

ABSTRACT
Quorum sensing (QS) is a process by which bacteria alter gene expression in response to cell density changes. In Vibrio species, at low cell density, the sigma 54-dependent response regulator LuxO is active and regulates the two QS master regulators AphA, which is induced, and OpaR, which is repressed. At high cell density the opposite occurs: LuxO is inactive, and therefore OpaR is induced while AphA is repressed. In Vibrio parahaemolyticus, a significant enteric pathogen of humans, the roles of these regulators in pathogenesis are less known. We examined deletion mutants of luxO, opaR, and aphA for in vivo fitness using an adult mouse model. We found that the luxO and aphA mutants were defective in colonization compared to levels in the wild type. The opaR mutant did not show any defect in vivo. Colonization was restored to wild-type levels in a luxO opaR double mutant and was also increased in an opaR aphA double mutant. These data suggest that AphA is important and that overexpression of opaR is detrimental to in vivo fitness. Transcriptome sequencing (RNA-Seq) analysis of the wild type and luxO mutant grown in mouse intestinal mucus showed that 60% of the genes that were downregulated in the luxO mutant were involved in amino acid and sugar transport and metabolism. These data suggest that the luxO mutant has a metabolic disadvantage, which was confirmed by growth pattern analysis using phenotype microarrays. Bioinformatics analysis revealed OpaR binding sites in the regulatory region of 55 carbon transporter and metabolism genes. Biochemical analysis of five representatives of these regulatory regions demonstrated direct binding of OpaR in all five tested. These data demonstrate the role of OpaR in carbon utilization and metabolic fitness, an overlooked role in the QS regulon.
KEYWORDS: Vibrio parahaemolyticus, carbon metabolism, intestinal colonization, quorum sensing
INTRODUCTION
Vibrio parahaemolyticus is the leading cause of bacterial seafood-borne gastroenteritis worldwide, resulting in mild to severe inflammatory gastroenteritis (1–5). The completed genome sequence of V. parahaemolyticus RIMD2210633, an O3:K6 serotype associated with pandemic disease, demonstrated the presence of two type III secretion systems (T3SS-1 and T3SS-2), one on each chromosome, which led to the identification of several effector proteins associated with inflammatory diarrhea (6–8). Studies have shown that T3SS-2 is the major contributing factor to enterotoxicity and that inflammatory diarrhea and intestinal epithelium cell disruption are dependent upon a functional T3SS-2 (9–12).
Much less is known about how V. parahaemolyticus initially colonizes and survives within the host gastrointestinal tract. This lack of knowledge is in part due to a lack of animal models to study colonization and infection in vivo. The development of a streptomycin-pretreated adult mouse model that removes microbiota colonization resistance and allows V. parahaemolyticus to colonize has uncovered a number of bacterial colonization factors required for colonization (13–15). Examination of a mutant deficient in the global regulator ToxR demonstrated a significant defect in intestinal colonization compared to that of the wild type (13). This study showed that ToxR was a negative regulator of the global regulator LeuO and a positive regulator of the T3SS-1 and the major outer membrane porin OmpU (13). OmpU was shown to be essential for resistance and tolerance to acid and bile salts, important abiotic stresses in vivo (13). The importance of ToxR for in vivo survival also was demonstrated in an infant rabbit model of infection (16). The alternative sigma factor RpoE, required for the cell envelope stress response, was shown to be essential for in vivo survival as deletion of the rpoE gene resulted in attenuation of mouse colonization (15). In contrast, analysis of a deletion mutant of sigma-54, rpoN, showed that in in vivo competition assays, the mutant colonized significantly more proficiently than the wild-type strain. The mechanism for the enhanced colonization of the rpoN mutant is unknown; however, it was shown that the rpoN mutant had a metabolic advantage over the wild type when grown in intestinal mucus and its components. Expression analysis showed that genes required for gluconate, ribose, and arabinose catabolism were induced in the rpoN mutant. These data suggested that specific carbon metabolism genes are negatively regulated by RpoN and that competitive carbon utilization could be an important colonization factor (14).
Quorum sensing (QS) is a process by which bacteria modulate gene expression in response to cell density changes and is mediated by autoinducers such as acyl homoserine lactone that act as extracellular signals (17–22). Quorum sensing has been studied in a number of Vibrio species, including V. parahaemolyticus, which contains the central conserved components of the quorum sensing pathway identified in Vibrio harveyi (Fig. 1) (22–34). In V. harveyi, quorum sensing regulation of gene expression is carried out by two quorum sensing master regulators, the low-cell-density (LCD) regulator AphA and the high-cell-density (HCD) regulator, LuxR. At LCD, when the autoinducer concentration is low, LuxO, the QS response regulator, is active and functions as an activator for sigma factor RpoN (σ54) that then aids in the transcription of five small RNAs termed quorum regulatory RNAs (qrr1 to qrr5) (35, 36). The bacterial Qrr small RNAs (sRNAs) are partially complementary to their target mRNAs and thereby regulate gene expression posttranscriptionally. They require the sRNA chaperone Hfq for their activity (36–38). Qrr sRNAs were identified first in V. cholerae where they were shown to repress HapR (the LuxR homologue) through direct base pairing (37). The Qrr sRNAs stabilize the mRNA of aphA and destabilize the mRNA of luxR. In addition, AphA represses luxR transcription, independent of the Qrr sRNAs (36, 39, 40). At HCD, LuxO is not active, and the qrr genes are not transcribed, leading to a constitutively expressed LuxR, which in turn represses the transcription of aphA (Fig. 1) (36, 39, 40). The V. parahaemolyticus genome contains each of the components described above, luxO, qrr1 to qrr5, aphA, and opaR, the luxR homologue in this species (25, 30, 33, 37, 41, 42). LuxR and OpaR regulate hundreds of genes in V. harveyi and V. parahaemolyticus, respectively (30, 39, 41). OpaR was shown to positively regulate capsule polysaccharide (CPS) production, competence, and type VI secretion system-2 (T6SS-2) production and to negatively regulate motility, biofilm, and T3SS-1 and T6SS-1 production (30, 32–34, 41–43). AphA was shown to regulate several genes in various Vibrio species, including genes involved in biofilm formation, motility, and virulence (39, 44–46). In V. harveyi, AphA coregulated a number of genes, including the T3SS apparatus along with LuxR (39). In V. parahaemolyticus, AphA is required for motility and biofilm formation, and an aphA mutant strain is avirulent in a murine infection model (42).
FIG 1.
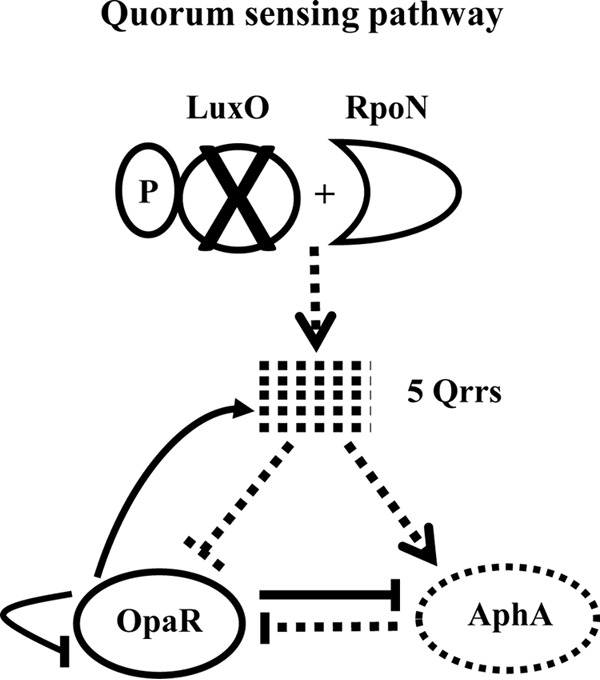
Quorum sensing pathway in V. parahaemolyticus. The sigma 54-dependent response regulator LuxO activates the transcription of the quorum regulatory RNAs (qrr1 to qrr5). These Qrr sRNAs bind to the mRNA of the QS master regulators, blocking the translation of OpaR and promoting the translation of AphA. OpaR and AphA repress the transcription of each other. In a luxO mutant, indicated by X, no qrr genes are transcribed, and this results in constitutive activation of OpaR and repression of aphA. Dashed lines indicate no transcripts made, dashed arrows indicate no regulation, solid lines indicate regulation, arrows indicate positive regulation, and hammers indicate negative regulation.
In this study, we determined the role of the QS regulators in V. parahaemolyticus intestinal colonization, the first step in pathogenesis. We constructed an luxO deletion mutant, and in vivo analysis showed that the mutant had a significant defect in a streptomycin pretreated adult mouse model of colonization. We determined whether the defect in the luxO mutant was through its regulation of the QS regulator OpaR or AphA by constructing deletions in each of these genes as well as double deletion mutants and examined the in vivo phenotypes. The aphA mutant was attenuated for colonization similar to the luxO mutant, whereas the opaR mutant showed no defect in colonization. The luxO opaR and opaR aphA double deletion mutant strains showed significantly increased colonization abilities compared to those of the single luxO and aphA deletion mutants. These results suggested that AphA is important for in vivo fitness, likely in part through its negative regulation of opaR, and that overexpression of opaR is detrimental. Comparative transcriptome analysis of the wild-type strain versus the luxO mutant grown in mouse intestinal mucus showed that 60% of genes downregulated in the luxO mutant were involved in metabolism. Using phenotype microarrays, we found significant differences in growth between the wild type and the luxO and aphA mutant strains in 25 carbon sources. Bioinformatics analysis identified putative OpaR binding sites in the regulatory regions of carbon metabolism and transporter genes. By using electrophoretic mobility shift assays (EMSAs), we show direct binding to five of these regulatory regions. Overall, the data demonstrate a direct role for the QS regulator OpaR in cell metabolism and suggest a mechanism for the in vivo phenotypes of the luxO and aphA mutants.
RESULTS
Deletion of luxO or aphA leads to a defect in intestinal colonization.
To determine the role of the QS regulators in V. parahaemolyticus pathogenesis, we examined each of the QS mutants for their ability to colonize the adult mouse intestine. We examined luxO, aphA, and opaR deletion mutant strains as well as luxO opaR and opaR aphA double deletion mutant strains in in vivo competition assays with the wild type using the streptomycin-pretreated adult mouse model of intestinal colonization (13, 14). Mice pretreated with streptomycin were orogastrically coinoculated with an equal mixture of the WBWlacZ strain (wild-type strain marked with lacZ and that allows blue-white screening) and each of the mutants. In these assays, the WBWlacZ strain significantly out-competed the luxO mutant, which had a competitive index (CI) of 0.27, indicating that deletion of luxO leads to reduced fitness in vivo (Fig. 2). In order to investigate whether the defect in the luxO mutant was through its regulation of the QS regulator AphA or OpaR, we constructed deletion mutants in each of these genes. The aphA mutant showed a significant defect in colonization similar to the luxO mutant, with a CI of 0.39 (Fig. 2). Both the luxO and aphA mutant strains grew at levels similar to those of the wild type in in vitro competition assays in lysogeny broth (LB) containing 3% NaCl (LBS), with CIs of 0.9 and 1.0, respectively. These data show that deletion of luxO or aphA affects colonization ability specifically (Fig. 2). The opaR mutant behaved similarly to the wild type in both in vitro and in vivo assays, with a CI of ∼1. In order to determine further the importance of each of these regulators in colonization, we examined double deletion mutant luxO opaR and opaR aphA strains. The deletion of opaR in the luxO and aphA mutants resulted in a significant increase in colonization ability compared to deletion of the single luxO and aphA genes. Colonization was restored to wild-type levels in the luxO opaR double mutant, which had a CI of 1.4. The opaR aphA mutant also had increased colonization compared to that of the aphA single mutant, with a CI of 0.7; however, this mutant still showed a defect in colonization compared to the wild-type level (Fig. 2). Taken together, these data demonstrate that overexpression of OpaR in the mutant compared to the wild-type level is detrimental and that AphA is required for in vivo colonization.
