

ABSTRACT
The literature presently lacks a population pharmacokinetic analysis of doxycycline. This study aimed to develop a population pharmacokinetic model of doxycycline plasma concentrations that could be used to assess the power of bioequivalence between Doryx delayed-release tablets and Doryx MPC. Doxycycline pharmacokinetic data were available from eight phase 1 clinical trials following single/multiple doses of conventional-release doxycycline capsules, Doryx delayed-release tablets, and Doryx MPC under fed and fasted conditions. A population pharmacokinetic model was developed in a stepwise manner using NONMEM, version 7.3. The final covariate model was developed according to a forward inclusion (P < 0.01) and then backward deletion (P < 0.001) procedure. The final model was a two-compartment model with two-transit absorption compartments. Structural covariates in the base model included formulation effects on relative bioavailability (F), absorption lag (ALAG), and the transit absorption rate (KTR) under the fed status. An absorption delay (lag) for the fed status (FTLAG2 = 0.203 h) was also included in the model as a structural covariate. The fed status was observed to decrease F by 10.5%, and the effect of female sex was a 14.4% increase in clearance. The manuscript presents the first population pharmacokinetic model of doxycycline plasma concentrations following oral doxycycline administration. The model was used to assess the power of bioequivalence between Doryx delayed-release tablets and Doryx MPC, and it could potentially be used to critically examine and optimize doxycycline dose regimens.
KEYWORDS: doxycycline, population pharmacokinetics
INTRODUCTION
Doxycycline is an oral tetracycline antimicrobial which possesses anti-inflammatory mechanisms that contribute to its use under conditions such as acne vulgaris (1). As an expanded-spectrum tetracycline, doxycycline has increased lipophilicity over its predecessors, with increased absorption and tissue penetration (2, 3). Doxycycline absorption occurs in the duodenum, with oral bioavailability (F) within the range of 73 to 95% (2, 3). Doxycycline also has improved absorption in the fed state and in the presence of dairy products compared to absorption of narrow-spectrum tetracyclines, to the point that delayed-release doxycycline tablets are marketed to be taken with or without food although some guidelines recommend taking the drug with food or milk to reduce stomach upset (2–4).
Mayne Pharma International currently has two marketed oral formulations available in the United States, a delayed-release doxycycline hyclate tablet (Doryx delayed-release tablets; here, Doryx tablets) and the recently launched Doryx MPC delayed-release tablets (here, Doryx MPC), both of which have modified acid resistance profiles compared to the profile of immediate-release oral dose forms (5). Modifying the acid resistance profile is expected to result in less release of doxycycline prior to the small intestine. The Doryx delayed-release tablet was developed due to an association between drug release in acid and the potential occurrence of esophageal and gastric irritation (5).
Despite the delayed-release properties of Doryx delayed-release tablets, some doxycycline release does occur in acid environments, potentially resulting in drug release in the stomach prior to gastric emptying. Accordingly, the Doryx MPC delayed-release tablet was developed with a further modified in vitro acid resistance profile. Based upon prior evidence, Doryx MPC delayed-release tablets have the potential to offer increased protection from esophageal and gastric irritation. Given the change in the release profile of Doryx MPC delayed-release tablets, it was anticipated that the formulation would demonstrate a reduction in the bioavailability compared to that of the Doryx delayed-release tablet on a milligram-per-milligram basis. To address this reduction in relative bioavailability (RELF), Doryx MPC delayed-release tablets were formulated to have a 20% higher doxycycline (hyclate) content (120 mg) than Doryx delayed-release tablets (100 mg).
Doxycycline is predominately cleared unchanged in the urine and feces through renal, biliary, and intestinal wall elimination, resulting in a half-life between 15 and 25 h (3). Previously, Beringer et al. (6) performed a compartmental pharmacokinetic analysis in 20 adults with cystic fibrosis dosed with multiple 40-mg, 100-mg, or 200-mg doses of oral doxycycline (formulation unspecified) and found a two-compartment model with a delay (lag) on first-order absorption to best describe the plasma concentration-time data. To our knowledge, there are no population pharmacokinetic analyses in the literature exploring doxycycline concentrations following oral administration. Such an analysis is essential to the development of a population pharmacokinetic-pharmacodynamic model that could be used to critically examine and optimize doxycycline dose regimens, which has been understudied to date (2). In the manuscript, phase I pharmacokinetic data collected by Mayne Pharma International following the administration of either the conventional-release doxycycline capsules (here, Doryx capsule), Doryx delayed-release tablets, or Doryx MPC delayed-release tablets were used to develop a population pharmacokinetic model. The primary aim of the study was to assess the equivalence of doxycycline plasma exposure following a 100-mg Doryx delayed-release tablet or a 120-mg Doryx MPC delayed-release tablet. The influence of the fasted/fed status was also assessed on the pharmacokinetics of the new formulation.
RESULTS
Study population and pharmacokinetic data.
There were 188 subjects in total enrolled across the eight phase 1 clinical trials. A summary of the study populations' demographics is represented in Table 1. All subjects with evaluable pharmacokinetic data (including partial data) were included in the analysis. In total, pharmacokinetic data were available from 178 participants, who received a total of 651 oral doxycycline doses in both single- and multidose studies, with either one or two treatment periods. Participants contributed 7,093 doxycycline plasma concentrations across these studies. Overall, 12.7% of the doxycycline plasma concentration data was missing, predominately at the beginning or end of the observation periods, where plasma concentrations were below the level of quantification (BLOQ). Models accounting for BLOQ-censored data were investigated using the YLO and M3 methods (7); however, these were characterized by unreliable minimization and covariance step status. Samples below the limit of quantitation were thus excluded a priori from the data set (M1 method). For the base and final model, BLOQ visual predictive checks (VPCs) were used to ensure that there were sufficient data to describe the elimination phase of doxycycline. There were no missing covariate data from the study.
TABLE 1.
Study population characteristics
| Parameter | Value for the parametera |
|---|---|
| Age (yr) | 28 (18–73) |
| Smoking status (no. of subjects) | |
| Smokers | 22 |
| Nonsmokers | 156 |
| BMI | 25.25 (18.1–34.5) |
| Weight (kg) | 75.9 (47.2–114.4) |
| FFM (kg) | 56.3 (32.3–80.1) |
| Sex (no. of subjects) | |
| Male | 120 |
| Female | 58 |
| Race (no. of subjects) | |
| Caucasian/White | 99 |
| Otherb | 79 |
All values are calculated as means (range) unless stated otherwise.
This category was used due to variability in the ethnicity categorization between the available studies. For example, studies 30, 40, 50, 60, and 80 categorized participants as White, Black, American Indian, or Hispanic/Latino. Studies 20 and 70 categorized participants as Caucasian, Black, Hispanic, and American Indian. Finally, study 10 categorized participants as White, Black, Asian, and other.
Base model.
