

ABSTRACT
Anaplasma marginale causes bovine anaplasmosis, a debilitating and potentially fatal tick-borne infection of cattle. Because A. marginale is an obligate intracellular organism, its adhesins that mediate entry into host cells are essential for survival. Here, we demonstrate that A. marginale outer membrane protein A (AmOmpA; AM854) contributes to the invasion of mammalian and tick host cells. AmOmpA exhibits predicted structural homology to OmpA of A. phagocytophilum (ApOmpA), an adhesin that uses key lysine and glycine residues to interact with α2,3-sialylated and α1,3-fucosylated glycan receptors, including 6-sulfo-sialyl Lewis x (6-sulfo-sLex). Antisera against AmOmpA or its predicted binding domain inhibits A. marginale infection of host cells. Residues G55 and K58 are contributory, and K59 is essential for recombinant AmOmpA to bind to host cells. Enzymatic removal of α2,3-sialic acid and α1,3-fucose residues from host cell surfaces makes them less supportive of AmOmpA binding. AmOmpA is both an adhesin and an invasin, as coating inert beads with it confers adhesiveness and invasiveness. Recombinant forms of AmOmpA and ApOmpA competitively antagonize A. marginale infection of host cells, but a monoclonal antibody against 6-sulfo-sLex fails to inhibit AmOmpA adhesion and A. marginale infection. Thus, the two OmpA proteins bind related but structurally distinct receptors. This study provides a detailed understanding of AmOmpA function, identifies its essential residues that can be targeted by blocking antibody to reduce infection, and determines that it binds to one or more α2,3-sialylated and α1,3-fucosylated glycan receptors that are unique from those targeted by ApOmpA.
KEYWORDS: Anaplasma, Ehrlichia, Lewis antigen, Lewis x, adhesin, bacterial invasion, intracellular bacteria, obligate intracellular bacteria, rickettsia, tick-borne pathogens
INTRODUCTION
Anaplasma marginale is a Gram-negative obligate intracellular bacterium and the etiologic agent of bovine anaplasmosis, a debilitating infection that is transmitted biologically by ticks, mechanically via fly bites or blood-contaminated fomites, and vertically from mother to calf (1–3). It is a febrile illness, the symptoms of which can include anemia, weight loss, abortion, decreased milk production, and death (1–3). Due to these clinical manifestations, its propensity to become a chronic infection, and the costs associated with treatment, bovine anaplasmosis results in a combined economic loss for the United States and South American cattle industries that exceeds one billion dollars annually (2). In sub-Saharan Africa, where livestock sustain the livelihood of the rural poor (4, 5), the disease can have devastating socioeconomic impacts. A. marginale is a member of the family Anaplasmataceae, which consists of veterinary and human obligate intracellular bacterial pathogens that reside within host cell-derived vacuoles. A. marginale predominantly infects erythrocytes in vivo. Detection of the bacterium colocalizing with the endothelial cell marker von Willebrand factor in tissue sections from an experimentally inoculated calf indicates it is also capable of infecting endothelial cells in vivo and might serve as a reservoir for infection (6). Moreover, endothelial cell lines are useful for studying A. marginale infection in vitro, as they are the only mammalian cell types in which continuous cultivation of these microbes has been achieved (7, 8). The immortalized tick cell line ISE6 is susceptible to A. marginale infection and supports its replication, making it a useful model for studying bacterium-tick cell interactions (9–11).
The pathogen exhibits a biphasic developmental cycle in which it transitions between an infectious dense-core (DC) form that mediates binding and entry and a noninfectious reticulate cell (RC) form that replicates by binary fission inside the A. marginale-occupied vacuole (AmV). Following replication, RCs reconvert to DCs that exit to invade naive host cells and thereby initiate new infections (7, 12). Because A. marginale is an obligate intracellular bacterium, adhesins that mediate binding and entry into host cells are essential for survival. Such key virulence factors, however, are poorly defined.
A. marginale expresses the surface protein OmpA (outer membrane protein A; AM854 in the St. Maries strain) (13) during infection of cattle (14–16). OmpA is highly conserved among A. marginale sensu stricto strains and isolates, exhibiting 99.6 to 100% identity (14). Clues pertaining to the role of A. marginale OmpA (AmOmpA) are provided by recent studies demonstrating the importance of OmpA proteins to cellular invasion by A. phagocytophilum and Ehrlichia chaffeensis, two Anaplasmataceae members that cause potentially fatal infections of humans and animals (17–19). Indeed, we discovered that A. phagocytophilum OmpA (ApOmpA) is one of a trio of adhesins that cooperatively function to mediate optimal bacterial binding to and invasion of host cells (17, 18, 20, 21). Recombinant ApOmpA binds to host cells, confers adhesiveness and invasiveness to inert beads, and acts as a competitive agonist to inhibit A. phagocytophilum infection in vitro (17, 18), confirming that it alone is sufficient to mediate binding and uptake. ApOmpA functionally depends on a lysine and a glycine in its essential linear binding domain that interacts with α2,3-sialic acid and α1,3-fucose of the Lewis antigen receptor, sialyl Lewis x (sLex; NeuAcα2,3Galβ1,4[Fucα1,3]GlcNac), on myeloid cells and 6-sulfo-sialyl Lewis x (6-sulfo-sLex; NeuAcα2,3Galβ1-4[Fucα1,3]HSO33,6GlcNac) on endothelial cells (17, 18). Antibodies raised against full-length ApOmpA or its 16-residue binding domain inhibit A. phagocytophilum infection of host cells (18). Likewise, antibodies against E. chaffeensis OmpA inhibit ehrlichial infection in vitro (19).
In this study, we demonstrate that AmOmpA is an adhesin that contributes to A. marginale infection of mammalian and tick host cells. The adhesin capability of AmOmpA depends on specific lysine and glycine residues located within an essential binding domain, the position of which is predicted to be structurally conserved with that of ApOmpA. It recognizes an α2,3-sialylated and α1,3-fucosylated glycan that is not 6-sulfo-sLex. Collectively, these data reveal the role of AmOmpA, identify its essential region that can be targeted by antibodies to inhibit infection, and underscore the conserved pathobiological importance of OmpA proteins to Anaplasma and Ehrlichia spp.
RESULTS
Molecular modeling reveals high predicted structural homology between AmOmpA and ApOmpA and delineates a putative binding domain.
Given the demonstrated roles of A. phagocytophilum and E. chaffeensis OmpA proteins in promoting infection of mammalian host cells (17, 19, 21), we sought to determine if AmOmpA performs a similar adhesin function for A. marginale. An alignment of ApOmpA and AmOmpA revealed that the two exhibit 52.33% sequence identity (17). Notably, one particular stretch where the two proteins exhibit considerable identity occurs between ApOmpA residues 59 and 74 (ApOmpA59–74; L59KGPGKKVILELVEQL74), which forms the essential binding domain (18), and AmOmpA53–68 (I53KGSGKKVLLGLVERM68) (identical and similar residues between the two peptides are denoted by boldface and underlined text, respectively). In our preceding study, molecular modeling of ApOmpA predicted that residues 59 to 74 form a surface-exposed alpha helix in which G61 and K64 help form a binding pocket that interacts with Lewis antigen receptors. This model proved useful for directing experiments that validated the functional essentiality of ApOmpA G61 and K64 (18). Therefore, as a first step in assessing the potential adhesin role of AmOmpA, molecular modeling of amino acids 19 to 236 (excluding the signal sequence) was performed using the PHYRE2 recognition server (www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi) (22), which predicts three-dimensional structures for protein sequences and threads the predicted models on known crystal structures. Threading the AmOmpA and ApOmpA tertiary models onto each other using PyMOL (pymol.org/educational) revealed that the two are very similar structurally and that the relative positions of the AmOmpA53–68 and ApOmpA59–74 alpha helices overlap (Fig. 1A and B; also see Movie S1 in the supplemental material). Moreover, the predicted tertiary locations of AmOmpA G55 and K58 overlay perfectly with ApOmpA G61 and K64, respectively (Fig. 1B). A space-filling model of AmOmpA indicated that G55, K58, and flanking residues form a binding pocket that is structurally analogous to that predicted for ApOmpA (Fig. 1C). ApOmpA and other microbial proteins that interact with sLex do so at cationic surface patches (18, 23–28). Consistent with this trend, using the APBS (29) plugin for PyMOL to calculate AmOmpA surface electrostatic values predicted that amino acids 19 to 67, which contain the region that is homologous to the sLex/6-sulfo-sLex binding domain of ApOmpA, have an overall cationic surface charge (Fig. 1D). Based on these data, we hypothesize that AmOmpA functions as an adhesin and that key amino acids within the stretch comprised of residues 53 to 68 are functionally essential.
FIG 1.

