to neutrophil-mediated killing, as indicated by the increased sensitivity to lysozyme and altered expression of a peptidoglycan amidase (murA) in a CpsY-lacking strain of this species (29). Our RNA-seq analysis indicates that expression of the murA homolog of S. iniae in GAS (Spy0500 [murA2]) is also influenced by GAS CpsY. We therefore examined whether the GAS (ι)cpsY mutant also displayed altered sensitivity to antimicrobials that attack different components of the bacterial cell wall. When grown in THY, the (ι)cpsY mutant displayed a sensitivity to multiple cell wall-targeting compounds similar to that of wild-type 5448, including carbenicillin (minimum bactericidal concentration [MBC] = 0.15 μg ml−1), bacitracin (MBC = 0.5 μg ml−1), nisin (MBC = 125 μg ml−1), and lysozyme (MBC = 2,350,000 U ml−1).
DISCUSSION
The cpsY gene was initially identified as being required for GAS survival in a transposon site hybridization (TraSH) screen in whole human blood (19). CpsY shares homology (83% identity) with a LysR family transcriptional regulator of S. mutans (MetR), as well as to proteins in S. iniae (CpsY) and S. agalactiae (MtaR) that regulate methionine transport (25), amino acid metabolism (26), and resistance to neutrophil-mediated killing and survival in vivo (27–29). This led us to hypothesize that CpsY functions as a transcriptional regulator of GAS metabolism required for survival in human host blood. Unlike with the CpsY homologs in S. iniae (28) and S. agalactiae (25), loss of CpsY function by insertional inactivation of the encoding gene (cpsY [Spy0701]) did not affect growth of GAS in human plasma or rich medium (THY). CpsY was also not required for GAS growth in C medium or CDM, as seen for MtaR in S. agalactiae, nor did supplementing media with methionine have any effect on the growth of either the (ι)cpsY mutant or the parental 5448 GAS strain. Finally, we found no significant differences in utilization of various carbohydrate sources by the 5448 strain and (ι)cpsY mutant. Together, these data suggest that the reduced fitness of a GAS mutant lacking CpsY in whole blood is not related to a broader metabolic defect.
We hypothesized that reduced survival of the (ι)cpsY mutant in whole blood is due to reduced resistance to immune cell activity, and/or decreased tolerance to cellular stresses encountered within these cells. Circulating neutrophils found abundantly in the bloodstream employ multiple mechanisms to kill invading bacterial pathogens, including the production of antimicrobial peptides, neutrophil extracellular traps (NETs), and phagocytosis of the pathogen. Within the phagocytic cell, engulfed pathogens must deal with production of reactive oxygen species, release of antibacterial granule components, and acidification (31, 32). The production of virulence factors by GAS to resist these mechanisms of killing is well documented (33), yet their coordination and regulation in different strain backgrounds is not fully understood (18). When we tested the resistance to challenge by phagocytes in an opsonophagocytic killing assay (OKA), survival of the (ι)cpsY mutant strain was significantly reduced across PMNs, monocytes, and neutrophil-like differentiated HL60 cells. Inhibiting phagocytosis and blocking opsonization by treating phagocytes with cytochalasin D and heat inactivating the plasma used in the assay rescued survival of the (ι)cpsY mutant strain. This indicates that CpsY function is required for GAS to effectively resist opsonophagocytic killing by innate immune cells. The fact that inhibition of phagocytosis rescues survival of the (ι)cpsY mutant suggests that extracellular killing mechanisms of neutrophils such as NETs are likely not involved in the CpsY-associated phenotype, though we did not specifically test for this. It appears that disruption of CpsY in GAS makes the pathogen more susceptible to phagocytosis and/or less capable of intracellular growth/survival within neutrophils. This is likely due to altered expression of a CpsY regulon that may include antiphagocytic factors and/or proteins necessary for resisting cellular stresses to sustain bacterial growth within host immune cells.
The observed susceptibility to opsonophagocytic killing led us to postulate that CpsY is required for virulence in an in vivo model of infection. We utilized both a subcutaneous (localized and disseminated) and an intraperitoneal (systemic) mouse model of infection to test for changes in virulence resulting from the loss of CpsY. The (ι)cpsY mutant strain did not show any decrease in virulence in either mouse model compared to both the 5448 wild-type parent and cpsYR rescue strain. We examined whether the (ι)cpsY mutant strain also displayed reduced growth in whole mouse blood, and no differences were observed between the mutant and wild-type strains in the mouse blood Lancefield assay. These results lead us to conclude that the role of CpsY in resistance to opsonophagocytic killing involves a mechanism of the innate immune cells of its human host that is absent from murine immune cells. Host restriction of GAS has been associated with differences in host innate immunity. Examination of Toll-like receptor (TLR) pathways of the innate immune response to GAS found that the TLR13 response in mice is specifically triggered by GAS rRNA and that the TLR13 molecule is absent in human macrophages, as is an equivalent route for GAS RNA recognition (34). Furthermore, there are other instances of GAS regulators that influence resistance to host immunity in a host-specific manner. FruR, the transcriptional repressor of the fruRBA operon encoding the fructose-specific EII of the GAS phosphoenolpyruvate phosphotransferase system (PTS), was shown to mediate innate immune evasion in human host blood but not in murine models of infection (22). Also, the human-specific antimicrobial cathelicidin LL-37 stimulates the CovRS/CsrRS two-component regulatory system-dependent expression of the capsule synthesis operon (hasABC), the interleukin 8 (IL-8) protease PrtS/ScpC, and the integrin-like/IgG protease Mac/IdeS (35). Induction of these virulence factors by the LL-37 peptide through CovRS/CsrRS resulted in a marked increase in GAS resistance to opsonophagocytic killing by human leukocytes.
Given its homology to transcriptional regulators of S. mutans (MetR), S. agalactiae (MtaR), and S. iniae, we also proposed that CpsY regulates expression of factors that, in the absence of CpsY, are not adequately expressed and thus fail to sustain survival of the (ι)cpsY strain in the presence of phagocytic immune cell challenge. We investigated whether there was any correspondence in the regulatory targets of GAS CpsY and its homologs in other streptococci, despite the clear differences in phenotypes that we observed. With the exception of a modest increase in artP expression during late logarithmic growth in enriched medium (THY), both RNA-seq and qRT-PCR analysis revealed that the transcript levels of known MetR/MtaR/CpsY regulon genes were unchanged in the (ι)cpsY GAS strain in all growth phases and under all medium conditions tested. These results support our finding that GAS CpsY activity does not reproduce the phenotypes observed for its homologs in other streptococci.