The base model of the available doxycycline plasma concentrations which best fit the model-building criteria was a two-compartment model with two-transit absorption compartments (Fig. 1). The model was allometrically scaled for fat-free mass (FFM), had a covariance matrix with between-subject variability for the parameters transit compartment absorption rate (KTR), apparent clearance (CL/F), apparent central volume (V/F), and apparent peripheral volume (VP1/F). These parameters are referred to as CL, V, and VP1 henceforth, albeit they are influenced by the true bioavailability (F). Between-occasion variability was included on KTR and CL. The model had a proportional and an additive residual error term. Structural covariates included in the base model were a formulation effect (for Doryx MPC, FMPC; for Doryx capsule, FCAP) on relative bioavailability (RELF), an absorption lag on the Doryx MPC and Doryx capsule formulations (ALAG), an absorption lag for the fed status (FTLAG2), and a food effect on KTR for the Doryx tablet and capsules (COVFED), as well as Doryx MPC (COVFED2). Between-study differences were also included on the F, KTR, CL, and V parameters. For the between-study differences on F, there were three groups apparent, composed of studies 10, 30, and 50 (reference group), studies 20, 70, and 80 (COVSTDF1), and studies 40 and 60 (COVSTDF2). For the between-study differences on the KTR parameter, two groups were apparent, composed of studies 10, 20, 70, and 80 (reference group) and studies 30, 40, 50, and 60 (COVSTDKTR). For the between-study differences on the CL and V parameters, two groups were apparent composed of study 10 (reference group) and studies 20, 30, 40, 50, 60, 70, and 80 (for CL, COVSTDCL; for V, COVSTDV). The parameter estimates for the base model can be found in File S1 in the supplemental material. Given that the diagnostic plots of the base model were consistent with an unbiased model and that the prediction-corrected VPC (pcVPC) showed a good description of the data (File S1), it was considered that the structural and population components of the model appropriately represented the data, allowing for confidence in the subsequent analysis of covariates and power analyses.
FIG 1.
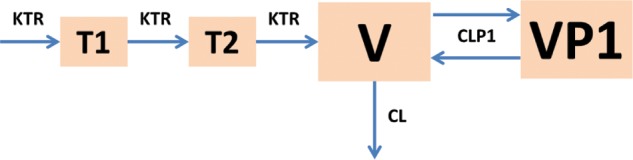
Structural components of the base model. CL, apparent clearance; V, apparent central distribution volume; CLP1, apparent intercompartmental clearance; VP1, peripheral distribution volume; KTR, transit absorption rate; T1 and T2, transit absorption compartments.
Final model.
The base model was screened for the potential inclusion of covariates listed in Table 2, which presents the drop in objective function value (OBJ), the number of parameters, and the significance (P value) upon each covariate screening. Of the screened covariates, only the fasted/fed effect on relative bioavailability (COVFEDF on RELF; OBJ drop of 41.837 for 1 parameter), sex effects on clearance (COVSEX on CL; OBJ drop of 25.661 for 1 parameter), and sex effects on volume (COVSEX on V; OBJ drop of 15.086 for 1 parameter) significantly decreased the OBJ at the level of a P value of <0.01 with acceptable parameter precision. A fasted/fed effect dependent on formulation was also explored on relative bioavailability (COVFEDF, effect with Doryx tablet and capsule; COVFEDFMPC, effect with Doryx MPC; OBJ drop of 43.739 for 2 parameters). Despite a significant drop in OBJ, the estimation of the COVFEDFMPC parameter was imprecise (percent standard error [%SE] of 63.3), and it was not included in the model.
TABLE 2.
Significance of screened covariates on the base model
| Covariate screened | No. of parameters added | OBJ dropa | P valueb |
|---|---|---|---|
| Fed status on RELFc | 2 | 43.739 | <0.01 |
| Fed status on RELF | 1 | 41.837 | <0.01 |
| Sex on CL | 1 | 25.661 | <0.01 |
| Sex on V | 1 | 15.086 | <0.01 |
| Tablet strength on RELF | 4 | 11.849 | >0.01 |
| Age on RELF | 1 | 5.291 | >0.01 |
| Smoker status on CL | 1 | 2.554 | >0.01 |
| Dose on CL | 1 | 2.484 | >0.01 |
| Age on KTR | 1 | 2.274 | >0.01 |
| Sex on RELF | 1 | 1.853 | >0.01 |
| Dose on RELF | 1 | 1.157 | >0.01 |
| Tablet strength on KTR | 4 | 1.129 | >0.01 |
| Formulation on KTR | 2 | 1.09 | >0.01 |
| Age on V | 1 | 0.982 | >0.01 |
| Sex on VP1 | 1 | 0.756 | >0.01 |
| Race on VP1 | 1 | 0.436 | >0.01 |
| Sex on KTR | 1 | 0.151 | >0.01 |
| Race on CL | 1 | 0.104 | >0.01 |
| Race on KTR | 1 | 0.075 | >0.01 |
| Race on RELF | 1 | 0.061 | >0.01 |
| Age on VP1 | 1 | 0.053 | >0.01 |
| Dose on KTR | 1 | 0.05 | >0.01 |
| Race on V | 1 | 0.019 | >0.01 |
| Age on CL | 1 | 0.011 | >0.01 |
The drop in the objective function value when the screened covariate is added to the base model.
The P value indicates whether upon screening the covariate tested resulted in a significant drop in the OBJ at a P value of <0.01 (6.63 units for one parameter).
Fed status effect on RELF dependent upon formulation (covariate %SE was 63.3, and it was not explored further).
On forward inclusion, sex effects on clearance (COVSEX on CL) were retained in the full covariate model after the OBJ was decreased by a further 25.492 compared to the model containing just a fasted/fed effect on relative bioavailability (COVFEDF on RELF). The inclusion of sex effects on volume (COVSEX on V) did not further improve the covariate model (OBJ drop of 1.05 units). The backward deletion of sex effects on clearance (COVSEX on CL) and fasted/fed effect on relative bioavailability (COVFEDF on RELF), respectively, increased the OBJ function by 25.492 and 41.668 units, and thus both were retained in the final covariate model at the level of a P value of <0.001. This model was therefore declared the final model as it passed the model-building criteria. The parameter estimates for the final model are shown in Table 3. The NONMEM control stream for the final model can be found in File S1.
TABLE 3.