AmOmpA and ApOmpA are structurally similar and exhibit conservation of glycine and lysine residues demonstrated to be important for adhesin function in ApOmpA. (A and B) The predicted tertiary structures for ApOmpA and AmOmpA are highly similar. (A) Presented is a static image from Movie S1 in which the predicted tertiary structures for ApOmpA (gray) and AmOmpA (blue) are overlaid to demonstrate their structural similarity. A PHYRE2 model of the mature sequence lacking signal peptide for each OmpA protein was generated, and the models were threaded onto each other using PyMOL. ApOmpA residues 59 to 74, which comprise the essential binding domain, are orange. AmOmpA residues 53 to 68, which form an alpha-helix that is similar in location to that formed by ApOmpA essential binding domain residues 59 to 74 and are therefore predicted to form the AmOmpA binding domain, are green. ApOmpA residues glycine 61 and lysine 64, which were previously confirmed to be essential for adhesin function, are red. (B) Zoom-in of the image presented in panel A. Note that the alpha helices formed by the essential binding domain of ApOmpA and the putative AmOmpA binding domain overlap. ApOmpA functionally essential residues glycine 61 and lysine 64 correspond to AmOmpA G55 and K58. (C) Space-filling model of AmOmpA. The putative binding domain encompassed by residues 53 to 68 is indicated in green and G55 and K58 are red. (D) Electrostatic surface map of A. marginale OmpA, as generated using the PyMol APBS plugin. Positive and negative charges are indicated by blue and red, respectively.
Antisera raised against AmOmpA and its putative binding domain inhibit infection of mammalian host cells.
To begin to test our hypothesis, we generated antisera against His-tagged mature AmOmpA and a peptide corresponding to its putative binding domain. For the binding domain peptide, one comprised of residues 50 to 67 was selected because it contains all of the residues that are likely to be critical for function, as described below, and has a higher Jameson-Wolfe antigenicity index score (30) than one corresponding to residues 53 to 68. Both antisera recognized a recombinant version of AmOmpA, AmOmpA50–67, and exhibited no to minimal cross-reactivity via Western blotting and enzyme-linked immunosorbent assay (ELISA) with glutathione S-transferase (GST) alone, recombinant ApOmpA proteins, or a His-tagged version of OmpA from Orientia tsutsugamushi (OtOmpA), an obligate intracellular bacterial pathogen that is in the order Rickettsiales with Anaplasma spp. (Fig. 2A to C). Screening A. marginale-infected RF/6A endothelial and tick embryonic ISE6 cells and A. phagocytophilum-infected promyelocytic HL-60 cells with anti-AmOmpA detected a band of the expected size for AmOmpA only in lysates of A. marginale-infected cells (Fig. 2D). Thus, AmOmpA and AmOmpA50–67 antisera exclusively recognize their target antigens. An additional observation gleaned from these data is that while A. phagocytophilum expresses OmpA during infection of mammalian but not tick cells (17), A. marginale expresses OmpA during infection of both host cell types.
FIG 2.
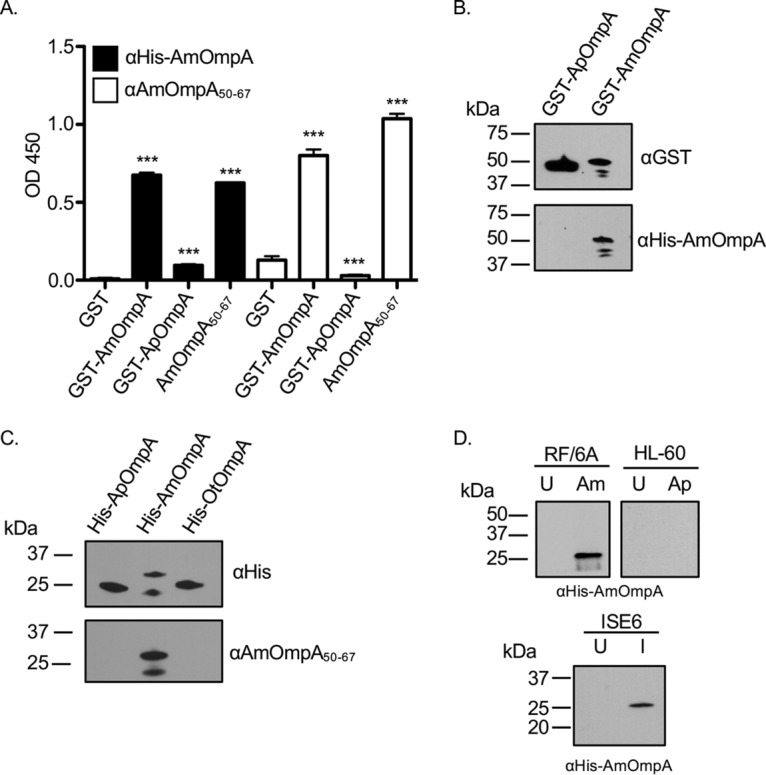
Antibodies raised toward AmOmpA are specific. (A) Wells coated with GST alone, GST-AmOmpA, GST-ApOmpA, or AmOmpA50–67 were screened with antibodies targeting mature AmOmpA or AmOmpA50–67. Results shown are the means ± standard deviations (SD) from triplicate samples. OD450, optical density at 450 nm. ***, P < 0.001. (B) GST-tagged ApOmpA and AmOmpA were subjected to Western blot analyses with anti-GST, anti-ApOmpA59–74, or anti-HisAmOmpA. (C) Western blot analyses of His-ApOmpA, His-AmOmpA, and His-OtOmpA using antibodies specific for the His tag, ApOmpA59–74, and AmOmpA50–67. (D) Rat anti-HisAmOmpA was used to screen Western-blotted A. marginale (Am)-infected (I) and uninfected (U) RF/6A, ISE6 whole-cell lysates, and A. phagocytophilum (Ap)-infected and uninfected HL-60 cell lysates.
The abilities of both antisera to inhibit A. marginale infection of mammalian host cells were evaluated next. A. marginale DC organisms were treated with heat-inactivated AmOmpA or AmOmpA50–67 antiserum prior to incubation with RF/6A cells. After 48 h, infection was assessed using immunofluorescence microscopy. Each antiserum reduced the percentage of infected cells by approximately 25% and decreased the number of AmVs per cells by approximately 40%, whereas preimmune serum had no effect (Fig. 3A to D). To ensure that the blocking effects achieved were specific and not due to steric hindrance, the experiments were repeated using fragment antigen binding (Fab fragment) portions of anti-AmOmpA and anti-AmOmpA50–67. Blocking achieved with the Fab fragments was identical to that achieved with intact antibodies (Fig. 3E to H). These data indicate that AmOmpA contributes to A. marginale infection of mammalian host cells. Moreover, the high similarity of the inhibitory effects achieved by anti-AmOmpA and anti-AmOmpA50–67 supports that residues within 50 to 67 are important for AmOmpA-mediated infection.
FIG 3.
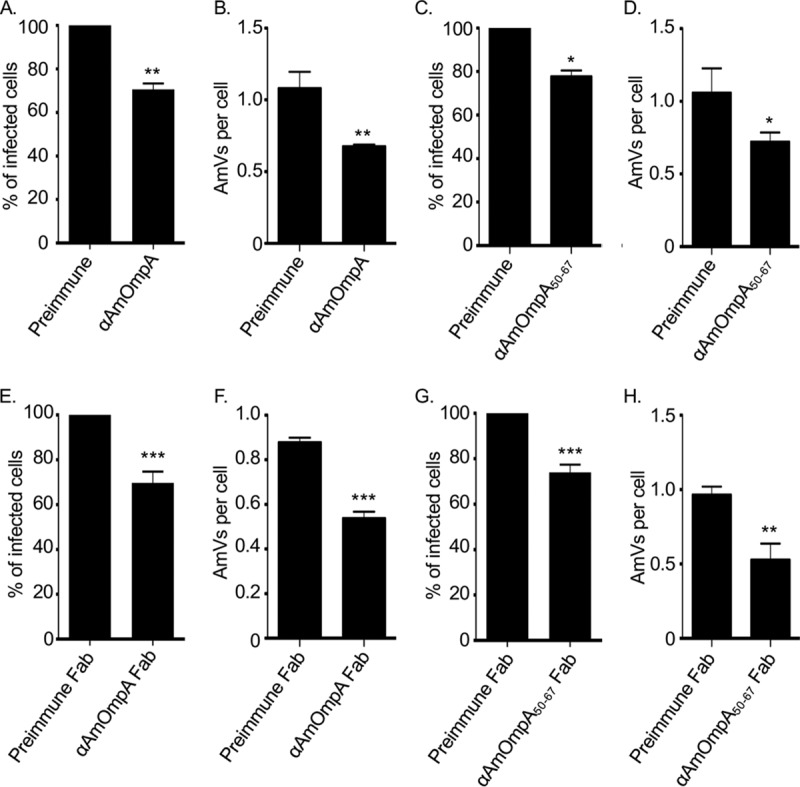
Antisera raised against AmOmpA and AmOmpA50–67 inhibit infection. A. marginale DC organisms were incubated with preimmune serum, antiserum specific for AmOmpA (A and B), AmOmpA50–67 (C and D), or Fab fragments thereof (E to H) for 1 h followed by incubation with RF/6A cells in the continued presence of sera for 2 h. Unbound bacteria were removed and the infection was allowed to proceed for 48 h, after which the host cells were fixed and examined using immunofluorescence microscopy to determine the percentages of infected cells (A, C, E, and G) and the number of AmVs per cell (B, D, F, and H). Results are the means ± SD from triplicate samples and are representative of three independent experiments with similar results. Statistically significant (*, P < 0.05; **, P < 0.005; ***, P < 0.001) values are indicated.