FIG 2.
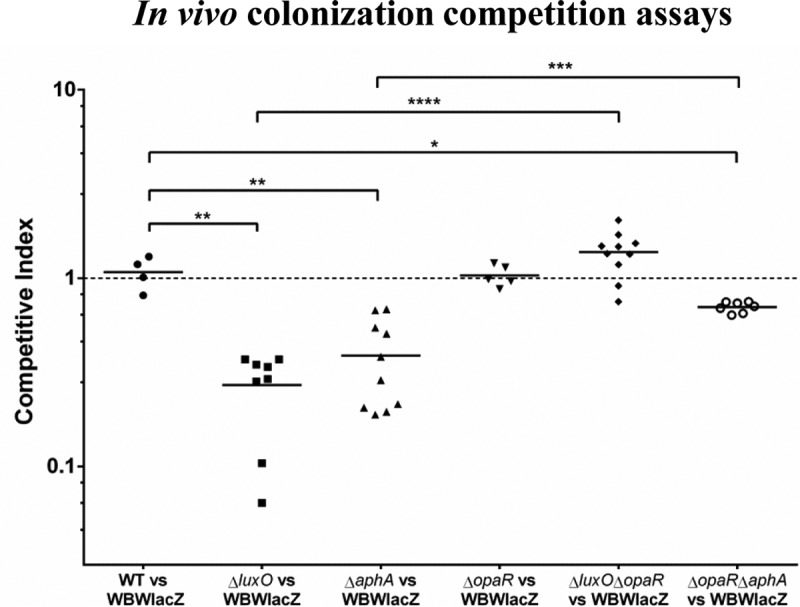
In vivo competition assays. A 1:1 mixed culture of WBWlacZ and deletion mutants was used to orogastrically infect streptomycin-pretreated adult mice. CFU numbers were calculated at 24 h postinfection from the entire gastrointestinal tracts using blue-white colony selection. Data are pooled from two separate experiments and reported as competitive indexes (CIs) for the luxO (n = 8), aphA (n = 10), opaR (n = 5), luxO opaR (n = 10), and opaR aphA (n = 7) mutants. The solid lines indicate the means. P values were calculated using a Welch's unpaired t test with a 95% confidence interval. Asterisks denote significant differences between the CIs of the mutant strains and the CI of the wild-type strain. *, P < 0.05; **, P < 0.01; ***, P < = 0.001 ****, P < 0.0001. WT, wild type.
The in vivo defect observed for the luxO mutant is in contrast to the superior colonization phenotype that we had previously showed for an rpoN deletion mutant (14). According to the quorum sensing pathway in V. parahaemolyticus, deletion of both luxO and rpoN should have the same effect on the expression of the two master regulators aphA and opaR. We determined the expression patterns of these regulators in luxO, opaR, aphA, and rpoN mutant strains grown to an optical density (OD) of 1.0 in M9 medium supplemented with glucose (M9G). Expression of opaR was significantly induced in the luxO mutant, with a 6.3-fold change in expression relative to that of the wild type. The expression of opaR was increased in both the rpoN and the aphA mutants but not to the same level as in the luxO mutant (Fig. 3A). Expression of aphA, although not significant, was reduced in the luxO mutant and was induced in the opaR mutant. The expression of aphA was not repressed in the rpoN mutant (Fig. 3B). Both RpoN and its activator LuxO are required for expression of the qrr genes since each of the genes qrr1 to qrr5 contains a conserved RpoN −12 and −24 promoter binding sequence, indicating that this sigma factor is involved in expression (35). To address why aphA was not repressed and opaR is not as highly expressed in the rpoN mutant compared to the level in the luxO mutant, we examined the expression patterns of qrr1 to qrr5 in both of these mutants under the same conditions as opaR expression. Quantitative PCR (qPCR) analysis showed that in the luxO mutant compared to wild-type strain, qrr2, qrr3, and qrr5 were repressed while qrr1 was unchanged (Fig. 3C). In the rpoN mutant, qPCR analysis showed that qrr1, qrr3, and qrr5 were repressed; however, qrr2 was not repressed compared to the level in the wild type (Fig. 3D). In both cases, qrr4 expression was either very low or altogether not detected. The most notable difference in expression patterns between the luxO and rpoN mutants was in qrr2. While qrr2 was significantly downregulated in the luxO mutant, its expression was not repressed in the rpoN mutant. We speculate that the differential expression of qrr2 in the rpoN mutant may explain the reduced level of opaR compared to that of the luxO mutant.
FIG 3.
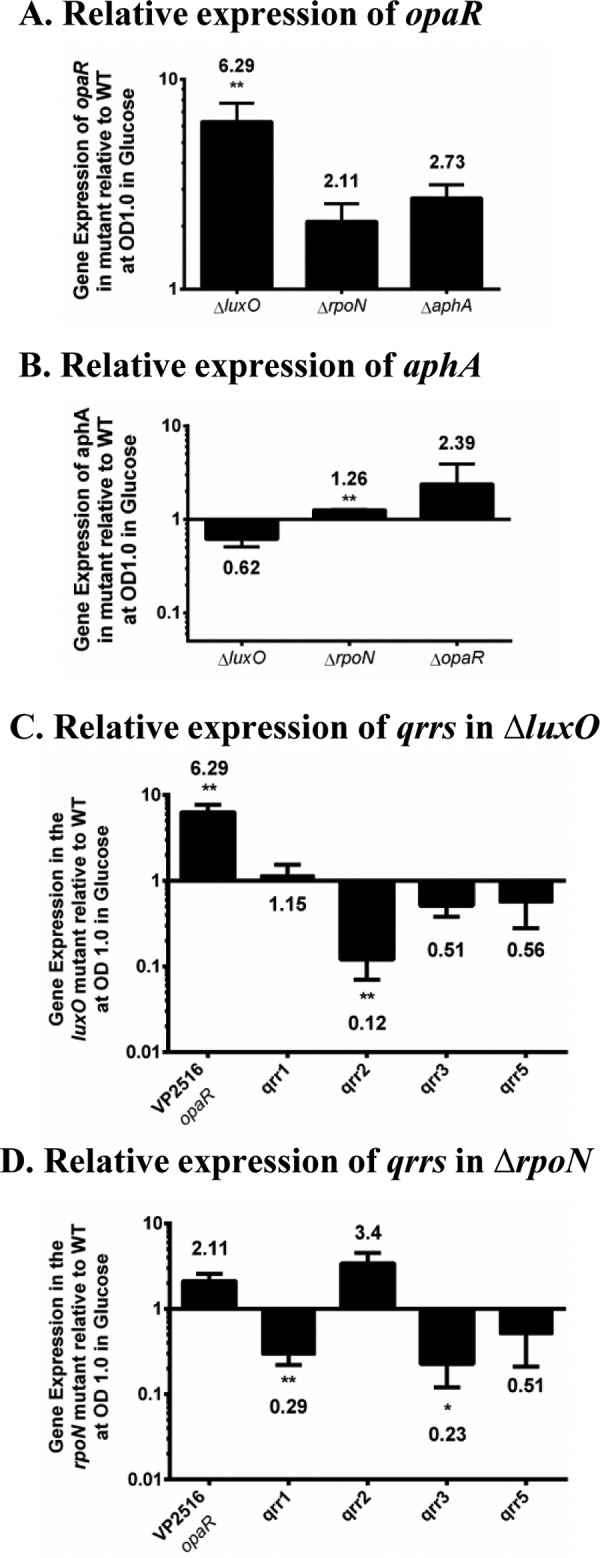
Expression analysis of quorum sensing master regulators and qrr genes. RNA was extracted from wild-type and mutant strains grown in M9 medium supplemented with glucose (M9G) to an OD of 1.0 and analyzed by qPCR in duplicate for each biological replicate. (A) Bars represent the expression of opaR normalized to expression of the 16S rRNA in the luxO, rpoN, and aphA mutants relative to that in wild-type cells. (B) Bars represent the expression of the aphA gene normalized to that of the 16S rRNA in the luxO, rpoN, and opaR mutants relative to that in wild-type cells. (C) Bars represent the expression of qrr1, qrr2, qrr3, and qrr5 normalized to that of the 16S rRNA in the ΔluxO mutant. (D) Bars represent the expression of the qrr1, qrr2, qrr3, and qrr5 genes normalized to that of the 16S rRNA in the ΔrpoN mutant. P values were calculated using an unpaired Student's t test with a 95% confidence interval. Asterisks denote significant differences in relative gene expression levels between mutants and the wild type. *, P < 0.05; **, P < 0.01.
The V. parahaemolyticus quorum sensing master regulators OpaR and AphA have been shown to regulate CPS production and biofilm formation (25, 30, 42, 47, 48). We examined these phenotypes in the QS regulator mutants investigated in this study. The luxO and aphA mutants produced rugose colonies similar to those of the wild type, indicating CPS production, which indicates that CPS production is not involved in the in vivo phenotype of these mutants (see Fig. S1A in the supplemental material). The luxO and aphA mutants produced amounts of biofilm similar to the amount of the wild type at the initial time points but were found to be defective at 24 h (Fig. S1B). Previously, we showed that an rpoN mutant had a defect in biofilm formation but had a superior colonization phenotype, suggesting that this is not the cause of the in vivo phenotypes of the luxO and aphA mutants.
RNA-Seq data and comparative analysis of gene expression in mouse intestinal mucus.