Parameter values for the final model
| Parameter | Symbol(s) | Description | Unit(s) | Parameter estimate | %SE | Shrinkage (%) | Eta P value |
|---|---|---|---|---|---|---|---|
| Population parameters | |||||||
| V | Θ3 | Apparent central distribution vol | liters/70 kg FFM | 55.2 | 7.8 | ||
| CL | Θ4 | Apparent clearance | liters/h/70 kg FFM | 4.63 | 5.0 | ||
| VP1 | Θ5 | Apparent peripheral distribution vol | liters/70 kg FFM | 49.8 | 3.5 | ||
| CLP1 | Θ6 | Apparent intercompartmental clearance | liters/h/70 kg FFM | 11.3 | 4.1 | ||
| KTR | Θ7 | Transit compartment rate constant | h−1 | 2 | 4.3 | ||
| F1MPC | Θ8 | Relative bioavailability of Doryx MPC compared to Doryx tablet | 0.863 | 4.9 | |||
| F1CAP | Θ9 | Relative bioavailability of Doryx capsule compared to Doryx tablet | 0.978 | 3.8 | |||
| COVFED | Θ10 | Effect of food on KTR for the Doryx tablet and capsules | −0.209 | 26.3 | |||
| COVFED2 | Θ11 | A food effect on KTR for Doryx MPC | −0.549 | 7.7 | |||
| ALAG1 | Θ12 | A lag on KTR for the Doryx MPC and Doryx Capsule formulations | h | 0.115 | 24.4 | ||
| FTLAG2 | Θ13 | A lag on KTR for the fed status | h | 0.203 | 14.2 | ||
| Between-study effects | |||||||
| COVSTDF1 | Θ14 | Effect of studies 20, 70 and 80 on relative bioavailability | 0.516 | 11.9 | |||
| COVSTDF2 | Θ15 | Effect of studies 40 and 60 on relative bioavailability | −0.146 | 19.9 | |||
| COVSTDKTR | Θ16 | Effect of studies 30, 40, 50, and 60 on KTR | −0.213 | 17.3 | |||
| COVSTDCL | Θ17 | Effect of studies 20, 30, 40, 50, 60, 70, and 80 on CL | −0.241 | 18.0 | |||
| COVSTDV | Θ18 | Fractional effect of studies 20, 30, 40, 50, 60, 70, and 80 on V | −0.245 | 26.7 | |||
| Covariate effects | |||||||
| COVFEDF | Θ19 | Effect of food on relative bioavailability | 0.105 | 33.2 | |||
| COVSEX | Θ20 | Effect of female sex on CL | 0.144 | 20.4 | |||
| Between-subject variability | |||||||
| BSV_CL (η1) | ω1 | Between subject variability on CL | %CVa | 19.3 | 6.4 | 9.1 | 0.959 |
| BSV_KTR (η2) | ω2 | Between subject variability on KTR | %CV | 28.2 | 8.3 | 14.6 | 0.891 |
| Population variability | |||||||
| PPV_VP1 (η3) | ω3 | Population variability on VP1 | %CV | 15.1 | 11.2 | 30.7 | 0.870 |
| PPV_V (η4) | ω4 | Population variability on V | %CV | 37.6 | 5.8 | 7.8 | 0.913 |
| Between-occasion variability | |||||||
| BOVCL1 (η5) | ω5 | Between occasion variability on CL (period 1) | %CV | 13.5 | 7.8 | 23.0 | 0.978 |
| BOVCL2 (η6) | ω6 | Between occasion variability on CL (period 2) | %CV | 13.5 | - | 25.3 | 0.853 |
| BOVKTR(η7) | ω7 | Between occasion variability on KTR (period 1) | %CV | 27.2 | 10.5 | 15.6 | 0.815 |
| BOVKTR(η8) | ω8 | Between occasion variability on KTR (period 2) | %CV | 27.2 | - | 9.2 | 0.888 |
| Covariance | |||||||
| Θ4, Θ7 | ω1, ω2 | Covariance between CL and KTR | 0.420 | ||||
| Θ4, Θ5 | ω1, ω3 | Covariance between CL and VP1 | 0.363 | ||||
| Θ4, Θ3 | ω1, ω4 | Covariance between CL and V | 0.720 | ||||
| Θ7, Θ5 | ω2, ω3 | Covariance between KTR and VP1 | −0.234 | ||||
| Θ7, Θ3 | ω2, ω4 | Covariance between KTR and V | 0.884 | ||||
| Θ5, Θ3 | ω3, ω4 | Covariance between VP1 and V | −0.089 | ||||
| Residual error | |||||||
| RUV_PROP | Θ1 | Proportional error | %CV | 19.6 | 2.5 | ||
| RUV_ADD | Θ2 | Additive error | μg/liter | 19.8 | 18.5 |
CV, coefficient of variation.
Figure 2 presents the key diagnostic plots for the final model following the administration of Doryx MPC, Doryx tablet, and Doryx capsules. The diagnostic plots appear consistent with an unbiased model (File S1 includes diagnostic plots for the final model facetted on key model covariates).
FIG 2.

Diagnostic plots for doxycycline concentrations following Doryx MPC (red), Doryx tablet (blue), and Doryx capsule (green) administration from the final model. Symbols are data points, the solid black line is a line of identity with slope 1 or 0, and the red lines are a loess-smooth of the data. CWRES, conditional weighted residuals.
The pcVPC of the final model provided a good description of the lower, median, and higher doxycycline concentrations, as represented by a good overlay of the 5th, median, and 95th percentiles of the observed prediction-corrected concentrations over the empirical 95% confidence intervals (CIs) of the prediction-corrected doxycycline concentrations from 200 simulations of the index data set (Fig. 3 and File S1). The BLOQ VPC also indicates that the final model provides a good description of doxycycline plasma concentration BLOQ throughout time within the database, with the percentage of the observed data BLOQ throughout time overlaying well with the 95% CI of the percentage of BLOQ data from the 200 simulations of the original data set (Fig. 4), indicating that the M1 method for handling BLOQ data was acceptable. Both the BLOQ VPC and pcVPC plots indicated that the structural, population, and covariate components of the model provided a good description of the data following single- or multidose Doryx MPC, Doryx tablet, or Doryx capsule under fed or fasted conditions, allowing for confidence in the subsequent simulations and power analyses.
FIG 3.
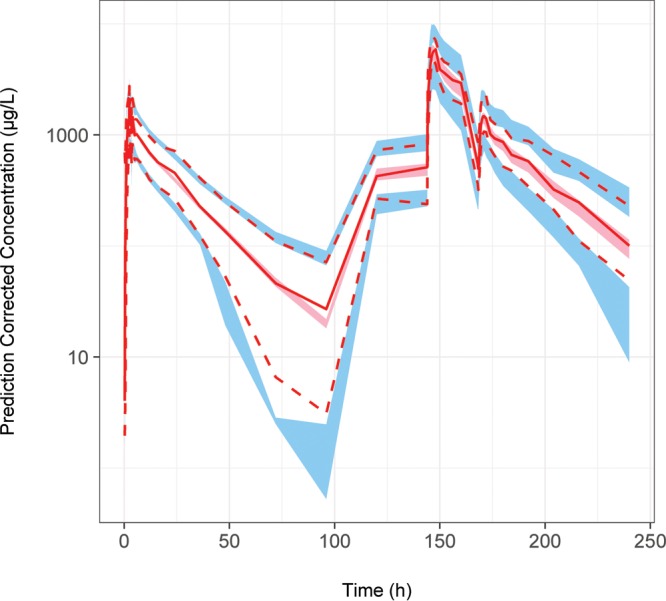
pcVPC of the final model for doxycycline concentrations. The prediction-corrected observed data are represented by the red solid line (median) and the red dashed lines (5th and 95th percentiles). The simulated prediction-corrected doxycycline concentrations are represented by the red shaded area (empirical 95% confidence interval of median) and the blue shaded areas (empirical 95% confidence intervals of 5th and 95th percentiles). The model predictions overlay the observed data with good agreement.
FIG 4.
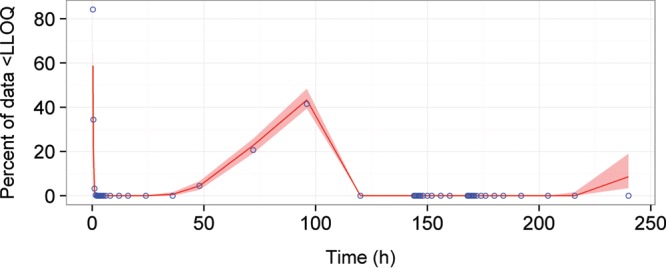
BLOQ VPC of the final model for doxycycline concentrations BLOQ (<LLOQ). The observed BLOQ data throughout time are represented by the blue circles. The simulated BLOQ data are represented by the 95% CI (red shaded area) and the median (solid red line). The predictions of the 95% CIs of the BLOQ throughout time from the model overlay the observed data with good agreement.
Power analysis.
The final model indicated that Doryx MPC had a relative bioavailability of 0.863 and an absorption lag of 0.115 h compared to a Doryx tablet for the studied population. In addition, the fed status reduced KTR by 54.9% for Doryx MPC and 20.9% for a Doryx tablet (plus the formulation nonspecific 0.203-h lag on absorption). Therefore, simulation and power analyses were used to interpret the chance of establishing bioequivalence between 120 mg of Doryx MPC and a reference 100-mg Doryx tablet in the fed and fasted states.