G55, K58, and K59 are critical for recombinant AmOmpA to bind to mammalian host cells.
To determine if AmOmpA exhibits adhesin activity and, if so, to define the importance of individual amino acid residues within the binding domain to such activity, His-tagged AmOmpA and versions thereof in which specific residues were mutated to alanine were assessed for the ability to bind to RF/6A cells using flow cytometry. ApOmpA binding domain residues G61 and K64, but not other binding domain residues, are functionally essential (18). Therefore, AmOmpA G55 and K58 were prioritized for substitution because they align both sequentially and in relative position in the predicted tertiary structure with ApOmpA G61 and K64. K54 and K59 were also replaced with alanine, since they immediately flank G55 and K58. D47 was replaced as a negative control because it lies outside the AmOmpA binding domain and corresponds to ApOmpA D53, which was previously shown to be functionally irrelevant (18). As expected, both His-AmOmpA and His-AmOmpAD47A bound to host cells (Fig. 4). K54 is dispensable for AmOmpA function, as His-AmOmpAK54A was uncompromised in its ability to bind to host cells. His-tagged AmOmpAG55A and AmOmpAK59 displayed modest and considerably more pronounced reductions in binding, respectively. Replacing K58 alone led to an increase in binding, and replacing it together with G55 did not further reduce binding compared to replacing G55 alone. However, replacing K58 together with K59 abolished binding. Overall, these observations demonstrate that AmOmpA adhesin function critically relies on G55, K58, and K59.
FIG 4.
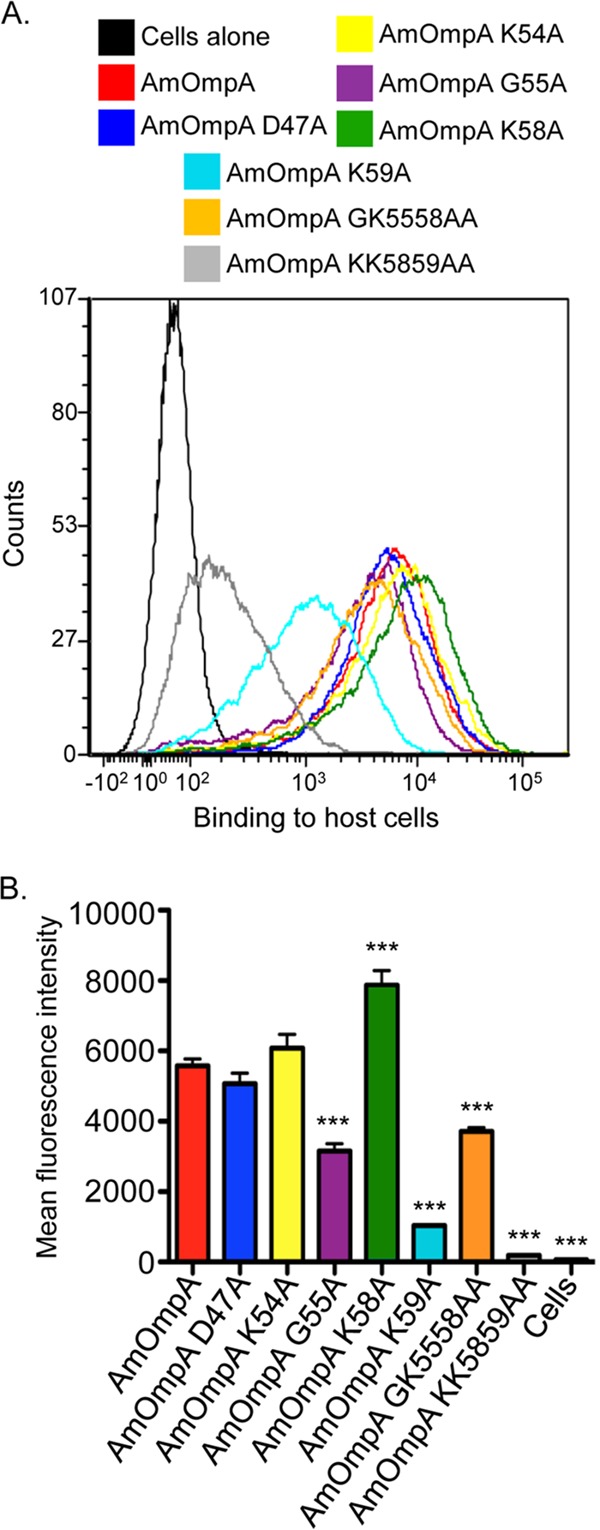
G55, K58, and K59 are critical for recombinant AmOmpA to optimally bind to mammalian host cells. RF/6A cells were incubated with His-tagged AmOmpA or versions thereof in which specific residues were replaced with alanine. The cells were successively incubated with His tag antibody and Alexa Fluor 488-conjugated anti-mouse IgG and analyzed by flow cytometry. (A) Representative histograms (A) and the mean fluorescence intensities ± SD from triplicate samples (B) are presented. Data are representative of three independent experiments with similar results. Statistically significant (***, P < 0.001) values compared to AmOmpA are indicated.
AmOmpA interacts with sialic acid and fucose on mammalian host cells.
Consistent with it being an adhesin that interacts with α2,3-sialylated and α1,3-fucosylated receptors on mammalian host cells, binding of recombinant ApOmpA to cell surfaces from which either sugar residue has been enzymatically removed is significantly reduced (17, 18). To determine if AmOmpA binds to α2,3-sialic acid or α1,3-fucose, His-tagged AmOmpA was incubated with RF/6A cells that had been treated with α2,3/6-sialidase or α1,3/4-fucosidase, respectively, and binding was assessed by immunofluorescence microscopy and flow cytometry. To verify the efficacy of the glycosidases, treated and untreated cells were screened with lectins that recognize fucose and sialic acid residues that are in the specific linkages of interest. AAL (Aleuria aurantia lectin) recognizes fucose residues that are in α1,3- and α1,6-linkages with N-acetylglucosamine (31, 32). MAL II (Maackia amurensis lectin II) recognizes sialic acid residues that are in α2,3-linkages with galactose. Fucosidase treatment abolished binding of AAL but not MAL II. Conversely, sialidase treatment prevented binding of MAL II but not AAL (Fig. 5A). Thus, the glycosidases effectively and specifically enzymatically removed their target sugar residues. His-AmOmpA binding to sialidase- and fucosidase-treated cells was similarly reduced compared to vehicle control-treated cells (Fig. 5A to E). Thus, AmOmpA utilizes both α2,3-sialic acid and α1,3-fucose for optimal adhesion to host cells.
FIG 5.

AmOmpA interacts with α2,3-sialic acid and α1,3-fucose on mammalian host cell surfaces. RF/6A cells were pretreated with α2,3/6-sialidase (A to C), α1,3/4-fucosidase (A, D, and E), or vehicle control (A to E). Glycosidase- and vehicle-treated cells were incubated with α1,3/6-fucose-specific lectin, AAL (A); α2,3-sialic acid-specific lectin, MALII (A); His-AmOmpA (A to E); or media (cells alone; A to E). The cells were fixed and screened using immunofluorescence microscopy (A) or flow cytometry (B to E). In panel A, green fluorescence corresponds to lectin or His-AmOmpA bound at cell surfaces. Host cell nuclei are stained blue by 4′,6-diamidino-2-phenylindole (DAPI). For flow-cytometric data (B to E), representative histograms showing His-AmOmpA binding to RF/6A cells are presented in panels B and D. Mean fluorescence intensities ± SD from triplicate samples are presented in panels C and E. Data shown are representative of three independent experiments with similar results. Statistically significant (***, P < 0.001) values are indicated.
AmOmpA-coated beads bind to and are internalized by endothelial cells.
The ability of His-AmOmpA to bind to host cells suggests that it exhibits adhesin function. Whether it also functions as an invasin is unknown. As a complementary approach to confirm its adhesin activity and to assess its capacity to function as an invasin, the ability of His-AmOmpA to confer adhesiveness and invasiveness to inert particles was assessed. His-AmOmpA was conjugated to red fluorescent microspheres that were 1.0 μm in diameter, which approximates the diameter of a typical A. marginale DC organism (0.8 ± 0.2 μm) (7). Nonphagocytic RF/6A endothelial cells were incubated with recombinant AmOmpA-coated or noncoated control beads and screened with AmOmpA antibody to determine the numbers of beads bound per cell. To measure bead internalization, the cells were incubated for an additional 7 h and trypsin was used to remove noninternalized beads prior to screening. Immunofluorescence microscopy confirmed that significantly more AmOmpA-coated beads bound to and were internalized by RF/6A cells than noncoated control beads (Fig. 6), thereby demonstrating that AmOmpA has the capacity to act as both an adhesin and invasin.