To begin to determine the mechanism of the in vivo luxO mutant colonization defect, we performed transcriptome sequencing (RNA-Seq) expression analysis of the wild-type and the luxO mutant strains. RNA was isolated from both strains grown to early exponential phase in M9 medium supplemented with mouse intestinal mucus as the sole carbon source. The LCD time point was chosen since it should show maximum differences in opaR expression levels between the wild type (low OpaR levels) and the luxO mutant (high OpaR levels). Sequencing resulted in greater than 10 million sequence reads obtained for each sample (Fig. S2A). Over 98% of the reads aligned to genomic features, including mRNA, tRNA, and sRNA, or to unannotated regions of the genome. The rRNA depletion procedure resulted in less than 0.5% of the reads aligning to these features in the genome (Fig. S2A and B). Differential expression analysis revealed that 106 genomic features and 102 features were downregulated and upregulated (>2-fold; adjusted P value [Padj] of <1 × 10−4), respectively, in the luxO mutant compared to wild-type expression levels (Fig. S3 and Tables S3 and S4). Of the total 208 differentially regulated features, 134 were from chromosome I, and 74 were from chromosome II (Tables S3 and S4). The 106 downregulated features were all annotated open reading frames (ORFs). The 102 upregulated features included 93 annotated ORFs, 3 small RNAs, and 6 tRNAs. The opaR gene was induced, and aphA was repressed in the luxO mutant compared to expression in the wild type, and this was confirmed by qPCR (Fig. 4A). Expression analysis of the qrr genes by qPCR showed that qrr5 was significantly downregulated in the luxO mutant (Fig. 4A). Twenty-one of the genes upregulated in the luxO mutant belonged to the T6SS-2 region on chromosome II (VPA1024 to VPA1044), which was previously shown to be positively regulated by OpaR (30). qPCR analysis of VPA1027 (hcp2) from the T6SS-2 cluster confirmed that in the luxO mutant this gene was induced (Fig. 4C). Furthermore, qPCR analysis of hcp1 (VP1393) from the T6SS-1 cluster and vopD (VP1656) from the T3SS-1 cluster showed that their expression levels were reduced compared to the level in the wild type (Fig. 4C). The most highly upregulated genes in the luxO mutant were genes for the replication and synthesis of the filamentous phage f237 (VP1550 to VP1562). Many genes within the class-1 integron region on chromosome I (VP1790 to VP1851) were also induced in the luxO mutant compared to the wild-type level (Table S3A). The majority of the genes within the f237 phage and the class-1 integron were categorized into the Clusters of Orthologous Groups (COG) classes S, function unknown, and R, general function prediction only (Fig. S4A). The T6SS-2 genes were classified into the COG class U, intracellular trafficking, secretion, and vesicular transport (Fig. S4A). Among the genes belonging to downregulated COG classes, it was most notable that 60% of the genes were classified into categories involved in metabolism and transport (Fig. S4B).
FIG 4.
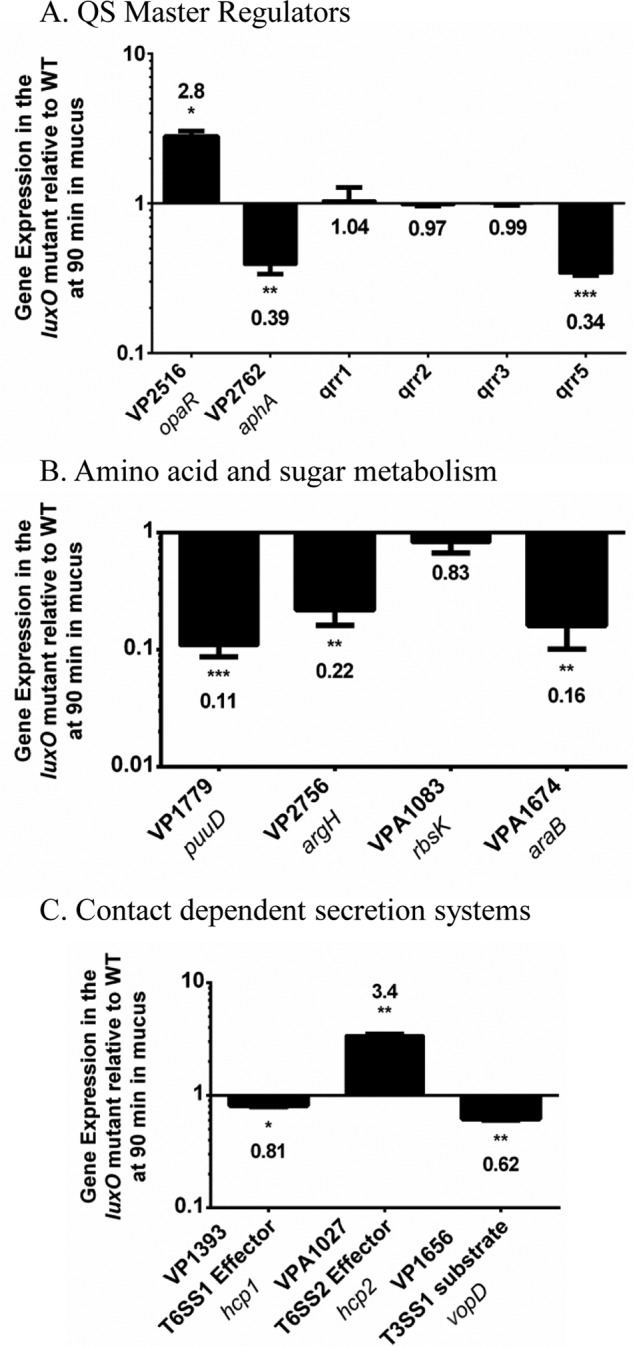
qPCR validations of RNA-Seq expression in the luxO mutant relative to that of the wild-type. Pre-Ribo-Zero-treated RNA from the wild type and the luxO mutant was used for cDNA synthesis, and expression was analyzed by qPCR in duplicate for each biological replicate. (A) Bars represent the expression of the opaR, aphA, qrr1, qrr2, qrr3, and qrr5 genes normalized to that of the 16S rRNA in the luxO mutant relative to that in wild-type cells. (B) Bars represent the expression levels of four metabolism genes normalized to expression of the 16S rRNA in the luxO mutant relative to the wild-type level. (C) Bars represent relative expression levels of the T3SS and T6SS genes normalized to the 16S rRNA level in the luxO mutant relative to that of wild-type cells. P values were calculated using an unpaired Student's t test with a 95% confidence interval. Asterisks denote significant differences in relative gene expression levels between the mutant and wild type. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Metabolism and transporter genes are downregulated in the luxO mutant.
Of the 106 genomic features downregulated in the luxO mutant, 64 genes were involved in transport and metabolism of amino acids, carbohydrate, and lipids (Fig. S4B and Table S4). Downregulated gene clusters that comprised amino acid transport and metabolism included arginine biosynthesis (VP2756 to VP2760) and transport (VPA0637 to VPA0639) (Fig. S4A), phenylalanine/tyrosine biosynthesis (VP0546 to VP0547 and VP0555), and histidine biosynthesis (VP1137 to VP1138). qPCR analysis confirmed the downregulation of VP2756 (argH), an ORF in the arginine biosynthesis (VP2756 to VP2760) pathway (Fig. 4B). The carbohydrate metabolism and transport genes downregulated in the luxO mutant included genes involved in d-mannitol metabolism (VPA0501 to VPA0502), d-galactose degradation (VP2397 to VP2400), and l-arabinose transport and metabolism (VPA1671 to VPA1677) (Table S4). qPCR analysis confirmed the downregulation of VPA1674 (araB), an ORF in the arabinose catabolism pathway (Fig. 4B). A region required for tetrathionate reductase synthesis (VP2012 to VP2016) was also repressed in the luxO mutant compared to the level in the wild type. Tetrathionate can be used as an electron donor that is produced in vertebrate intestinal mucosa from thiosulfate by the action of tetrathionate reductase (49). VP1771 to VP1779 and VP1781 to VP1782 are two operons involved in polyamine putrescine utilization, and all ORFs within this region were downregulated in the luxO mutant, which was confirmed by qPCR analysis of VP1779 (puuD) (Fig. 4B). ORFs VP1447 to VP1451 are homologues of genes required for the synthesis of a putative anaerobic dimethyl sulfoxide reductase, and these genes were all downregulated in the luxO mutant. There were eight putative transcription regulators downregulated in the luxO mutant compared to wild-type levels: VP0358 (DeoR), VP1778 (PuuR), VP3009 (AraC/XylS family), VPA0053 (TetR family), VPA0251 (LysR family), VPA0717 (LysR family), VPA0883 (LysR family), and VPA1678 (AraC/XylS family). In addition, genes for compatible solute biosynthesis were also downregulated: two genes in the ectoine biosynthesis pathway (VP1721 to VP1720) and two genes involved in betaine biosynthesis (VPA1112) and transport (VPA1111) (Table S4).
Growth comparisons of wild-type and mutant strains on different carbon sources.
Our RNA-Seq data suggest that the luxO strain could be at a metabolic disadvantage, given the downregulation of many metabolism and transporter genes. Thus, the growth patterns of the wild-type and QS mutants were examined in 190 carbon sources to determine whether there were differences among the strains. A total of 71 different carbon sources were utilized by wild-type V. parahaemolyticus (Fig. S5). There was a total of 33 substrates in which the luxO mutant showed a defect compared to growth of the wild type; for seven of these substrates only the luxO mutant exhibited a growth defect. The aphA mutant showed a significant growth defect on 30 substrates; the defect was unique to the aphA mutant on five of these substrates while the luxO mutant also showed a growth defect with 13 of these carbon sources (Fig. S5). Thus, there were 25 carbon substrates on which the luxO and aphA mutants showed a growth defect whereas the opaR mutant did not show a defect on these substrates. These 25 carbon sources included six amino acids or their derivatives, four dipeptides, seven sugars, two nucleosides, one tricarboxylic acid (TCA) cycle metabolite, and five miscellaneous carbon sources (Fig. S5). The most significant growth defects in the luxO mutant were found with growth on amino acids and their dipeptide derivatives: l-aspartic acid, glycyl-l-aspartic acid, glycyl-l-glutamic acid, glycyl-l-proline acid, l-serine, and l-threonine (Fig. S5). The luxO and aphA mutants also showed notable defects in growth on l-glutamic acid and l-arginine, two TCA cycle metabolites (pyruvic acid and α-keto-glutaric acid), and two polysaccharides (glycogen and inulin). We confirmed many of these growth defects by examining the growth pattern of the QS mutants in 12 of the carbon sources (Fig. S6). The opaR mutant did not show a defect in growth on these carbon sources and grew at levels similar to those of the wild type (Fig. S6). These data suggest that the QS regulators may play a key role in regulation of cell metabolism. In addition, considering that many of these carbon sources are components of intestinal mucus, which is the primary carbon source available for the bacteria in vivo, this disadvantage could contribute to these mutants being out-competed by the wild type in vivo.
Deletion of opaR leads to increased metabolic fitness in intestinal mucus and its components.
Intestinal mucus is composed of glycoproteins, known as mucins, that are comprised of 80% oligosaccharide and 20% protein. The main sugars in mucin include fucose, galactose, mannose, sialic acid, N-acetylglucosamine, and N-acetyl galactosamine, as well as arabinose, ribose, gluconate, galacturonate, and glucuronate. The three main amino acids that make up the protein core of mucin are serine, threonine, and proline (50–54). From genomic analysis, we know that V. parahaemolyticus cannot utilize fucose, sialic acid, or galacturonate and that only clinical strains can utilize arabinose. We examined growth of the luxO, aphA, and opaR mutants on M9 medium supplemented with individual mucus sugars as the sole carbon sources (Fig. S6). These data demonstrate that the luxO and aphA mutants had significantly longer lag phases than the wild type when they were grown on these substrates. These longer lag phases indicate that these mutants would be at a significant disadvantage in comparison to the wild-type strain at utilizing mucus as a carbon source.