Plots of the median and 90% CI of doxycycline concentrations from 1,000 simulated studies of 28 subjects are provided in File S1, following the administration of the crossover study designs indicated previously. Table 4 presents the percentage of the 1,000 simulated studies displaying equivalent values of the area under the concentration-time curve at time (t) 0 or infinity (AUC0t and AUCinf, respectively) maximum concentration of unbound drug in plasma (Cmax), and time to maximum concentration of drug (Tmax) for the crossover studies including 12, 24, 28, and 36 subjects. Table 4 also presents the percentage of simulated studies deemed bioequivalent (BE), which occurs when AUC0t and Cmax (BE0t) or AUCinf and Cmax (BEinf) were equivalent for a given study. For the studies which were deemed bioequivalent, Table 5 indicates the percentage of those that included 1 within the 90% CI of the ln-transformed test-to-reference ratio for AUC0t and Cmax or for AUCinf and Cmax.
TABLE 4.
Model-predicted bioequivalence results calculated using the ANOVA methoda
| Study size and dosingb | % AUC0t | % AUCinf | % Cmax | % Tmax | % BE0t | % BEinf |
|---|---|---|---|---|---|---|
| 12 participants per study | ||||||
| Doryx MPC vs Doryx tablet (Fasted) | 89.8 | 87.9 | 88.1 | 15.7 | 79.2 | 77.8 |
| Doryx MPC vs Doryx tablet (Fed) | 90.5 | 86.7 | 7.5 | 0 | 6 | 5.6 |
| Doryx MPC vs Doryx MPC (Fed/Fasted) | 62.4 | 60.5 | 0 | 0 | 0 | 0 |
| Doryx tablet vs Doryx tablet (Fed/Fasted) | 65.2 | 61.1 | 12.2 | 5 | 8.6 | 8.1 |
| 24 participants per study (1,000 simulated studies) | ||||||
| Doryx MPC vs Doryx tablet (Fasted) | 99.7 | 99.6 | 99.8 | 58.6 | 99.5 | 99.4 |
| Doryx MPC vs Doryx tablet (Fed) | 99.6 | 99.5 | 7.6 | 0 | 7.4 | 7.3 |
| Doryx MPC vs Doryx MPC (Fed/Fasted) | 88 | 85.9 | 0 | 0 | 0 | 0 |
| Doryx tablet vs Doryx tablet (Fed/Fasted) | 88.4 | 86 | 18.4 | 6.3 | 16.8 | 16.3 |
| 28 participants per study | ||||||
| Doryx MPC vs Doryx tablet (Fasted) | 99.8 | 99.7 | 99.8 | 67.4 | 99.6 | 99.5 |
| Doryx MPC vs Doryx tablet (Fed) | 99.8 | 99.6 | 8.9 | 0 | 8.8 | 8.7 |
| Doryx MPC vs Doryx MPC (Fed/Fasted) | 92 | 90.6 | 0 | 0 | 0 | 0 |
| Doryx tablet vs Doryx tablet (Fed/Fasted) | 93.6 | 91.2 | 19 | 4.7 | 17.8 | 17.7 |
| 36 participants per study | ||||||
| Doryx MPC vs Doryx tablet (Fasted) | 100 | 100 | 100 | 82 | 100 | 100 |
| Doryx MPC vs Doryx tablet (Fed) | 100 | 100 | 9.1 | 0 | 9.1 | 9.1 |
| Doryx MPC vs Doryx MPC (Fed/Fasted) | 95.8 | 95.1 | 0 | 0 | 0 | 0 |
| Doryx tablet vs Doryx tablet (Fed/Fasted) | 96.4 | 95.3 | 21.2 | 5.8 | 20.8 | 20.7 |
Percent AUCinf, AUC0t, Cmax, and Tmax indicates, respectively, the power percentage for a study to display equivalence of AUCinf, AUC0t, Cmax, and Tmax for the crossover designs indicated. Percent BE0t and BEinf indicates, respectively, the power percentage for a study to display bioequivalence via AUC0t and Cmax and via AUCinf and Cmax for the crossover designs indicated.
For the studies, 120 mg of Doryx MPC and 100-mg Doryx tablets were used. For each group, 1,000 simulated studies were performed.
TABLE 5.
BE0t and BEinf studies that included 1 with the 90% CI of ln-transformed test-to-reference ratios for AUC0t, AUCinf, and Cmaxa
| Study size and dosingb | No. of BE0t studies from the 1,000 simulated replicates | % of BE0t studies that included 1 within the 90% CI of the test-to-reference ratio for AUC0t and Cmax | No. of BEinf studies from the 1,000 simulated replicates | % of BEinf studies that included 1 within the 90% CI of the test-to-reference ratio for AUCinf and Cmax |
|---|---|---|---|---|
| 12 participants per study | ||||
| Doryx MPC vs Doryx tablet (Fasted) | 792 | 84.85 | 778 | 85.35 |
| Doryx MPC vs Doryx tablet (Fed) | 60 | 56.67 | 56 | 51.79 |
| Doryx MPC vs Doryx MPC (Fed/Fasted) | 0 | 0 | ||
| Doryx tablet vs Doryx tablet (Fed/Fasted) | 86 | 44.19 | 81 | 43.21 |
| 24 participants per study | ||||
| Doryx MPC vs Doryx tablet (Fasted) | 995 | 64.02 | 994 | 65.59 |
| Doryx MPC vs Doryx tablet (Fed) | 74 | 4.05 | 73 | 4.11 |
| Doryx MPC vs Doryx MPC (Fed/Fasted) | 0 | 0 | ||
| Doryx tablet vs Doryx tablet (Fed/Fasted) | 168 | 4.17 | 163 | 4.91 |
| 28 participants per study | ||||
| Doryx MPC vs Doryx tablet (Fasted) | 996 | 60.84 | 995 | 61.91 |
| Doryx MPC vs Doryx tablet (Fed) | 88 | 3.41 | 87 | 4.60 |
| Doryx MPC vs Doryx MPC (Fed/Fasted) | 0 | 0 | ||
| Doryx tablet vs Doryx tablet (Fed/Fasted) | 178 | 1.12 | 177 | 1.13 |
| 36 participants per study | ||||
| Doryx MPC vs Doryx tablet (Fasted) | 1000 | 55.10 | 1000 | 57.50 |
| Doryx MPC vs Doryx tablet (Fed) | 91 | 1.10 | 91 | 1.10 |
| Doryx MPC vs Doryx MPC (Fed/Fasted) | 0 | 0 | ||
| Doryx tablet vs Doryx tablet (Fed/Fasted) | 208 | 0 | 207 | 0 |
BE0t indicates studies that were equivalent for both AUC0t and Cmax. BEinf indicates studies that were equivalent for both AUCinf and Cmax.
For the studies, 120 mg of Doryx MPC and 100-mg Doryx tablets were used. For each group, 1,000 simulated studies were performed.
In File S1 the mean of the geometric mean, 10th percentile, and 90th percentile for the ln-transformed test-to-reference ratios for AUC0t, AUCinf, Cmax, and Tmax, as calculated using analysis of variance (ANOVA), are presented following the simulation of 1,000 replicate studies including 12, 24, 28, and 36 subjects. As expected, increasing the number of subjects within the studies was associated with a higher power for displaying bioequivalence. Table 4 indicates that for a study containing 36 subjects, the power of bioequivalence between 120 mg of Doryx MPC and a 100-mg Doryx tablet under fasted conditions was 100% when BE0t and BEinf were assessed, respectively. For a study containing 28 subjects, values for the power of bioequivalence between 120 mg of Doryx MPC and the 100-mg Doryx tablet under fasted conditions were 99.6% and 99.5% when BE0t and BEinf were assessed, respectively.