FIG 6.
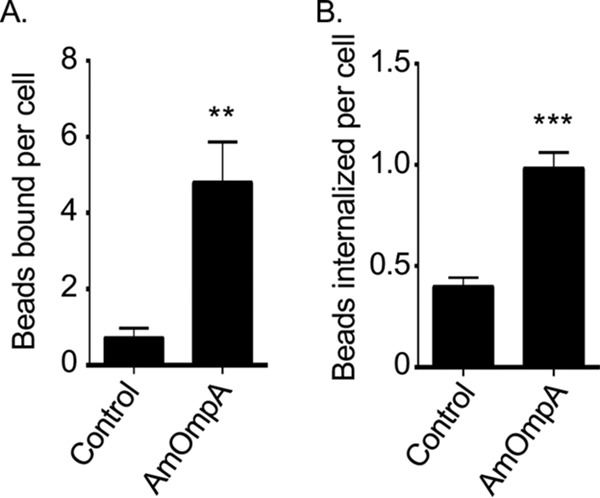
AmOmpA-coated beads bind to and are internalized by endothelial cells. Red fluorescent His-AmOmpA-coated microspheres (AmOmpA beads) were incubated with RF/6A endothelial cells. (A) Binding was assessed by immunofluorescence microscopy after 1 h. (B) To assess internalization, cells were treated with trypsin after 8 h, washed, adhered to coverslips, fixed, and screened with an anti-His tag antibody by immunofluorescence microscopy. Results are the means ± SD and are representative of three independent experiments done in triplicate with similar results. Statistically significant (**, P < 0.005; ***, P < 0.001) values are indicated.
AmOmpA and ApOmpA recognize different but structurally similar receptors on endothelial cells.
Recombinant ApOmpA binding to the 6-sulfo-sLex receptor competitively inhibits A. phagocytophilum infection of RF/6A cells (18). Because AmOmpA binding to RF/6A cells involves recognition of α2,3-sialic acid and α1,3-fucose, because AmOmpA and ApOmpA each bind to RF/6A cells, and because of the homologies between the two proteins' binding domains, we rationalized that they might recognize the same or structurally similar receptors on endothelial cells. If so, then recombinant forms of AmOmpA and ApOmpA should competitively antagonize A. marginale infection of RF/6A cells to comparable degrees. Indeed, preincubating the host cells with GST-tagged AmOmpA and ApOmpA led to similar reductions in the percentage of infected cells and the mean number of AmVs per cell (Fig. 7). To determine if AmOmpA interacts with 6-sulfo-sLex on RF/6A cells, His-AmOmpA binding to the host cells treated with the 6-sulfo-sLex-specific monoclonal antibody G72 (33) was assessed. This antibody was previously confirmed to bind to RF/6A cell surfaces and thereby inhibit recombinant ApOmpA adhesion (18). Monoclonal antibodies CSLEX1 and KM93, recognizing sLex (34, 35), which is poorly expressed on RF/6A cells, and IgM served as negative and isotype controls, respectively. None of the antibodies inhibited His-AmOmpA binding (Fig. 8A). Likewise, G72 was ineffective at inhibiting A. marginale infection of RF/6A cells (Fig. 8B and C). Taken together, these data and the results presented above indicate that both recombinant AmOmpA and native AmOmpA on the A. marginale surface recognize an α2,3-sialylated and α1,3-fucosylated receptor on endothelial cells that is distinct from the ApOmpA endothelial receptor, 6-sulfo-sLex.
FIG 7.
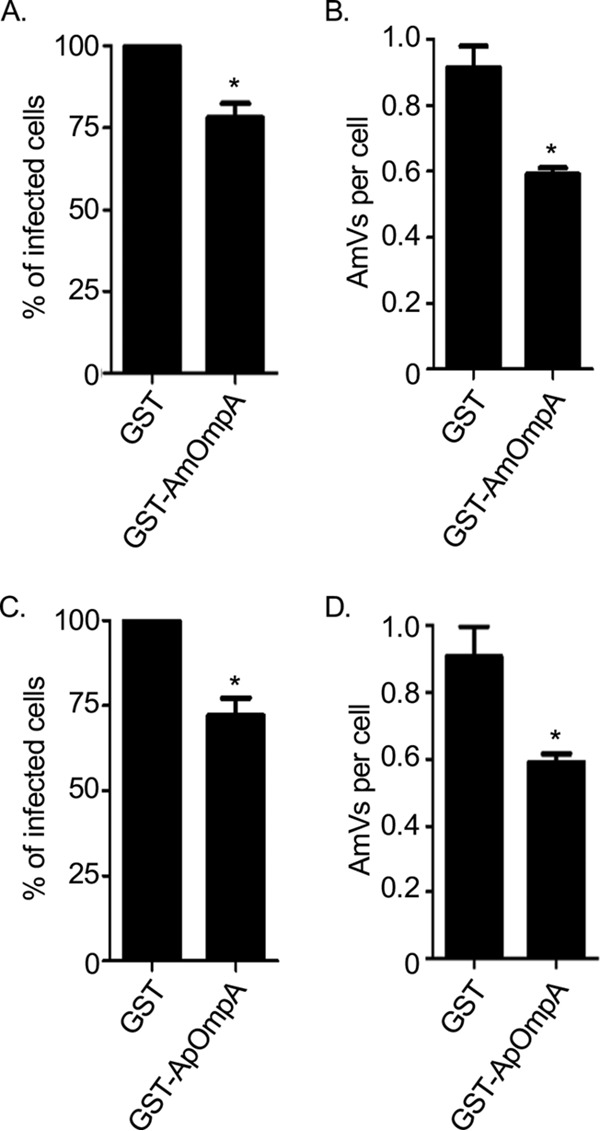
Recombinant AmOmpA and ApOmpA competitively inhibit A. marginale infection of endothelial cells. RF/6A cells were incubated with GST alone, GST-AmOmpA (A and B), or GST-ApOmpA (C and D) proteins for 1 h. A. marginale DC organisms were then added and incubated with the cells in the presence of recombinant protein for 2 h. After washing to remove unbound bacteria, host cells were incubated for 48 h and subsequently examined by immunofluorescence microscopy to determine the percentage of infected cells (A and C) and AmVs per cell (B and D). Results are the means ± SD from triplicate samples and are representative of three independent experiments with similar results. Statistically significant (*, P < 0.05) values are indicated.
FIG 8.

6-Sulfo-sLex is dispensable for recombinant AmOmpA binding to RF/6A cell surfaces and for A. marginale infection. (A) RF/6A cells were incubated with CSLEX1, KM93, G72, or IgM control for 1 h, followed by the addition of His-OmpA. Unbound recombinant protein was then washed away. Flow cytometry was used to detect bound His-AmOmpA. Cells alone served as a negative control. The histogram is representative of three independent experiments done in triplicate. (B and C) RF/6A endothelial cells were pretreated with IgM or G72. These cells were then incubated with DC A. marginale organisms for 2 h, after which unbound bacteria were removed. Cells were examined after 48 h by immunofluorescence microscopy to determine the percentage of infected cells (B) and AmVs per cell (C).
AmOmpA contributes to A. marginale infection of tick cells in a manner that is dependent on residues 50 to 67.
Because A. marginale also infects tick cells, the relevance of AmOmpA to A. marginale infection of ISE6 cells was examined. Treating DC organisms with heat-inactivated AmOmpA or AmOmpA50–67 antiserum prior to incubation with ISE6 cells significantly reduced the percentage of infected cells and number of AmVs per cell by comparable degrees as observed for RF/6A cells (Fig. 9A to D). Thus, AmOmpA contributes to A. marginale infection of tick cells and requires amino acids 50 to 67 to optimally do so. Also, GST-tagged AmOmpA and ApOmpA competitively antagonized A. marginale infection of ISE6 cells (Fig. 9E to H), suggesting that both recognize either the same or a structurally similar receptor on tick cells that A. marginale engages as part of its infection strategy. Sialic acids are rare in invertebrates and have not been detected in Ixodes scapularis, but α1,3-fucose residues are important for A. phagocytophilum to colonize these ticks (36). An evaluation of whether AmOmpA binding involves recognition of α1,3- or α1,4-fucose residues on ISE6 cells could not be attempted because α1,3/4-fucosidase treatment failed to reduce AAL binding, indicating that ISE6 cell surfaces have an abundance of fucose residues that exist in α1,6 or other linkages that would not be cleaved by α1,3/4-fucosidase.
FIG 9.