In order to assess whether the metabolic fitness effects could account for the defect of the luxO and aphA mutants in vivo, we performed in vitro competition assays in M9 medium supplemented with mucus (M9M) or M9 medium with individual mucus sugars as sole carbon sources (Fig. 5). We observed that the luxO mutant was out-competed in intestinal mucus, with a CI of 0.6 (Fig. 5), and in the mucus sugars gluconate, ribose, and arabinose, with CIs of 0.3, 0.24, and 0.25, respectively (Fig. 5). The aphA mutant was also significantly out-competed by the wild-type strain in intestinal mucus, with a CI of 0.59, and in the mucus sugars ribose and arabinose, with CIs of 0.67 and 0.66, respectively (Fig. 5). In addition, the competitive indices for the opaR mutant in in vitro competition assays showed that the mutant significantly out-competed the wild-type strain in intestinal mucus, with a CI of 1.6 (Fig. 5). The opaR mutant also significantly out-competed the wild type in the individual mucus sugars mannose, ribose, and arabinose, with CIs of 1.6, 1.6, and 1.5, respectively (Fig. 5). Overall, the in vitro metabolic assays suggest not only that the presence of aphA is important to the cells, in part due to its regulation of opaR, but also that induced expression of opaR can have a detrimental effect. While constitutive expression of opaR results in a fitness defect (luxO mutant), deletion of opaR provides a fitness advantage (opaR mutant) in vitro.
FIG 5.
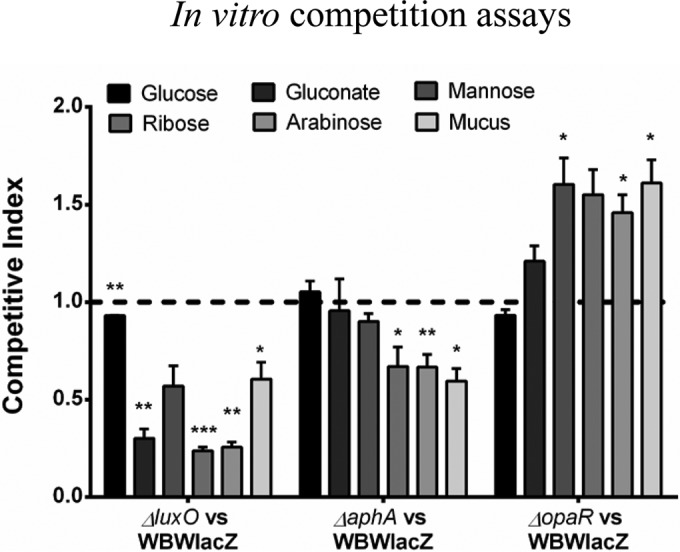
In vitro competition assays of the V. parahaemolyticus wild-type strain and the QS luxO, opaR, and aphA mutants. In vitro competition assays between the WBWlacZ strain and the mutant strains in mucus and in individual mucus sugars were performed. P values were calculated using an unpaired Student's t test with a 95% confidence interval. Asterisks denote significant differences in CIs between the mutant strains and the wild type. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
OpaR binding sites in the promoter regions of carbon transport and metabolism genes.
Both the luxO and aphA mutants have constitutively expressed opaR and repressed aphA genes. We wanted to determine whether the observed in vitro growth phenotypes were due to direct or indirect regulation of metabolism and transporter genes by OpaR. First, we performed bioinformatics analysis to identify putative binding sites for OpaR in the promoter regions of 89 metabolism and transporter genes. We chose genes that were involved in the transport and metabolism of carbon sources that showed different growth patterns between the wild type and mutants in the phenotypic arrays. The MOODS (Motif Occurrence Detection Suite) tool was used to identify OpaR and AphA binding sites using the consensus sequence and position frequency matrix identified by Zhang et al. (33) and Sun et al. (55). In this analysis, we identified 55 promoter regions with strong binding sites (P < = 0.005 and >90% probability) for OpaR (Table S5). Interestingly, the same analysis for an AphA consensus binding sequence identified only nine putative AphA binding sites, suggesting that OpaR is the main QS regulator of metabolism (Table S5). Of the 55 putative OpaR binding sites, 28 OpaR binding sites were in promoter regions of operons, and 27 binding sites were in single gene promoter regions. These included genes for arabinose, ribose, glucose, maltose, trehalose, mannitol, mannose, glycogen, glycerol, cellobiose, and sperimidine/putrescine transport and/or metabolism. Putative OpaR binding sites were identified also in genes for general amino acid transport (ORFs VP0008 to VP0006 and VP1620) and thymidine, uridine, serine, aspartate, fumarate, glutarate, arginine, histidine, phenylalanine, tyrosine, and tryptophan transport and/or metabolism. In contrast, of the 89 promoter regions examined for AphA binding sites, only nine promoter regions showed strong binding sites (probability >90%), which included mannose (VPA1424 to VPA1425), arabinose (VPA1673 to VPA1671), and glycogen (VPA1620) transport and/or metabolism genes (Table S6).
In order to validate the bioinformatics analysis of putative OpaR binding sites, we purified OpaR to homogeneity and performed EMSAs on five representative target promoter regions. The five targets were comprised of the amino acid transporter promoter region of VP0008 to VP0006, the polyamine putrescine cluster (VP1779 to VP1771), two sugar transporter promoter regions (mannitol, VPA0500 to VPA0501; ribose, VPA1087 to VPA1084), and a promoter region for a mannose transport and metabolism cluster (VPA1424 to VPA1425). The glucose-specific phosphotransferase system (PTS) VPA1667 probe was included as a negative control as it did not have any predicted OpaR binding sites. (Fig. 6). For the promoter region PVP0008, OpaR bound to the 323-bp DNA probe with increasing concentrations of the OpaR protein (0 to 2.7 μM). Similarly, promoter regions PVP1779 (244 bp), PVPA1087 (333 bp), PVPA0500 (360 bp), and PVPA1424 (350 bp) were bound by OpaR in a concentration-dependent manner (Fig. 6). The negative DNA control target remained unbound by OpaR at the highest concentrations tested (Fig. 6). These data demonstrate that OpaR binding is specific and that OpaR regulates the expression of these targets, indicating that it plays a direct role in cell metabolism.
FIG 6.
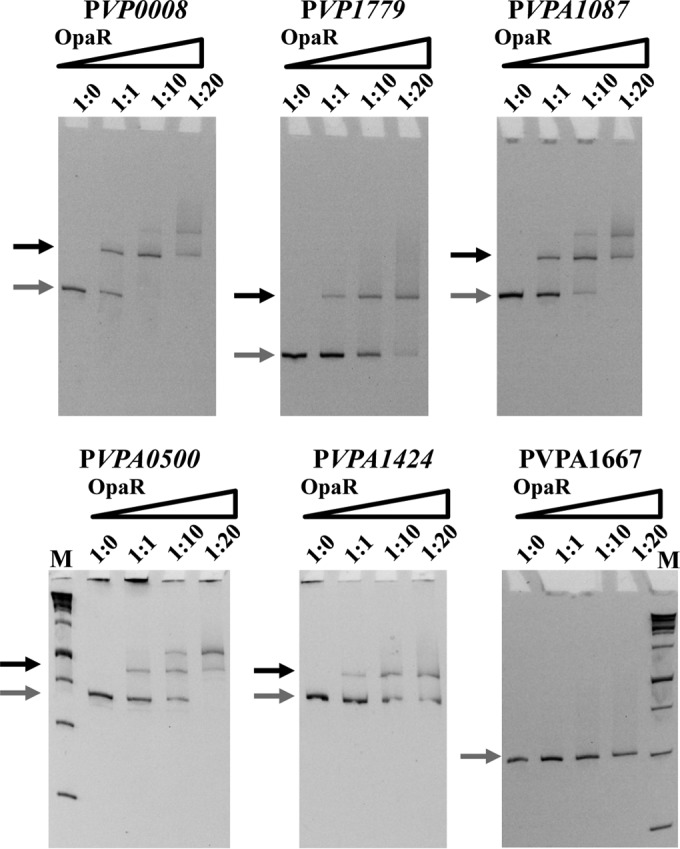
OpaR binding sites in regulatory regions of metabolism and transporter genes. Electrophoretic mobility shift assays (EMSAs) were performed using various concentrations of OpaR (0 to 3.7 μM) incubated with DNA substrates corresponding to the promoter regions of the following genes, starting upstream of the translational ATG start site: VP0008 (PVP0008: 323-bp DNA probe), VP1779 (PVP1779; 244-bp DNA probe), VPA1087 (PVPA1087; 333-bp DNA probe), VPA0500 (PVPA0500; 360-bp DNA probe), and VPA1424 (PVPA1424; 350-bp DNA probe). The DNA fragment from ORF VPA1667 (PVPA1667; 308-bp DNA probe) was used as a negative control. Binding of OpaR was shown for all sites that were identified to contain putative OpaR binding sites by bioinformatics. Ratios indicated above each gel image indicate ratios of DNA to the protein concentrations used in each well. Gray arrows represent unbound DNA, and black arrows represent bound DNA. M, marker.
DISCUSSION
In this study, we examined the role of QS regulators in V. parahaemolyticus pathogenesis, specifically, their role in intestinal colonization. The in vivo colonization data show that the QS regulators are essential for efficient colonization. RNA-Seq transcriptome data between the luxO mutant and wild-type cells showed global gene expression differences. A striking feature of these data is the number of genes involved in cell metabolism and transport that were downregulated in the luxO mutant compared to levels in the wild type. These included genes required for transport and metabolism of substrates present in intestinal mucus, one of the main nutrient sources in vivo. The gene expression data suggested that the luxO mutant could have metabolic defects based on the downregulation of key metabolism genes. Competition for intestinal nutrients and the ability to utilize intestinal mucus as a carbon and energy source have been shown to be important for successful colonization of intestinal bacteria (56–59). Phenotypic array data showed that the deletion of luxO resulted in metabolic defects, with the mutant demonstrating a defect in growth on a number of carbon sources. This was also shown to be the case for the aphA mutant, which showed a growth defect compared to growth of the wild type on 25 carbon sources. The carbon substrates on which the mutants showed defects were comprised of nearly equal numbers of sugars, organic acids, and peptides, indicating that not just one pathway was affected. Some of the most significant growth defects were in the utilization of amino acids and amino acid derivatives, key intermediates in central metabolism. The luxO mutant also had a defect in in vitro competition assays in mucus. This is not too surprising, given that mucus is made of mainly the glycoprotein mucin and is therefore rich in amino acids as well as sugars (51–54). Neither the aphA nor the opaR mutant demonstrated a dramatic in vitro metabolic fitness effect compared to that of the luxO mutant although each had slightly different growth patterns in one or two carbon sources. One scenario to explain these differences is that in the luxO mutant, aphA expression is down as is qrr expression, and opaR expression is highly induced. In the aphA mutant, opaR is induced, but so too should be the qrr genes, which are negatively regulated by AphA in V. harveyi. Thus, knocking out both aphA and qrr expression is more detrimental than just knocking out aphA alone. This suggests that there may be dual regulation of genes involved in metabolism and/or additional roles for the Qrr sRNAs.