The power analysis was also used to assess the effect of food on equivalence power. For a study containing 28 subjects, the power of bioequivalence between 120 mg of Doryx MPC and a 100-mg Doryx tablet under fed conditions was 8.8% and 8.7% when BE0t and BEinf were assessed, respectively, which is in stark contrast to the results in the fasted state. Despite this, for a study containing 28 individuals, the equivalence power values for AUC0t and AUCinf when 120 mg of Doryx MPC was compared to a 100-mg Doryx tablet under fed conditions were 99.8% and 99.6%, respectively; this is comparable to the results in the fasted state where the equivalence power values were 99.8% and 99.7%, respectively. Furthermore, in a study containing 28 subjects for the crossover design of 120 mg of Doryx MPC under fasted/fed conditions, the power of equivalence values for AUC0t, AUCinf, and Cmax were 92%, 90.6%, and 0%, respectively. This result was similar to a study containing 28 subjects for the crossover design of a 100-mg Doryx tablet under fasted/fed conditions, where the power of equivalence values for AUC0t, AUCinf, and Cmax were 93.6%, 91.2%, and 19%, respectively.
Shiny application.
A Shiny application for the final model can be accessed at https://isop.shinyapps.io/DoxycyclineSimulation/. A detailed description of how to use the application can also be found at this address, accompanied by the equations for the final model. The application has been designed to allow users to assess the influence of covariates (formulation, fed/fasted status, and gender) on doxycycline concentrations. The effect of covariates can be evaluated in three different doxycycline dosing regimens: (i) a single dose of 120 mg of Doryx MPC versus a 100-mg Doryx tablet over 96 h; (ii) multiple dose (standard infection) using 120 mg of Doryx MPC every 12 h on day 1 and then 120 mg every 24 h on days 2 to 7 or using a 100-mg Doryx tablet every 12 h on day 1 and then every 24 h on days 2 to 7; multiple dose (severe infection) using 120 mg of Doryx MPC every 12 h for 7 days or using a 100-mg Doryx tablet every 12 h for 7 days.
DISCUSSION
A population pharmacokinetic model was developed to describe doxycycline plasma concentrations following a single dose of Doryx MPC or single/multiple doses of Doryx tablets or Doryx capsules using data from eight pharmacokinetic trials using healthy human subjects. Of particular interest was capturing any bioavailability and absorption profile differences between the Doryx MPC and Doryx tablet formulations, as well as quantitating the effect of food on the two formulations.
To our knowledge, this is the first population pharmacokinetic analysis of doxycycline plasma concentrations following oral doxycycline dosing. Previously Beringer et al. (6) performed a compartmental pharmacokinetic analysis in 20 adults with cystic fibrosis dosed with multiple 40-mg, 100-mg, or 200-mg doses of oral doxycycline (formulation unspecified) and found a two-compartment model with a lag on first-order absorption to best describe the plasma concentration-time data. Similar results were found here, albeit the present study is a population analysis based on extensive single- and multidose oral data, and a two-transit absorption compartment model performed considerably better than a delay (lag) on simple first-order absorption.
In the literature, there are few data on dose linearity or the effect of multiple doses on doxycycline pharmacokinetics (2). In the study of Beringer et al. (6) noncompartmental pharmacokinetic analysis indicated that a less than dose-proportional relationship may be present in a small cystic fibrosis population dosed with multiple 40-mg, 100-mg, or 200-mg doses of oral doxycycline. The mean exponent values of the power model used to determine dose nonproportionality were 0.75 and 0.74 for Cmax and AUCinf, respectively. However, the 95% CI of the exponents did not support dose nonproportionality as they included 1, suggesting that dose proportionality could not be excluded. Dose proportionality of pharmacokinetics was supported in this population analysis as demonstrated by the nonsignificance of dose effects on RELF, KTR, and CL that were assessed in the covariate screening stage of this large population analysis. Given that the diagnostic plots of the base and final models were consistent with an unbiased model and that the pcVPCs showed a good description of the lower, median, and higher doxycycline concentrations following single- or multidose Doryx MPC, Doryx tablet, or Doryx capsule under fed or fasted conditions, it was considered that the structural and population components of our model appropriately represented the data, supported dose linearity, and allowed for confidence in the subsequent power analyses.
In this study, ANOVA was implemented to calculate the 90% CI of the ln-transformed data (8; J. Gordon and H. Kemmler, presented at the Training Workshop on Pharmaceutical Quality, Good Manufacturing Practice and Bioequivalence, White Sands, NM, 23 August 2006). ANOVAs can account for varying standard deviations within the ln-transformed AUCinf, AUC0t, Cmax, or Tmax values of the test and reference formulations and can account for period and sequence effects which are encouraged by regulatory authorities (9). Increasing the number of subjects within a given bioequivalence study evidently increased the power of the study and tightened the CI of the ln-transformed test-to-reference ratios of each exposure metric (see File S1 in the supplemental material). The results of the power analysis also support the bioequivalence of a 120-mg dose of Doryx MPC and a 100-mg Doryx tablet in the fasted state. This is reflected by the bioequivalence power values being 99.6% and 99.5% when BE0t and BEinf are assessed, respectively, for a study of 28 participants. Of the studies deemed bioequivalent, the percentages that included 1 (the value of no difference) within the 90% CI of the ln-transformed test-to-reference ratios both for AUC0t and Cmax and for AUCinf and Cmax were 60.8% and 61.9%, respectively. This is of interest as it may highlight a potential flaw with current bioequivalence guidelines and may support the value of updating guidelines to include a value of no difference within the 90% CI, as has been discussed previously (10). In the fed state for a study containing 28 subjects, the values for the bioequivalence power between 120 mg of Doryx MPC and a 100-mg Doryx tablet were 8.8% and 8.7% when BE0t and BEinf were assessed, respectively; this is in stark contrast to the results in the fasted state. Nonetheless, for a study containing 28 individuals the equivalence power values for AUC0t and AUCinf when 120 mg of Doryx MPC is compared to a 100-mg Doryx tablet under fed conditions were 99.8% and 99.6%, respectively; this is comparable to the results in the fasted state where the equivalence power values were 99.8% and 99.7%, respectively. Therefore, changes to the power of bioequivalence values between the two formulations in the fed state are the result of differences in the equivalence of Cmax values. Clinically, this is unlikely to be of importance as previous studies have indicated that the AUC/MIC ratio is the key to doxycycline efficacy (11).
The final model included a lag on the start of absorption for the Doryx MPC formulation (and the Doryx capsule) in comparison to that of the Doryx tablet formulation (ALAG = 0.115), consistent with the Doryx MPC formulation being developed to reduce the release of doxycycline within the esophagus and stomach, thus delaying the start of dissolution. Furthermore, the final model indicated that the relative bioavailability of Doryx MPC was 86.3% of that of a Doryx tablet. This is consistent with Mayne Pharma International's hypothesis that the altered release profile of the Doryx MPC formulation would result in a reduction of the relative bioavailability of the formulation (data on file with Mayne Pharma). The relative bioavailability of a Doryx capsule was indicated to be 97.8% of a Doryx tablet, which is consistent with the two formulations having very little difference in bioavailability. Furthermore, the final model included a lag on absorption for the fed status (FTLAG2 = 0.203 h). The fed status also resulted in formulation-dependent differences in the absorption KTR parameter, where KTR decreased by 20.9% in the fed status for the Doryx tablet and Doryx capsule formulations and decreased by 54.9% for the Doryx MPC formulation. Lowering the KTR parameter also has the effect of adding a further delay to doxycycline reaching systemic circulation due to the increased time taken for the drug to move through the absorption transit compartments. The larger decrease in KTR for the Doryx MPC formulation over the Doryx tablet in the fed status is not surprising, given that the fed status will likely increase the transit time of the formulation within the stomach, effectively increasing the time until Doryx MPC dissolution will begin and thus reach systemic circulation. Irrespective of formulation, the fed status was observed to decrease relative bioavailability by 10.5% during the covariate assessment stage.