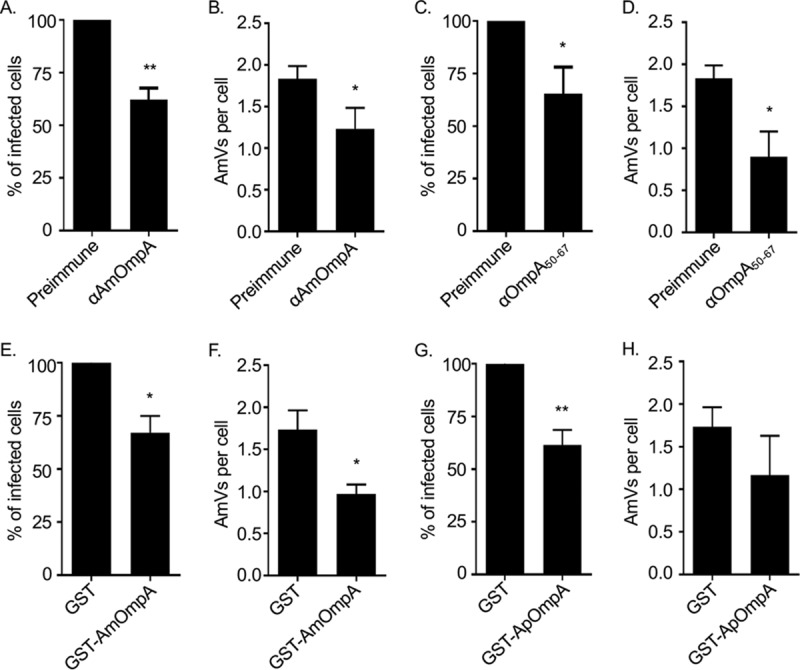
AmOmpA contributes to A. marginale infection of tick cells. (A to D) Antisera raised against AmOmpA and AmOmpA50–67 inhibit infection. A. marginale DC organisms were incubated with preimmune serum or antiserum specific for AmOmpA (A and B) or AmOmpA50–67 (C and D) for 1 h, followed by incubation with ISE6 cells in the continued presence of sera for 5 h. Unbound bacteria were removed and the infection was allowed to proceed for 72 h, after which the host cells were fixed and examined using immunofluorescence microscopy to determine the percentages of infected cells (A and C) and the number of AmVs per cell (B and D). (E to H) Recombinant AmOmpA and ApOmpA competitively inhibit A. marginale infection of tick cells. ISE6 cells were incubated with GST alone (E to H), GST-AmOmpA (E and F), or GST-ApOmpA (G and H) for 1 h. A. marginale DC organisms were then added and incubated with the cells in the presence of recombinant protein for 5 h. After washing to remove unbound bacteria, host cells were incubated for 72 h and subsequently examined by immunofluorescence microscopy to determine the percentage of infected cells (E and G) and AmVs per cell (F and H). Results are the means ± SD from triplicate samples and are representative of three independent experiments with similar results. Statistically significant (*, P < 0.05; **, P < 0.005) values are indicated.
DISCUSSION
Identifying A. marginale adhesins, delineating their functional domains, and determining the host cell determinants to which they bind not only will augment fundamental understanding of A. marginale pathobiology but also could benefit development of novel approaches for protecting against bovine anaplasmosis. Here, we determined that AmOmpA contributes to A. marginale invasion of mammalian host cells. Its binding domain lies within amino acids 50 to 67, as AmOmpA50–67 antibody inhibited bacterial infection of RF/6A cells. This region is homologous both in sequence and predicted structural location to the ApOmpA binding domain (18). Moreover, the positions of two of the three AmOmpA amino acids determined to be essential for adhesin function, G55 and K58, are identical to those of ApOmpA functionally essential residues G61 and K64. Whereas AmOmpA K59 is important for function, analogous ApOmpA K65 is not, which may at least partially account for the disparity between the two proteins' abilities to recognize 6-sulfo-sLex versus an unidentified α2,3-sialylated and α1,3-fucosylated glycan. G55 and K59 are conserved among OmpA proteins of Anaplasma spp., while K58 is conserved among those of Anaplasma and Ehrlichia spp. (17). Replacing only K58 with alanine resulted in no loss of AmOmpA function. However, the importance of K58 became apparent when it and K59 were replaced with alanine, as AmOmpA KK5859AA binding to host cells was nearly abolished. Given its demonstrated role in AmOmpA and ApOmpA function, K58 might contribute to the adhesin capabilities of all Anaplasma and Ehrlichia species OmpA proteins. Our findings presented here, together with a previous report that E. chaffeensis OmpA contributes to infection of monocytic cells (19), prompt us to propose that ehrlichial OmpA proteins also are adhesins that contribute to cellular invasion and do so by recognizing sialylated and fucosylated glycans in a manner that involves the conserved lysine.
AmOmpA G55, K58, and K59 are predicted to form a cationic binding pocket. This is likely critical for OmpA to recognize negatively charged fucose and sialic acid, as positively charged patches of numerous microbial sialic acid binding proteins have been shown to be important for receptor binding (18, 23–28). Indeed, recombinant AmOmpA proteins in which G55 or K59 had been replaced with alanine were modestly and pronouncedly compromised, respectively, in their abilities to bind to host cells. Recombinant AmOmpA in which K58 and K59 were both mutated to alanine was devoid of adhesin capability. This additive reduction in binding is presumably due to the large net loss in positive charge in the binding domain.
Recombinant ApOmpA and AmOmpA competitively antagonize A. marginale infection of RF/6A cells to comparable degrees, and AmOmpA binding to cells from which α2,3-sialic acid or α1,3-fucose have been removed is compromised. Together, these findings indicate that one or more sialylated and fucosylated glycans recognized by AmOmpA are important for A. marginale cellular invasion. However, our hypothesis that the AmOmpA endothelial cell receptor was the same as that bound by ApOmpA, 6-sulfo-sLex (18), proved incorrect. 6-Sulfo-sLex antibody G72 did not affect recombinant AmOmpA binding to or A. marginale infection of host cells, suggesting that AmOmpA engages a distinct sialylated and fucosylated glycan. Support for this premise comes from the fact that although ApOmpA preferentially recognizes 6-sulfo-sLex, G72 inhibits but does not abrogate recombinant ApOmpA binding to host cells (18). This indicates that ApOmpA is also able to recognize other sialylated and fucosylated glycans, potentially the AmOmpA primary endothelial cell receptor, which could explain why recombinant ApOmpA but not G72 inhibits recombinant AmOmpA binding to RF/6A cells. The differential preferences of the two OmpA proteins for similar but distinct receptors could be related to the tropism of A. phagocytophilum and A. marginale for neutrophils and erythrocytes, respectively. Given that ApOmpA binds distinct but structurally related receptors on myeloid and endothelial cells, the same could be true of the receptors that AmOmpA binds on erythrocytes and endothelial cells. A second possibility is that AmOmpA binds to a receptor that is shared by red blood and endothelial cells. A third scenario is that AmOmpA is exclusively important for A. marginale infection of endothelial cells. However, because of the data presented here and the fact that the bacterium agglutinates bovine erythrocytes in a sialic acid- and fucose-dependent manner (37), we favor the first two models.
AmOmpA by itself functions as both an adhesin and an invasin, as demonstrated by the ability of His-AmOmpA to confer adhesiveness and invasiveness to inert beads. However, by itself it does so inefficiently, as only 25% of the bound His-AmOmpA beads internalized. Similarly, competitively inhibiting A. marginale infection using recombinant AmOmpA or antiserum targeting AmOmpA or AmOmpA50–67 reduces infection by only 25%. Because A. marginale uses multiple surface proteins to mediate binding and entry (38–41), compensatory actions of other adhesins likely facilitate infection when AmOmpA is blocked. Likewise, targeting ApOmpA alone achieves only a partial reduction in A. phagocytophilum infection (18). It would be worthwhile to confirm if targeting other A. marginale adhesins in concert with AmOmpA abrogates infection of mammalian host cells in a manner similar to that achieved for A. phagocytophilum by blocking ApOmpA together with the adhesins Asp14 and AipA (18).
ISE6 tick cell culture is an acceptable model for studying A. marginale infection of tick cells (9, 10). Using this cell line, we discovered that AmOmpA is also important for A. marginale infection of tick cells and that the same AmOmpA50–67 domain that is key for the bacterium to optimally invade RF/6A cells is also critical for tick cell infection. This finding, combined with the observation that recombinant AmOmpA and ApOmpA competitively antagonize A. marginale infection of tick cells to comparable degrees, suggests that AmOmpA recognizes the same or a structurally similar receptor on the tick cell surface. A notable discrepancy between AmOmpA and ApOmpA is that the former is expressed during growth in ISE6 cells, while the latter is not (17). Why then does recombinant ApOmpA bind to and antagonize A. marginale infection of ISE6 cells? The answer might lie in the fact that A. phagocytophilum expresses ApOmpA while in a mammalian host and would therefore be present on the bacterium's surface when introduced into the tick by the acquisition bloodmeal (17). As A. phagocytophilum requires an α1,3-fucosylated receptor to colonize its tick vector (36), ApOmpA could be linked to this ability.