A transcriptome study of Pseudomonas aeruginosa over 10 years ago demonstrated that the QS-activated regulon was overrepresented by genes involved in intermediate central metabolism (60). They showed that in the QS-repressed regulon carbohydrate utilization and nutrient transport genes were the most abundant representatives (60). A more recent study in the same species demonstrated a global impact of QS on the metabolome and proposed that QS plays a key role in metabolic rewiring of the cell under certain conditions (61). A study examining the targets of LuxR homologues in Brucella, an intracellular pathogen, identified a large number of proteins involved in metabolic pathways, such as central metabolism or amino acid metabolism, respiration, and transport of amino acids and sugars that were under the control of QS regulators (62). QS control of metabolic pathways that affect fitness has also been shown in Burkholderia species (63). Hwang and colleagues showed that QS regulates oxalate synthesis to counteract alkalization of the growth medium and was essential for fitness (63, 64). More recently, this same group has demonstrated that the QS master regulator QsmR downregulates glucose transport, substrate-level and oxidative phosphorylation, and nucleotide biosynthesis, acting as a metabolic brake on individuals as the population increases (64). Evidence for a role for LuxR in metabolism also comes from Vibrio species. In V. harveyi, it was demonstrated that the argA, purM, lysE, and rluA promoter regions were LuxR-dependent genes involved in arginine and purine biosynthesis, amino acid efflux, and pseudouridine synthesis, respectively (65). In addition, it was demonstrated in V. fischeri that QS AinS signaling is essential for control of the acetate switch and that this regulation is mediated through the LuxR homologue LitR (66). A recent study in V. cholerae showed that the QS LuxR homologue HapR regulated chitin metabolism that provided predator grazing resistance in biofilms. Sun et al. showed that 19 of 22 genes involved in GlcNAc catabolism were repressed in an hapR mutant compared to levels in the wild type (67).
QS regulators are required for both population-level and individual control of gene expression, which corresponds to stationary and early exponential growth phases, respectively, during which availability of the type and amount of nutrients is very different. Thus, the involvement of QS regulators controlling expression of transporter and metabolism genes makes biological sense. The luxO and aphA mutants had an in vivo defect that correlates with reduced metabolic fitness. In both of these mutants OpaR is highly expressed, which suggests that OpaR could be a direct or indirect negative regulator of cell metabolism. Our RNA-Seq data revealed that in the luxO mutant, 60% of downregulated genes were involved in transport and metabolism. In addition, in the luxO mutant a number of regulators that could be involved in regulation of metabolism were also downregulated. These included genes belonging to the LysR family of proteins, which have been shown to regulate a diverse set of genes including those involved in metabolism (68), and the AraC/XylS family of transcriptional regulators, which are predominantly involved in the regulation of carbon metabolism (69). Interestingly, we also found that the DNA-binding protein Fis (VP2885) was slightly upregulated in the luxO mutant (1.62-fold; Padj < 0.0001). Fis is a known global regulator of metabolism. In Salmonella enterica, Fis was shown to negatively regulate genes contributing to metabolism in the mammalian gut (70). In addition, the Hfq-binding sRNA Spot 42 was upregulated in the V. parahaemolyticus luxO mutant. In Escherichia coli, Spot 42 plays an essential role as a regulator in carbohydrate metabolism and uptake, and its expression is activated by glucose and inhibited by cyclic AMP (cAMP) receptor protein (CRP). Spot 42 was shown to be a negative regulator of metabolism of many sugars in both E. coli and Vibrio (Allivibrio) salmonicida (71–74). A second sRNA, VrrA was also induced in the V. parahaemolyticus luxO mutant. In V. cholerae, studies showed that a vrrA mutant had a 5-fold-increased ability to colonize infant mice (75). VrrA downregulates outer membrane proteins OmpA and OmpT, the stationary-phase survival factor Vrp, and biofilm matrix protein RbmC in V. cholerae (76, 77).
To determine the possible extent of direct regulation of cell metabolism and transport by the QS regulators, we performed bioinformatics analysis and examined 89 regulatory regions of metabolism genes for the presence of putative OpaR and AphA binding sites. We identified 55 loci that contained strong putative OpaR binding sites and only 9 putative AphA binding sites. From the 55 loci with putative OpaR binding sites, we chose five representatives to examine further using EMSAs: a general amino acid transporter (VP0008 to VP0006), ribose (VPA1087 to VPA1084), and mannose (VPA1424 to VPA1425) metabolism and transporter, mannitol (VPA0500 to VPA0501), and putrescine metabolism (VP1771 to VP1779) regulatory regions. We investigated the regulatory region of the general amino acid transporter since both microarray and RNA-Seq analysis showed repression of these genes by OpaR (30, 41). Our binding analysis confirmed that, indeed, OpaR does bind to the regulatory region of this operon. We examined the regulatory regions of ribose and mannose since these sugars are important mucus sugars, and we observed growth defects in these sugars in the luxO and aphA mutants compared to growth in the wild type. We investigated binding to the regulatory region of the transporter of sugar alcohol mannitol because it had one of the strongest putative OpaR binding sites. The polyamine putrescine cluster VP1779 to VP1771 is involved in putrescine metabolism and contains genes that shuttle into multiple metabolic pathways. All the genes in this pathway were downregulated in the luxO mutant compared to levels in the wild type, and this was confirmed by qPCR while EMSAs showed binding of OpaR to the regulatory region. These data indicate that OpaR is a negative regulator of putrescine metabolism. Thus, EMSAs demonstrated binding to all five regulatory regions and no binding to the negative control using the highest concentration of OpaR. These data demonstrate a direct role for OpaR in cell metabolism and suggest that this role may be more prevalent than previously appreciated.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
All the strains and plasmids used in this study are listed in Table S1 in the supplemental material. A streptomycin-resistant strain of V. parahaemolyticus O3:K6, clinical isolate RIMD2210633, was used throughout this study (13, 78). For competition experiments a β-galactosidase-positive strain of RIMD2210633, named WBWlacZ, was used. Colonies of the WBWlacZ strain appear blue on an X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) plate in comparison to the wild type and its isogenic mutants that appear white on the plate, thus allowing for a blue-white screen in a competition experiment. The WBWlacZ strain was previously shown to behave identically to the wild type in vitro and in vivo (13, 14). Unless stated otherwise, all V. parahaemolyticus strains were grown in lysogeny broth (LB) containing 3% NaCl (LBS) (Fischer Scientific, Pittsburgh, PA) at 37°C with aeration. For growth studies, M9 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 3% NaCl was used and different carbon sources were added. For genetic manipulations, an Escherichia coli diaminopimelic acid (DAP) auxotroph, β2155 λpir, was used. The E. coli β2155 λpir strain was cultured in LB medium supplemented with 0.3 mM DAP (Sigma-Aldrich). When required, antibiotics were used at the following concentrations: streptomycin, 200 μg/ml; chloramphenicol, 25 μg/ml; ampicillin, 100 μg/ml.
Construction of V. parahaemolyticus RIMD2210633 quorum sensing deletion mutants.
Splicing by overlap extension (SOE) PCR with homologous recombination (79) was used to construct in-frame nonpolar deletions in VP2099 (luxO), VP2762 (aphA), VP2516 (opaR), and luxO opaR and opaR aphA double deletion mutants as previously described by this group (13–15, 78, 80). Primers were designed to the QS response regulator luxO and the two QS master regulators opaR and aphA using the V. parahaemolyticus RIMD2210633 genome sequence as the template. All primers used in the study are listed in Table S2, and SOE PCR was performed to obtain a 75-bp truncated version of the 1,362-bp luxO gene, a 39-bp truncated version of the 615-bp opaR gene, and a 48-bp truncated version of the 540-bp aphA gene. All mutants were confirmed by PCR analysis, and mutations were verified to be in frame by sequencing.
In vivo competition assays.
All experiments involving mice were approved by the University of Delaware Institutional Animal Care and Use Committee. Male C57BL/6 mice, aged 6 to 10 weeks, were housed under specific-pathogen-free conditions in standard cages in groups (4 or 5 per group) and provided standard mouse feed and water ad libitum. Streptomycin pretreatment and inoculations were performed as previously described (13, 14). Briefly, 24 h before bacterial inoculations by oral gavage, mice were fasted for 4 h and then administered 20 mg of streptomycin per animal orogastrically; food and water were then immediately returned. Four hours prior to inoculation, food and water were removed. Water was restored immediately upon inoculation, and food was restored at 2 h postinfection. The V. parahaemolyticus strain used for in vivo experiments is the β-galactosidase knock-in designated WBWlacZ, which allows for blue-white colony screening (13–15). Overnight cultures were diluted 1:50 with LBS-streptomycin medium and grown for 4 h at 37°C with aeration. An aliquot of the 4-h culture was pelleted and resuspended in phosphate-buffered saline (PBS) to a final concentration of ∼1 × 1010 CFU/ml. A 1-ml aliquot of each deletion mutant strain was combined with 1 ml of the WBWlacZ strain, yielding a bacterial suspension of ∼1 × 1010 CFU/ml at a ratio of 1:1 CFU of the mutant to WBWlacZ strain. Mice were inoculated with 100 μl of the appropriate bacterial suspension. An aliquot of the inoculum was serially diluted and plated onto LBS plates with streptomycin and X-Gal in order to determine the exact ratio of CFU in the inoculum. For in vitro competition assays, a 100-μl aliquot of the in vivo inoculum was added to 5 ml of LBS, grown at 37°C with aeration for 24 h, and serially diluted and plated. The mice were sacrificed at 24 h postinfection, and the gastrointestinal tract was harvested and suspended in 8 ml of sterile PBS, homogenized mechanically, serially diluted, plated on LBS plates containing 120 μg/ml X-Gal, and incubated at 37°C overnight. The competitive index (CI) for the in vivo and in vitro assays was determined with the following equation: CI = ratio out(mutant/wild type)/ratio in(mutant/wild type). A CI of >1 indicates that the test strain has the ability to out-compete the wild-type strain, while a CI of <1 indicates that the test strain is less fit than the wild-type strain.
CPS production and biofilm assays.
Capsule polysaccharide (CPS) production was examined using heart infusion (HI) (Remel, Lenexa, KS) plates containing 1.5% agar, 2.5 mM CaCl2, and 0.25% Congo red dye. Single colonies were inoculated onto the surface of the plates and were incubated at 30°C for 36 h before images were taken. Biofilm formation was examined using a crystal violet assay. Briefly, overnight cultures of V. parahaemolyticus were diluted 1:40 into LBS medium and grown statically in 96-well strip plates at 37°C for 3, 6, 12, and 24 h. After static incubation, the culture was decanted from each well, and the well was washed once with sterile phosphate-buffered saline (PBS). Crystal violet was added into each well, and the plate was incubated at room temperature for 30 min. The crystal violet was decanted out, and the well was washed with sterile PBS. The PBS was then decanted out, and crystal violet that had stained the adherent cells was solubilized completely in dimethyl sulfoxide (DMSO). The optical density at 595 nm (OD595) was measured to quantify the amount of biofilm formed.