During the covariate assessment stage, female sex was observed to increase doxycycline clearance by 14.4%. Although the clinical significance of this result is unknown, this sex effect has not been identified previously (2, 3). Previously, the impact of age on the pharmacokinetics of doxycycline was investigated in 25 elderly patients (over the age of 65), with findings indicating that concentrations were higher than those of a younger population (12). In this study, age was not observed to influence doxycycline pharmacokinetics; however, the involvement of older individuals in this study was limited. The literature also indicated a minimal impact of renal function on doxycycline pharmacokinetics (2), albeit this was not assessed in this study as all participants were required to have normal renal function.
The analysis presented here is the first to explore a population pharmacokinetic model of doxycycline concentrations following oral administration, which is an important step toward a population pharmacokinetic-pharmacodynamic model that could be used to critically examine and optimize doxycycline dose regimens. Although in vitro studies indicate that low concentrations of doxycycline kill in a time-dependent kinetic manner while higher concentrations kill in a concentration-dependent kinetic manner (13), at present this area has been insufficiently explored (2). Albeit this study is a step toward this, it is of note that this study presents a population pharmacokinetic model of doxycycline concentrations following the oral administration of doxycycline concentrations in healthy individuals, and the effect of disease status or sickness on doxycycline pharmacokinetics was not investigated here as all participants were healthy volunteers. Therefore, the translation of this model to the clinical arena may require additional work.
In conclusion, a population pharmacokinetic model was developed to describe doxycycline plasma concentrations following a single dose of Doryx MPC or single/multiple doses of Doryx tablets or Doryx capsules in the fasted/fed states. Using the developed model, the power of establishing bioequivalence between a 100-mg Doryx tablet and the newly developed 120-mg Doryx MPC formulation was assessed in the fed or fasted state. The power analysis indicated that the fed/fasted state had minimal effect on the high likelihood of AUCinf and AUC0t equivalence between 120 mg of Doryx MPC and a 100-mg Doryx tablet. However, given the anticipated delay in the release of the Doryx MPC formulation within the esophagus and stomach, it was not surprising that the power of Cmax equivalence was low between 120 mg of Doryx MPC and a 100-mg Doryx tablet under the fed condition, which was not comparable to the results under the fasted condition. In this study, a Shiny application that performs simulations of the final model was developed, which could be used to examine the marginally lower Cmax of 120 mg of Doryx MPC in the fed state compared to that of the 100-mg Doryx tablet.
MATERIALS AND METHODS
Pharmacokinetic data.
Pharmacokinetic data were available from eight phase 1 clinical trials conducted by Mayne Pharma International. These studies assessed doxycycline plasma concentrations following oral administration of either a 100-mg conventional release doxycycline hyclate capsule, a 75/100/150- or 200-mg Doryx delayed-release tablet, or a 120-mg Doryx MPC delayed-release tablet in healthy subjects. The conventional release doxycycline hyclate capsules are referred to as Doryx capsules, the Doryx delayed-release tablets are referred to as Doryx tablets, and Doryx MPC delayed-release tablets (formulation code MP336) are referred to as Doryx MPC.
The available phase 1 clinical trials included six single-dose two-treatment period studies assessing the influence of formulation or the fed/fasted state on doxycycline concentrations. Two trials were multidose studies, with one a single-treatment period study and the other a two-treatment period study assessing doxycycline plasma concentrations following Doryx capsule or Doryx tablet administration. All studies were conducted in the United States. All studies that included two-treatment periods were randomized crossover studies that had a 14-day washout after the completion of the first plasma sampling period. The design and dosing schedules are summarized in Table 6. The available studies were assigned a local identifier during model development (10, 20, 30, 40, 50, 60, 70, and 80), as indicated in Table 6, and are referred to by these numbers in the manuscript.
TABLE 6.
Phase 1 clinical trial data available for analysis
| Study no. | Protocol no. | No. of subjects recruited | No. of subjects with concn data | Dose type | Fed/fasted study | No. of treatment periods/subject | Dosing schedule (condition) | LLOQ (μg/ml) |
|---|---|---|---|---|---|---|---|---|
| 10 | 11450707 | 28 | 28 | Single | No | 2 | 120 mg of Doryx MPC vs 100-mg Doryx tablet | 0.1 |
| 20 | PR-08302.0 | 18 | 18 | Single | Yes | 2 | 100-mg Doryx tablet (fed/fasted) | 0.025 |
| 30 | PR-02308.0 | 18 | 17 | Single | Yes | 2 | 200-mg Doryx tablet (fed/fasted) | 0.05 |
| 40 | PR-06410 | 20 | 19 | Multiple | No | 1 | 200-mg Doryx tablet | 0.05 |
| 50 | PR-02707 | 26 | 25 | Single | No | 2 | 150-mg Doryx tablet vs two 75-mg Doryx tablets | 0.05 |
| 60 | PR-02108.1 | 26 | 23 | Single | No | 2 | 200-mg Doryx tablet vs two 100-mg Doryx tablets | 0.05 |
| 70 | PR-01402.0 | 24 | 20 | Multiple | No | 2 | 100-mg Doryx tablet vs 100-mg Doryx capsule | 0.025 |
| 80 | MYP-P2-991 | 28 | 28 | Single | Yes | 2 | 120 mg of Doryx MPC (fed/fasted) | 0.01 |
Studies 10, 20, 30, 50, 60, and 80 were single-dose studies, and plasma samples were acquired at nominal times approximate to 0, 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 10, 12, 16, 24, 36, 48, 72, and 96 h after the dose on day 1 of that period. Study 40 was a single-treatment period multidose study, where participants were dosed seven times at 24-h intervals, and plasma samples were collected at nominal times of 0, 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, 24, 144, 144.25, 144.5, 145, 145.5, 146, 146.5, 147, 148, 150, 152, 156, 160, 168, 180, 192, 216, and 240 h after the dose on day 1. Study 70 was a two-treatment period multidose study, where doses were given at 0, 72, 96, 120, 144, and 168 h after the start of treatment, and plasma samples were collected at nominal times of 0, 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, 72, 96, 120, 144, 168, 168.25, 168.5, 169, 169.5, 170, 170.5, 171, 172, 174, 176, 180, 184, 192, 204, 216, and 240 h after the dose on day 1. Deviations between nominal sampling times and actual sample times were recorded, and the actual times were used during model development. Covariates describing fed status, formulation, sex, age, race, weight, body mass index (BMI), smoking status, and study number were available for each individual.
Doxycycline plasma concentrations were measured using validated high-performance liquid chromatographic methods, with the lower limits of quantification (LLOQ) of the assays indicated in Table 6. All subjects with evaluable pharmacokinetic data were included in the analysis. All studies were conducted according to the International Conference of Harmonisation (ICH) guidelines for good clinical practice (14), the Declaration of Helsinki on the ethical conduct of medical research (15), and applicable regulatory requirements. Each subject signed and dated a written informed consent before study participation.
Modeling software.
Modeling was performed using a Dell PowerEdge R910 server with 4-by-10 core Xeon 2.26-Ghz processors running the Windows Server 2008 R2 Enterprise 64-bit operating system. Model development employed nonlinear mixed-effects modeling using NONMEM, version 7.3 (16), with the Wings for NONMEM interface (http://wfn.sourceforge.net) and Intel Visual Studio Fortran compiler. Data manipulation and postrun processing of NONMEM output were conducted using the R data analysis language (version 3.1.1 or greater) and the packages ggplot2, GGally, foreign, tidyr, Hmisc, gdata, doBy, plyr, grid, stringr, and npde (17–29).
General modeling strategy.