A. marginale subsp. centrale is used as a live vaccine against bovine anaplasmosis in some parts of the world, but this results in unreliable protection, as immunity is not uniform against all strains and outbreaks have occurred in immunized populations. Moreover, it is not USDA approved, has a high production cost, and carries the risks of vaccine-induced disease and transmission of known and unknown pathogens (1, 2). Immunization with an A. marginale outer membrane fraction protects against bacteremia and disease after experimental challenge. However, this outer membrane preparation contains over 20 proteins and is difficult to concoct and standardize (14). Accordingly, it has been proposed that a recombinant subunit vaccine would be ideal, as it would reduce time and cost of production (2) in addition to being safer. Given their essential roles, adhesins of obligate intracellular bacteria are rational targets for such subunit vaccines. In three independent genome-wide screens using in vivo immunization, one potential A. marginale target identified was AmOmpA (16). The conserved roles that OmpA proteins play in Anaplasma and Ehrlichia species infection of host cells (17–19) signify them as important targets to consider for incorporation into vaccines that would protect against the several infectious diseases that pathogens in these genera cause.
In fact, the role of AmOmpA as a potential vaccinogen against bovine anaplasmosis was recently explored. Immunizing cows against recombinant AmOmpA elicited a humoral immune response, achieving a mean IgG titer of 540 with a median of 300. However, the immunized animals were not protected against A. marginale infection but instead developed bacteremias that were higher than those of animals injected with adjuvant-only controls or A. marginale outer membranes. When a panel of sera from 10 cows immunized with recombinant protein or outer membranes, all of which detected recombinant full-length AmOmpA, were evaluated for immunoreactivity against AmOmpA19–68, only one had a titer of 80 while the other nine had titers that were less than 10 (14). Thus, even though full-length AmOmpA is immunogenic, the region that carries its functional domain is not. The poor immunogenicity of this AmOmpA region is consistent with it being designated a subdominant epitope and our findings that A. phagocytophilum infection does not yield a strong anti-ApOmpA humoral immune response (14, 16, 17). This makes sense from a pathobiological standpoint: an adhesin that is important for invasion and thus survival of an obligate intracellular bacterium would presumably be counterproductive if its functional domain were highly immunogenic. Moreover, our data presented here for AmOmpA and previously for ApOmpA (18) demonstrate that OmpA-targeting antibodies are effective, at least in vitro, only if they specifically block the binding domain. Armed with the knowledge generated here that AmOmpA residues G55, K58, and K59 are functionally essential, it would be prudent to examine whether immunizing against the adhesin's newly identified binding domain, ideally together with those of other adhesins and using carrier proteins and/or adjuvants to enhance immunogenicity, elicits a protective immune response against A. marginale infection in vivo.
MATERIALS AND METHODS
Cultivation of uninfected and A. marginale-infected host cell lines.
Uninfected and A. marginale (St. Maries strain)-infected RF/6A rhesus monkey choroidal endothelial cells (CRL-1780; American Type Culture Collection, Manassas, VA) and Ixodes scapularis embryonic ISE6 cells were cultured as described previously (8, 42, 43). Both host cell types infected with A. marginale were originally donated by Ulrike Munderloh (University of Minnesota, Minneapolis, MN).
Site-directed mutagenesis and recombinant protein production.
AmOmpA nucleotides 60 to 708, which encode residues 21 to 236 lacking the signal sequence (mature AmOmpA), were PCR amplified using primers containing the BamHI and NotI restriction sites (5′-GATCGGATCCCTTTTCAGCAAGGAAAAGGTCGGGATG-3′ and 5′-ATCGGCGGCCGCCTATTCAGGCGCGACCACTCC-3′ [boldface indicates extra nucleotides upstream of restriction sites; restriction sites are underlined]). The sequence integrity of the resulting PCR product was verified, after which it was digested and ligated into pGEX4T1 (GE Healthcare Bio-Sciences, Pittsburgh, PA) that had been digested with BamHI and NotI. GST-AmOmpA was expressed and purified by glutathione Sepharose affinity chromatography as previously described (44). AmOmpA genes encoding proteins with D47, K54, G55, K58, and/or K59 replaced with alanine were synthesized by Genewiz (South Plainfield, NJ). Plasmids encoding His-tagged mature wild-type AmOmpA and site-directed versions thereof were generated by amplifying wild-type and mutant AmOmpA sequences using primers 5′-GACGACGACAAAATGCTTTTCAGCAAGGAAAA-3′ and 5′-GAGGAGAAGCCCGGTTACTATTCAGGCGCGA-3′ (boldface indicates ligase-independent cloning [LIC] tails) and annealing the amplicons into the pET46 Ek/LIC vector (Novagen, EMD Millipore, Darmstadt, Germany) per the manufacturer's instructions. His-OmpA proteins were expressed and purified by immobilized metal affinity chromatography as previously described (45). GST-ApOmpA, His-ApOmpA, and His-OtOmpA (Orientia tsutsugamushi OmpA) have been previously described (17, 18, 46).
Antibodies, reagents, Western blotting, and ELISA.
His-AmOmpA was used to immunize mice, and the resulting antiserum was collected as previously described (46). New England Peptide (Garner, MA) generated serum against the AmOmpA putative binding domain as follows. A peptide corresponding to AmOmpA residues 50 to 67 (AmOmpA50–67) was synthesized, conjugated to keyhole limpet hemocyanin, and used to immunize rabbits, and the resulting serum was affinity purified. Antiserum against A. phagocytophilum OmpA59–74 has been previously described (18). Each antiserum's specificity was determined by ELISA using GST, GST-AmOmpA, GST-ApOmpA, and AmOmpA50–67 as immobilized antigens and the TMB substrate kit (Thermo Scientific, Waltham, MA) by following the manufacturer's instructions or by Western blotting as previously described (47). Each antiserum's concentration was determined using the Bradford assay. Fragments of antibody binding (Fab) of mouse anti-AmOmpA and rabbit anti-AmOmpA50–67 were generated using the Fab preparation kit (Pierce, Rockford, IL). Fab concentrations were determined based on absorbance at 280 nm. Monoclonal antibody AnaF16C1, which recognizes A. marginale major surface protein 5 (48) and was used to detect the bacterium in indirect immunofluorescence microscopy assays, was provided by Beverly Hunter and Guy Palmer (Washington State University, Pullman, WA). sLex antibodies CSLEX1 (BD Biosciences, San Jose, CA) and KM93 (Millipore, Darmstadt, Germany) were obtained commercially. 6-Sulfo-sLex antibody G72 was described previously (34). Alexa Fluor 488-conjugated anti-His tag secondary antibody and Alexa Fluor 488-conjugated streptavidin were obtained from Invitrogen (Carlsbad, CA). Biotinylated AAL and MAL II were obtained from Vector Laboratories (Burlingame, CA). Glycosidases used in this study were α2,3/6-sialidase (Sigma-Aldrich, St. Louis, MO) and α1,3/4-fucosidase (Clontech, Mountain View, CA). Lectins and glycosidases were used as previously described (17, 18).
Molecular modeling of AmOmpA.
To obtain a putative tertiary AmOmpA protein structure, the mature AmOmpA sequence was threaded onto solved crystal structures of proteins with similar sequences using the PHYRE2 server (www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi) (22) as previously described (17, 18). Amino acids 29 to 154 (58% of the mature AmOmpA sequence) were modeled with greater than 90% confidence to known structures for similar proteins (PDB entries 2AIZ [Haemophilus influenzae OmpP6 peptidoglycan-associated lipoprotein, or PAL], 4G4X [Acinetobacter baumannii PAL], 4B5C [Burkholderia pseudomallei PAL], 2HQS [Escherichia coli PAL], and 2L26 [OmpA-like domain of Mycobacterium tuberculosis ArfA]). The remainder of the protein lacked sufficient homology to any experimentally derived structure but could be modeled using the Poing method (22), which was performed as part of the PHYRE2 analyses. To generate the overlay, PHYRE2 models from mature ApOmpA and mature AmOmpA were threaded onto each other using PyMOL (pymol.org/educational). Mature AmOmpA surface electrostatic values were calculated using the PyMOL adaptive Poisson-Boltzman solver (APBS) plugin for PyMOL (29).
Binding of recombinant proteins to host cells.
RF/6A cells were incubated with 4 μM recombinant His-tagged AmOmpA proteins in culture media for 1 h in a 37°C incubator supplemented with 5% CO2 and a humidified atmosphere. Binding was assessed via flow cytometry or immunofluorescence microscopy as previously described (18). Spinning-disk confocal microscopy was accomplished using an Olympus BX51 microscope affixed with a disk-spinning unit (Olympus, Center Valley, PA). A BD FACSCanto II flow cytometer (BD, Franklin Lakes, NJ) was used at the Virginia Commonwealth University (VCU) Flow Cytometry and Imaging Shared Resource Facility. Post-data acquisition analyses were performed using the FCS Express 4 software package (De Novo Software, Los Angeles, CA). In some cases, cells were pretreated with α2,3-sialidase (5 μg/ml), α1,3/4-fucosidase (10 μU/ml), CSLEX1 (10 μg/ml), KM93 (10 μg/ml), or G72 (10 μg/ml) prior to the addition of AmOmpA.