Moreover, our results indicate that CpsY is a pleiotropic regulator of GAS that influences expression of genes markedly different from those regulated by CpsY homologs of related streptococcal species. RNA-seq analysis of cultures grown in THY and RPMI 1640 supplemented with human plasma (OKA medium) revealed two small and distinct medium-associated sets of differentially expressed genes in the (ι)cpsY mutant. Genes involved in modifying the cell surface of GAS dominate the OKA medium-associated CpsY regulon. The reduced expression of genes encoding the cell wall-anchored M protein (emm1) and cell wall-modifying enzymes (mur1.2 and sibA [cdhA]) in the (ι)cpsY mutant suggests that CpsY induces expression of these factors during phagocyte challenge. M protein is a known antiphagocytic factor of GAS (36–38) that is also involved in resistance to antimicrobial peptides (39). Likewise, modification of the cell wall is important for immune cell evasion in multiple streptococcal species (8, 40–42). Cell wall-modifying enzymes have been linked to the expression of antiphagocytic factors in GAS. Eliminating d-alanine esterification of surface lipoteichoic acid resulted in reduced expression of emm transcripts in 5448 and a related covS mutant strain (8004) (41). The underlying mechanism was not described, but Cox et al. speculated that unidentified regulatory pathways may be responsible. Whether CpsY-mediated direct or indirect regulation of emm1, mur1.2, and sibA (cdhA) is part of a mechanism linking cell wall modification and antiphagocytic factor expression in GAS is being investigated.
The manganese-specific efflux pump encoded by mntE (Spy0980) is another known immune evasion factor of GAS (43) identified as differentially expressed in the absence of CpsY. Increased transcript levels of mntE in the (ι)cpsY mutant suggest that CpsY represses expression of the efflux pump. Turner et al. showed that deletion of mntE reduces GAS resistance to neutrophils and to oxidative stress (43). Their results indicate that these phenotypes are due to loss of manganese homeostasis, which disrupts regulation of the peroxide-responsive repressor PerR and its respective regulon. However, expression of PerR or its regulon was unaffected according to our RNA-seq analysis (data not shown). Mn2+-dependent regulation of PerR is sensitive to ion flux across the bacterial membrane, and multiple targets of PerR regulation exhibit PerR-independent expression, so it is possible that our RNA-seq analysis failed to capture CpsY-related effects on mntE-dependent regulation of PerR.
RNA-seq analysis also identified increased transcript levels of Spy0500 in the absence of CpsY. The M5005_Spy0500 gene (here called murA2) encodes a putative N-acetylmuramoyl-l-alanine amidase that is 78% identical to the transcript identified by Allen and Neely as regulated by CpsY and annotated as murA in S. iniae (29). The gene annotated in M1T1 5448 as murA was not differentially expressed in the (ι)cpsY mutant. In S. iniae, deletion of cpsY produces a strain (ΔcpsY) with increased expression of murA, a result that mirrors our observations on expression of the Spy0500 transcript in the (ι)cpsY mutant. Loss of CpsY-dependent repression of murA in S. iniae reduced the proportion of cross-linked muropeptide and altered the cell wall structure of the bacterium (29). However, overexpression of murA did not replicate the muropeptide profile or the reduced neutrophil resistance phenotype observed in the ΔcpsY strain. This result in S. iniae and the variety of transcripts we have identified as differentially expressed in the (ι)cpsY strain suggest that CpsY-associated resistance to neutrophils could be due to pleiotropic effects of CpsY.
In contrast to case with the OKA medium-associated CpsY regulon, an entirely distinct set of transcripts was differentially expressed in the (ι)cpsY mutant strain when it was grown in rich medium (THY). Most relevant to immune cell resistance was the induced set of known virulence factors of GAS (i.e., speB, spd, nga [spn], prtS [SpyCEP], and sse). Expression of the SpeB cysteine protease promotes colonization and aids in immune cell evasion (44–46), whereas downregulation of SpeB expression is linked to neutrophil-dependent selection of invasive GAS strains (47–49). Streptococcal nucleases like the one encoded by spd enhance GAS evasion of the innate immune response (50) by degrading neutrophil extracellular traps (NETs) (51). The NAD-glycohydrolase encoded by nga (spn) promotes survival and escape from the phagolysosome of phagocytic cells (52, 53) and induces immune cell death (54). Coordinate regulation of these factors is crucial to resistance to host immunity, and our analysis suggests that CpsY does participate in this regulation during in vitro growth.
Another intriguing finding was the CpsY-associated differential expression of the fatty acid biosynthetic pathway. Recent studies have linked FabT-mediated regulation of fatty acid synthesis in GAS to virulence and intrahost genetic variation in invasive disease (55). In the (ι)cpsY mutant strain, the majority of the fabKDGFZ-accABCD operon was repressed (Table 1), as was the enoyl coenzyme A (enoyl-CoA) hydratase-encoding phaB. Conversely, the acetaldehyde-CoA/alcohol dehydrogenases encoded by adhA (adhP) and adh2 (adhE) showed increased expression. Our qRT-PCR analysis confirmed the differential expression of adh2 (adhE) in the (ι)cpsY mutant, but not that of fabK and phaB. The greater differential expression of adh2 (adhE) than of fabK and phaB, indicated by the log2 fold change in expression obtained from RNA-seq analysis, suggests that this discrepancy could be due to the difference in sensitivity between RNA-seq and qRT-PCR analysis. The fabKDGFZ-accABCD operon and phaB constitute the majority of the unsaturated fatty acid biosynthesis pathway of GAS, whereas adhA (adhP) and adh2 (adhE) participate in fatty acid degradation. The potential influence of CpsY on fatty acid biosynthesis merits further study.