The base model was developed in a stepwise manner. Pharmacokinetic models were coded using the ADVAN5 subroutines of NONMEM. Linear kinetic models with 1, 2, and 3 compartments were evaluated, with absorption models including simple first-order absorption and “transit compartment” models with up to five absorption transit compartments assessed. The parameter KTR (transit absorption rate) was used to describe the first-order movement through these absorption compartments. The first-order conditional estimation (FOCE) method was used to fit models. The base model was selected on mechanistic plausibility (e.g., if the assessed drug is known to be cleared by the kidneys, renal function would be assessed as a covariate in the base model), visual inspection of goodness-of-fit diagnostic plots, the precision of parameter estimates (%SE of <30% for fixed, of <50% for random, and of <51.2% for covariate effects parameters), and the lowest value of the Akaike information criterion (AIC; equation 1). The base model was also required to pass the covariance step.
| (1) |
where OBJ is objective function value.
Unless stated otherwise, population parameter variability (PPV) was represented using an exponential error model:
| (2) |
where Pj is the individual value for the parameter in the jth individual, TVP is the typical population value of P, and ηj is an independent random variable with a mean of zero and variance ω2.
Models with and without covariance for P were investigated using the Omega BLOCK functionality of NONMEM. Population parameters were tested for between-occasion variability (BOV), where pharmacokinetic data were available from two treatment periods. Total PPV was defined as follows:
| (3) |
where the variance of η represents between-subject variability (BSV) and the variance of κ represents BOV. OCC1 was a flag that has a value of 1 when period equals 1 and is zero otherwise. OCC2 was a flag that has value 1 when period equals 2 and is zero otherwise, etc. κ is a random variable with mean zero and variance ω2 that is equal for all occasions (via the Omega SAME structure in NONMEM, thus assuming that BOV was the same for all occasions).
A combined proportional (θprop) and additive residual (θadd) error model of the doxycycline concentrations (Cs) was used, where estimation of a theta was employed, and epsilon was fixed to zero:
| (4) |
where Cij is the ith concentration measured in the jth individual, Cij(pred) is the model-predicted Cij, and θprop and θadd are parameters representing the proportional and additive residual errors, respectively. The influence of the additive component of the error was also examined by a comparison with a proportional-only error model. The YLO/M2 method (30) and Beal's M3 method (7) for censored concentrations below the limit of quantification (BLOQ) were also investigated.
All base models were investigated with allometric scaling, initially through total body weight (TBW) referenced to 70 kg and fixed exponents of 0.75 for clearance parameters and 1 for volumes (31). Allometric scaling to fat-free mass (FFM) (32) and normal-fat mass (NFM) (33) were also considered. Structural covariates controlling the bioavailability differences between formulations and fed status, as well as KTR differences between formulations and fed status, were investigated early and, where appropriate, were incorporated within the base model. Given the extensive washout between dosing periods and that all period 2 predose samples were BLOQ, the event identification (EVID) = 4 functionality of NONMEM was utilized to reset all compartment amounts to 0 at the start of a new dosing period.
During the final stage of base model development, between-study differences on the population parameter values were investigated. This was important due to the sampling time differences between the studies and a tendency for subtle changes in pharmacokinetic parameters to occur from one study to another (34), thus affecting the visual predictive check subset by study. As guided by Laporte-Simitsidis et al. (34) for a pooled analysis containing less than 20 studies, a fixed study-effect method was used where the effect of each study was represented within a binary relationship.
Prediction-corrected visual predictive checks (pcVPCs) were used to assess the appropriateness of the candidate base and final models (35). For the pcVPCs, the median, 5th, and 95th percentiles of the prediction-corrected observations were compared against the empirical 95% confidence intervals (CIs) of the median, 5th, and 95th percentiles of prediction-corrected doxycycline concentrations from 200 simulations of the index (original) data set. The predictive performance of the model was considered acceptable if the median, 5th, and 95th percentiles of the prediction-corrected observed data lay inside the CIs of the prediction-corrected simulated data for the majority of the time. In addition to the pcVPC, the percentage of the observed data BLOQ throughout time was compared against the 95% CI of the percentage of BLOQ data from the 200 simulations of the original data set (BLOQ VPC). The predictive performance of the model was considered acceptable if the BLOQ data percentage of the original data was within the predicated CI for the simulation data for the majority of time.
Covariate model building.
The base model was screened for the influence of covariates based upon physiological plausibility and prior knowledge of the factors considered to influence doxycycline plasma concentrations (Table 7). The effects of continuous covariates on a parameter were represented as a power function referenced to the median value of the covariate in the data, while the effects of categorical covariates on a parameter were represented by a binary relationship. The effects of doxycycline dose and tablet strength were also investigated through linear functions referenced to the median value in the data.
TABLE 7.
Biologically guided covariate screening plan
| Parameter | Covariates screened |
|---|---|
| Relative bioavailability (RELF) | Fed status,a dose, tablet strength, sex, age, race |
| Transit absorption rate (KTR) | Formulation, dose, tablet strength, sex, age, race |
| Central compartment vol (V) | Sex, age, race |
| Clearance (CL) | Dose, sex, age, race, smoker status |
| Peripheral compartment vol (VP1) | Sex, age, race |
Fed status effect on RELF was also assessed for a difference between formulations.
The potential inclusion of covariates on the base model were selected based upon a significant decrease in OBJ at the level of a P value of <0.01 (6.63 units for one parameter). The final model was developed via a forward inclusion/backward deletion process, whereby each covariate (significant on screening) was sequentially added, starting with the covariate that caused the largest drop in the OBJ. A covariate(s) was retained only if there was a significant decrease in OBJ at a the level of a P value of <0.01. The final model was further subjected to a backward elimination procedure at the level of a P value of <0.001 (10.8 units for one parameter), starting with the least significant covariate from the forward inclusion step.
Simulations and power analysis.
The final model determined from the doxycycline data was used to perform simulations and power analyses of bioequivalence, according to the following randomized crossover designs: 120 mg of Doryx MPC compared to a 100-mg Doryx tablet (fasted), 120 mg of Doryx MPC compared to a 100-mg Doryx tablet (fed), 120 mg of Doryx MPC compared to 120 mg of Doryx MPC (fed/fasted), and a 100-mg Doryx tablet compared to a 100-mg Doryx tablet (fed/fasted).
For the simulations and power analyses, the between-study effect parameters identified in the final model were set to the mean value of the eight studies. Studies including 12, 24, 28, and 36 subjects were simulated 1,000 times, and noncompartmental analysis (NCA) pharmacokinetic exposure parameters (AUC0t, AUCinf, Cmax, and Tmax) were calculated. These NCA metrics were calculated for the simulated test and reference product concentrations using an R script. NCA metrics below the 2.5th and above the 97.5th percentiles were not used for summary statistics as they present as outliers, and their inclusion was considered to produce summary statistics that are not representative of model predictions. For each simulated crossover study, NCA metric equivalence and bioequivalence was assessed through the use of an analysis of variance (ANOVA) method using the aov function in R (8, 36; Gordon and Kemmler, training workshop presentation). For the 1,000 simulated studies of 28 subjects, the median and 90% CI of the predicted doxycycline concentrations were plotted (see File S1 in the supplemental material). The percentage of bioequivalent studies was interpreted as the power of the study design for establishing bioequivalence between the two formulations, given the trial design and dose selection used in that simulation. Additionally, the studies that were deemed bioequivalent were assessed for whether the 90% CI of ln-transformed test-to-reference ratios for AUC0t, AUCinf, and Cmax included 1.
Shiny application.
The final model was incorporated within a Web application that allows users not familiar with the details of population modeling to perform simulations using the model (37). The application is built using the Shiny package for R, and simulations are performed using the mrgsolve package (38, 39).