Competitive inhibition of A. marginale infection.
A. marginale-infected RF/6A cells that were >90% infected and beginning to lyse were sonicated to destroy host cells and RC organisms but left DC organisms intact. Cellular debris was removed by two successive 5-min centrifugation steps at 1,000 × g. A. marginale DC bacteria were pelleted by centrifugation at 5,000 × g for 10 min. For competitive inhibition assays using antiserum and RF/6A cells, A. marginale DC organisms were incubated with AmOmpA antiserum (200 μg/ml), AmOmpA50–67 antiserum (200 μg/ml), or Fab fragments thereof (200 μg/ml) for 1 h, after which bacteria were incubated with host cells at a multiplicity of infection (MOI) of approximately 1 in the continued presence of antibodies for 2 h. Preimmune rat or rabbit serum (200 μg/ml) was used as a negative control. Unbound bacteria were removed and infection was allowed to proceed for 48 h. To determine if recombinant OmpA proteins could antagonize A. marginale infection, RF/6A cells were incubated with GST-AmOmpA, GST-ApOmpA, or GST alone (4 μM) for 1 h, after which A. marginale DC organisms were added and incubated with the host cells in the continued presence of recombinant protein for 2 h. Unbound bacteria and proteins were removed and the infection was allowed to proceed for 48 h. Experiments that assessed if antibodies targeting AmOmpA or recombinant OmpA proteins could inhibit A. marginale infection of ISE6 cells were performed identically to those just described, except that A. marginale organisms were incubated with ISE6 cells for 5 h before unbound bacteria were removed, the infection was allowed to proceed for 72 h, and the MOI achieved was approximately 1.7. At the endpoint of each experiment, cells were analyzed by spinning-disk confocal microscopy to determine the percentage of infected cells and number of AmVs per cell (17, 20).
OmpA-coated bead uptake assay.
His-AmOmpA was conjugated to red fluorescent sulfate-modified 1.0-μm-diameter microfluospheres (Life Technologies, Carlsbad, CA) as described previously (18). Coated and uncoated beads were incubated with RF/6A cells in culture medium at a bead-to-cell ratio of 500:1. Binding and internalization of the beads were assessed by spinning-disk confocal microscopy as described previously (18).
Statistical analysis.
The Student t test or one-way analysis of variance (ANOVA) was performed using the Prism 5.0 software package (GraphPad, San Diego, CA). Statistical significance was set to a P value of <0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank Beverly Hunter and Guy Palmer of The Paul G. Allen School for Global Animal Health at Washington State University for providing monoclonal antibody AnaF16C1, Ulrike Munderloh of The University of Minnesota for providing A. marginale (St. Maries strain) and uninfected and A. marginale-infected ISE6 cells, Joao Pedra and Dana Shaw of The University of Maryland for helpful discussions, and Chelsea Cockburn and Andrea Beyer for critical review of the manuscript.
This study was supported by NIH grant R01 AI072683 (to J.A.C.) and a grant from the Stephen and Alexandra Cohen Foundation (to R.T.M.). The VCU Massey Cancer Center Flow Cytometry Shared Resource Facility is supported in part by funding from NIH-NCI Cancer Center support grant P30 CA016059.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00968-16.
REFERENCES
- 1.Kocan KM, de la Fuente J, Blouin EF, Coetzee JF, Ewing SA. 2010. The natural history of Anaplasma marginale. Vet Parasitol 167:95–107. doi: 10.1016/j.vetpar.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 2.Suarez CE, Noh S. 2011. Emerging perspectives in the research of bovine babesiosis and anaplasmosis. Vet Parasitol 180:109–125. doi: 10.1016/j.vetpar.2011.05.032. [DOI] [PubMed] [Google Scholar]
- 3.Aubry P, Geale DW. 2011. A review of bovine anaplasmosis. Transbound Emerg Dis 58:1–30. doi: 10.1111/j.1865-1682.2010.01173.x. [DOI] [PubMed] [Google Scholar]
- 4.Herrero M, Thornton PK, Gerber P, Reid RS. 2009. Livestock, livelihoods and the environment: understanding the trade-offs. Curr Opin Environ Sustain 1:111–120. doi: 10.1016/j.cosust.2009.10.003. [DOI] [Google Scholar]
- 5.Valbuena D, Erenstein O, Homann-Kee Tui S, Abdoulaye T, Claessens L, Duncan AJ, Gérard B, Rufino MC, Teufel N, van Rooyen A, van Wijk MT. 2012. Conservation agriculture in mixed crop–livestock systems: scoping crop residue trade-offs in sub-Saharan Africa and South Asia. Field Crops Res 132:175–184. doi: 10.1016/j.fcr.2012.02.022. [DOI] [Google Scholar]
- 6.Carreno AD, Alleman AR, Barbet AF, Palmer GH, Noh SM, Johnson CM. 2007. In vivo endothelial cell infection by Anaplasma marginale. Vet Pathol 44:116–118. doi: 10.1354/vp.44-1-116. [DOI] [PubMed] [Google Scholar]
- 7.Munderloh UG, Lynch MJ, Herron MJ, Palmer AT, Kurtti TJ, Nelson RD, Goodman JL. 2004. Infection of endothelial cells with Anaplasma marginale and A. phagocytophilum. Vet Microbiol 101:53–64. doi: 10.1016/j.vetmic.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Truchan HK, Cockburn CL, Hebert KS, Magunda F, Noh SM, Carlyon JA. 2016. The pathogen-occupied vacuoles of Anaplasma phagocytophilum and Anaplasma marginale interact with the endoplasmic reticulum. Front Cell Infect Microbiol 6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammac GK, Pierle SA, Cheng X, Scoles GA, Brayton KA. 2014. Global transcriptional analysis reveals surface remodeling of Anaplasma marginale in the tick vector. Parasit Vectors 7:193. doi: 10.1186/1756-3305-7-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramabu SS, Ueti MW, Brayton KA, Baszler TV, Palmer GH. 2010. Identification of Anaplasma marginale proteins specifically upregulated during colonization of the tick vector. Infect Immun 78:3047–3052. doi: 10.1128/IAI.00300-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zivkovic Z, Blouin EF, Manzano-Roman R, Almazan C, Naranjo V, Massung RF, Jongejan F, Kocan KM, de la Fuente J. 2009. Anaplasma phagocytophilum and Anaplasma marginale elicit different gene expression responses in cultured tick cells. Comp Funct Genomics 2009:1–9. doi: 10.1155/2009/705034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blouin EF, Kocan KM. 1998. Morphology and development of Anaplasma marginale (Rickettsiales: Anaplasmataceae) in cultured Ixodes scapularis (Acari: Ixodidae) cells. J Med Entomol 35:788–797. doi: 10.1093/jmedent/35.5.788. [DOI] [PubMed] [Google Scholar]
- 13.Brayton KA, Kappmeyer LS, Herndon DR, Dark MJ, Tibbals DL, Palmer GH, McGuire TC, Knowles DP Jr. 2005. Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. Proc Natl Acad Sci U S A 102:844–849. doi: 10.1073/pnas.0406656102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ducken DR, Brown WC, Alperin DC, Brayton KA, Reif KE, Turse JE, Palmer GH, Noh SM. 2015. Subdominant outer membrane antigens in Anaplasma marginale: conservation, antigenicity, and protective capacity using recombinant protein. PLoS One 10:e0129309. doi: 10.1371/journal.pone.0129309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noh SM, Brayton KA, Brown WC, Norimine J, Munske GR, Davitt CM, Palmer GH. 2008. Composition of the surface proteome of Anaplasma marginale and its role in protective immunity induced by outer membrane immunization. Infect Immun 76:2219–2226. doi: 10.1128/IAI.