Together, the results of our investigation indicate that CpsY has a role in GAS resistance to the innate immune cells of its human host. Furthermore, GAS CpsY appears to regulate a small number of genes of varied functions whose expression CpsY influences fairly modestly. OKA medium represents the environment influencing the GAS transcriptome during immune cell challenge in our studies and thus we believe most accurately identifies the CpsY regulon relevant to resistance to opsonophagocytic killing. There was no overlap between the THY and OKA medium-associated regulons, and whether the CpsY regulon identified in THY reflects any CpsY-dependent gene regulation relevant to GAS pathogenesis is an interesting question to explore. Our investigation also indicates that the GAS CpsY regulon is markedly different from that of related streptococcal species, as the only overlap with the known MetR/MtaR/CpsY regulons of other streptococci consisted of just two genes (artP and murA2 [Spy0500]) under specific medium and growth conditions. Additionally, the role of GAS CpsY in resistance to immune cells is specific to human innate immune cells. The source of this specificity is unknown at this time and a research question we are actively pursuing.
Given that our data indicate that the CpsY regulon is medium dependent, CpsY potentially influences expression of its regulon in response to signaling by either a metabolic or immunogenic molecule. The CovRS/CsrRS-mediated induction of GAS virulence factors by the LL-37 antimicrobial peptide is an example of the latter (35). LysR family transcriptional regulators like CpsY have a C-terminal cofactor-binding domain (reviewed in reference 56), which in various family members has been shown to bind metabolites like homocysteine in other streptococci (57) or signaling molecules like 4-hydroxy-2-heptylquinone in Pseudomonas aeruginosa (58) or to sense redox changes in Escherichia coli (59). Another possible source of specificity of the CpsY regulon for human immune cells is that the CpsY regulon is required for optimal resistance to immune factors exclusively present in human immune cells or absent from immune cells of murine models of infection. For example, murine neutrophils do not express defensins, and the murine immune response lacks several signaling factors like CD4 on macrophages, Fc receptors like FcαR1 (CD89) on neutrophils, and FcγRIIA and FcγRIIC (60). Investigation of the source of specificity for immune cell opsonophagocytic killing resistance of the CpsY regulon and the mechanism of CpsY-mediated gene regulation is an important avenue of research for understanding GAS pathogenesis.
MATERIALS AND METHODS
Bacterial strains and media.
Streptococcus pyogenes (GAS) strain 5448 (61) is an M1T1 strain isolated from an invasive infection. The genome of strain 5448 was used as a reference genome, and the gene nomenclature used for this study corresponds to the MGAS5005 genome (62). GAS bacteria were cultured in either Todd-Hewitt medium supplemented with 0.2% yeast extract (THY), C medium, or chemically defined medium supplemented with 0.5% glucose (CDM). Escherichia coli strain DH5α (hsdR17 recA1 gyrA endA1 relA1) was used as the host for plasmid constructions. All E. coli strains were grown in Luria-Bertani (LB) broth. Antibiotics were used at the following concentrations: spectinomycin (Sp) at 100 μg ml−1 for both E. coli and GAS and kanamycin (Km) at 50 μg ml−1 for E. coli and 300 μg ml−1 for GAS. Growth of GAS was assayed by measuring absorbance using a Klett-Summerson colorimeter (A filter) and expressed in Klett units. Alternatively, overnight cultures of GAS (10 ml) were grown in THY and adjusted to an optical density at 600 nm (OD600) of 0.2 in saline, and 50-μl aliquots were added to individual wells of a 24-well plate (Corning/Costar) containing CDM with a given carbohydrate.
Construction of (ι)cpsY and rescue strains.
The (ι)cpsY mutant strain and its rescue strain were constructed in the 5448 background using the pSinS/pHlpK mutagenesis system (63) for stable plasmid integration into the GAS chromosome. A 345-nucleotide (nt) internal fragment of cpsY (M5005_Spy0701) was PCR amplified from GAS 5448 gDNA using the primers oIISpy0701F and oIISpy0701R (Table S1), digested with BamHI, and cloned into the pSinS suicide plasmid to obtain pSinS-CpsY. The pSinS-CpsY construct was introduced into pHlpK-containing GAS cells, and transformants were selected in the presence of Km and Sp at a permissive temperature (30°C) to allow replication of the two plasmids. Chromosomal integration of the mutagenic plasmid was achieved by culturing the clone at a nonpermissive temperature (37°C) in the presence of Sp only. GAS clones that were Sp resistant and Km sensitive were selected, and insertional inactivation of cpsY by chromosomal integration of the pSinS-CpsY construct was confirmed by PCR analysis and sequencing using the primers oSISpy0701V1 and oSISpy0700V2 (Table S1). The rescue strain was obtained by reintroduction of the pHlpK plasmid into the (ι)cpsY mutant and two overnight passages at 30°C without antibiotic selection to allow excision of the integrated plasmid. Following passage, Sp- and Km-sensitive clones were selected for at 37°C and reconstitution of the cpsY wild-type allele was confirmed by PCR sequencing using primers oSISpy0701V1 and oSISpy0700V2 (Table S1).
Bacterial growth and antimicrobial sensitivity assays.
Growth and antimicrobial sensitivity assays employed mid-exponential-phase bacterial cultures (OD600 = 0.4) of either the wild type (5448) or mutant (ι)cpsY or rescue (cpsYR) strains of GAS. Bacteria were washed twice in phosphate-buffered saline (PBS), normalized to 1 × 103 CFU ml−1, and incubated at 37°C in 5% CO2 unless otherwise indicated. For the assessment of GAS survival in human plasma, normalized bacterial cultures were used to inoculate plasma isolated from heparinized blood of volunteer donors. Inoculated plasma was suspended 1:1 in RPMI 1640 cell culture medium (HyClone; SH30027) supplemented with 2.05 mM l-glutamine and incubated in 48-well flat-bottom plates; at the desired time points, samples were taken for plating on THY agar to obtain viable CFU counts. For the assessment of the (ι)cpsY strain response to methionine, CDM was supplemented with 50 μg ml−1 of methionine.
Carbohydrate metabolic profiles were determined using API 50 CH strips (bioMérieux). Strains were cultured overnight on Trypticase soy agar with 5% sheep blood plates (TSA II; Becton Dickinson Diagnostic), resuspended in 1 ml of saline, and vortexed for 3 min. Strains were then diluted to a final OD600 of 0.14 in 10 ml of API 50 CHL medium (bioMérieux) and cell suspension added to each of the 50 cupules, representing one carbon source each, in addition to a negative control. Strains were incubated covered at 37°C in 5% CO2 for 48 h. Utilization scores were determined at 24 h and 48 h. A “+” was given if the cupule changed from purple to yellow, indicating complete utilization. A “+/−” was given if there was a partial color change, indicating partial utilization. A “−” was given if there was no color change at all, indicating no utilization.