Supplementary Material
ACKNOWLEDGMENTS
All the pharmacokinetic studies used in the analysis were sponsored by Mayne Pharma International. S.M. is an employee at Mayne Pharma International. R.N.U., D.J.R.F., and A.M.H. were commercially contracted by Mayne Pharma International to conduct the analyses presented in the manuscript.
We acknowledge that the Australian Centre for Pharmacometrics is an initiative of the Australian Government as part of the National Collaborative Research Infrastructure Strategy.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02401-16.
REFERENCES
- 1.Holmes NE, Charles PGP. 2009. Safety and efficacy review of doxycycline. Clin Med Ther 1:471. [Google Scholar]
- 2.Agwuh KN, MacGowan A. 2006. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J Antimicrob Chemother 58:256–265. doi: 10.1093/jac/dkl224. [DOI] [PubMed] [Google Scholar]
- 3.Saivin S, Houin G. 1988. Clinical pharmacokinetics of doxycycline and minocycline. Clin Pharmacokinet 15:355–366. doi: 10.2165/00003088-198815060-00001. [DOI] [PubMed] [Google Scholar]
- 4.Australian Medicines Handbook Pty., Ltd. 2014. Australian medicines handbook 2014. Australian Medicines Handbook Pty., Ltd., Adelaide, South Australia. [Google Scholar]
- 5.Mayne Pharma. 2015. Doryx–doxycycline hyclate tablet, delayed release. Prescribing information. Mayne Pharma, Salisbury South, South Australia, Australia: https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=99cf2de6-e0a3-42f2-9929-d33e107af948&type=display. [Google Scholar]
- 6.Beringer PM, Owens H, Nguyen A, Benitez D, Rao A, D'Argenio DZ. 2012. Pharmacokinetics of doxycycline in adults with cystic fibrosis. Antimicrob Agents Chemother 56:70–74. doi: 10.1128/AAC.05710-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beal SL. 2001. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 28:481–504. [DOI] [PubMed] [Google Scholar]
- 8.Rani S, Pargal A. 2004. Bioequivalence: an overview of statistical concepts. Indian J Pharmacol 36:209–216. [Google Scholar]
- 9.Center for Drug Evaluation and Research. 2001. Statistical approaches to establishing bioequivalence. Food and Drug Administration, Rockville, MD. [Google Scholar]
- 10.Danish Medicines Agency. 2012. Bioequivalence and labelling of medicines with regard to generic substitution. Danish Medicines Agency, Copenhagen, Denmark. [Google Scholar]
- 11.Craig WA. 2002. Pharmacodynamics of antimicrobials: general concepts and applications. Marcel Dekker, New York, NY. [Google Scholar]
- 12.Böcker R, Mühlberg W, Platt D, Estler C-J. 1986. Serum level, half-life and apparent volume of distribution of doxycycline in geriatric patients. Eur J Clin Pharmacol 30:105–108. doi: 10.1007/BF00614205. [DOI] [PubMed] [Google Scholar]
- 13.Cunha BA, Domenico P, Cunha CB. 2000. Pharmacodynamics of doxycycline. Clin Microbiol Infect 6:270–273. doi: 10.1046/j.1469-0691.2000.00058-2.x. [DOI] [PubMed] [Google Scholar]
- 14.European Medicines Agency. 1996. Note for guidance on good clinical practice (CPMP/ICH/135/95). European Medicines Agency, London, United Kingdom. [Google Scholar]
- 15.World Medical Association. 1997. World medical association declaration of Helsinki: recommendations guiding physicians in biomedical research involving human subjects. JAMA 277:925–926. doi: 10.1001/jama.1997.03540350075038. [DOI] [PubMed] [Google Scholar]
- 16.Beal SL, Sheiner LB, Boeckmann AJ, Bauer RJ (ed). 2013. NONMEM 7.3.0 users guides (1989–2013). ICON Development Solutions, Hanover, MD. [Google Scholar]
- 17.Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer, New York, NY. [Google Scholar]
- 18.Højsgaard S, Halekoh U, Robison-Cox J, Wright K, Leidi AA. 2014. doBy: groupwise summary statistics, LSmeans, general linear contrasts, various utilities. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=doBy. [Google Scholar]
- 19.Wickham H. 2011. The split-apply-combine strategy for data analysis. J Stat Softw 40:1–29. [Google Scholar]
- 20.Wickham H. 2007. Reshaping data with the {reshape} package. J Stat Softw 21:1–20. [Google Scholar]
- 21.Wickham H. 2014. scales: scale functions for graphics. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=scales. [Google Scholar]
- 22.Comets E, Brendel K, Nguyen THT, Mentre F. 2012. npde: normalised prediction distribution errors for nonlinear mixed-effect models, version 2.0. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=npde. [Google Scholar]
- 23.Wickham H. 2012. stringr: make it easier to work with strings. R package version 0.6.2. R Project for Statistical Computing, Vienna, Austria: http://CRAN.R-project.org/package=stringr. [Google Scholar]
- 24.Auguie B. 2012. gridExtra: functions for grid graphics. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=gridExtra. [Google Scholar]
- 25.Warnes GR, Bolker B, Gorjanc G, Grothendieck G, Korosec A, Lumley T, MacQueen D, Magnusson A, Rogers J. 2015. gdata, version 2.17.0. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=gdata. [Google Scholar]
- 26.Harrell FE., Jr 2016. Hmisc: Harrell miscellaneous. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=Hmisc. [Google Scholar]
- 27.Wickham H. 2016. tidyr easily tidy data with “spread ()” and “gather ()” functions, version 0.4.1. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=tidyr. [Google Scholar]
- 28.R Development Core Team. 2015. R-CRAN foreign version 0.8.66. R Project for Statistical Computing, Vienna, Austria. [Google Scholar]
- 29.Schloerke B, Crowley J, Cook D, Hofmann H, Wickham H, Marbach M, Thoen E, Elberg A, Larmarange J. 2014. GGally, version 0.5.0. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=GGally. [Google Scholar]
- 30.Byon W, Fletcher CV, Brundage RC. 2008. Impact of censoring data below an arbitrary quantification limit on structural model misspecification. J Pharmacokinet Pharmacodyn 35:101–116. doi: 10.1007/s10928-007-9078-9. [DOI] [PubMed] [Google Scholar]
- 31.Anderson B, Holford N. 2008. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 48:303–332. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 32.Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. 2005. Quantification of lean bodyweight. Clin Pharmacokinet 44:1051–1065. doi: 10.2165/00003088-200544100-00004. [DOI] [PubMed] [Google Scholar]
- 33.Anderson BJ, Holford NH. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24:25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- 34.Laporte-Simitsidis S, Girard P, Mismetti P, Chabaud S, Decousus H, Boissel J-P. 2000. Inter-study variability in population pharmacokinetic meta-analysis: when and how to estimate it? J Pharm Sci 89:155–167. doi: 10.1002/(SICI)1520-6017(200002)89:2<155::AID-JPS3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 35.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. 2011. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13:143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.R Development Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 37.Wojciechowski J, Hopkins AM, Upton RN. 2015. Interactive pharmacometric applications using R and the Shiny package. CPT Pharmacometric Syst Pharmacol 4:e00021. doi: 10.1002/psp4.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.R Studio, Inc. 2015. shiny: Web application framework for R. R package version, 0.10.1. R Project for Statistical Computing, Vienna, Austria: https://CRAN.R-project.org/package=shiny. [Google Scholar]
- 39.Baron KT, Hindmarsh AC, Petzold LR, Gillespie B, Margossian C, Metrum Research Group LLC. 2016. Simulation from ODE-based population PK/PD and systems pharmacology models. Metrum Research Group, Tariffville, CT. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.