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmer GH, Brown WC, Noh SM, Brayton KA. 2012. Genome-wide screening and identification of antigens for rickettsial vaccine development. FEMS Immunol Med Microbiol 64:115–119. doi: 10.1111/j.1574-695X.2011.00878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ojogun N, Kahlon A, Ragland SA, Troese MJ, Mastronunzio JE, Walker NJ, Viebrock L, Thomas RJ, Borjesson DL, Fikrig E, Carlyon JA. 2012. Anaplasma phagocytophilum outer membrane protein A interacts with sialylated glycoproteins to promote infection of mammalian host cells. Infect Immun 80:3748–3760. doi: 10.1128/IAI.00654-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seidman D, Hebert KS, Truchan HK, Miller DP, Tegels BK, Marconi RT, Carlyon JA. 2015. Essential domains of Anaplasma phagocytophilum invasins utilized to infect mammalian host cells. PLoS Pathog 11:e1004669. doi: 10.1371/journal.ppat.1004669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Z, Miura K, Popov VL, Kumagai Y, Rikihisa Y. 2011. Insights into the CtrA regulon in development of stress resistance in obligatory intracellular pathogen Ehrlichia chaffeensis. Mol Microbiol 82:1217–1234. doi: 10.1111/j.1365-2958.2011.07885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kahlon A, Ojogun N, Ragland SA, Seidman D, Troese MJ, Ottens AK, Mastronunzio JE, Truchan HK, Walker NJ, Borjesson DL, Fikrig E, Carlyon JA. 2013. Anaplasma phagocytophilum Asp14 is an invasin that interacts with mammalian host cells via its C terminus to facilitate infection. Infect Immun 81:65–79. doi: 10.1128/IAI.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seidman D, Ojogun N, Walker NJ, Mastronunzio J, Kahlon A, Hebert KS, Karandashova S, Miller DP, Tegels BK, Marconi RT, Fikrig E, Borjesson DL, Carlyon JA. 2014. Anaplasma phagocytophilum surface protein AipA mediates invasion of mammalian host cells. Cell Microbiol 16:1133–1145. doi: 10.1111/cmi.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 23.Chung MC, Wines BD, Baker H, Langley RJ, Baker EN, Fraser JD. 2007. The crystal structure of staphylococcal superantigen-like protein 11 in complex with sialyl Lewis X reveals the mechanism for cell binding and immune inhibition. Mol Microbiol 66:1342–1355. doi: 10.1111/j.1365-2958.2007.05989.x. [DOI] [PubMed] [Google Scholar]
- 24.Rademacher C, Bru T, McBride R, Robison E, Nycholat CM, Kremer EJ, Paulson JC. 2012. A Siglec-like sialic-acid-binding motif revealed in an adenovirus capsid protein. Glycobiology 22:1086–1091. doi: 10.1093/glycob/cws073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermans SJ, Baker HM, Sequeira RP, Langley RJ, Baker EN, Fraser JD. 2012. Structural and functional properties of staphylococcal superantigen-like protein 4. Infect Immun 80:4004–4013. doi: 10.1128/IAI.00764-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein PE, Boodhoo A, Armstrong GD, Heerze LD, Cockle SA, Klein MH, Read RJ. 1994. Structure of a pertussis toxin-sugar complex as a model for receptor binding. Nat Struct Biol 1:591–596. doi: 10.1038/nsb0994-591. [DOI] [PubMed] [Google Scholar]
- 27.Dormitzer PR, Sun ZY, Wagner G, Harrison SC. 2002. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J 21:885–897. doi: 10.1093/emboj/21.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varghese JN, McKimm-Breschkin JL, Caldwell JB, Kortt AA, Colman PM. 1992. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins 14:327–332. doi: 10.1002/prot.340140302. [DOI] [PubMed] [Google Scholar]
- 29.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jameson BA, Wolf H. 1988. The antigenic index: a novel algorithm for predicting antigenic determinants. Comput Appl Biosci 4:181–186. [DOI] [PubMed] [Google Scholar]
- 31.Chandrasekaran EV, Chawda R, Rhodes JM, Locke RD, Piskorz CF, Matta KL. 2003. The binding characteristics and utilization of Aleuria aurantia, Lens culinaris and few other lectins in the elucidation of fucosyltransferase activities resembling cloned FT VI and apparently unique to colon cancer cells. Carbohydr Res 338:887–901. doi: 10.1016/S0008-6215(03)00021-1. [DOI] [PubMed] [Google Scholar]
- 32.Yamashita K, Kochibe N, Ohkura T, Ueda I, Kobata A. 1985. Fractionation of L-fucose-containing oligosaccharides on immobilized Aleuria aurantia lectin. J Biol Chem 260:4688–4693. [PubMed] [Google Scholar]
- 33.Akahori T, Yuzawa Y, Nishikawa K, Tamatani T, Kannagi R, Miyasaka M, Okada H, Hotta N, Matsuo S. 1997. Role of a sialyl Lewis (x)-like epitope selectively expressed on vascular endothelial cells in local skin inflammation of the rat. J Immunol 158:5384–5392. [PubMed] [Google Scholar]
- 34.Dohi T, Nemoto T, Ohta S, Shitara K, Hanai N, Nudelman E, Hakomori S, Oshima M. 1993. Different binding properties of three monoclonal antibodies to sialyl Le (x) glycolipids in a gastric cancer cell line and normal stomach tissue. Anticancer Res 13:1277–1282. [PubMed] [Google Scholar]
- 35.Fukushima K, Hirota M, Terasaki PI, Wakisaka A, Togashi H, Chia D, Suyama N, Fukushi Y, Nudelman E, Hakomori S. 1984. Characterization of sialosylated Lewisx as a new tumor-associated antigen. Cancer Res 44:5279–5285. [PubMed] [Google Scholar]
- 36.Pedra JH, Narasimhan S, Rendic D, DePonte K, Bell-Sakyi L, Wilson IB, Fikrig E. 2010. Fucosylation enhances colonization of ticks by Anaplasma phagocytophilum. Cell Microbiol 12:1222–1234. doi: 10.1111/j.1462-5822.2010.01464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGarey DJ, Allred DR. 1994. Characterization of hemagglutinating components on the Anaplasma marginale initial body surface and identification of possible adhesins. Infect Immun 62:4587–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de la Fuente J, Garcia-Garcia JC, Blouin EF, Kocan KM. 2001. Differential adhesion of major surface proteins 1a and 1b of the ehrlichial cattle pathogen Anaplasma marginale to bovine erythrocytes and tick cells. Int J Parasitol 31:145–153. doi: 10.1016/S0020-7519(00)00162-4. [DOI] [PubMed] [Google Scholar]
- 39.de la Fuente J, Garcia-Garcia JC, Barbet AF, Blouin EF, Kocan KM. 2004. Adhesion of outer membrane proteins containing tandem repeats of Anaplasma and Ehrlichia species (Rickettsiales: Anaplasmataceae) to tick cells. Vet Microbiol 98:313–322. doi: 10.1016/j.vetmic.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Garcia JC, de la Fuente J, Bell-Eunice G, Blouin EF, Kocan KM. 2004. Glycosylation of Anaplasma marginale major surface protein 1a and its putative role in adhesion to tick cells. Infect Immun 72:3022–3030. doi: 10.1128/IAI.72.5.3022-3030.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGarey DJ, Barbet AF, Palmer GH, McGuire TC, Allred DR. 1994. Putative adhesins of Anaplasma marginale: major surface polypeptides 1a and 1b. Infect Immun 62:4594–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang B, Ojogun N, Ragland SA, Carlyon JA. 2012. Monoubiquitinated proteins decorate the Anaplasma phagocytophilum-occupied vacuolar membrane. FEMS Immunol Med Microbiol 64:32–41. doi: 10.1111/j.1574-695X.2011.00873.x. [DOI] [PubMed] [Google Scholar]
- 43.Beyer AR, Truchan HK, May LJ, Walker NJ, Borjesson DL, Carlyon JA. 2015. The Anaplasma phagocytophilum effector AmpA hijacks host cell SUMOylation. Cell Microbiol 17:504–519. doi: 10.1111/cmi.12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Troese MJ, Kahlon A, Ragland SA, Ottens AK, Ojogun N, Nelson KT, Walker NJ, Borjesson DL, Carlyon JA. 2011. Proteomic analysis of Anaplasma phagocytophilum during infection of human myeloid cells identifies a protein that is pronouncedly upregulated on the infectious dense-cored cell. Infect Immun 79:4696–4707. doi: 10.1128/IAI.05658-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller DP, McDowell JV, Bell JK, Marconi RT. 2011. Crystallization of the factor H-binding protein, FhbB, from the periopathogen Treponema denticola. Acta Crystallogr Sect F Struct Biol Cryst Commun 67:678–681. doi: 10.1107/S1744309111011298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beyer AR, VieBrock L, Rodino KG, Miller DP, Tegels BK, Marconi RT, Carlyon JA. 2015. Orientia tsutsugamushi strain Ikeda ankyrin repeat-containing proteins recruit SCF1 ubiquitin ligase machinery via poxvirus-like F-box motifs. J Bacteriol 197:3097–3109. doi: 10.1128/JB.00276-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carlyon JA, Chan WT, Galan J, Roos D, Fikrig E. 2002. Repression of rac2 mRNA expression by Anaplasma phagocytophila is essential to the inhibition of superoxide production and bacterial proliferation. J Immunol 169:7009–7018. doi: 10.4049/jimmunol.169.12.7009. [DOI] [PubMed] [Google Scholar]
- 48.Visser ES, McGuire TC, Palmer GH, Davis WC, Shkap V, Pipano E, Knowles DP Jr. 1992. The Anaplasma marginale msp5 gene encodes a 19-kilodalton protein conserved in all recognized Anaplasma species. Infect Immun 60:5139–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.