RNA extraction, Illumina sequencing, and quantitative real-time PCR (qPCR).
Vibrio parahaemolyticus wild-type and mutant strains were grown for 4 h in LBS medium and then diluted 1:50 into M9 medium supplemented with mouse intestinal mucus as the sole carbon source, and the cells were grown statically. We examined early-exponential-phase cultures that were grown for 1.5 h, considering that the low-cell-density condition would restrict opaR levels in the wild type, thereby allowing us to observe the greatest difference in opaR levels between the luxO mutant and wild type. Total RNA was extracted from cells obtained by centrifugation at the end of 1.5 h using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. The RNA samples were then quantified using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA). The samples were treated with Turbo DNase (Invitrogen) according to the manufacturer's instructions. For each sample of the wild type and mutant, RNA samples from two independent cultures were pooled. NanoDrop quantifications were used to ensure equal representation of RNAs from both biological replicates. Then, 3 μg of RNA was used for rRNA depletion using a Ribo-Zero rRNA removal kit for Gram-negative bacteria (Illumina, San Diego, CA). Libraries for each sample were prepared from 100 ng of rRNA-depleted RNA using an Illumina TruSeq Stranded mRNA kit (Illumina). Sequencing was performed at the University of Delaware Sequencing and Genotyping Center on a HiSeq 2500 platform to yield 51-base single-end reads.
For qPCR validations of the RNA-Seq, 500 ng of pre-Ribo-Zero-treated RNA was used as a template for cDNA synthesis. cDNA was synthesized using Superscript III reverse transcriptase (RT) (Invitrogen) according to the manufacturer's instructions using 500 ng of RNA template and priming with 200 ng of random hexamers. cDNA samples were then diluted 1:25 and used for quantitative real-time PCR (qPCR). To analyze expression of the wild-type and mutant strains at HCD, cells were grown for 4 h in LBS medium and were then diluted 1:50 into M9 medium containing 3% NaCl and supplemented with glucose (M9G) and grown to an OD of 1.0. Total RNA was extracted using the TRIzol extraction protocol detailed above. A total of 500 ng of the DNase-treated RNA samples was used as the template for cDNA synthesis. cDNA samples were then diluted 1:25 or 1:10 and used for qPCR. Fast SYBR green master mix or PowerUp SYBR green master mix (Life Technologies, Carlsbad, CA) was used for qPCR, and samples were run on an Applied Biosystems 7500 fast real-time PCR system or QuantStudio 6 Flex real-time PCR system (Applied Biosystems, Foster City, CA). Each experiment was performed in duplicate with at least two biological replicates. Primers used for the qPCRs are listed in Table S2. Data were analyzed using Applied Biosystems software. Expression levels of each gene, as determined by their cycle threshold (CT) values, were normalized using the 16S rRNA housekeeping gene to correct for sampling errors. Differences in the ratios of gene expression levels were determined using the ΔΔCT method (81).
RNA-Seq analysis.
Raw 51-base reads were filtered to remove adaptor-only sequences and low-quality reads using the FASTX Toolkit. Filtered reads were aligned to the V. parahaemolyticus RIMD2210633 genome (NCBI RefSeq accession numbers NC_004603.1 for chromosome 1 and NC_004605.1 for chromosome 2) using the Burrows-Wheeler Aligner (BWA.aln), version 0.7.7. Gene annotations were obtained from Ensembl Bacteria, the Rfam database, the Bacterial Small Regulatory RNA Database (BSRD), and RAST. Numbers of reads aligning to each genomic position were calculated using Htseq, version 0.6.1. Differential expression analysis was performed on obtained read counts using DESeq2, version 1.4.5. Differentially expressed genes were categorized using the Clusters of Orthologous Groups (COG) obtained from the Integrated Microbial Genomes (IMG) database.
Growth analysis and in vitro competition assays.
Strains were grown overnight in M9G medium at 37°C with aeration. For the Biolog PM1 and PM2A phenotype microarrays (Biolog, Inc., Hayward, CA), overnight cultures were then diluted 1:50 into fresh M9G medium and allowed to grow for 4 h. These cultures were pelleted by centrifugation for 10 min at 4,000 × g, washed twice with PBS, and then diluted 1:50 into fresh M9 medium supplemented with 3% NaCl, and 100 μl was then added to each well of the Biolog plate. Plates were incubated at 37°C with intermittent shaking for 1 min during every hour. Optical densities at 595 nm were taken hourly for a total of 24 h using a Tecan Sunrise microplate reader and Magellan plate reader software (Tecan Systems Inc., San Jose, CA). Growth characteristics were analyzed by calculating the area under the curve using Origin, version 8.5, software. The area under the curve for the blank well was subtracted from each well to perform the analysis. For growth curves in individual mucus sugars and amino acids, 4-h cultures were pelleted, washed, and diluted 1:40 in M9G (10 mM) medium or M9 medium with d-gluconate (10 mM), d-mannose (10 mM), d-ribose (10 mM), l-arabinose (10 mM), d-galactose (10 mM), d-glucosamine (10 mM), pyruvic acid (10 mM), d-trehalose (10 mM), fructose (10 mM), l-glutamic acid (5 mM), or l-aspartic acid (30 mM). Mouse intestinal mucus was extracted as described previously (14, 15). Mouse gastrointestinal tracts were harvested and then flushed with PBS to remove intestinal contents. Mucus was collected and pooled by gently scraping the surface walls of the intestine using a spatula or blunted blade. The collected mucus was suspended in PBS and vortexed until homogenized. The mucus solution was then centrifuged at 500 × g for 10 min, and the supernatant was collected. Protein concentration was determined using a Bradford assay. Approximately 30 μg/ml of protein was used in M9 medium for experiments involving mucus (14, 82, 83). Each experiment was performed in triplicate with at least two biological replicates. In vitro competition assays in mucus and mucus sugars were performed with inocula prepared as described for in vivo competition assays. A 100-μl aliquot of the inoculum was added to 5 ml of M9 minimal medium supplemented with 10 mM individual mucus sugars or 30 μg/ml of intestinal mucus and grown at 37°C with aeration for 24 h, serially diluted, and plated. The competitive index for each assay was calculated as detailed above.
Bioinformatics analysis of OpaR and AphA binding sites.
The consensus binding sequence and position frequency matrix were obtained for OpaR (33) and AphA (55). The position frequency matrix was then used to identify potential binding sites using the MOODS (Motif Occurrence Detection Suite) algorithm (version 1.0.2.1) (84, 85). The upstream intergenic sequence for the first gene of each operon was obtained from the NCBI database and used to identify putative binding sites. Operon information was obtained from the DOOR2 prokaryotic operon database (86) and was confirmed using the Integrative Genomics Viewer (IGV) (87) with the RNA-Seq sequence data. The MOODS tool returned a log-odds score for each putative binding site that was then used to access probability of binding.
Purification of OpaR.
OpaR was purified using a method previously described (88). Briefly, opaR was cloned into the pProEX HTa expression plasmid (Invitrogen) in which an N-terminal 6×His tag is fused to opaR, separated by a tobacco etch virus (TEV) protease cleavage site. The primer pair SfoIVP2516Fwd/SacIVP2516Rev (Table S2) was used to amplify opaR (VP2516) from the V. parahaemolyticus RIMD2210633 genome using Accura HiFidelity Polymerase (Lucigen, Middleton, WI) according to the manufacturer's instructions. The opaR PCR product was gel cut purified using a NucleoSpin Gel and PCR cleanup kit (Macherey-Nagel) and cloned into pJET1.2 using the blunt-end ligation protocol. This was transformed into E. coli DH5α using a standard CaCl2 transformation protocol. Plasmid DNA was isolated, restriction digested, and ligated into pProEX HTa plasmid. The ligation product was transformed into E. coli DH5α, and plasmid DNA was isolated and confirmed by sequencing before being transformed into E. coli BL21(DE3) using a standard CaCl2 method. The pProExHtaOpaR plasmid was expressed in E. coli BL21(DE3). A volume of 10 ml of overnight culture was inoculated into 1 liter of LB broth at 37°C and induced with 0.5 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) at an OD600 of 0.5. Growth continued overnight at 18°C. Cells were harvested by centrifugation (5,000 × g for 20 min at 4°C) and resuspended in immobilized metal affinity chromatography (IMAC) wash buffer (50 mM sodium phosphate, 200 mM NaCl, 20 mM imidazole, pH 7.6) supplemented with the protease inhibitors phenylmethanesulfonyl fluoride (PMSF; 1 mM) and benzamidine (1 mM). Bacterial cells were lysed on ice using a high-pressure homogenizer (EmulsiFlex-C5; Avestin, Ottawa, Canada). Cell debris was removed by centrifugation (15,000 × g for 1 h at 4°C). The supernatant was passed through a column containing 5 ml of Profinity IMAC resin (Bio-Rad Laboratories, Hercules, CA). The column was washed with 10 column volumes (CV) of IMAC wash buffer. The fusion protein, 6×His-OpaR, was eluted with three CV of IMAC elution buffer (50 mM sodium phosphate, 200 mM NaCl, 500 mM imidazole, pH 7.6). A hexahistidine-tagged TEV protease was added to the eluent in a 1:10 molar ratio (TEV protease/6×His-OpaR), and the cleavage reaction proceeded overnight at 4°C. The cleavage mixture was centrifuged, adjusted to 20 mM imidazole, and subjected to IMAC using Profinity IMAC resin to remove the His-tagged TEV protease and any remaining uncleaved fusion protein. The flowthrough and one CV of wash with IMAC wash buffer contained OpaR. The fractions were combined and concentrated, and the buffer was exchanged for that of the electrophoretic mobility shift assay binding buffer (10 mM Tris, 150 mM KCl, 0.1 mM dithiothreitol, 0.1 mM EDTA, 5% polyethylene glycol [PEG], pH 7.4). Identity of the protein was confirmed by mass spectrometry, and its purity was determined to be higher than 95% by SDS-PAGE.
Electrophoretic mobility shift assays.
DNA probes VP0008 (amino acid transport), VP1779 (putrescine metabolism), VPA1087 (ribose transport), VPA0500 (mannitol PTS transporter), VPA1424 (mannose metabolism), and negative-control VPA1667 (glucose-specific PTS transporter) were PCR amplified using Accura HiFidelity Polymerase in 50-μl reaction mixtures using the corresponding primer sets listed in Table S2, with V. parahaemolyticus DNA as the template. PCR products were separated on a 1% agarose gel, and bands excised from the gel were purified using a NucleoSpin Gel and PCR cleanup kit (Macherey-Nagel). Purified DNA probes were quantified using a NanoDrop spectrophotometer. Various concentrations of purified OpaR were incubated with 30 ng of target DNA in binding buffer (10 mM Tris, 150 mM KCl, 0.1 mM dithiothreitol, 0.1 mM EDTA, 5% PEG, pH 7.4) for 20 min at room temperature, and 10 μl was loaded onto a prerun (200 V for 2 h at 4°C) 6% native acrylamide gel. The gel was run at 200 V for 3 h in 1× Tris-acetate-EDTA (TAE) buffer at 4°C. Following electrophoresis, gels were stained in an ethidium bromide bath (0.5 μg/ml) for 20 min, washed with water, and imaged.