GAS sensitivity to antimicrobial compounds was determined as previously described (29), with some modifications. Briefly, working concentrations of the following antimicrobials (Sigma) were prepared in THY broth: carbenicillin (3 μg ml−1), bacitracin (4 μg ml−1), and nisin (40 μg ml−1). Aliquots of antimicrobial stock solutions (500 μl) were added to the last column of a 48-well flat-bottom plate and serially diluted 1:2 into 500 μl of THY broth containing normalized culture. For lysozyme treatments, wells were prepared with increasing concentrations of chicken egg white lysozyme (46,900 U mg−1; Sigma), from 6.25 mg ml−1 to 50 mg ml−1 in a 500-μl total volume of THY broth. Plates were incubated overnight, and samples were taken for plating on THY agar at 5 h and 16 h to determine viable CFU counts. The minimum bactericidal concentration (MBC) was scored as the lowest antimicrobial concentration with no detectable colonies on THY agar after incubation for 5 h.
Bactericidal assays in whole blood.
The ability of GAS strains to survive in heparinized whole human blood was tested using the Lancefield blood bactericidal assay as previously described (19, 30). Briefly, strains were grown to early mid-exponential phase (OD600 ∼ 0.15) and serially diluted in saline. A 50-μl volume of a 10−4 dilution (ca. 50 to 200 CFU) was added to 500 μl of fresh whole human blood and rotated for 3 h at 37°C. The multiplication factor (MF) was calculated by dividing CFU obtained from blood challenge by initial CFU inoculated. Data are presented as percent growth in blood corresponding to the MF of the mutant divided by the MF of the wild type × 100.
The bactericidal assay was adapted to use whole mouse blood as follows: 8- to 10-week-old female CD-1 mice (Charles River Laboratories) were anesthetized with ketamine, and a blood volume of ≥500 μl was withdrawn by terminal cardiac puncture and the blood immediately heparinized. A 30-μl volume of GAS diluted mid-exponential-phase culture (ca. 30 to 150 CFU) was added to 300 μl of fresh whole mouse blood and incubated as indicated above, and the MF was calculated. For the MF of each strain in murine blood obtained from multiple mice (n = 4 to 6), the MF was normalized to the MF of the wild-type strain (5448). All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Maryland, College Park (protocol R-16-05). Results of bactericidal assays were tested for significance by unpaired t test comparing the (ι)cpsY mutant or cpsYR rescue strain to the wild-type 5448 strain.
Cell culture.
Neutrophils (polymorphonuclear leukocytes [PMNs]) and monocytes were isolated from heparinized blood of nonimmune volunteer donors using Polymorphprep (Axis-Shield) and Ficoll Paque PLUS (GE Healthcare), respectively, as per the manufacturer's instructions. Contaminating red blood cells were removed by treatment with red blood cell lysis solution (Epicentre) and washing with Dulbecco's modified PBS (HyClone). Isolated PMNs and monocytes were both maintained in RPMI 1640 cell culture medium (HyClone) supplemented with 2.05 mM l-glutamine and 20% plasma from donor blood for the duration of the assay.
HL60 cells (Sigma) were maintained as indicated in the UAB-GBS-OPA protocol of Nahm and Burton (version E.02 [64]). Briefly, HL60 cells line were cultured in RPMI 1640 medium supplemented with 2.05 mM l-glutamine and 10% FBS (HyClone). Low-passage HL60 cells were differentiated into neutrophil-like cells for opsonophagocytic killing assays by supplementing culture medium with 1 μM all-trans retinoic acid (ATRA) in dimethyl sulfoxide (DMSO; 3-mg ml−1 stock solution). PMNs, monocytes, and HL60 cells were maintained at 37°C in 5% CO2.
OKAs.
Isolated PMNs or monocytes or differentiated HL60 cells were seeded at a density of 106 ml−1 in 24-well plates with RPMI 1640 medium supplemented with 2.05 mM l-glutamine. Wild-type GAS 5448 and mutant (ι)cpsY strains from overnight cultures were diluted into fresh THY and grown to mid-log phase (OD600 ∼ 0.4). For opsonophagocytic killing assays (OKAs), bacteria were opsonized prior to phagocyte challenge by suspension in donor plasma for 30 min at 37°C. GAS organisms were then added by centrifugation (500 × g for 5 min in a Sorvall/Heraeus 6445 rotor) to seeded phagocytes to the desired multiplicity of infection (MOI; an MOI of 0.1 was used unless otherwise indicated) at a final volume of 1 ml of RPMI 1640 medium (HyClone)–2.05 mM l-glutamine plus 20% donor plasma. Phagocyte-challenged bacteria were incubated at 37°C in 5% CO2 for 30 min or 2 h. For nonphagocytic killing assays, opsonophagocytosis of GAS was prevented by the addition of cytochalasin D (10 μg ml−1) to PMNs, monocytes, or HL60 cells 10 min prior to bacterial challenge, as well as resuspension of GAS in heat-inactivated donor plasma as required prior to addition to seeded phagocytes. GAS organisms were also incubated in RPMI 1640–2.05 mM l-glutamine plus 20% donor plasma in the absence of PMNs, monocytes, or differentiated HL60 cells for the purpose of survival comparison.
Following incubation, viable GAS were harvested by collecting well supernatants as well as neutrophils. Phagocytes were immediately lysed by resuspension in sterile H2O and the intracellular contents pelleted and recombined with corresponding well supernatants for plating on THY agar to obtain total viable bacterial counts after overnight incubation at 37°C in 5% CO2. Resistance of GAS to opsonophagocytic killing was assessed by comparing CFU obtained from plating of viable bacteria isolated from killing assays to CFU obtained from GAS incubation in cell culture media in the absence of phagocytes [(CFU obtained in PMNs/CFU obtained in media) × 100]. The percentages of viable bacteria recovered presented are normalized to that of wild-type 5448 bacteria, set to 100%, under the condition tested. Data presented are the results of at least 3 biological replicates, each performed in triplicate, and results were tested for significance by one-way analysis of variance (ANOVA) with Dunnett's multiple-comparison test for comparison to the wild-type strain (5448).
In vivo mouse models of GAS infection.
All animal experiments were approved by the IACUC at the University of Maryland, College Park (protocol R-16-05). An overnight GAS culture was used to inoculate 80 ml of THY and incubated statically at 37°C to late-logarithmic phase (Klett value, 90 to 100). A bacterial suspension in saline of approximately 4 × 109 CFU ml−1, as determined by microscope counts and verified by plating for viable colonies, was used to infect 5- to 6-week-old female CD-1 mice (Charles River Laboratories). For the systemic infection model, the mice were injected intraperitoneally (i.p.) with 100 μl of the bacterial suspension in saline (4 × 108 CFU/mouse). Mice were monitored three times daily over a period not exceeding 72 h for morbidity. For the subcutaneous infection model, the mice were anesthetized with ketamine, fur was removed from an ∼3-cm2 area of the haunch with Nair (Carter Products), and 100 μl of a cell suspension in saline (3 × 108 CFU/mouse) was injected under the skin. Mice were monitored twice daily for 7 days and were euthanized by CO2 asphyxiation upon signs of morbidity. Lesions were recorded photographically at 24, 48, and 72 h postinfection and measured using ImageJ software. Lesion size data were analyzed using GraphPad Prism (GraphPad Software) and tested for significance using an unpaired two-tailed t test with 99% confidence. Survival data were assessed by Kaplan-Meier survival analysis and tested for significance by log rank test. Data shown represent values for 10 mice for each strain.
RNA isolation and qRT-PCR analysis.
Quantitative real-time PCR (qRT-PCR) was performed as follows. Total RNA was isolated from GAS grown in enriched medium (THY) to early and mid- and late-logarithmic phase or to mid-logarithmic phase in THY (Klett value, 55 to 65), resuspended in RPMI 1640–2.05 mM l-glutamine plus 20% plasma isolated from heparinized whole blood from human donors and incubated for 2 h as described above. RNA was isolated using the Direct-zol RNA MiniPrep kit (Zymo Research) with a modified procedure to improve GAS cell disruption, as described previously (22). Briefly, cells were resuspended in 700 μl of TRIzol supplemented with ca. 300 mg of acid-washed glass beads (Sigma) and disrupted by vortexing for 5 min. Beads were collected by brief centrifugation, and the cell lysate was used for RNA purification as recommended by the manufacturer. Total RNA (5 μg) was subjected to DNase I treatment with the Turbo DNase-free kit (Life Technologies) to avoid gDNA contamination. Subsequently, 25 ng of DNase-treated total RNA was added to SYBR green master mix (Applied Biosystems) with 6.5 μl of each gene-specific real-time primer from a 20 nM stock (Table S1) using the one-step protocol on a Light Cycler 480 (Roche). Real-time primers were designed using the interactive tool Primer3 (http://biotools.umassmed.edu/bioapps/primer3_www.cgi). The levels shown represent ratios of the experimental/wild-type levels relative to gyrA transcripts as the internal control. Standard error was calculated from three biological replicates, and differences over 2-fold in expression were considered significant.
RNA-seq and data analysis.
For RNA sequencing (RNA-seq), bacteria were grown in enriched medium (THY) either to late logarithmic phase (Klett value, 85 to 95) or to mid-logarithmic phase (Klett value, 55 to 65), resuspended in RPMI 1640–2.05 mM l-glutamine plus 20% plasma isolated from human blood donors and incubated for 2 h as described above. Following growth, cells were resuspended 1:2 in RNAprotect bacterial reagent (Qiagen) and incubated at room temperature for 5 min to stabilize RNA. Total RNA was extracted using the Direct-zol RNA miniprep kit (Zymo Research) as described above. RNA samples were treated with the Turbo DNase-free kit (Life Technologies) to avoid genomic DNA (gDNA) contamination. A total of 5 μg of DNase-treated RNA was subjected to rRNA removal using the Ribo-Zero Magnetic kit (Epicentre) for Gram-positive bacteria, and rRNA-depleted RNA was then purified with an RNAClean XP kit (Agencourt). Sample quality was assessed using a Bioanalyzer 2100 (Agilent), and sample quantity was determined using a NanoDrop 8000 spectrophotometer (Thermo Scientific).
RNA-seq directional libraries were generated using the ScriptSeq v2 RNA-seq library preparation kit (Illumina) according to the manufacturer's recommendations. Briefly, 45 ng of rRNA-depleted RNA was fragmented and used for reverse transcription with random primers containing a 5′-tagging sequence. The 5′-tagged cDNA was then modified at its 3′ end by a terminal-tagging reaction to generate di-tagged, single-stranded cDNA that was then purified using the AMPure system (Agencourt). The purified di-tagged cDNA was used as a template to generate second-strand cDNA containing Illumina adaptor sequences, to incorporate index barcodes, and to amplify the library by limited-cycle PCR. The resulting RNA-seq libraries were purified using the AMPure system (Agencourt), and RNA-seq library quality was verified as described above. Rapid-run 100-bp single-read DNA sequencing was then performed at the Institute for Bioscience and Biotechnology Research (IBBR) Sequencing Facility at the University of Maryland, College Park, using the Illumina HiSeq 1500 platform. Data were generated in the standard Sanger FastQ format.
Read quality was measured using FastQC (65), filtered and trimmed using trimmomatic (66), and mapped against the GAS 5448 genome at the National Center for Biotechnology Information (accession number CP008776) using bowtie (67), bowtie2 (68), and tophat (69), with options to allow one mismatch and randomly map multihit reads. The resulting alignments were converted to sorted BAM alignments (70) and counted (71) by coding and intergenic region. Initial visualizations of the sequencing mapping were performed using the Integrative Genomics Viewer (IGV) (72). Differential expression analyses were performed following size factor and quantile normalization of read counts, and batch effect estimation was taken into account by including date in the Limma (73) statistical model. The resulting metrics of expression were visualized using circos (74) as well as tested for ontology enrichment using KEGG (75), goseq (76), clusterProfiler (77), GOstats (78), and topGO (79). Correlation coefficients for RNA-seq were determined by plotting the log2 value of the array on the x axis to the log2 value of the quantitative real-time PCR on the y axis. Linear regression was used to determine the line of best fit, and the resulting R2 value was calculated, which represented the fitness of the data.