Accession number(s).
The sequence data have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under accession number GSE93547.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nathan McDonald, Abish Regmi, Joe Borowski, Gwen Gregory, and Aoife Boyd for reviewing the manuscript and helpful discussions.
This work was supported in part by a National Science Foundation grant DEB-0844409 to E.F.B. and departmental funds. The Delaware COBRE program in part supported this research with grants from the National Institute of General Medical Sciences under awards P30 GM110758-02 and P20 GM104316.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00930-16.
REFERENCES
- 1.Hondo S, Goto I, Minematsu I, Ikeda N, Asano N, Ishibashi M, Kinoshita Y, Nishibuchi N, Honda T, Miwatani T. 1987. Gastroenteritis due to Kanagawa negative Vibrio parahaemolyticus. Lancet i:331–332. [DOI] [PubMed] [Google Scholar]
- 2.Daniels NA, MacKinnon L, Bishop R, Altekruse S, Ray B, Hammond RM, Thompson S, Wilson S, Bean NH, Griffin PM, Slutsker L. 2000. Vibrio parahaemolyticus infections in the United States, 1973–1998. J Infect Dis 181:1661–1666. doi: 10.1086/315459. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin JB, DePaola A, Bopp CA, Martinek KA, Napolilli NP, Allison CG, Murray SL, Thompson EC, Bird MM, Middaugh JP. 2005. Outbreak of Vibrio parahaemolyticus gastroenteritis associated with Alaskan oysters. N Engl J Med 353:1463–1470. doi: 10.1056/NEJMoa051594. [DOI] [PubMed] [Google Scholar]
- 4.Nair GB, Ramamurthy T, Bhattacharya SK, Dutta B, Takeda Y, Sack DA. 2007. Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin Microbiol Rev 20:39–48. doi: 10.1128/CMR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qadri F, Alam MS, Nishibuchi M, Rahman T, Alam NH, Chisti J, Kondo S, Sugiyama J, Bhuiyan NA, Mathan MM, Sack DA, Nair GB. 2003. Adaptive and inflammatory immune responses in patients infected with strains of Vibrio parahaemolyticus. J Infect Dis 187:1085–1096. doi: 10.1086/368257. [DOI] [PubMed] [Google Scholar]
- 6.Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A, Kubota Y, Kimura S, Yasunaga T, Honda T, Shinagawa H, Hattori M, Iida T. 2003. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet 361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- 7.Ham H, Orth K. 2012. The role of type III secretion system 2 in Vibrio parahaemolyticus pathogenicity. J Microbiol 50:719–725. doi: 10.1007/s12275-012-2550-2. [DOI] [PubMed] [Google Scholar]
- 8.O'Boyle N, Boyd A. 2014. Manipulation of intestinal epithelial cell function by the cell contact-dependent type III secretion systems of Vibrio parahaemolyticus. Front Cell Infect Microbiol 3:114. doi: 10.3389/fcimb.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiyoshi H, Kodama T, Iida T, Honda T. 2010. Contribution of Vibrio parahaemolyticus virulence factors to cytotoxicity, enterotoxicity, and lethality in mice. Infect Immun 78:1772–1780. doi: 10.1128/IAI.01051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, Honda T. 2004. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun 72:6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pineyro P, Zhou X, Orfe LH, Friel PJ, Lahmers K, Call DR. 2010. Development of two animal models to study the function of Vibrio parahaemolyticus type III secretion systems. Infect Immun 78:4551–4559. doi: 10.1128/IAI.00461-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritchie JM, Rui H, Zhou X, Iida T, Kodoma T, Ito S, Davis BM, Bronson RT, Waldor MK. 2012. Inflammation and disintegration of intestinal villi in an experimental model for Vibrio parahaemolyticus-induced diarrhea. PLoS Pathog 8:e1002593. doi: 10.1371/journal.ppat.1002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitaker WB, Parent MA, Boyd A, Richards GP, Boyd EF. 2012. The Vibrio parahaemolyticus ToxRS regulator is required for stress tolerance and colonization in a novel orogastric streptomycin-induced adult murine model. Infect Immun 80:1834–1845. doi: 10.1128/IAI.06284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitaker WB, Richards GP, Boyd EF. 2014. Loss of sigma factor RpoN increases intestinal colonization of Vibrio parahaemolyticus in an adult mouse model. Infect Immun 82:544–556. doi: 10.1128/IAI.01210-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haines-Menges B, Whitaker WB, Boyd EF. 2014. Alternative sigma factor RpoE is important for Vibrio parahaemolyticus cell envelope stress response and intestinal colonization. Infect Immun 82:3667–3677. doi: 10.1128/IAI.01854-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubbard TP, Chao MC, Abel S, Blondel CJ, Abel Zur Wiesch P, Zhou X, Davis BM, Waldor MK. 2016. Genetic analysis of Vibrio parahaemolyticus intestinal colonization. Proc Natl Acad Sci U S A 113:6283–6288. doi: 10.1073/pnas.1601718113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuqua WC, Winans SC, Greenberg EP. 1994. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol 176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardman AM, Stewart GS, Williams P. 1998. Quorum sensing and the cell-cell communication dependent regulation of gene expression in pathogenic and non-pathogenic bacteria. Antonie Van Leeuwenhoek 74:199–210. doi: 10.1023/A:1001178702503. [DOI] [PubMed] [Google Scholar]
- 19.Hastings JW, Nealson KH. 1977. Bacterial bioluminescence. Annu Rev Microbiol 31:549–595. doi: 10.1146/annurev.mi.31.100177.003001. [DOI] [PubMed] [Google Scholar]
- 20.Bassler BL. 1999. How bacteria talk to each other: regulation of gene expression by quorum sensing. Curr Opin Microbiol 2:582–587. doi: 10.1016/S1369-5274(99)00025-9. [DOI] [PubMed] [Google Scholar]
- 21.Bassler BL, Wright M, Showalter RE, Silverman MR. 1993. Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol Microbiol 9:773–786. doi: 10.1111/j.1365-2958.1993.tb01737.x. [DOI] [PubMed] [Google Scholar]
- 22.Ng WL, Bassler BL. 2009. Bacterial quorum-sensing network architectures. Annu Rev Genet 43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyamoto CM, Dunlap PV, Ruby EG, Meighen EA. 2003. LuxO controls luxR expression in Vibrio harveyi: evidence for a common regulatory mechanism in Vibrio. Mol Microbiol 48:537–548. doi: 10.1046/j.1365-2958.2003.03453.x. [DOI] [PubMed] [Google Scholar]
- 24.Gray KM, Passador L, Iglewski BH, Greenberg EP. 1994. Interchangeability and specificity of components from the quorum-sensing regulatory systems of Vibrio fischeri and Pseudomonas aeruginosa. J Bacteriol 176:3076–3080. doi: 10.1128/jb.176.10.3076-3080.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarter LL. 1998. OpaR, a homolog of Vibrio harveyi LuxR, controls opacity of Vibrio parahaemolyticus. J Bacteriol 180:3166–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. 2002. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell 110:303–314. doi: 10.1016/S0092-8674(02)00829-2. [DOI] [PubMed] [Google Scholar]
- 27.Jaques S, McCarter LL. 2006. Three new regulators of swarming in Vibrio parahaemolyticus. J Bacteriol 188:2625–2635. doi: 10.1128/JB.188.7.2625-2635.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milton DL. 2006. Quorum sensing in vibrios: complexity for diversification. Int J Med Microbiol 296:61–71. doi: 10.1016/j.ijmm.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 29.Hammer BK, Bassler BL. 2003. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol 50:101–104. doi: 10.1046/j.1365-2958.2003.03688.x. [DOI] [PubMed] [Google Scholar]
- 30.Gode-Potratz CJ, McCarter LL. 2011. Quorum sensing and silencing in Vibrio parahaemolyticus. J Bacteriol 193:4224–4237. doi: 10.1128/JB.00432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henke JM, Bassler BL. 2004. Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus. J Bacteriol 186:3794–3805. doi: 10.1128/JB.186.12.3794-3805.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Zhou D, Mao P, Zhang Y, Hou J, Hu Y, Li J, Hou S, Yang R, Wang R, Qiu J. 2013. Cell density- and quorum sensing-dependent expression of type VI secretion system 2 in Vibrio parahaemolyticus. PLoS One 8:e73363. doi: 10.1371/journal.pone.0073363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Qiu Y, Tan Y, Guo Z, Yang R, Zhou D. 2012. Transcriptional regulation of opaR, qrr2-4 and aphA by the master quorum-sensing regulator OpaR in Vibrio parahaemolyticus. PLoS One 7:e34622. doi: 10.1371/journal.pone.0034622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou D, Yan X, Qu F, Wang L, Zhang Y, Hou J, Hu Y, Li J, Xin S, Qiu J, Yang R, Mao P. 2013. Quorum sensing modulates transcription of cpsQ-mfpABC and mfpABC in Vibrio parahaemolyticus. Int J Food Microbiol 166:458–463. doi: 10.1016/j.ijfoodmicro.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Lilley BN, Bassler BL. 2000. Regulation of quorum sensing in Vibrio harveyi by LuxO and sigma-54. Mol Microbiol 36:940–954. doi: 10.1046/j.1365-2958.2000.01913.x. [DOI] [PubMed] [Google Scholar]
- 36.Tu KC, Bassler BL. 2007. Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev 21:221–233. doi: 10.1101/gad.1502407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. 2004. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 38.Bardill JP, Zhao X, Hammer BK. 2011. The Vibrio cholerae quorum sensing response is mediated by Hfq-dependent sRNA/mRNA base pairing interactions. Mol Microbiol 80:1381–1394. doi: 10.1111/j.1365-2958.2011.07655.x. [DOI] [PubMed] [Google Scholar]
- 39.van Kessel JC, Rutherford ST, Shao Y, Utria AF, Bassler BL. 2013. Individual and combined roles of the master regulators AphA and LuxR in control of the Vibrio harveyi quorum-sensing regulon. J Bacteriol 195:436–443. doi: 10.1128/JB.01998-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutherford ST, van Kessel JC, Shao Y, Bassler BL. 2011. AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev 25:397–408. doi: 10.1101/gad.2015011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kernell Burke A, Guthrie LT, Modise T, Cormier G, Jensen RV, McCarter LL, Stevens AM. 2015. OpaR controls a network of downstream transcription factors in Vibrio parahaemolyticus BB22OP. PLoS One 10:e0121863. doi: 10.1371/journal.pone.0121863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Ling Y, Jiang H, Qiu Y, Qiu J, Chen H, Yang R, Zhou D. 2013. AphA is required for biofilm formation, motility, and virulence in pandemic Vibrio parahaemolyticus. Int J Food Microbiol 160:245–251. doi: 10.1016/j.ijfoodmicro.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 43.Salomon D, Klimko JA, Orth K. 2014. H-NS regulates the Vibrio parahaemolyticus type VI secretion system 1. Microbiology 160:1867–1873. doi: 10.1099/mic.0.080028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gu D, Liu H, Yang Z, Zhang Y, Wang Q. 2016. Chromatin immunoprecipitation sequencing technology reveals global regulatory roles of low-cell-density quorum-sensing regulator AphA in the pathogen Vibrio alginolyticus. J Bacteriol 198:2985–2999. doi: 10.1128/JB.00520-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skorupski K, Taylor RK. 1999. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol Microbiol 31:763–771. doi: 10.1046/j.1365-2958.1999.01215.x. [DOI] [PubMed] [Google Scholar]