Accession number(s).
Raw reads were deposited with the Sequence Read Archive (SRA) at the National Center for Biotechnology Information (Bioproject PJRNA351857).
Supplementary Material
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00925-16.
REFERENCES
- 1.Stetzner ZW, Li D, Feng W, Liu M, Liu G, Wiley J, Lei B. 2015. Serotype M3 and M28 group A streptococci have distinct capacities to evade neutrophil and TNF-alpha responses and to invade soft tissues. PLoS One 10:e0129417. doi: 10.1371/journal.pone.0129417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reglinski M, Sriskandan S. 2014. The contribution of group A streptococcal virulence determinants to the pathogenesis of sepsis. Virulence 5:127–136. doi: 10.4161/viru.26400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kansal RG, Datta V, Aziz RK, Abdeltawab NF, Rowe S, Kotb M. 2010. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J Infect Dis 201:855–865. doi: 10.1086/651019. [DOI] [PubMed] [Google Scholar]
- 4.Mayfield JA, Liang Z, Agrahari G, Lee SW, Donahue DL, Ploplis VA, Castellino FJ. 2014. Mutations in the control of virulence sensor gene from Streptococcus pyogenes after infection in mice lead to clonal bacterial variants with altered gene regulatory activity and virulence. PLoS One 9:e100698. doi: 10.1371/journal.pone.0100698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham MR, Smoot LM, Migliaccio CA, Virtaneva K, Sturdevant DE, Porcella SF, Federle MJ, Adams GJ, Scott JR, Musser JM. 2002. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc Natl Acad Sci U S A 99:13855–13860. doi: 10.1073/pnas.202353699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nordenfelt P, Grinstein S, Bjorck L, Tapper H. 2012. V-ATPase-mediated phagosomal acidification is impaired by Streptococcus pyogenes through Mga-regulated surface proteins. Microbes Infect 14:1319–1329. doi: 10.1016/j.micinf.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Liu G, Feng W, Li D, Liu M, Nelson DC, Lei B. 2015. The Mga regulon but not deoxyribonuclease Sda1 of invasive M1T1 group A Streptococcus contributes to in vivo selection of CovRS mutations and resistance to innate immune killing mechanisms. Infect Immun 83:4293–4303. doi: 10.1128/IAI.00857-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voyich JM, Sturdevant DE, Braughton KR, Kobayashi SD, Lei B, Virtaneva K, Dorward DW, Musser JM, DeLeo FR. 2003. Genome-wide protective response used by group A Streptococcus to evade destruction by human polymorphonuclear leukocytes. Proc Natl Acad Sci U S A 100:1996–2001. doi: 10.1073/pnas.0337370100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hertzén E, Johansson L, Kansal R, Hecht A, Dahesh S, Janos M, Nizet V, Kotb M, Norrby-Teglund A. 2012. Intracellular Streptococcus pyogenes in human macrophages display an altered gene expression profile. PLoS One 7:e35218. doi: 10.1371/journal.pone.0035218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agrahari G, Liang Z, Mayfield JA, Balsara RD, Ploplis VA, Castellino FJ. 2013. Complement-mediated opsonization of invasive group A Streptococcus pyogenes strain AP53 is regulated by the bacterial two-component cluster of virulence responder/sensor (CovRS) system. J Biol Chem 288:27494–27504. doi: 10.1074/jbc.M113.494864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sjöholm K, Karlsson C, Linder A, Malmstrom J. 2014. A comprehensive analysis of the Streptococcus pyogenes and human plasma protein interaction network. Mol Biosyst 10:1698–1708. doi: 10.1039/C3MB70555B. [DOI] [PubMed] [Google Scholar]
- 12.Loof TG, Deicke C, Medina E. 2014. The role of coagulation/fibrinolysis during Streptococcus pyogenes infection. Front Cell Infect Microbiol 4:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsatsaronis JA, Hollands A, Cole JN, Maamary PG, Gillen CM, Ben Zakour NL, Kotb M, Nizet V, Beatson SA, Walker MJ, Sanderson-Smith ML. 2013. Streptococcal collagen-like protein A and general stress protein 24 are immunomodulating virulence factors of group A Streptococcus. FASEB J 27:2633–2643. doi: 10.1096/fj.12-226662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janulczyk R, Pallon J, Bjorck L. 1999. Identification and characterization of a Streptococcus pyogenes ABC transporter with multiple specificity for metal cations. Mol Microbiol 34:596–606. doi: 10.1046/j.1365-2958.1999.01626.x. [DOI] [PubMed] [Google Scholar]
- 15.Bates CS, Montanez GE, Woods CR, Vincent RM, Eichenbaum Z. 2003. Identification and characterization of a Streptococcus pyogenes operon involved in binding of hemoproteins and acquisition of iron. Infect Immun 71:1042–1055. doi: 10.1128/IAI.71.3.1042-1055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahesh S, Nizet V, Cole JN. 2012. Study of streptococcal hemoprotein receptor (Shr) in iron acquisition and virulence of M1T1 group A streptococcus. Virulence 3:566–575. doi: 10.4161/viru.21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lei B, Liu M, Voyich JM, Prater CI, Kala SV, DeLeo FR, Musser JM. 2003. Identification and characterization of HtsA, a second heme-binding protein made by Streptococcus pyogenes. Infect Immun 71:5962–5969. doi: 10.1128/IAI.71.10.5962-5969.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vega LA, Malke H, McIver KS. 2016. Virulence-related transcriptional regulators of Streptococcus pyogenes, p 270–303. In Ferretti JJ, Stevens DL, Fischetti VA (ed), Streptococcus pyogenes: basic biology to clinical manifestations. University of Oklahoma Health Sciences Center, Oklahoma City, OK. [PubMed] [Google Scholar]
- 19.Le Breton Y, Mistry P, Valdes KM, Quigley J, Kumar N, Tettelin H, McIver KS. 2013. Genome-wide identification of genes required for fitness of group A Streptococcus in human blood. Infect Immun 81:862–875. doi: 10.1128/IAI.00837-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malke H, McShan WM, Ferretti JJ. 2009. Integration of metabolic and virulence pathways in serological group A and C streptococci: nutritional status meets virulence, p 12–25. In Manger K, Klöcking H-P (ed), Symbiosen—wissenschaftliche Wechselwirkungen zu gegenseitigem Vorteil, vol 39 Sonderschriften, Erfurt, Germany. [Google Scholar]
- 21.Cusumano ZT, Watson ME Jr, Caparon MG. 2014. Streptococcus pyogenes arginine and citrulline catabolism promotes infection and modulates innate immunity. Infect Immun 82:233–242. doi: 10.1128/IAI.00916-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valdes KM, Sundar GS, Vega LA, Belew AT, Islam E, Binet R, El-Sayed NM, Le Breton Y, McIver KS. 2016. The fruRBA operon is necessary for group A streptococcal growth in fructose and for resistance to neutrophil killing during growth in whole human blood. Infect Immun 84:1016–1031. doi: 10.1128/IAI.01296-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Efstratiou A, Lamagni T. 2016. Epidemiology of Streptococcus pyogenes, p 465–477. In Ferretti JJ, Stevens DL, Fischetti VA (ed), Streptococcus pyogenes: basic biology to clinical manifestations. University of Oklahoma Health Sciences Center, Oklahoma City, OK. [PubMed] [Google Scholar]
- 24.Sperandio B, Gautier C, McGovern S, Ehrlich DS, Renault P, Martin-Verstraete I, Guedon E. 2007. Control of methionine synthesis and uptake by MetR and homocysteine in Streptococcus mutans. J Bacteriol 189:7032–7044. doi: 10.1128/JB.00703-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shelver D, Rajagopal L, Harris TO, Rubens CE. 2003. MtaR, a regulator of methionine transport, is critical for survival of group B Streptococcus in vivo. J Bacteriol 185:6592–6599. doi: 10.1128/JB.185.22.6592-6599.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryan JD, Liles R, Cvek U, Trutschl M, Shelver D. 2008. Global transcriptional profiling reveals Streptococcus agalactiae genes controlled by the MtaR transcription factor. BMC Genomics 9:607. doi: 10.1186/1471-2164-9-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koskiniemi S, Sellin M, Norgren M. 1998. Identification of two genes, cpsX and cpsY, with putative regulatory function on capsule expression in group B streptococci. FEMS Immunol Med Microbiol 21:159–168. doi: 10.1111/j.1574-695X.1998.tb01162.x. [DOI] [PubMed] [Google Scholar]
- 28.Allen JP, Neely MN. 2011. The Streptococcus iniae transcriptional regulator CpsY is required for protection from neutrophil-mediated killing and proper growth in vitro. Infect Immun 79:4638–4648. doi: 10.1128/IAI.05567-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen JP, Neely MN. 2012. CpsY influences Streptococcus iniae cell wall adaptations important for neutrophil intracellular survival. Infect Immun 80:1707–1715. doi: 10.1128/IAI.00027-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lancefield RC. 1957. Differentiation of group A streptococci with a common R antigen into three serological types, with special reference to the bactericidal test. J Exp Med 106:525–544. doi: 10.1084/jem.106.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okumura CY, Nizet V. 18 June 2014. Subterfuge and sabotage: evasion of host innate defenses by invasive gram-positive bacterial pathogens. Annu Rev Microbiol doi: 10.1146/annurev-micro-092412-155711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, Benarafa C, Roos D, Skokowa J, Hartl D. 2015. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog 11:e1004651. doi: 10.1371/journal.ppat.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwinn LA, Nizet V. 2007. How group A Streptococcus circumvents host phagocyte defenses. Future Microbiol 2:75–84. doi: 10.2217/17460913.2.1.75. [DOI] [PubMed] [Google Scholar]
- 34.Fieber C, Janos M, Koestler T, Gratz N, Li XD, Castiglia V, Aberle M, Sauert M, Wegner M, Alexopoulou L, Kirschning CJ, Chen ZJ, von Haeseler A, Kovarik P. 2015. Innate immune response to Streptococcus pyogenes depends on the combined activation of TLR13 and TLR2. PLoS One 10:e0119727. doi: 10.1371/journal.pone.0119727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gryllos I, Tran-Winkler HJ, Cheng MF, Chung H, Bolcome R III, Lu W, Lehrer RI, Wessels MR. 2008. Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc Natl Acad Sci U S A 105:16755–16760. doi: 10.1073/pnas.0803815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitnack E, Beachey EH. 1982. Antiopsonic activity of fibrinogen bound to M protein on the surface of group A streptococci. J Clin Invest 69:1042–1045. doi: 10.1172/JCI110508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horstmann RD, Sievertsen HJ, Knobloch J, Fischetti VA. 1988. Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc Natl Acad Sci U S A 85:1657–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horstmann RD, Sievertsen HJ, Leippe M, Fischetti VA. 1992. Role of fibrinogen in complement inhibition by streptococcal M protein. Infect Immun 60:5036–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LaRock CN, Dohrmann S, Todd J, Corriden R, Olson J, Johannssen T, Lepenies B, Gallo RL, Ghosh P, Nizet V. 2015. Group A streptococcal M1 protein sequesters cathelicidin to evade innate immune killing. Cell Host Microbe 18:471–477. doi: 10.1016/j.chom.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramos-Sevillano E, Urzainqui A, Campuzano S, Moscoso M, Gonzalez-Camacho F, Domenech M, Rodriguez de Cordoba S, Sanchez-Madrid F, Brown JS, Garcia E, Yuste J. 2015. Pleiotropic effects of cell wall amidase LytA on Streptococcus pneumoniae sensitivity to the host immune response. Infect Immun 83:591–603. doi: 10.1128/IAI.02811-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cox KH, Ruiz-Bustos E, Courtney HS, Dale JB, Pence MA, Nizet V, Aziz RK, Gerling I, Price SM, Hasty DL. 2009. Inactivation of DltA modulates virulence factor expression in Streptococcus pyogenes. PLoS One 4:e5366. doi: 10.1371/journal.pone.0005366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lecours MP, Gottschalk M, Houde M, Lemire P, Fittipaldi N, Segura M. 2011. Critical role for Streptococcus suis cell wall modifications and suilysin in resistance to complement-dependent killing by dendritic cells. J Infect Dis 204:919–929. doi: 10.1093/infdis/jir415. [DOI] [PubMed] [Google Scholar]
- 43.Turner AG, Ong CL, Gillen CM, Davies MR, West NP, McEwan AG, Walker MJ. 2015. Manganese homeostasis in group A Streptococcus is critical for resistance to oxidative stress and virulence. mBio 6:e00278-15. doi: 10.1128/mBio.00278-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukomski S, Burns EH Jr, Wyde PR, Podbielski A, Rurangirwa J, Moore-Poveda DK, Musser JM. 1998. Genetic inactivation of an extracellular cysteine protease (SpeB) expressed by Streptococcus pyogenes decreases resistance to phagocytosis and dissemination to organs. Infect Immun 66:771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Terao Y, Mori Y, Yamaguchi M, Shimizu Y, Ooe K, Hamada S, Kawabata S. 2008. Group A streptococcal cysteine protease degrades C3 (C3b) and contributes to evasion of innate immunity. J Biol Chem 283:6253–6260. doi: 10.1074/jbc.M704821200. [DOI] [PubMed] [Google Scholar]