- 46.Yang M, Frey EM, Liu Z, Bishar R, Zhu J. 2010. The virulence transcriptional activator AphA enhances biofilm formation by Vibrio cholerae by activating expression of the biofilm regulator VpsT. Infect Immun 78:697–703. doi: 10.1128/IAI.00429-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Enos-Berlage JL, Guvener ZT, Keenan CE, McCarter LL. 2005. Genetic determinants of biofilm development of opaque and translucent Vibrio parahaemolyticus. Mol Microbiol 55:1160–1182. [DOI] [PubMed] [Google Scholar]
- 48.Guvener ZT, McCarter LL. 2003. Multiple regulators control capsular polysaccharide production in Vibrio parahaemolyticus. J Bacteriol 185:5431–5441. doi: 10.1128/JB.185.18.5431-5441.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu YW, Denkmann K, Kosciow K, Dahl C, Kelly DJ. 2013. Tetrathionate stimulated growth of Campylobacter jejuni identifies a new type of bi-functional tetrathionate reductase (TsdA) that is widely distributed in bacteria. Mol Microbiol 88:173–188. doi: 10.1111/mmi.12176. [DOI] [PubMed] [Google Scholar]
- 50.Chang DE, Smalley DJ, Tucker DL, Leatham MP, Norris WE, Stevenson SJ, Anderson AB, Grissom JE, Laux DC, Cohen PS, Conway T. 2004. Carbon nutrition of Escherichia coli in the mouse intestine. Proc Natl Acad Sci U S A 101:7427–7432. doi: 10.1073/pnas.0307888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clamp JR, Fraser G, Read AE. 1981. Study of the carbohydrate content of mucus glycoproteins from normal and diseased colons. Clin Sci (Lond) 61:229–234. doi: 10.1042/cs0610229. [DOI] [PubMed] [Google Scholar]
- 52.Conway T, Krogfelt KA, Cohen PS. 1 December 2004, posting date The life of commensal Escherichia coli in the mammalian intestine. EcoSal Plus 2014 doi: 10.1128/ecosalplus.8.3.1.2. [DOI] [PubMed] [Google Scholar]
- 53.Corazziari ES. 2009. Intestinal mucus barrier in normal and inflamed colon. J Pediatr Gastroenterol Nutr 48(Suppl 2):S54–S55. doi: 10.1097/MPG.0b013e3181a117ea. [DOI] [PubMed] [Google Scholar]
- 54.Fabich AJ, Jones SA, Chowdhury FZ, Cernosek A, Anderson A, Smalley D, McHargue JW, Hightower GA, Smith JT, Autieri SM, Leatham MP, Lins JJ, Allen RL, Laux DC, Cohen PS, Conway T. 2008. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun 76:1143–1152. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun F, Zhang Y, Wang L, Yan X, Tan Y, Guo Z, Qiu J, Yang R, Xia P, Zhou D. 2012. Molecular characterization of direct target genes and cis-acting consensus recognized by quorum-sensing regulator AphA in Vibrio parahaemolyticus. PLoS One 7:e44210. doi: 10.1371/journal.pone.0044210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conway T, Cohen PS. 2015. Commensal and pathogenic Escherichia coli metabolism in the gut. Microbiol Spectr 3(3):MBP-000602014. doi: 10.1128/microbiolspec.MBP-0006-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maltby R, Leatham-Jensen MP, Gibson T, Cohen PS, Conway T. 2013. Nutritional basis for colonization resistance by human commensal Escherichia coli strains HS and Nissle 1917 against E. coli O157:H7 in the mouse intestine. PLoS One 8:e53957. doi: 10.1371/journal.pone.0053957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stecher B, Hardt WD. 2011. Mechanisms controlling pathogen colonization of the gut. Curr Opin Microbiol 14:82–91. doi: 10.1016/j.mib.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 59.Donaldson GP, Lee SM, Mazmanian SK. 2016. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14:20–32. doi: 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schuster M, Lostroh CP, Ogi T, Greenberg EP. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol 185:2066–2079. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davenport PW, Griffin JL, Welch M. 2015. Quorum sensing is accompanied by global metabolic changes in the opportunistic human pathogen Pseudomonas aeruginosa. J Bacteriol 197:2072–2082. doi: 10.1128/JB.02557-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uzureau S, Lemaire J, Delaive E, Dieu M, Gaigneaux A, Raes M, De Bolle X, Letesson JJ. 2010. Global analysis of quorum sensing targets in the intracellular pathogen Brucella melitensis 16 M. J Proteome Res 9:3200–3217. doi: 10.1021/pr100068p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goo E, Majerczyk CD, An JH, Chandler JR, Seo YS, Ham H, Lim JY, Kim H, Lee B, Jang MS, Greenberg EP, Hwang I. 2012. Bacterial quorum sensing, cooperativity, and anticipation of stationary-phase stress. Proc Natl Acad Sci U S A 109:19775–19780. doi: 10.1073/pnas.1218092109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.An JH, Goo E, Kim H, Seo YS, Hwang I. 2014. Bacterial quorum sensing and metabolic slowing in a cooperative population. Proc Natl Acad Sci U S A 111:14912–14917. doi: 10.1073/pnas.1412431111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyamoto CM, Meighen EA. 2006. Involvement of LuxR, a quorum sensing regulator in Vibrio harveyi, in the promotion of metabolic genes: argA, purM, lysE and rluA. Biochim Biophys Acta 1759:296–307. doi: 10.1016/j.bbaexp.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 66.Studer SV, Mandel MJ, Ruby EG. 2008. AinS quorum sensing regulates the Vibrio fischeri acetate switch. J Bacteriol 190:5915–5923. doi: 10.1128/JB.00148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun S, Tay QX, Kjelleberg S, Rice SA, McDougald D. 2015. Quorum sensing-regulated chitin metabolism provides grazing resistance to Vibrio cholerae biofilms. ISME J 9:1812–1820. doi: 10.1038/ismej.2014.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maddocks SE, Oyston PC. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 69.Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. 1997. Arac/XylS family of transcriptional regulators. Microbiol Mol Biol Rev 61:393–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kelly A, Goldberg MD, Carroll RK, Danino V, Hinton JC, Dorman CJ. 2004. A global role for Fis in the transcriptional control of metabolism and type III secretion in Salmonella enterica serovar Typhimurium. Microbiology 150:2037–2053. doi: 10.1099/mic.0.27209-0. [DOI] [PubMed] [Google Scholar]
- 71.Beisel CL, Storz G. 2011. Discriminating tastes: physiological contributions of the Hfq-binding small RNA Spot 42 to catabolite repression. RNA Biol 8:766–770. doi: 10.4161/rna.8.5.16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beisel CL, Storz G. 2011. The base-pairing RNA spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli. Mol Cell 41:286–297. doi: 10.1016/j.molcel.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hansen GA, Ahmad R, Hjerde E, Fenton CG, Willassen NP, Haugen P. 2012. Expression profiling reveals Spot 42 small RNA as a key regulator in the central metabolism of Aliivibrio salmonicida. BMC Genomics 13:37. doi: 10.1186/1471-2164-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moller T, Franch T, Udesen C, Gerdes K, Valentin-Hansen P. 2002. Spot 42 RNA mediates discoordinate expression of the E. coli galactose operon. Genes Dev 16:1696–1706. doi: 10.1101/gad.231702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Song T, Mika F, Lindmark B, Liu Z, Schild S, Bishop A, Zhu J, Camilli A, Johansson J, Vogel J, Wai SN. 2008. A new Vibrio cholerae sRNA modulates colonization and affects release of outer membrane vesicles. Mol Microbiol 70:100–111. doi: 10.1111/j.1365-2958.2008.06392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sabharwal D, Song T, Papenfort K, Wai SN. 2015. The VrrA sRNA controls a stationary phase survival factor Vrp of Vibrio cholerae. RNA Biol 12:186–196. doi: 10.1080/15476286.2015.1017211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Song T, Sabharwal D, Wai SN. 2010. VrrA mediates Hfq-dependent regulation of OmpT synthesis in Vibrio cholerae. J Mol Biol 400:682–688. doi: 10.1016/j.jmb.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 78.Whitaker WB, Parent MA, Naughton LM, Richards GP, Blumerman SL, Boyd EF. 2010. Modulation of responses of Vibrio parahaemolyticus O3:K6 to pH and temperature stresses by growth at different salt concentrations. Appl Environ Microbiol 76:4720–4729. doi: 10.1128/AEM.00474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 80.Kalburge SS, Whitaker WB, Boyd EF. 2014. High-salt preadaptation of Vibrio parahaemolyticus enhances survival in response to lethal environmental stresses. J Food Prot 77:246–253. doi: 10.4315/0362-028X.JFP-13-241. [DOI] [PubMed] [Google Scholar]
- 81.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cohen PS, Laux DC. 1995. Bacterial adhesion to and penetration of intestinal mucus in vitro. Methods Enzymol 253:309–314. doi: 10.1016/S0076-6879(95)53026-6. [DOI] [PubMed] [Google Scholar]
- 83.Leatham MP, Stevenson SJ, Gauger EJ, Krogfelt KA, Lins JJ, Haddock TL, Autieri SM, Conway T, Cohen PS. 2005. Mouse intestine selects nonmotile flhDC mutants of Escherichia coli MG1655 with increased colonizing ability and better utilization of carbon sources. Infect Immun 73:8039–8049. doi: 10.1128/IAI.73.12.8039-8049.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Korhonen J, Martinmaki P, Pizzi C, Rastas P, Ukkonen E. 2009. MOODS: fast search for position weight matrix matches in DNA sequences. Bioinformatics 25:3181–3182. doi: 10.1093/bioinformatics/btp554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pizzi C, Rastas P, Ukkonen E. 2011. Finding significant matches of position weight matrices in linear time. IEEE/ACM Trans Comput Biol Bioinform 8:69–79. doi: 10.1109/TCBB.2009.35. [DOI] [PubMed] [Google Scholar]
- 86.Mao F, Dam P, Chou J, Olman V, Xu Y. 2009. DOOR: a database for prokaryotic operons. Nucleic Acids Res 37:D459–D463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carpenter MR, Rozovsky S, Boyd EF. 2015. Pathogenicity island cross talk mediated by recombination directionality factors facilitates excision from the chromosome. J Bacteriol 198:766–776. doi: 10.1128/JB.00704-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.