- 46.Hung CH, Tsao N, Zeng YF, Lu SL, Chuan CN, Lin YS, Wu JJ, Kuo CF. 2012. Synergistic effects of streptolysin S and streptococcal pyrogenic exotoxin B on the mouse model of group A streptococcal infection. Med Microbiol Immunol 201:357–369. doi: 10.1007/s00430-012-0241-6. [DOI] [PubMed] [Google Scholar]
- 47.Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. 2000. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun 68:6362–6369. doi: 10.1128/IAI.68.11.6362-6369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cole JN, McArthur JD, McKay FC, Sanderson-Smith ML, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D, Kotb M, Nizet V, Chhatwal GS, Walker MJ. 2006. Trigger for group A streptococcal M1T1 invasive disease. FASEB J 20:1745–1747. doi: 10.1096/fj.06-5804fje. [DOI] [PubMed] [Google Scholar]
- 49.Li J, Liu G, Feng W, Zhou Y, Liu M, Wiley JA, Lei B. 2014. Neutrophils select hypervirulent CovRS mutants of M1T1 group A Streptococcus during subcutaneous infection of mice. Infect Immun 82:1579–1590. doi: 10.1128/IAI.01458-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, Bailey JR, Parnell MJ, Hoe NP, Adams GG, Deleo FR, Musser JM. 2005. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A 102:1679–1684. doi: 10.1073/pnas.0406641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V. 2006. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16:396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 52.Bastiat-Sempe B, Love JF, Lomayesva N, Wessels MR. 2014. Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages. mBio 5:e01690-14. doi: 10.1128/mBio.01690-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma O, O'Seaghdha M, Velarde JJ, Wessels MR. 2016. NAD+-glycohydrolase promotes intracellular survival of group A Streptococcus. PLoS Pathog 12:e1005468. doi: 10.1371/journal.ppat.1005468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chandrasekaran S, Caparon MG. 2016. The NADase-negative variant of the Streptococcus pyogenes toxin NAD(+) glycohydrolase induces JNK1-mediated programmed cellular necrosis. mBio 7:e02215-15. doi: 10.1128/mBio.02215-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eraso J, Olsen RJ, Beres SB, Kachroo P, Porter AR, Nasser W, Bernard PE, DeLeo F, Musser JM. 18 November 2016. Genomic landscape of intrahost variation in group A Streptococcus: repeated and abundant mutational inactivation of the fabT gene encoding a regulator of fatty acid synthesis. Infect Immun doi: 10.1128/IAI.00608-16. [DOI] [PMC free article] [PubMed]
- 56.Maddocks SE, Oyston PC. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 57.Kovaleva GY, Gelfand MS. 2007. Transcriptional regulation of the methionine and cysteine transport and metabolism in streptococci. FEMS Microbiol Lett 276:207–215. doi: 10.1111/j.1574-6968.2007.00934.x. [DOI] [PubMed] [Google Scholar]
- 58.Cao H, Krishnan G, Goumnerov B, Tsongalis J, Tompkins R, Rahme LG. 2001. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc Natl Acad Sci U S A 98:14613–14618. doi: 10.1073/pnas.251465298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farr SB, Kogoma T. 1991. Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol Rev 55:561–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mestas J, Hughes CC. 2004. Of mice and not men: differences between mouse and human immunology. J Immunol 172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 61.Chatellier S, Ihendyane N, Kansal RG, Khambaty F, Basma H, Norrby-Teglund A, Low DE, McGeer A, Kotb M. 2000. Genetic relatedness and superantigen expression in group A Streptococcus serotype M1 isolates from patients with severe and nonsevere invasive diseases. Infect Immun 68:3523–3534. doi: 10.1128/IAI.68.6.3523-3534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, Musser JM. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis 192:771–782. doi: 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 63.Le Breton Y, Belew AT, Valdes KM, Islam E, Curry P, Tettelin H, Shirtliff ME, El-Sayed NM, McIver KS. 2015. Essential genes in the core genome of the human pathogen Streptococcus pyogenes. Sci Rep 5:9838. doi: 10.1038/srep09838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nahm MH, Burton RL. December 2014. Protocol for multiplexed opsonophagocytic killing assay (UAB-MOPA) for antibodies against Streptococcus pneumoniae, version E.02. University of Alabama, Birmingham, AL. [Google Scholar]
- 65.Andrews S. 2010. FASTQC. A quality control tool for high throughput sequence data. Babraham Bioinformatics, Cambridge, United Kingdom: http://www.bioinformatics.babraham.ac.uk/projects/fastqc. [Google Scholar]
- 66.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B. 2010. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics Chapter 11:Unit 11.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. 1999. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Young MD, Wakefield MJ, Smyth GK, Oshlack A. 2010. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu G, Wang LG, Han Y, He QY. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beissbarth T, Speed TP. 2004. GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics 20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 79.Alexa A, Rahnenfuhrer J, Lengauer T. 2006. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 80.Pancholi V, Boel G, Jin H. 2010. Streptococcus pyogenes Ser/Thr kinase-regulated cell wall hydrolase is a cell division plane-recognizing and chain-forming virulence factor. J Biol Chem 285:30861–30874. doi: 10.1074/jbc.M110.153825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chang JC, LaSarre B, Jimenez JC, Aggarwal C, Federle MJ. 2011. Two group A streptococcal peptide pheromones act through opposing Rgg regulators to control biofilm development. PLoS Pathog 7:e1002190. doi: 10.1371/journal.ppat.1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.